

Abstract
The structure activity relationships and molecular modeling of the uracil nucleotide-activated P2Y6 receptor have been studied. A series of UDP analogues bearing substitutions of the ribose moiety, the uracil ring, and the diphosphate group was synthesized and assayed for activity at the human P2Y6 receptor. The uracil ring was modified at the 4-position, with the synthesis of 4-substituted-thiouridine-5′-diphosphate analogues, as well as at positions 3 and 5. The effect of modifications at the level of the phosphate chain was studied by preparing a cyclic 3′,5′-diphosphate analogue, a 3′-diphosphate analogue and several dinucleotide diphosphates. 5-Iodo-UDP 32 (EC50 0.15 μM) was equipotent to UDP, while substitutions of the 2′-hydroxyl (amino, azido) greatly reduce potency. 2- and 4-Thio analogues, 20 and 21, respectively, were also relatively potent in comparison to UDP. However, most other modifications greatly reduced potency. Molecular modeling indicates that the β-phosphate of 5′-UDP and analogs is essential for the establishment of electrostatic interactions with two of the three conserved cationic residues of the receptor. Among 4-thioether derivatives, a 4-ethylthio analogue 23 displayed an EC50 of 0.28 μM, indicative of favorable interactions predicted for a small 4-alkylthio moiety with the aromatic ring of Y33 in TM1. The activity of analogue 19 in which the ribose was substituted with a 2-oxabicyclohexane ring in a rigid (S) conformation (P= 126°, 1′-exo) was consistent with molecular modeling. These results provide a better understanding of molecular recognition at the P2Y6 receptor and will be helpful in designing selective and potent P2Y6 receptor ligands
Keywords: G protein-coupled receptor, nucleotides, thionucleotides, phospholipase C, pyrimidines, homology modeling
Introduction
P2 nucleotide receptors consist of two families: G protein-coupled receptors designated P2Y, which include 8 subtypes (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11-P2Y14), and ligand-gated cation channels designated P2X, which include 7 subtypes (P2X1-P2X7). All of these subtypes have been cloned and functionally characterized.1-10 The family of P2Y receptors can be further divided in two different subgroups based on overall sequence similarity, coupling to specific G proteins, and second messenger responses. The P2Y1-like subgroup (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11) is Gq-coupled and stimulates phospholipase C (PLC), and the P2Y12-like subgroup (P2Y12, P2Y13, P2Y14) is Gi coupled and inhibits adenylyl cyclase (AC).11 The distribution of P2Y receptors is broad, and certain of these are of therapeutic interest including antithrombotic therapy, modulation of the immune system and cardiovascular system, inflammation, pain, diabetes, and treatment of cystic fibrosis and other pulmonary diseases.12-15 Whereas P2X receptors tend to be activated principally by adenine nucleotides, P2Y receptors are activated by adenine and/or uracil nucleotides. The P2Y2 receptor is activated by both uridine 5′-triphosphate (UTP) and adenosine 5′-triphosphate (ATP), the P2Y4 receptor by UTP, the P2Y14 by UDP-glucose, and the P2Y6 receptor by uridine 5′-diphosphate (UDP 1, Chart 1).16
Chart 1.

Structures of UDP and various analogues and the reported EC50 values in stimulation of PLC through the recombinant P2Y6 receptor (human, unless noted).16,22,23,24,35 The EC50 for INS48823 is similar to UDP.18
The P2Y6 receptor is distributed in various tissues including lung, heart, thymus, aorta, bone, spleen, digestive tract, placenta and brain. This receptor has been implicated in protection against apoptosis induced by TNFα,17 enhancement of osteoclast survival through NF-κB activation,18 regulation of electrolyte transport in the airways,19 and production of pro-inflammatory cytokines and chemokines,20,21 and growth and contraction of vascular muscles.22
The SAR (structure activity relationship)of nucleotides in activating the human P2Y6 receptor has been probed (Chart 1). 5-Br-UDP 2, UDP-β-S 3 and the dinucleotide triphosphate INS48823 (P1-((2-benzyl-1,3-dioxolo-4-yl)uridine 5′) P3-(uridine 5′) triphosphate) 4 are potent and/or stable agonists of the P2Y6 receptor.5,18,19,22 We have reported that the South (S) conformation of the ribose moiety is preferred for ligand recognition by the P2Y6 receptor.23,35 The conformationally-constrained (N)-methanocarba (mc) derivative 5 was inactive, and the (S)-methanocarba derivative 6 was moderately potent in activating the P2Y6 receptor. Replacement of the uracil moiety with other nucleobases 7–10 greatly reduced the potency at the human P2Y6 receptor.24 In order to investigate more extensively the SAR at the P2Y6 receptor, we synthesized a series of UDP analogues, where modifications at the level of uracil moiety, ribose ring and/or phosphate chain have been introduced. Molecular modeling was carried out to predict sites of interaction of the ligands with the P2Y6 receptor and to provide hypotheses for the design of additional analogues.
To facilitate the comparison among receptors, throughout this paper we use the GPCR residue indexing system, as explained in detail elsewhere.11
Results
Chemical Synthesis
Analogues of UDP 1 with modifications in the ribose ring, uracil moiety, and phosphate chain, as well as dinucleotides (Table 1), were synthesized. The ribose ring was modified by removal of the hydroxyl group at the 3′ position 13, replacement of the hydroxyl at the 2′ position with various functional groups, 15-18, and with the substitution of a 2-oxabicyclohexane ring (2-OBH) in place of the ribose ring,25 which fixed the sugar moiety in a rigid (S) conformation, 19. The uracil ring was modified at the 2 and 4 positions, with the synthesis of 2-thiouridine diphosphate 20 and 4-substituted-thiouridine diphosphates analogues 22-30, and at the 3 and 5 positions with 31 and 32 respectively. Finally, the synthesis of 2′-deoxy-2′-aminouridine 3′-diphosphate 17, the cyclic 3′,5′-diphosphate 33 and the dinucleotides 34-37 as byproducts has allowed us to study modifications at the level of the phosphate chain. All the nucleotide analogues were prepared in their ammonium or triethyl ammonium salt form according to the methods shown in Schemes 1-5 and tested in functional assays of the P2Y6 receptor (Table 1). The nucleotide analogues were characterized using nuclear magnetic resonance (NMR, 1H, 13C, 31P and COSY), and high-resolution mass spectrometry.
Table 1.
In vitro pharmacological data for UDP, 1, and its analogues in the stimulation of PLC at recombinant human P2Y6 receptors expressed in 1321N1 astrocytoma cells. Unless noted: R1, R3, R5, R6 = H; R2, R4 = O; and R7, R8 = OH.

| |||
|---|---|---|---|
| Compound | Modification | Structure | EC50 at hP2Y6 receptor, μMa |
| 1 | (= UDP) | 0.013 ± 0.004 | |
| 11 | (= UMP) | NE | |
| Ribose-modified | |||
| 12 | 2′-deoxy | R7 = H | 1.72 ± 0.76 |
| 13 | 3′-deoxy | R8 = H | 2.5 ± 0.8 |
| 14 | (= 2′-deoxyuridine-bisphosphate) | c | NE |
| 15 | 2 ′-deoxy-2′-azido | R7 = N3 | 1.5 ± 0.4 |
| 16 | 2′-deoxy-2′-amino | R7 = NH2 | 3.9 ± 0.9 |
| 17 | 2′-deoxy-2′-amino 3′-diphosphate | c | NE |
| 18 | 2′-fluoro-2′-deoxyara | R7 = H, R6 = F | 5.5 ± 0.5 |
| 5 | (N)-methanocarba | d | NE |
| 6 | (S)-methanocarba-2′-deoxy | d | 0.23 ± 0.05 |
| 19 | 3′,4′-cyclopropylb | c | 3.5 ± 0.8 |
| Uracil-modified | |||
| 20 | 2-thio | R2 = S | 0.06 ± 0.01 |
| 21 | 4-thio | R4 = S | 0.08 ± 0.04 |
| 22 | 4-methylthio | R4 = SCH3 | 2.3 ± 0.6 |
| 23 | 4-ethylthio | R4 = SCH2CH3 | 0.28 ± 0.02 |
| 24 | 4-allylthio | R4 = SCH2-CH=CH2 | 0.56 ± 0.09 |
| 25 | 4-benzylthio | R4 = SCH2C6H5 | 0.80 ± 0.05 |
| 26 | 4-carboxymethylthio | R4 = SCH2CO2H | 1.7 ± 0.3 |
| 27 | 4-carboxamido-methylthio | R4 = SCH2CONH2 | 18 ± 9 |
| 28 | 4-carboxyethylthio | R4 = S(CH2)2COOH | 1.1 ± 0.2 |
| 29 | 4-carboxypropylthio | R4 = S(CH2)3COOH | 8.8 ± 2.0 |
| 30 | 4-hexylthio | R4 = S(CH2)5CH3 | 4.9 ± 0.2 |
| 31 | 3-methyl | R3 = CH3 | 3.3 ± 1.0 |
| 32 | 5-iodo | R5 = I | 0.015 ± 0.002 |
| Phosphate-and ribose modified | |||
| 33 | Cyclic 3′,5′-diphosphate and 2′-ara-F | c | NE |
| Dinucleotides | |||
| 34 | Up2U | R1 = uridine-5′- | 2.3 ± 0.9 |
| 35 | U(4-ethylthio) p2U(4-ethylthio) | R1 = 4-SCH2CH3-uridine-5′-, R4 = SCH2CH3 | <50% at 10 μM |
| 36 | U(4-benzylthio) p2U(4-benzylthio) | R1 = 4-SCH2C6H5-uridine-5′-, R4 = SCH2C6H5 | <50% at 10 μM |
| 37 | Cp2C | R1 = cytidine-5′-, R4 = NH | NE |
Agonist potencies were calculated using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). EC50 values (mean ± standard error) represent the concentration at which 50% of the maximal effect is achieved. Relative efficacies (%) were determined by comparison with the effect produced by a maximal effective concentration of reference agonist (UDP) in the same experiment. If no maximal effect is given, then 100% efficacy was achieved.
oxabicyclohexane ring system (2-OBH)25.
