

Abstract
2, N6, and/or 5′ substituted adenosine derivatives were synthesized via alkylation of 2-oxypurine nucleosides leading to 2-aralkylether derivatives. 2-(3-(Indolyl)ethyloxy)adenosine 17 was found to be a potent agonist of the human A2BAR in both binding and cAMP assays. Simplification, altered connectivity and mimicking of the indole ring of 17 failed to maintain A2BAR potency. Introduction of N6-ethyl or N6-guanidino substitution, shown to favor A2BAR potency, failed to enhance potency in the 2-(3-(indolyl)ethyloxy)adenosine series. Indole 5″- or 6″-halo substitution was favored at the A2BAR, but a 5′-N-ethylcarboxyamide did not further enhance potency. 2-(3″-(6″-Bromoindolyl)ethyloxy)adenosine 28 displayed an A2BAR EC50 value (nM) of 128, i.e. more potent than the parent 17 (299) and similar to 5′-N-ethylcarboxamidoadenosine (140). 28 was a full agonist at A2B and A2AARs and a low efficacy partial agonist at A1 and A3ARs. Thus, we have identified and optimized 2-(2-arylethyl)oxo moieties in AR agonists that enhance A2BAR potency and selectivity.
Keywords: G protein-coupled receptor, nucleosides, adenylate cyclase, purines, receptor binding, indole
INTRODUCTION
There are four subtypes of adenosine receptors (ARs): A1, A2A, A2B, and A3.1 Three of these subtypes already possess selective and potent agonists and antagonists.2 Only the A2BAR remains without a selective agonist. It should be noted, however, that highly potent and selective antagonists have been reported for this subtype.3–8 A2BAR antagonists are directed toward clinical use for treating asthma and/or diabetes. Conversely, a selective A2BAR agonist would provide a useful pharmacological probe for exploring the role of receptor activation. Activation of the A2BAR is known to induce angiogenesis,9 reduce vascular permeabilization,10 increase production of the anti-inflammatory cytokine IL-10,11 increase chloride secretion in epithelial cells,12–14 and increase release of inflammatory mediators from human and canine mast cells.15,16 A2BAR agonists have been proposed for the treatment of septic shock17 and cystic fibrosis,18 and cardiac,19 pulmonary,20 and kidney19 diseases associated with remodeling and hyperplasia. Thus, such an agonist may be useful in preventing restenosis. The A2BAR mediates vasodilation in the corpus cavernosum of rabbit and agonists, therefore, may be useful in treating erectile dysfunction.21 Recently Yang et al., describe a proinflammatory phenotype resulting from deletion of the gene encoding the A2BAR in the mouse, suggesting that activation of the A2BAR can have anti-inflammatory effects.22 Activation of the A2BAR also promotes postconditioning salvage of ischemic myocardium.23
Modulation of A2BAR potency has been achieved through structural changes at several sites on the adenosine molecule. R-PIA (R-N6-(phenylisopropyl)adenosine) 1 was one of the earliest potent agonists to be extensively investigated at ARs and was one of the nucleosides used initially to distinguish the low affinity A2B and high affinity A2AARs.24 It was found to activate the A2BAR with a micromolar potency and later was noted to bind to the A3AR with nearly the same affinity as at the A2AAR.
For several decades NECA (5′-N-ethylcarboxamidoadenosine) 2 was considered to be the most potent known agonist at the A2BAR, with an EC50 of 140 nM.25–27 A survey of structurally diverse adenosine derivatives as agonists of the human A2BAR failed to identify a lead that surpassed the potency of 2.28 Cristalli and coworkers have explored the SAR (structure-activity relationship) of 2-substituted 5′-uronamide adenosine derivatives such as (S)-PHP-NECA 4 (EC50 = 220 nM) as potent but relatively nonselective agonists of the A2BAR.29–31
Recent reports provided new insights into the SAR of agonists of the A2BAR. A three-fold enhancement in potency at the A2BAR was achieved by combining 2 with the N6-guanidino modification in 3.32 This structural change produced a three-fold gain in potency at the A3AR, a 300-fold loss of potency at the A2AAR, and no change at the A1AR. Also, while 2-ether derivatives of adenosine were characterized as potent A2AAR agonists, specific 2-(2-arylethyloxy) ethers were also noted to be particularly potent at the A2BAR.26 In the present study we characterized the SAR of potent A2BAR agonists based on compound 17. In particular, 5″ or 6″-functionalization on the indole moiety of 17 led to the achievement of new AR agonists with enhanced potency at the A2BAR reduced potency at other AR subtypes. Moreover, we have used molecular modeling of the human A2BAR31 to propose a mode of docking of the potent 2-substituted derivatives.
RESULTS
Chemical Synthesis
The structures of the target adenosine 2-ether derivatives 5 – 40 appear in Chart 1 and Table 1. Compounds 5 – 7 and 9 – 16 were studied previously at the human A2BAR.26 Routes to the synthetic intermediates for the 2-ether component are shown in Schemes 1 and 2. The novel 2-alkoxyadenosines 8 and 17 – 36, N6-guanidino-2-(3-indolyl)ethyloxy)adenosine derivatives 37 and 38, N6-ethyl-2-(3-indolyl)ethyloxy)adenosine 39 and 5′-N-ethylcarboxamido-2-(3-indolyl)ethyloxyadenosines 40 were prepared via alkylation of 2-oxypurine nucleosides using arylalkyl/alkyl iodides as shown in Schemes 3 – 5.
Chart 1.