NE - no effect at 10 μM
![]()
see Chart 1.
Scheme 1.

Preparation of ribose- and uracil-modified UDP analogues. Reagents and conditions: (i) (1) POCl3, proton sponge, PO(OMe)3, 0 °C; (2) (Bu3NH+)2PO4H, Bu3N, DMF, 0 °C; (ii) CF3COOEt, DIEA, DMF, rt. (iii) H2, Pd/C, MeOH, rt.
Scheme 5.
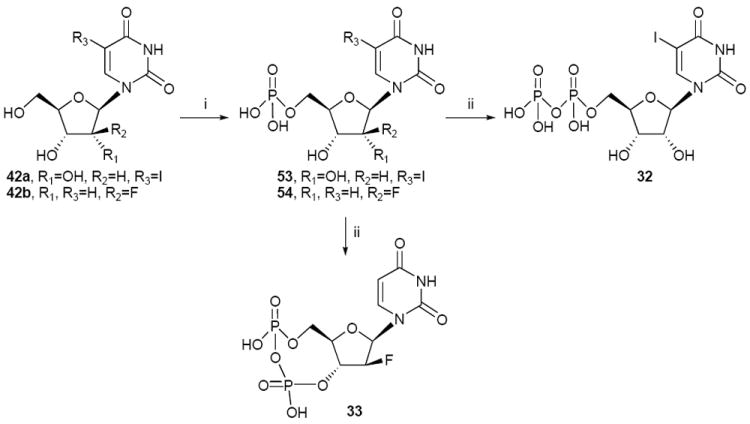
Preparation of 5-halo and phosphate- and ribose-modified UDP analogues. Reagents and conditions: (i) POCl3, proton sponge, PO(OMe)3, 0 °C; (ii) (1) 1,1’- carbonyldiimidazole, DMF, rt; (2) Et3N 5% in MeOH, rt; (3) (Bu3NH+)2PO4H2-, Bu3N, DMF, rt.
The nucleotide diphosphates were obtained following two different methods of phosphorylation. The one-pot method using a sequential reaction with phosphorous oxychloride and phosphoric acid tributylammonium salt26 provided the 5′-diphosphate analogues 13, 15, 16, 18-20 in moderate yields (Schemes 1, 2). The phosphorylation of the 2′-amino-2′-deoxyuridine 40 using the one-pot method provided the 3′-diphosphate derivative 17. The 1H NMR of compound 17 shows peaks at 5.01 ppm (H-3′) and 3.89 ppm (H-5′), when the chemical shifts of 5′-diphosphate uridine analogues are usually at 4.65-4.35 ppm (H-3′) and 4.40-4.15 ppm (H-5′). These results combined with 31P NMR and HRMS indicate that for 17 the diphosphate group is in 3′ position. The synthesis of 2′-deoxy-2′-aminouridine-5′-diphosphate 16 was accomplished after the acetylation of the amino group in 40 with a trifluoroacetyl group to obtain 41. Phosphorylation of the 2′-trifluoroacetylamino derivative 41 followed by an in situ deprotection of the 2′-amine provided the 5′-diphosphate analogue 16. Compound 16 was also obtained by reduction of the 2′-deoxy-2′-azidouridine-5′-diphosphate 15 with H2 and Pd/C. The NMR spectrum for 16 shows peaks at 4.61 ppm (H-3′) and 4.24, 4.16 ppm (H-5′) in accordance with others 5′-diphosphates derivatives. The preparation of compounds 28, 30 and 32 was accomplished by another method, i.e. activation of the 5′-monophosphate by reaction with 1,1′-carbonyldiimidazole followed by the addition of phosphoric acid tributylammonium salt23 to provide the corresponding diphosphate (Schemes 4, 5).
Scheme 2.

Preparation of 2-OBH UDP analogue. Reagents and conditions: (i) (1) POCl3, proton sponge, PO(OMe)3, 0 °C; (2) (Bu3NH+)2PO4H, Bu3N, DMF, 0 °C.
Scheme 4.

Preparation of 4-substituted-thio-UDP analogues. Procedure C. Reagents and conditions: (i) (1) 0.25 M NaOH, MeOH, rt; (2) R1X, DMF, 90 °C; (ii) (1) 1,1’-carbonyldiimidazole, DMF, rt; (2) Et3N 5% in H2O/MeOH 1/1, rt; (3) (Bu3NH+)2PO4H, Bu3N, DMF, rt; (iii) 0.25 M NaOH, H2O, rt.
The synthesis of various 4-substituted-thiouridine nucleosides and nucleotides from uridine was described previously,27-29 and various methods of S-alkylation were applied to a purine nucleoside, 2-thioadenosine.26,30 By adapting these alkylation procedures, we have developed a more direct synthesis of 4-substituted-thiouridine nucleotides starting with the commercially available 4-thiouridine 5′-diphosphate (4-thio-UDP) 21 that allowed us to obtain the final compounds in only two steps (Scheme 3).
Scheme 3.

Preparation of 4-substituted-thio-UDP analogues. Procedure B. Reagents and conditions: (i) (1) 0.25 M NaOH, MeOH, rt; (2) RX, DMF, rt; (ii) 0.25 M NaOH, H2O, rt.
The treatment of 4-thio-UDP with 0.25 M NaOH in methanol for 2 h at room temperature gave the corresponding sodium thiolate salt, which appeared as a yellow solid after the solvent was lyophilized. The nucleotide diphosphate sodium salt was selectively S-alkylated by reacting with an excess of the corresponding alkyl halide at room temperature and using dimethylformamide as solvent. Compounds 22-27, and 29 were obtained following this procedure. Because of stability problems the ester derivatives 45 and 46 were directly subjected to hydrolysis without purification by treatment with 0.25 M NaOH at room temperature to afford the corresponding acid derivatives 26 and 29, respectively.
Nucleoside 5′-diphosphates are generally less stable than the corresponding monophosphates. For this reason, for alkyl halides in which heating is necessary for the alkylation reaction, the starting material was the commercially available 4-thiouridine 5′-monophosphate (4-thio-UMP) 47 (Scheme 4). After the formation of the sodium thiolate salt of the 4-thio-UMP, the thio group of the uracil moiety was alkylated following the same procedure as in Scheme 3. The alkylthio monophosphates obtained were converted to the corresponding diphosphates through a phosphoryl imidazolide intermediate, which reacted with phosphoric acid tributylammonium salt.23 Compounds 28 and 30 were obtained following this procedure. The ester derivatives 48 and 49, because of stability problems, were directly subjected to next reaction without purification.
The nucleotides were selectively S-alkylated using the procedures described above. The 13C NMR spectrum of the amide derivative 27 shows the peak corresponding to the methylene of the acetamide at 33.0 ppm, typical of a carbon-sulfur bond.
Pharmacological Activity
Activation of PLC by the nucleotide derivatives was studied in 1321N1 astrocytoma cells stably expressing the human P2Y6 receptor.16,31 Removal of the 2′- or 3′-hydroxyl group of UDP led to a >100-fold decrease in potency in 12 and 13, respectively Substitution of the 2′-hydroxyl group of UDP with an azido or amino groups in 15, and 16, respectively, greatly reduced potency. The P2Y6 receptor is highly selective for 5′-diphosphate derivatives. Thus, uridine 5′-monophosphate 11, 2′-deoxyuridine 3′,5′-bisphosphate 1432 and a uridine 3′-diphosphate derivative 17 were inactive as either agonists or antagonists.
Rigid ring systems also were used to determine the preferred ribose conformation at the P2Y6 binding site. As previously reported, a strong preference for the (S) conformation was indicated (cf. 6 vs. 5). Another rigid ring system (2-OBH) in 19 was tolerated at the P2Y6 binding site.
Among uracil modified compounds, 2-thio-UDP 20 and 4-thio-UDP 21 were prepared and tested in order to probe the effect of the electronegativity as well as the size of the exocyclic atoms at positions 2 and 4. Both derivatives were five-fold less potent than the parent compound UDP. Among uncharged alkylated derivatives of 21, the highest potency was observed with the S-ethyl analogue 23, which was 20-fold less potent than UDP. In the series of carboxyalkylthio ethers, the highest potency was observed with the S-carboxyethylthio analogue 28. Substitution at the 3-position in 31 markedly decreased potency, while halogenation of the 5-position in the iodo analogue 32 resulted in a molecule that was equal in potency to UDP. By analogy, a 5-bromo analogue was previously reported to be roughly equipotent to UDP.16
The cyclic 3′,5′-diphosphate 33, which can be considered a closely related cyclized analogue of bisphosphate 14, was inactive at the P2Y6 receptor. The dinucleotide 34 was moderately potent at the P2Y6 receptor. 4-Thioether modification at both nucleobases in 35 and 36 reduced potency. A 4-O or 4-S was required for P2Y6 receptor activation, since the corresponding NH analogue 37 was inactive.
Within a series of uracil dinucleotide 5′,5′-polyphosphates, the triphosphates were the most potent and selective at the human P2Y6 receptor. However, dinucleotide 5′,5′-diphosphates also showed selectivity for the P2Y6 receptor.33,34 Therefore, several dinucleotide diphosphates 34–37, including 4-alkylthio analogues, were compared in this study. Only the simple Up2U 34 displayed substantial μmolar potency at the P2Y6 receptor.
Molecular Modeling
Docking studies of selected compounds were conducted at the model of the membrane-embedded human P2Y6 receptor.35 The binding mode of each studied compound was obtained by subjecting the complex to two distinct conformational searches, performed sequentially. These searches were intended to exhaustively explore the possible docking poses of the ligand by means of rotations and translations of the small molecule within the binding pocket. Simultaneously, the conformation of the residues lining the pocket was rearranged to minimize the energy of interaction with the ligand.
A schematic representation of the entire structure of the P2Y6 receptor complexed with 32 is given in Figure 1a, while Figure 1b shows the details of the receptor-ligand interactions. In analogy with our previous P2Y6-UDP complex,35 the diphosphate moiety of 32 is coordinated by three cationic residues from transmembrane domain (TM) 3, TM6, and TM7, namely 3.29, 6.55, and 7.39. The O at position 2 and the NH at position 3 of the uracil ring are H-bonded with S291(7.43) and Y33(1.33), respectively. Consistently with the equipotency of 32 and UDP, the 5-iodo group does not show any sort of steric or electronic incompatibility with the features of the P2Y6 binding pocket. Figure 1c shows compound 6 docked into the putative binding pocket of the P2Y6 receptor model. The locked puckering of the pseudoribose ensures that the phosphate and the nucleobase moiety of 6 maintain the optimal geometry for the interaction with the P2Y6 binding pocket. The donation of an H-bond from the NH at position 3 of the uracil ring to Y33(1.39), even though not essential for the recognition of the nucleotides, apparently contributes to enhanced potency at the P2Y6 receptor. Compound 31, which is incapable of donating an H-bond to Y33(1.39) due to the methyl substituent at the N3 position (Figure 1d), exhibited a 250-fold lower potency than UDP. A decreased partial negative atomic charge on the N3 might contribute to the decreased potency of 4-thio-UDP (21), as a consequence of a weaker H-bond donation to Y33(1.39). Moreover, all the 4-thio substituted compounds, which are incapable of donating an H-bond to Y33(1.39), exhibited lower potency than the parent compound 4-thio-UDP (21). Among these molecules, 4-ethylthio-UDP (23), 4-allylthio-UDP (24), and 4-benzylthio-UDP (25) showed the highest potency. Molecular modeling results suggest that the hydrophobic substituents at position 4 of the uracil ring establish favorable interactions with the aromatic ring of Y33(1.39) and with an hydrophobic pocket located between TM1 and TM2 (Figure 1e). The substitution at the 4-position with carboxyalkylthioethers (26, 28, 29) is also well tolerated by the receptor. These target structures were designed on the basis of molecular modeling results to engage an electrostatic interaction with K204(7.36) (Figure 1f).
Figure 1.

Molecular models of the complexes of the human P2Y6 receptor with UDP analogues 32, (a and b, 5-iodo), 6 (c, (S)-methanocarba), 31 (d, 3-methyl), 23 (e, 4-ethylthio), and 28 (f, 4-carboxyethylthio). In all cases, the diphosphate moiety of the nucleotide is coordinated by three cationic residues from TM3 (3.29), TM6 (6.55), and TM7 (7.39). The donation of an H-bond from the NH at position 3 of the uracil ring to Y33(1.39) seems to contribute to higher potency in the activation of the P2Y6 receptor. To facilitate the comparison, in the six panels of this figure the receptor maintains the same orientation relative to the plane of the membrane.
In the schematic representation of the P2Y6 receptor complexed with Cpd. 32 (a), the tube represents the backbone of the receptor and is colored according to residue positions, with a spectrum of colors that ranges from red (N-terminus) to purple (Cterminus): TM1 is in orange, TM2 in ochre, TM3 in yellow, TM4 in green, TM5 in cyan, TM6 in blue, TM7 in purple.
Discussion
As a result of mutagenesis and molecular modeling studies, we proposed that nucleotides bind to the P2Y receptor with the sugar moiety accommodated between TM3 and TM7, the base pointing toward TM1 and TM2, and the polyphosphate moiety pointing in the direction of TM6 (Figure 1a).11,35,36 According to this model, the electrostatic interaction of the phosphate moiety with three cationic residues from TM3, TM6, and TM7 (3.29, 6.55, and 7.39) is fundamental for the recognition of nucleotides. Our current modeling data (Figure 1) suggest that the β-phosphate of UDP and related analogs is fundamental for the establishment of electrostatic interactions with two of the three cationic residues, namely K259(6.55) and R287(7.39). Not surprisingly, compounds 11, 14, 17, and 33, which lack a β-phosphate linked through the 5′-position of ribose, were inactive at the P2Y6 receptor.
The puckering of the ribose ring, described by the phase angle of pseudorotation (P, Figure 2), is of fundamental importance for the biological activity of nucleosides and nucleotides. We recently discovered that the (S) conformation of the ribose moiety is preferred for ligand recognition by the P2Y6 receptor.35 Our molecular modeling studies suggested that a pure (S) conformation (P= 180°, 2′-endo, 3′-exo) would confer to the phosphate and nucleobase moieties of UDP the most favorable orientation for establishment of interactions with the residues from TM1, TM3, TM6, and TM7. Compound 6, which is locked in the (S) conformation (P= -162°, 3′-exo) by a (S)-methanocarba (mc) ring, proved to be about 10 times more potent than 12. Conversely, 5, locked in the (N) conformation by a (N)-methanocarba ring, was found completely inactive. Compound 19, which is locked in a (S) conformation (P= 126°, 1′-exo) by a 2-OBH ring system, but is further away from pure (S) than 6, maintained agonist activity at the P2Y6 receptor but was less potent than the corresponding flexible analogues. The proposed conformation of 19 was calculated using quenched molecular dynamics and energy minimization. The cyclopropane ring, fused between C3′ and C4′, forces the C1′ into the exo conformation in order for the six-membered ring to adopt a pseudoboat conformation.
Figure 2.
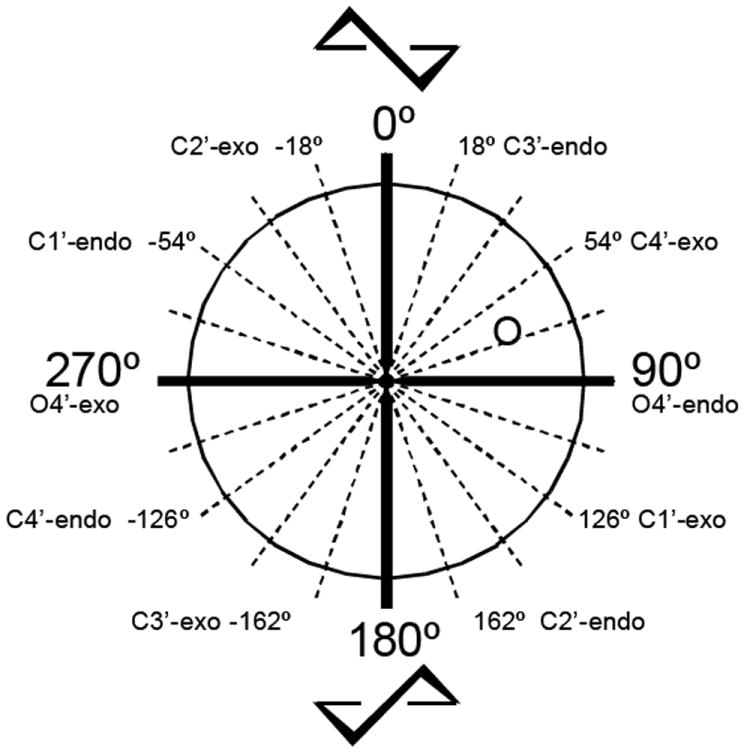
Graphical representation of the pseudorotational cycle,48 which defines all of the possible ribose ring conformations. The phase angle of pseudorotation (P) describes the geometry of the ribose puckering. The (N)-methanocarba ring system adopts a C2′-exo conformation and the (S)-methanocarba ring system a C3′-exo conformation. The 2-OBH ring system maintains a C1′-exo conformation, in the (S) region.
Table 2 shows the relative EC50 values with respect to the native ligands at three P2Y receptors for the same changes in the base or nucleotide, applied to the 5′-diphosphate derivative (P2Y6 receptor) and to the 5′-triphosphate derivative (P2Y2 and P2Y4 receptors). In most cases, there is a parallel reduction of potency at P2Y2 and P2Y4 receptors. However, there tended to be a greater loss of potency at the P2Y6 receptor, for example for 2′-ribose modifications. The removal of the 2′- or 3′-hydroxyl group was more poorly tolerated at the P2Y6 receptor than at the two other subtypes. The reduction of potency of the 2′-deoxyarabino-2′-fluoro analogue 18 was particularly pronounced at the P2Y6 receptor. The low potency of the 2′-amino analogue 16 is in sharp contrast with the high potency of the corresponding triphosphate at the P2Y2 receptor.37 The 2- and 4-thio modifications tended to preserve potency at all subtypes, but most effectively at the P2Y2 and P2Y4 receptors. Strikingly, the 5-iodo modification preserves potency most effectively at the P2Y6 receptor, and 5-halo substitution of the uracil ring may therefore be a basis for achieving selectivity at this subtype.
Table 2.
Relative effects of parallel structural modifications of uracil nucleotide analogues on their potency in the stimulation of PLC at recombinant human P2Y2, P2Y4, and P2Y6 receptors. The EC50 ratios at the P2Y6 receptors are derived from the data in Table 1 for derivatives of the native agonist, i.e. 5′-diphosphates. The EC50 ratios of the corresponding 5′-triphosphates at human P2Y2 and P2Y4 receptors expressed in astrocytoma cells are derived from published data,23,37 unless noted. In each case, the endogenous agonist (UDP at P2Y6 receptors, UTP at P2Y2 and P2Y4 receptors) is assigned the value of 1.