Chemical structures of selected adenosine derivatives 1 – 4 previously used as pharmacological reference compounds for characterization of the A2BAR and novel adenosine derivatives 8, 17 – 40. The remaining adenosine derivatives in the series, 5 – 7 and 9 – 16 (structures in Table 1) were reported previously.26
Table 1.
Potency of various adenosine derivatives in activation of the human A2BAR expressed in CHO cells, with values expressed either the EC50 (nM) or the percent stimulation at 1 μM (in parentheses). For comparison, binding affinities of the adenosine derivatives at human A1, A2A, and A3ARs expressed in CHO cells (expressed as Ki value or percent displacement at 10 μM) and maximal agonist effects at 10 μM at the A3AR. Values for compounds 5 – 7 and 9 – 16 are from ref. 25.
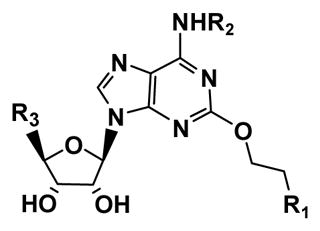 5 – 40, R2= H, R3 = CH2OH, unless noted | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | Name/Substitution | EC50 at A2BAR (nM) (or% activation) | Ki at A1AR (nM) or % inhib.a | Ki at A2AAR (nM) or % inhib.a | Ki at A3AR (nM) or % inhib.a | Efficacy, A3AR % |
| Reference Agonists | ||||||
| 1 | R-PIA | 1680±500 | 2.0±0.3 | 884±188 | 8.7±0.9 | 102±6 |
| 2 | NECA | 140±19 | 6.8±2.4 | 2.2±0.6 | 16.0±5.4 | 100 |
| 3b | 6-guanidino-NECA | 54.5±13.3 | 7.0±1.0 | 628±39 | 5.1±1.3 | 100 |
| 4c | (S)-PHP-NECA | 220 | 2.1 | 2.0 | 0.75 | |
|
| ||||||
| 2-Ethers | ||||||
| 5d | R1 = H | (−1%) | 2640±540 | 360±139 | 568±205 | 99±4 |
| 6d |

|
(36%) | (36%) | 579±250 | 578±182 | 52±3 |
| 7d |
|
3490±1490 | 221±57 | 9.3±2.9 | 54.2±14.3 | 71±3 |
| 8 |

|
(29%) | 960±95 | 500±50 | 66 ± 3 | 42±8 |
| 9d |
|
(3%) | 2060 ± 630 | 519 ± 41 | 352 ± 66 | 37±8 |
| 10d |

|
(13%) | 1560 ± 250 | 413 ± 37 | 312 ± 47 | 18±8 |
| 11d |

|
(12%) | 331±22 | 58.1±24.9 | 77.8±13.5 | 45±5 |
| 12d |

|
(64%) | 312 ± 24 | 69.3 ± 4.7 | 119 ± 8 | 50±7 |
| 13d |
|
(11%) | 467±100 | 56.8±16.3 | 112±16 | 74±5 |
| 14d |