| Compound Number of 5′-Diphosphate | Modification (as di- or triphosphate) | Structuredd | EC50 Ratio of 5′-Diphosphate/UDP, hP2Y6 Receptor | EC50 Ratio of 5′-Triphosphate/UTP, hP2Y2 Receptor | EC50 Ratio of 5′-Triphosphate/UTP, hP2Y4 Receptor |
|---|---|---|---|---|---|
| Substitution of base moiety | |||||
| 1 | UDP, UTP | d | 1 | 1 | 1 |
| 7 | CDP, CTP | d | 1000a | 110 | NA |
| 8 | ADP, ATP | d | >7000 | 1.7 | NA |
| 9 | GDP | d | 500a | 54 | 90 |
| 10 | IDP | d | 400a | 13b | 9.1b |
| Ribose modifications | |||||
| 5 | (N)-d methanocarba | d | >7000 | 2.0c | 1.7c |
| 12 | 2′-deoxy | R7 = H | 130 | 22 | 26 |
| 13 | 3′-deoxy | R8 = H | 190 | 17e | 42e |
| 15 | 2′-azido-2′-deoxy | R7 = N3 | 120 | 100 | 15 |
| 16 | 2′-amino-2′-deoxy | R7 = NH2 | 300 | 1.3 | 16 |
| 18 | 2′-deoxy-arabino-2′-fluoro- | R7 = H, R6 = F | 420 | 11 | 7.1 |
| Uracil modifications | |||||
| 20 | 2-thio | R2 = S | 4 | 0.71 | 4.8 |
| 21 | 4-thio | R4 = S | 6 | 0.53 | 0.32 |
| 31 | 3-methyl | R3 = CH3 | 250 | 24 | 47 |
| 32 | 5-iodo | R5 = I | 1.1 | 17 | 55 |
Conclusions
The P2Y receptor family is unusual in comparison to other pharmacologically-defined clusters of GPCR sequences in that they respond to varied and diverse nucleotide ligands. Within the same phylogenetic cluster are receptors for other anionic ligands: lysophosphatidylserine (GPR34), succinic acid (GPR91), and α-keto-glutarate (GPR80).1,11 The challenge for the medicinal chemist is to understand the structural basis for the P2Y ligand selectivities and to use that information to design much needed pharmacological probes to act as selective agonists and antagonists. We have recently focused on the influence of the ribose conformation on subtype selectivity. In this study of SAR at the human P2Y6 receptor following diverse structural changes of UDP, we have identified various modifications that deselect for this receptor subtype in relation to other uracil nucleotide-responsive receptors and several modifications (i.e. 5-iodo and the (S)-mc ring) that provide P2Y6 selectivity. Combinations of these various modifications should achieve higher potency and selectivity, and in conjunction with molecular modeling and receptor docking studies should lead to high affinity agonists that are highly selective for the P2Y6 receptor.
Experimental Section
Chemical Synthesis
2′-Deoxyuridine-5′-diphosphate (12) was purchased from Jena Bioscience USA (San Diego, CA). 3′-Deoxyuridine-5′-triphosphate was purchased from TriLink BioTechnologies (San Diego, CA). 3′-Deoxyuridine (38) was purchased from T.R.C., Inc. (North York, Ontario, Canada). 2′-Azido-2′-deoxyuridine (39), and 2′-amino-2′-deoxyuridine (40) were purchased from CMS Chemicals Ltd. (Oxfordshire, UK). 2′-Arafluoro-2′-deoxyuridine (42b) was purchased from R.I. Chemical, Inc. (Orange, CA). 2-Thiouridine (43) was purchased from Berry & Associates, Inc (Dexter, MI). Compounds 21, 47, 53 and reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Compounds 5, 6, 14, 31 were synthesized in our laboratory as described,23,32,35,37. Compounds 34, 37 and 44 were synthesized as reported.25,38,39
1H NMR spectra were obtained with a Varian Gemini 300 spectrometer using D2O as a solvent. The chemical shifts are expressed as relative ppm from HOD (4.78 ppm). 13C NMR spectra were recorded at room temperature by use of Varian XL 300 spectrometer (75 MHz); methanol-d4 was used as an external standard. 31P NMR spectra were recorded at room temperature by use of Varian XL 300 spectrometer (121.42 MHz); orthophosphoric acid (85%) was used as an external standard. The complete assignment of the signals was performed by COSY experiments obtained with a Varian XL 300 spectrometer.
Purity of compounds was checked using a Hewlett–Packard 1100 HPLC equipped with a Luna 5 μ RP-C18(2) analytical column (250 X 4.6 mm; Phenomenex, Torrance, CA). System A: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 20 min, then isocratic for 2 min; the flow rate was 1 mL/min. System B: linear gradient solvent system: 10 mM TEAA (triethylammonium acetate)-CH3CN from 100:0 to 85:15 in 20 min, then isocratic for 2 min; the flow rate was 1 mL/min. System C: linear gradient solvent system: 10 mM TEAA-CH3CN from 90:10 to 60:40 in 20 min, then isocratic for 2 min; the flow rate was 1 mL/min. Purity of compounds 13, 15-17, 20, 25, 29, 32, 50 and 51 in system B or C was checked using a Zorbax Eclipse 5 μm XDB-C18 analytical column (250 X 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Peaks were detected by UV absorption with a diode array detector. All derivatives tested for biological activity showed >98% purity in the HPLC systems.
High-resolution mass measurements were performed on Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectometer coupled with a Waters HPLC system. Purification of the nucleotide analogues for biological testing was carried out on (diethylamino)ethyl (DEAE)-A25 Sephadex columns as described below. Compounds 13, 15-17 were additionally purified by HPLC using system D (10 mM TEAA-CH3CN from 100:0 to 90:10 in 30 min, then isocratic for 2 min; the flow rate was 2 mL/min) with a Luna 5 μ RP-C18(2) semipreparative column (250 X 10.0 mm; Phenomenex, Torrance, CA).
General Procedure for the Preparation of Nucleoside 5′-Diphosphates
Procedure A. A solution of the corresponding nucleoside (0.04-0.19 mmol) and Proton Sponge (1.5 equiv) in trimethyl phosphate (2 mL) was stirred for 10 min at 0 °C. Then phosphorous oxychloride (2 equiv) was added dropwise, and the reaction mixture was stirred for 2 hours at 0 °C. A mixture of tributylamine (9 equiv) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (6 equiv) was added at once. This salt was prepared by mixing tributylamine (0.16 mL, 0.66 mmol) and phosphoric acid (34 mg, 0.35 mmol) in DMF (1 mL). After 6 min 0.2 M triethylammonium bicarbonate solution (3 mL) was added, and the clear solution was stirred at room temperature for 45 min. The latter was lyophilized overnight. The residue was purified by ion-exchange column chromatography using a Sephadex-DEAE A-25 resin with a linear gradient (0.01-0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase. The corresponding nucleoside diphosphates were collected, frozen and lyophilized as the ammonium salts.
(2R,3R,5S)-1-(5-(Diphosphoryloxymethyl)-3-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione, Triethylammonium salt (13)
Procedure A Compound 13 (2 mg, 7%) was obtained as a white solid from 38 (10 mg, 0.04 mmol). 1H NMR (D2O) δ 8.04 (d, J=8.1 Hz, 1H), 5.94 (d, J=8.1 Hz, 1H), 5.85 (d, J=1.5 Hz, 1H), 4.68 (m, 1H), 4.55 (m, 1H), 4.35 (m, 1H), 4.14 (m, 1H), 2.25 (m, 1H), 2.10 (m, 1H); 31P NMR (D2O) δ -10.33 (d, J=20.8 Hz), -10.79 (d, J=19.6 Hz); HRMS m/z found 386.9995 (M – H+)-. C9H13N2O11P2 requires 386.9995; HPLC (System A) 13.7 min (98%), (System B) 6.7 min (98%).
(2R,3R,4S,5R)-1-(3-Azido-5-(diphosphoryloxymethyl)-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione, Triethylammonium salt (15)
Procedure A Compound 15 (12 mg, 35%) was obtained as a white solid from 39 (32 mg, 0.12 mmol). 1H NMR (D2O) δ 7.99 (d, J=8.1 Hz, 1H), 6.04 (d, J=5.1 Hz, 1H), 5.99 (d, J=8.1 Hz, 1H), 4.63 (m, 1H), 4.40 (m, 1H), 4.29 (m, 1H), 4.25 (m, 2H); 31P NMR (D2O) δ -9.63, -10.81; HRMS m/z found 427.9993 (M – H+)-. C9H12N5O11P2 requires 428.0009; HPLC (System A) 13.9 min (99%), (System B) 9.8 min (99%).
(2R,3R,4S,5R)-N-(2-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-3-yl)-2,2,2-trifluoroacetamide (41)
To a solution of 40 (20 mg, 0.08 mmol) in DMF (1 mL) was added DIEA (0.04 mL, 0.25 mmol) and ethyltrifluoroacetate (0.03 mL, 0.25 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was removed in vacuo, and the residue was purified by preparative thin layer chromatography (CH2Cl2-MeOH, 85:15) to afford 41 (24 mg, 86%) as a white solid. 1H NMR (CD3OD) δ 8.01 (d, J=8.1 Hz, 1H), 6.13 (d, J=7.8 Hz, 1H), 5.73 (d, J=8.1 Hz, 1H), 4.60 (m, 1H), 4.31 (m, 1H), 4.01 (m, 1H), 3.77 (m, 2H); HRMS m/z found 338.0587 (M – H+)-. C11H11N3O6F3 requires 338.0600.
(2R,3R,4S,5R)-1-(3-Amino-5-(diphosphoryloxymethyl)-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (16)
Procedure A Compound 16 in the triethylammonium salt (7 mg, 17%) was obtained as a white solid from 41 (20 mg, 0.06 mmol). Alternatively, compound 16 (0.6 mg, 75%) was obtained by treatment of a solution of the triethylammonium salt of compound 15 (1.5 mg, 0.002 mmol) in MeOH (1 mL) with 10% Pd/C (0.5 mg) and H2 at atmospheric pressure for 1 h at room temperature. 1H NMR (D2O) δ 7.99 (d, J=7.5 Hz, 1H, H-6 pyrimidine), 6.12 (d, J=7.2 Hz, 1H, H-2), 6.00 (d, J=7.5 Hz, 1H, H-5 pyrimidine), 4.61 (m, 1H, H-4), 4.36 (m, 1H, H-5), 4.24 (m, 1H, HCHO), 4.16 (m, 1H, HCHO), 3.88 (m, 1H, H-3); 31P NMR (D2O) δ -6.77, -10.57; HRMS m/z found 402.0126 (M – H+)-. C9H14N3O11P2 requires 402.0104; HPLC (System A) 6.0 min (99%), (System B) 7.1 min (99%).
(2R,3R,4S,5R)-1-(3-Amino-4-(diphosphoryloxy)-5-(hydroxymethyl)-tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione, Triethylammonium salt (17)
Procedure A Compound 17 (6 mg, 8%) was obtained as a white solid from 40 (25 mg, 0.10 mmol). 1H NMR (D2O) δ 7.88 (d, J=8.1 Hz, 1H, H-6 pyrimidine), 6.29 (d, J=6.9 Hz, 1H, H-2), 5.91 (d, J=8.1 Hz, 1H, H-5 pyrimidine), 5.01 (m, 1H, H-4), 4.46 (m, 1H, H-5), 4.14 (m, 1H, H-3), 3.89 (m, 2H, CH2O); 13C NMR (D2O) δ 167.3, 152.9, 142.4, 103.9, 87.9, 87.5, 74.7, 62.0, 56.3; 31P NMR (D2O) δ -7.73 (d, J=22.0 Hz), -11.16 (d, J=22.0 Hz); HRMS m/z found 402.0094 (M – H+)-. C9H14N3O11P2 requires 402.0104; HPLC (System A) 10.6 min (98%), (System B) 6.7 min (98%).
(2R,3S,4R,5R)-1-(5-(Diphosphoryloxymethyl)-3-fluoro-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione, Ammonium salt (18)
Procedure A Compound 18 (2.1 mg, 32%) was obtained as a white solid from 42b (9 mg, 0.04 mmol). 1H NMR (D2O) δ 7.79 (d, J=7.9 Hz, 1H), 6.36 (m, 1H), 5.93 (d, J=7.9 Hz, 1H), 5.53 (apparent dt, JH-F=69.0 Hz, 1H), 4.42 (m, 1H), 4.19 (m, 3H); 31P NMR (D2O) δ -8.89, -10.81; HRMS m/z found 404.9892 (M – H+)-. C9H12N2O11P2F requires 404.9900; HPLC (System A) 15.1 min (99%), (System B) 10.9 min (99%).
(1S,3R,4R,5S)-1-(4-Hydroxy-1-(diphosphoryloxymethyl)-2-oxabicyclo[3.1.0]hexan-3-yl)pyrimidine-2,4(1H,3H)-dione, Ammonium salt (19)
Procedure A Compound 19 (4 mg, 31%) was obtained as a white solid from 44 (10 mg, 0.04 mmol). 1H NMR (D2O) δ 7.80 (d, J=8.1 Hz, 1H), 5.79 (d, J=8.1 Hz, 1H), 5.60 (d, J=8.1 Hz, 1H), 4.65 (m, 1H), 4.07(m, 1H), 3.67(m, 1H), 1.81 (m, 1H), 1.37 (m, 1H), 0.84 (m, 1H); 31P NMR (D2O) δ -7.08, -10.55; HRMS m/z found 398.9985 (M – H+)-. C10H13N2O11P2 requires 398.9995; HPLC (System A) 14.5 min (99%), (System B) 10.2 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one, Ammonium salt (20)
Procedure A Compound 20 (21 mg, 23%) was obtained as a white solid from 38 (50 mg, 0.19 mmol). 1H NMR (D2O) δ 8.19 (d, J=8.1 Hz, 1H), 6.65 (d, J=2.7 Hz, 1H), 6.27 (d, J=8.1 Hz, 1H), 4.45 (m, 1H), 4.32 (m, 4H); 31P NMR (D2O) δ -9.35 (d, J=20.8 Hz), -10.84 (d, J=20.8 Hz); HRMS m/z found 418.9712 (M – H+)-. C9H13N2O11P2S requires 418.9715; HPLC (System A) 15.0 min (98%), (System B) 8.4 min (98%).
General Procedure for the Preparation of 4-Substituted-Thio-UDP Analogues
Procedure B. To a suspension of 4-thiouridine 5′-diphosphate (5 mg, 0.01 mmol) in MeOH (1 mL) was added 0.25 M NaOH (0.14 mL). The reaction mixture was stirred at room temperature for 2 h and then the solvent was eliminated under high vacuum. The 4-thio-UDP sodium salt obtained was suspended in dry DMF (1.5 mL) and an excess of the corresponding alkyl halide (50 equiv) was added. The reaction mixture was stirred at room temperature for 24 h, the progress of the reaction was monitored by analytical HPLC (System A). After removal of the solvent, the residue was purified using the same method than in procedure A, and the corresponding nucleotide diphosphates were obtained as the ammonium salts.
Procedure C. To a solution of 4-thiouridine 5′-monosphate (10 mg, 0.03 mmol) in MeOH (1 mL) was added 0.25 M NaOH (0.13 mL). The reaction mixture was stirred at room temperature for 2 h and then the solvent was eliminated under high vacuum. The 4-thio-UMP sodium salt obtained was suspended in dry DMF (2 mL) and an excess of the corresponding alkyl halide (50 equiv) was added. The reaction mixture was stirred at 90 °C for 4-8 h, the progress of the reaction was monitored by analytical HPLC (System A). The solvent was removed in vacuo to obtain the corresponding 4-substituted-thio-UMP as a crude solid, which then was used directly in the next step without further purification. To a solution of the crude solid containing 4-substituted-thio-UMP in DMF (1.5 mL) was added 1,1′-carbonyldiimidazole (26 mg, 0.16 mmol). The reaction mixture was stirred at room temperature for 6 h. Then 5% triethylamine solution in 1/1 water/methanol (3 mL) was added and stirring was continued at room temperature for additional 2 h. After removal of the solvent, the residue was dried in high vacuum and dissolved in DMF (1 mL). To this solution was successively added tributylamine (0.11 mL, 0.46 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (0.4 mL). The reaction mixture was stirred at room temperature for 2 days and then 0.2 M triethylammonium bicarbonate was added. After removal of the solvent, the residue was purified using the same method than in procedure A, and the corresponding nucleotide diphosphates were obtained as the ammonium salts.
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-4-(methylthio)pyrimidin-2(1H)-one, Ammonium salt (22)
Procedure B Compound 22 (7 mg, 80%) was obtained as a white solid from 21 (10 mg, 0.02 mmol) and using iodomethane as alkylating agent. 1H NMR (D2O) δ 8.25 (d, J=7.1 Hz, 1H), 6.76 (d, J=7.1 Hz, 1H), 5.94 (d, J=2.4 Hz, 1H), 4.41 (m, 1H), 4.32 (m, 4H), 2.54 (s, 3H); 13C NMR (D2O) δ 182.4, 156.8, 142.0, 106.4, 91.9, 83.8, 76.0, 69.6, 65.0, 14.0; 31P NMR (D2O) δ -7.15 (d, J=23.1 Hz), -10.67 (d, J=23.1 Hz); HRMS m/z found 432.9888 (M – H+)-. C10H15N2O11P2S requires 432.9872; HPLC (System A) 14.9 min (99%), (System B) 11.1 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-4-(ethylthio)pyrimidin-2(1H)-one, Ammonium salt (23)
Procedure B Compound 23 (2 mg, 44%) was obtained as a white solid using iodoethane as alkylating agent and stirring the reaction mixture at 70 °C for 4 h. 1H NMR (D2O) δ 8.24 (d, J=7.4 Hz, 1H), 6.75 (d, J=7.4 Hz, 1H), 5.94 (d, J=2.4 Hz, 1H), 4.41 (m, 1H), 4.32 (m, 4H), 3.15 (q, J=7.5 Hz, 2H), 1.36 (t, J=7.5 Hz, 3H); 31P NMR (D2O) δ -7.49, -10.73; HRMS m/z found 447.0032 (M – H+)-. C11H17N2O11P2S requires 447.0028; HPLC (System A) 14.7 min (99%), (System B) 14.5 min (99%).
(2R,3R,4S,5R)-4-(Allylthio)-1-(3,4-dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one, Ammonium salt (24)
Procedure B Compound 24 (3.4 mg, 73%) was obtained as a white solid using allyl bromide as alkylating agent. 1H NMR (D2O) δ 8.26 (d, J=7.5 Hz, 1H), 6.76 (d, J=7.5 Hz, 1H), 5.99 (m, 1H), 5.93 (m, 1H), 5.40 (d, J=17.1 Hz, 2H), 5.23 (d, J=10.2 Hz, 2H), 4.43 (m, 1H), 4.32 (m, 4H), 3.84 (d, J=6.6 Hz, 2H); 31P NMR (D2O) δ -6.08 (d, J=21.9 Hz), -10.55 (d, J=21.9 Hz); HRMS m/z found 459.0039 (M – H+)-. C12H17N2O11P2S requires 459.0028; HPLC (System A) 16.0 min (99%), (System B) 16.2 min (99%).
(2R,3R,4S,5R)-4-(Benzylthio)-1-(3,4-dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one, Ammonium salt (25)
Procedure B Compound 25 (3.3 mg, 65%) was obtained as a white solid using benzyl bromide as alkylating agent. 1H NMR (D2O) δ 8.21 (d, J=7.2 Hz, 1H), 7.52 (m, 2H), 7.39 (m, 3H), 6.73 (d, J=7.2 Hz, 1H), 5.95 (d, J=2.7 Hz, 1H), 4.46 (s, 2H), 4.34 (m, 4H), 4.23 (m, 1H); 31P NMR (D2O) δ -9.41, -10.93; HRMS m/z found 509.0171 (M – H+)-. C16H19N2O11P2S requires 509.0185; HPLC (System A) 16.8 min (98%), (System C) 7.9 min (98%).
(2R,3R,4S,5R)-2-(1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-ylthio)acetic acid, Ammonium salt (26)
Procedure B Compound 45 (HPLC (System A) 15.6 min) was obtained as a crude solid using methyl bromoacetate as alkylating agent. The residue that contained compound 45 was dissolved in H2O (1 mL) and 0.25 M NaOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 2 h. After removal of the solvent, the residue was purified following the general procedure to give compound 26 (2.4 mg, 49% from 21) as a white solid. 1H NMR (D2O) δ 8.21 (d, J=7.1 Hz, 1H), 6.75 (d, J=7.1 Hz, 1H), 5.96 (d, J=2.4 Hz, 1H), 4.35 (m, 4H), 4.25 (m, 1H), 3.88 (s, 2H); 31P NMR (D2O) δ -10.37 (d, J=20.6 Hz), -10.92 (d, J=20.6 Hz); HRMS m/z found 476.9773 (M – H+)-. C11H15N2O13P2S requires 476.9770; HPLC (System A) 18.3 min (99%), (System B) 9.0 min (99%)
(2R,3R,4S,5R)-2-(1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-ylthio)acetamide, Ammonium salt (27)
Procedure B Compound 27 (3.8 mg, 79%) was obtained as a white solid using iodoacetamide as alkylating agent. 1H NMR (D2O) δ 8.31 (d, J=7.4 Hz, 1H), 6.79 (d, J=7.4 Hz, 1H), 5.92 (d, J=1.5 Hz, 1H), 4.42 (m, 1H), 4.32 (m, 4H), 3.96 (s, 2H); 13C NMR (D2O) δ 177.9, 173.9, 156.0, 141.9, 105.8, 91.2, 83.0, 75.1, 68.3, 63.8, 33.0; 31P NMR (D2O) δ -7.63, -10.66 (d, J=21.9 Hz); HRMS m/z found 475.9935 (M – H+)-. C11H16N3O12P2S requires 475.9930; HPLC (System A) 14.6 min (99%), (System B) 8.6 min (99%).
(2R,3R,4S,5R)-3-(1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-ylthio)propanoic acid, Ammonium salt (28)
Procedure C Compound 49 (HPLC (System A) 16.0 min) was obtained as a crude solid using methyl 3-bromopropionate and stirring at 90 °C for 8h in the alkylation reaction. The residue that contained compound 49 was dissolved in H2O (1 mL) and 0.25 M NaOH (2 mL) was added. The reaction mixture was stirred at room temperature for 6 h. After removal of the solvent, the residue was purified following the general procedure to give compound 28 (2.8 mg, 20% from 47) as a white solid. 1H NMR (D2O) δ 8.21 (d, J=7.2 Hz, 1H), 6.76 (d, J=7.2 Hz, 1H), 5.99 (d, J=2.7 Hz, 1H), 4.38 (m, 4H), 4.26 (m, 1H), 3.39 (t, J=6.9 Hz, 2H), 2.73 (t, J=6.9 Hz, 2H); 31P NMR (D2O) δ -10.39, -10.84; HRMS m/z found 490.9951 (M – H+)-. C12H17N2O13P2S requires 490.9927; HPLC (System A) 18.5 min (99%), (System B) 9.6 min (99%).
(2R,3R,4S,5R)-4-(1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-ylthio)butanoic acid, Ammonium salt (29)
Procedure B Compound 46 (HPLC (System A) 16.4 min) was obtained as a crude solid using methyl 4-iodobutyrate as alkylating agent and stirring the reaction mixture at 50 °C for 20 h. The residue that contained compound 46 was dissolved in H2O (2 mL) and 0.25 M NaOH (2 mL) was added. The reaction mixture was stirred at room temperature for 2 h. After removal of the solvent, the residue was purified following the general procedure to give compound 29 (2.0 mg, 16% from 21) as a white solid. 1H NMR (D2O) δ 8.19 (d, J=6.8 Hz, 1H), 6.75 (d, J=6.8 Hz, 1H), 5.96 (m, 1H), 4.35 (m, 4H), 4.24 (m, 1H), 3.19 (d, J=7.1 Hz, 1H), 2.40 (d, J=7.5 Hz, 1H), 1.99 (m, 2H); 31P NMR (D2O) δ -10.22, -10.98; HRMS m/z found 505.0096 (M – H+)-. C13H19N2O13P2S requires 505.0083; HPLC (System A) 18.2 min (99%), (System B) 9.2 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(monophosphoryloxymethyl)tetrahydrofuran-2-yl)-4-(hexylthio)pyrimidin-2(1H)-one, Ammonium salt (50)
To a solution of 4-thiouridine 5′-monosphate (10 mg, 0.03 mmol) in MeOH (1 mL) was added 0.25 M NaOH (0.13 mL). The reaction mixture was stirred at room temperature for 2 h and then the solvent was eliminated under high vacuum. The 4-thio-UMP sodium salt obtained was suspended in dry DMF (2 mL) and 1-iodohexane (0.19 mL, 1.28 mmol) was added. The reaction mixture was stirred at 90 °C for 4 h, the progress of the reaction was monitored by analytical HPLC (System A). The solvent was removed in vacuo to obtain compound 50 as a crude solid, which then was used directly in the next step without further purification. For characterization purposes a small portion of compound 50 was purified by ion-exchange column chromatography as described in procedure A. 1H NMR (D2O) δ 8.21 (d, J=7.5 Hz, 1H), 6.73 (d, J=7.5 Hz, 1H), 5.97 (m, 1H), 4.34 (m, 3H), 4.29 (m, 1H), 4.14 (m, 1H), 3.18 (t, J=7.1 Hz, 2H), 1.74 (m, 2H), 1.46 (m, 2H), 1.33 (m, 4H), 0.89 (t, J=6.9 Hz, 3H); 31P NMR (D2O) δ 0.60; HRMS m/z found 423.0995 (M – H+)-. C15H24N2O8PS requires 423.0991; HPLC (System A) 15.1 min (99%), (System C) 13.7 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-4-(hexylthio)pyrimidin-2(1H)-one, Ammonium salt (30)
To a solution of the crude solid containing compound 50 in DMF (1.5 mL) was added 1,1′-carbonyldiimidazole (26 mg, 0.16 mmol). The reaction mixture was stirred at room temperature for 6 h. Then 5% triethylamine solution in 1/1 water/methanol (3 mL) was added and stirring was continued at room temperature for additional 2 h. After removal of the solvent, the residue was dried in high vacuum and dissolved in DMF (1 mL). To this solution was successively added tributylamine (0.11 mL, 0.46 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (0.4 mL). The reaction mixture was stirred at room temperature for 2 days and then 0.2 M triethylammonium bicarbonate was added. After removal of the solvent, the residue was purified using the same method than in procedure A to afford 30 (3.4 mg, 21% from 47) as a white solid. 1H NMR (D2O) δ 8.21 (d, J=7.4 Hz, 1H), 6.75 (d, J=7.4 Hz, 1H), 5.96 (d, J=2.7 Hz, 1H), 4.36 (m, 4H), 4.25 (m, 1H), 3.17 (t, J=7.2 Hz, 2H), 1.73 (m, 2H), 1.45 (m, 2H), 1.32 (m, 4H), 0.88 (t, J=7.1 Hz, 3H); 31P NMR (D2O) δ -9.67, -10.83; HRMS m/z found 503.0606 (M – H+)-. C15H25N2O11P2S requires 503.0654; HPLC (System A) 18.9 min (99%), (System C) 13.8 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(diphosphoryloxymethyl)tetrahydrofuran-2-yl)-5-iodopyrimidine-2,4(1H,3H)-dione, Ammonium salt (32)
To a solution of 53 (8 mg, 0.02 mmol) in DMF (2 mL) was added 1,1′-carbonyldiimidazole (16 mg, 0.10 mmol). The reaction mixture was stirred at room temperature for 6 h. Then 5% triethylamine solution in 1/1 water/methanol (2 mL) was added and stirring was continued at room temperature for additional 2 h. After removal of the solvent, the residue was dried in high vacuum and dissolved in DMF (2 mL). To this solution was successively added tributylamine (0.07 mL, 0.29 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (0.3 mL). The reaction mixture was stirred at room temperature for 3 days and then 0.2 M triethylammonium bicarbonate (2 mL) was added, and stirred for 30 min. After removal of the solvent, the residue was purified using the same method than in procedure A to afford 32 (1.8 mg, 19%) as a white solid. 1H NMR (D2O) δ 8.28 (s, 1H), 5.93 (d, J=4.2 Hz, 1H), 4.42 (m, 2H), 4.25 (m, 3H); 31P NMR (D2O) δ -6.89, -10.60; HRMS m/z found 528.8901 (M – H+)-. C9H12N2O12P2I requires 528.8910; HPLC (System A) 14.7 min (99%), (System B) 8.8 min (99%).
(2R,3S,4R,5R)-1-(3-Fluoro-4-hydroxy-5-(monophosphoryloxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione, Ammonium salt (54)
A solution of 2′-fluoro-2′-deoxy-β-D-arabinofuranosyluridine (15 mg, 0.06 mmol) and proton sponge (26 mg, 0.12 mmol) in trimethyl phosphate (2 mL) was stirred for 10 min at 0 °C. Then phosphorous oxychloride was added dropwise (0.01mL, 0.12 mmol), and the reaction mixture was stirred for 2 hours at 0 °C. Then 0.2 M triethylammonium bicarbonate solution (7 mL) was added, and the clear solution was stirred at room temperature for 45 min. The latter was lyophilized overnight. The residue was purified using the same method than in procedure A to afford 54 (7 mg, 60%) as a white solid. 1H NMR (D2O) δ 7.90 (d, J=8.1 Hz, 1H), 6.31 (dd, J=15.9, 4.2 Hz, 1H), 5.89 (d, J=8.1 Hz, 1H), 5.21 (apparent dt, JH-F=51.6 Hz, 1H), 4.52 (apparent dt, JH-F=19.2 Hz, 1H), 4.13 (m, 3H); 31P NMR (D2O) δ 4.01; HRMS m/z found 325.0240 (M – H+)-. C9H11N2O8FP requires 325.0237; HPLC (System A) 10.1 min (99%), (System B) 6.2 min (99%).
(2R,3S,4R,5R)-1-(3-Fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione cyclic-4,5-diphosphate, Ammonium Salt (33)
To a solution of 54 (5 mg, 0.02 mmol) in DMF (2 mL) was added 1,1′-carbonyldiimidazole (12 mg, 0.08 mmol). The reaction mixture was stirred at room temperature for 8 h. Then 5% triethylamine in methanol was added and stirring was continued at room temperature for additional 2 h. After removal of the solvent, the residue was dried in high vacuum and dissolved in DMF (1 mL). To this solution was successively added tributylamine (0.06 mL, 0.25 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (0.23 mL). The reaction mixture was stirred at room temperature for 3 days and then 0.2 M triethylammonium bicarbonate (2 mL) was added, and stirred for 30 min. After removal of the solvent, under reduced pressure at 50 °C, the residue was purified using the same method than in procedure A to afford 33 (1.6 mg, 29%) as a white solid. 1H NMR (D2O) δ 7.78 (d, J=8.1 Hz, 1H), 6.35 (dd, J=13.1, 5.3 Hz, 1H), 5.92 (d, J=8.1 Hz, 1H), 5.50 (apparent dt, JH-F=52.2 Hz, 1H), 4.47 (m, 1H), 4.25 (m, 3H); 31P NMR (D2O) δ -7.15, -10.37; HRMS m/z found 386.9783 (M – H+)-. C9H10N2O10P2F requires 386.9795; HPLC (System A) 13.2 min (99%), (System B) 8.6 min (99%).
(2R,3R,4S,5R)-1-(3,4-Dihydroxy-5-(monophosphoryloxymethyl)tetrahydrofuran-2-yl)-4-(ethylthio)pyrimidin-2(1H)-one, Ammonium salt (51)
To a solution of 4-thiouridine 5′-monosphate (10 mg, 0.03 mmol) in MeOH (1 mL) was added 0.25 M NaOH (0.13 mL). The reaction mixture was stirred at room temperature for 2 h and then the solvent was eliminated under high vacuum. The 4-thio-UMP sodium salt obtained was suspended in dry DMF (2 mL) and iodoethane (0.1 mL, 1.28 mmol) was added. The reaction mixture was stirred at 70 °C for 2 h, the progress of the reaction was monitored by analytical HPLC (System A). The solvent was removed in vacuo to obtain compound 51 as a crude solid, which then was used directly in the next step without further purification. For characterization purposes a small portion of compound 51 was purified by ion-exchange column chromatography as described in procedure A. 1H NMR (D2O) δ 8.26 (d, J=7.4 Hz, 1H), 6.73 (d, J=7.4 Hz, 1H), 5.96 (m, 1H), 4.33 (m, 3H), 4.24 (m, 1H), 4.09 (m, 1H), 3.15 (q, J=7.4 Hz, 2H), 1.36 (t, J=7.4 Hz, 3H); 31P NMR (D2O) δ 1.48; HRMS m/z found 367.0377 (M – H+)-. C11H16N2O8PS requires 367.0365; HPLC (System A) 8.5 min (98%), (System B) 6.7 min (98%).
P1,P2-Bis-5-[(2R,3R,4S,5R)-1-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-4-(ethylthio)pyrimidin-2(1H)-one]pyrophosphate, Ammonium salt, (4-SEt)Up2(4-SEt)U (35)
To a solution of the crude solid containing compound 51 in DMF (1.5 mL) was added 1,1′-carbonyldiimidazole (26 mg, 0.16 mmol). The reaction mixture was stirred at room temperature for 6 h. Then 5% triethylamine solution in 1/1 water/methanol (3 mL) was added and stirring was continued at room temperature for additional 2 h. After removal of the solvent, the residue was dried in high vacuum and dissolved in DMF (1 mL). To this solution was successively added tributylamine (0.11 mL, 0.46 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (0.4 mL). The reaction mixture was stirred at room temperature for 2 days and then 0.2 M triethylammonium bicarbonate was added. After removal of the solvent, the residue was purified using the same method than in procedure A to afford 35 (5 mg, 52% from 47) as a white solid. 1H NMR (D2O) δ 8.10 (d, J=7.4 Hz, 2H), 6.57 (d, J=7.4 Hz, 2H), 5.95 (d, J=2.7 Hz, 2H), 4.45 (m, 2H), 4.28 (m, 8H), 3.10 (q, J=7.5 Hz, 4H), 1.35 (t, J=7.5 Hz, 6H); 31P NMR (D2O) δ -10.23; HRMS m/z found 717.0711 (M – H+)-. C22H31N4O15P2S2 requires 717.0703; HPLC (System A) 14.9 min (99%), (System C) 6.8 min (99%).
P1,P2-Bis-5-[(2R,3R,4S,5R)-4-(benzylthio)-1-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one]pyrophosphate, Ammonium salt, (4-SBn)Up2(4-SBn)U (36)
Procedure C Compound 36 (5 mg, 45% from 47) was obtained as a white solid using benzyl bromide and stirring overnight at room temperature in the alkylation reaction. 1H NMR (D2O) δ 8.11 (d, J=7.2 Hz, 2H), 7.47 (m, 4H), 7.37 (m, 6H), 6.55 (d, J=7.2 Hz, 2H), 5.92 (d, J=2.4 Hz, 2H), 4.31 (m, 14H); 31P NMR (D2O) δ -10.81; HRMS m/z found 841.0991 (M – H+)-. C32H35N4O15P2S2 requires 841.1016; HPLC (System A) 19.2 min (99%), (System C) 16.8 min (99%).
Assay of P2Y6 receptor-stimulated PLC activity
A stable cell line expressing the human P2Y6 receptor in 1321N1 human astrocytoma cells was generated as previously described in detail.16 Agonist-induced [3H]inositol phosphate production was measured in 1321N1 cells grown to confluence on 48-well plates. Twelve h before the assay, the inositol lipid pool of the cells was radiolabeled by incubation in 200 μL of serum-free inositol-free Dulbecco’s modified Eagle’s medium, containing 0.4 μCi of myo-[3H]inositol. No changes of medium were made subsequent to the addition of [3H]inositol. On the day of the assay, cells were challenged with 50 μL of the five-fold concentrated solution of receptor agonists in 200 mM Hepes (N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid), pH 7.3, containing 50 mM LiCl for 20 min at 37 °C. Incubations were terminated by aspiration of the drug-containing medium and addition of 450 μL of ice-cold 50 mM formic acid. After 15 min at 4 °C, samples were neutralized with 150 μL of 150 mM NH4OH. [3H]Inositol phosphates were isolated by ion exchange chromatography on Dowex AG 1-X8 columns as previously described.31
Data Analysis
Agonist potencies (EC50 values) were obtained from concentration-response curves by non-linear regression analysis using the GraphPad software package Prism (GraphPad, San Diego, CA). All experiments were performed in triplicate assays and repeated at least three times. The results are presented as mean ± SEM from multiple experiments or in the case of concentration effect curves from a single experiment carried out with triplicate assays that were representative of results from multiple experiments.
Molecular Modeling
Molecular mechanics calculations have been carried out by means of the Discover3 module of InsightII,40 using the CFF91 forcefield.41 The receptor model used for the docking experiments was the P2Y6/UDP complex which we previously built11 and optimized by means of a molecular dynamics (MD) simulation in an explicit fully hydrated lipid bilayer.35
Before performing the docking experiments, the ligands were first fully optimized by means of quenched molecular dynamics followed by energy minimization. An NVT (constant-volume/constant-temperature) molecular dynamic simulation was carried out at constant temperature of 300 K for 10 ps, using a time step of 1 fs. During the simulation, snapshots of the system were taken at regular intervals of 1 ps. The structures in each snapshot were energy minimized (BFGS Newton method) until an RMSD (root mean squared) of 0.00001 kcal/mol/Å on the gradient was reached. For each ligand, the conformation showing the lowest energy was promoted to the next phase of the study.
Subsequently, the ligands were flexibly superimposed to the bound conformation of 1, as derived from our MD simulation.35 The flexible superimpositions have been carried out by means of the FieldFit program of the Search Compare module of InsightII,40 giving equal weight to the steric and the electrostatic factors. The bound conformation of 1 was kept rigid during the calculation, while the structures to be superimposed were fully flexible. An alignment of the molecules based on their dipole and quadruple moments was used as the starting position.
The docking experiments were carried out by means of the Monte Carlo minimization approach implemented in the Affinity module of InsightII.40 The binding site was defined as all the residues within a distance of 6 Å from the ligand. Full flexibility was granted to the ligands and to the residues of the binding site. The scaling factor for the van der Waals term was set at 0.1, while the coulombic term was set at 1. The maximum movement in each random translation and rotation of the ligand were set at 0.1 Å and 1° respectively.
After the docking procedure, the receptor-ligand complexes were optimized by means of a Monte Carlo Multiple Minimum (MCMM)42 conformational search as implemented in MacroModel.43,44 The search was performed on the ligand and the residues located within a distance of 6 Å from the ligand, while the remaining residues were conformationally frozen. The following parameters were employed for the conformational search: number of steps = 100; number of structures to save for each search = 100; energy window for saving structures = 1000.0 KJ/mol. The calculations were conducted with the MMFFs force field,45 using water as implicit solvent (GB/SA model as implemented in MacroModel)46 and a molecular dielectric constant of 1. For the energy minimizations the Polak-Ribier Conjugate Gradient was used with a convergence threshold on the gradient of 0.05 kJ/Å/mol.
Supplementary Material
Acknowledgments
Dr. Pedro Besada and Dr. Dae-Hong Shin thank the Cystic Fibrosis Foundation, Bethesda, MD, for financial support. Mass spectral measurements were carried out by Dr. John Lloyd and NMR by Wesley White (NIDDK). We thank Kanna Palaniappan for proofreading this manuscript. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. This work was supported by National Institutes of Health grants GM38213 and HL34322 to T.K. Harden.
Footnotes
Supporting Information Available: Coordinates of the complex of the human P2Y6 with compound 32 (5-iodoUDP) are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Towards a revised nomenclature for P1 and P2 receptors. Trends Pharmacol Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.North RA, Barnard EA. Nucleotide receptors. Curr Opin Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- 3.von Kügelgen I, Wetter A. Molecular pharmacology of P2Y-receptors. Naunyn Schmiedeberg’s Arch Pharmacol. 2000;362:310–323. doi: 10.1007/s002100000310. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson KA, Costanzi S, Ohno M, Joshi BV, Besada P, Xu B, Tchilibon S. Molecular recognition at purine and pyrimidine nucleotide (P2) receptors. Curr Top Med Chem. 2004;4:805–819. doi: 10.2174/1568026043450961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Kügelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacology Therapeutics. 2006;110:415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Lustig KD, Shiau AK, Brake AJ, Julius D. Expression cloning of an ATP receptor from mouse neuroblasoma cells. Proc Natl Acad Sci USA. 1993;90:5113–5117. doi: 10.1073/pnas.90.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Communi D, Pirotton S, Parmentier M, Boeynaems JM. Cloning and functional expression of a human uridine nucleotide receptor. J Biol Chem. 1995;270:30849–30852. doi: 10.1074/jbc.270.52.30849. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen T, Erb L, Weisman GA, Marchese A, Heng HHQ. Cloning, expression, and chromosomal localization of the human uridine nucleotide receptor gene. J Biol Chem. 1995;270:30845–30848. doi: 10.1074/jbc.270.52.30845. [DOI] [PubMed] [Google Scholar]
- 9.Chang K, Hanaoka K, Kumada M, Takuwa Y. Molecular cloning and functional analysis of a novel P2 nucleotide receptor. J Biol Chem. 1995;270:26152–26158. doi: 10.1074/jbc.270.44.26152. [DOI] [PubMed] [Google Scholar]
- 10.Communi D, Suarez Gonzalez N, Detheux M, Brezillon S, Lannoy V, Parmentier M, Boeynaems JM. Identification of a novel human ADP receptor coupled to Gi. J Biol Chem. 2001;276:41479–41485. doi: 10.1074/jbc.M105912200. [DOI] [PubMed] [Google Scholar]
- 11.Costanzi S, Mamedova L, Gao Z-G, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaver SR. P2Y receptors: biological advances and therapeutic opportunities. Curr Opin Drug Disc. 2001;4:665–670. [PubMed] [Google Scholar]
- 13.Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC. Pharmacology of INS37217 [P(1)-(uridine 5’)-P(4)-(2’-deoxycytidine 5’)tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- 14.Burnstock G. Purinergic P2 receptors as targets for novel analgesics. Pharmacology Therapeutics, Therapeutics. 2006;110:433–454. doi: 10.1016/j.pharmthera.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson KA, Jarvis MF, Williams M. Perspective: Purine and pyrimidine (P2) receptors as drug targets. J Med Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholas RA, Lazarowski ER, Watt WC, Li Q, Harden TK. Uridine nucleotide selectivity of three phospholipase C-activating P2 receptors: identification of a UDP-selective, a UTP-selective, and an ATP- and UTP-specific receptor. Mol Pharmacol. 1996;50:224–229. [PubMed] [Google Scholar]
- 17.Kim SG, Soltysiak KA, Gao ZG, Chang TS, Chung E, Jacobson KA. Tumor necrosis factor α-induced apoptosis in astrocytes is prevented by the activation of P2Y6, but not P2Y4 nucleotide receptors. Biochem Pharmacol. 2003;65:923–931. doi: 10.1016/s0006-2952(02)01614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korcok J, Raimundo LN, Du X, Sims SM, Dixon SJ. P2Y6 nucleotide receptors activate NF-κB and increase survival of osteoclasts. J Biol Chem. 2005;280:16909–16915. doi: 10.1074/jbc.M410764200. [DOI] [PubMed] [Google Scholar]
- 19.Schreiber R, Kunzelmann K. Purinergic P2Y6 receptors induce Ca2+ and CFTR dependent Cl- secretion in mouse trachea. Cell Physiol Biochem. 2005;16:99–108. doi: 10.1159/000087736. [DOI] [PubMed] [Google Scholar]
- 20.Cox MA, Gomes B, Palmer K, Du K, Wiekowski M, Wilburn B, Petro M, Chou C-C, Desquitado C, Schwarz M, Lunn C, Lundell D, Narula SK, Zavodny PJ, Jenh C-H. The pyrimidinergic P2Y6 receptor mediates a novel release of proinflammatory cytokines and chemokines in monocytic cells stimulated with UDP. Biochem Biophys Res Commun. 2005;330:467–473. doi: 10.1016/j.bbrc.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Khine AA, Del Sorbo L, Vaschetto R, Voglis S, Tullis E, Slutsky AS, Downey GP, Zhang H. Human neutrophil peptides induce interleukin-8 production via P2Y6 signaling pathway. Blood. 2006;107:2936–2942. doi: 10.1182/blood-2005-06-2314. [DOI] [PubMed] [Google Scholar]
- 22.Hou M, Harden TK, Kuhn CM, Baldetorp B, Lazarowski E, Pendergast W, Möller S, Edvinsson L, Erlinge D. UDP acts as a growth factor for vascular smooth muscle cells by activation of P2Y6 receptors. Am J Physiol Heart Circ Physiol. 2002;282:H784–H792. doi: 10.1152/ajpheart.00997.2000. [DOI] [PubMed] [Google Scholar]
- 23.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. Methanocarba modification of uracil and adenine nucleotides: High potency of Northern ring conformation at P2Y1, P2Y2, P2Y4 and P2Y11, but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robaye B, Boeynaems JM, Communi D. Slow desensitization of the human P2Y6 receptor. Eur J Pharmacol. 1997;329:231–236. [PubMed] [Google Scholar]
- 25.Gagneron J, Gosselin G, Mathe C. Synthesis of nucleoside analogues bearing the five naturally occurring nucleic acid bases built on a 2-oxabicylo[3.1.0]hexane scaffold. J Org Chem. 2005;70:6891–6897. doi: 10.1021/jo051047b. [DOI] [PubMed] [Google Scholar]
- 26.Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA. Identification of potent, selective P2Y-purinoceptor agonists: Structure activity relationships for 2-thioether derivatives of adenosine-5′-triphosphate. J Med Chem. 1993;36:3937–3946. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaver SR, Pendergast W, Siddiqi SM, Yerxa BR, Croom DK, Dougherty RW, James MK, Jones AC, Rideout JL. 4-Substituted uridine 5′-triphosphates as agonists of the P2Y2 purinergic receptor. Nucleosides & Nucleotides. 1997;16:1099–1102. [Google Scholar]
- 28.Aviño A, Guimil Garcia R, Eritja R. Synthesis of oligoribonucleotides containing 4-thiouridine using the convertible nucleoside approach and the 1-(2-fluorophenyl)-4-methoxypiperidin-4-yl group. Nucleosides, Nucleotides & Nucleic Acids. 2004;23:1767–1777. doi: 10.1081/NCN-200034044. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y-Z, Zheng Q, Swann PF. Synthesis of DNA containing modified bases by post-synthetic substitution. Synthesis of oligomers containing 4-substituted thymine: O4-alkylthymine, 5-methylcytosine, N4-dimethylamino-5-methylcytosine, and 4-thiothymine. J Org Chem. 1992;57:3839–3845. [Google Scholar]
- 30.Fischer B, Chulkin A, Boyer JL, Harden KT, Gendron FP, Beaudoin AR, Chapal J, Hillaire-Buys D, Petit P. 2-Thioether 5’-O-(1-thiotriphosphate)adenosine derivatives as new insulin secretagogues acting through P2Y-receptors. J Med Chem. 1999;42:3636–3646. doi: 10.1021/jm990158y. [DOI] [PubMed] [Google Scholar]
- 31.Brown HA, Lazarowski ER, Boucher RC, Harden TK. Evidence that UTP and ATP regulate phospholipase C through a common extracellular 5′-nucleotide receptor in human airway epithelial cells. Mol Pharmacol. 1991;40:648–655. [PubMed] [Google Scholar]
- 32.Nandanan E, Camaioni E, Jang S-Y, Kim YC, Cristalli G, Herdewijn P, Secrist JA, III, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. Structure activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists. J Med Chem. 1999;42:1625–1638. doi: 10.1021/jm980657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pendergast W, Yerxa BR, Douglass JG, 3rd, Shaver SR, Dougherty RW, Redick CC, Sims IF, Rideout RL. Synthesis and P2Y receptor activity of a series of uridine dinucleoside 5’-polyphosphates. Bioorg Med Chem Lett. 2001;11:157–60. doi: 10.1016/s0960-894x(00)00612-0. [DOI] [PubMed] [Google Scholar]
- 34.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR. Structure–activity relationships of dinucleotides: Potent and selective agonists of P2Y receptors. Purinergic Signalling. 2005;1:183–191. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y6 receptor: Molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J Med Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno M, Costanzi S, Kim HS, Kempeneers V, Vastmans K, Herdewijn P, Maddileti S, Gao Z-G, Harden TK, Jacobson KA. Nucleotide analogues containing 2-oxa-bicyclo[2.2.1]heptane and L-α-threofuranosyl ring systems: interactions with P2Y receptors. Bioorg Med Chem. 2004;12:5619–5630. doi: 10.1016/j.bmc.2004.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson KA, Costanzi S, Ivanov AA, Tchilibon S, Besada P, Gao Z-G, Maddileti S, Harden TK. Structure activity and molecular modeling analyses of ribose- and base-modified uridine 5′-triphosphate analogues at the human P2Y2 and P2Y4 receptors. Biochem Pharmacol. 2006;71:540–549. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christie SMH, Elmore DT, Kenner GW, Todd AR, Weymouth FJ. Syntheses of P1P2-diadenosine-5’ and P1P2-diuridine-5’ pyrophosphates. J Chem Soc. 1953:2947–2953. [Google Scholar]
- 39.Baddiley J, Buchanan JG, Fawcett CP. Syntheses of cytidine diphosphate ribitol. J Chem Soc. 1959:2192–2196. [Google Scholar]
- 40.InsightII, version 2000.1, Accelrys (former MSI) San Diego, CA: [Google Scholar]
- 41.Maple JR, Hwang MJ, Stockfisch TP, Dinur U, Waldman M, Ewig CS, Hagler AT. Derivation of class II force fields. 1. Methodology and quantum force field for the alkyl functional group and alkane molecules. J Comput Chem. 1994;15:162. [Google Scholar]
- 42.Chang G, Guida W, Still WC. An internal coordinate Monte-Carlo method for searching conformational space. J Am Chem Soc. 1989;111:4379–4386. [Google Scholar]
- 43.Macromodel, version 9.1. Schrödinger L.L.C; [Google Scholar]
- 44.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. MacroModel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
- 45.Halgren TA. MMFF VII. Characterization of mmff94, mmff94s and other widely available force fields for conformational energies and for intermolecular interaction energies and geometries. J Comput Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 46.Still WC, Tempczyk A, Hawley RC, Hendrickson T. A general treatment of solvation for molecular mechanics. J Am Chem Soc. 1990;112:6127. [Google Scholar]
- 47.Kennedy C, Qi AD, Herold CL, Harden TK, Nicholas RA. ATP, an agonist at the rat P2Y4 receptor, is an antagonist at the human P2Y4 receptor. Mol Pharmacol. 2000;57:926–931. [PubMed] [Google Scholar]
- 48.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.