|
1440±70 | 141±51 | 16.1±7.0 | 130±8 | 45±9 |
| 15d |

|
1780±260 | 174±20 | 10.9±4.8 | 93.3±16.8 | 80±5 |
| 16d |

|
(9%) | 280±72 | 13.3±4.1 | 101±34 | 62±15 |
| 17b |

|
299±45 | 148±19 | 45.0±11.6 | 232±54 | 17±3 |
| 18 |

|
(43%) | (39±6%) | 2670±630 | 1340±230 | 0±3 |
| 19 |

|
2580 | 218±55 | 95±18 | 104±40 | 75±1 |
| 20 |
|
(49%) | 197±47 | 373±71 | 513±84 | 37±8 |
| 21 |

|
(32%) | 47±2% | 2570±670 | 622±19 | 30±1 |
| 22 |

|
767 | 150±50 | 370±80 | 490±60 | −1±1 |
| 23 |

|
2180 | 130±40 | 390±110 | 296±8 | −4±1 |
| 24b |

|
365±73 | 358±1 | 502±32 | 234±24 | 1±2 |
| 25 |

|
1870 | 350±60 | 900±200 | 110±20 | −5±2 |
| 26 |

|
(30%) | (38±2%) | (54±8%) | 8730±340 | −1±5 |
| 27 |

|
216±59 | 145±6 | 29.3±13.7 | 92.3±7.9 | 2±5 |
| 28b |

|
128±32 | 253±3 | 150±20 | 90±15 | 20±1 |
| 29 |

|
(16%) | 443±82 | 39.7±14.4 | 260±19 | −5±1 |
| 30 |

|
(43%) | 210±19 | 252±109 | 142±17 | −8±5 |
| 31 |

|
(48%) | 579±88 | (64±2%) | 599±3 | 13±4 |
| 32 |

|
(24%) | 20±2% | (26±4%) | (66±5%) | 3±2 |
| 33 |

|
1250 | 1820±330 | 1400±300 | 360±50 | −3±3 |
| 34 |
|
896 | 310 ± 90 | 450±8 | 120 ± 20 | 24±3 |
| 35 |

|
(0%) | (18±4%) | 3870±497 | 2070 ± 700 | 3±2 |
| 36 |

|
(0%) | (41±5%) | (46±2%) | 1920 ± 470 | 13±4 |
| 37 |
 R2 = guanidino |
(40%) | 73.6 ± 8.0 | 277 ± 74 | 90 ± 10 | 58±8 |
| 38 |
 R2 = guanidino |
(42%) | 52±2% | 344 ± 72 | 457 ± 40 | 24±7 |
| 39 |
 R2 = Et |
3270 | 640±110 | 40±4% | 30±10 | 54±12 |
| 40 |
 R3 = CONHEt |
989 | 190±20 | 250±30 | 110±20 | 102±2 |
All experiments were performed using adherent CHO cells stably transfected with cDNA encoding a human AR. Percent activation of the human A2B or A3AR was determined at 10 μM. Binding at A1, A2A and A3ARs was carried out as described in Experimental Procedures. The A3 receptor activation results were from three separate experiments. The Ki and EC50 values from the present study are expressed as mean±s.e.m., N = 3–5.
Compounds 3, MRS3218; 17, MRS3534; 24, MRS3854; 28, MRS3997.
Data from reference 26.
Scheme 1.

Reagents and conditions: (a) TsCl, NaH, THF, 0°C - room temperature; (b) NaI, DMF, 60°C; (c) (i) TsOH-H2O, MeOH, 60°C (ii) LAH, THF, 4°C - room temperature; (d) EtOH, reflux; (e) oxalyl chloride, Et2O; (f) LAH, THF, reflux; (g) I2, PPh3, imidazole, benzene.
Scheme 2.

Reagents and conditions: (a) i. TsCl, pyridine, CH2Cl2; ii. 3-butyn-1-ol, Pd(PPh3)2Cl2, CuI, Et3N, DMF, 70°C; (b) I2, PPh3, imidazole, Et2O-MeCN, room temperature; (c) LAH, THF, 0°C - room temperature
Scheme 3.

Reagents and conditions: (a) R1CH2CH2-I (47 – 49, 59 – 61, 74 – 77, 85, 86, 1-iodo-3-phenylpropane, 89, 94, 95 and 99, respectively), Cs2CO3, DMF, room temperature; (b) saturated NH3 in EtOH, 120°C; (c) deprotection of tosyl group of 18, 119 – 121, 26, 32, 122 – 126, 21 and 127; KOH, MeOH 70 – 90°C.
Scheme 5.

Reagents and conditions: (a) 2,2-dimethoxypropane, p-TsOH-H2O, DMF; (b) KMnO4, KOH, H2O; (c) PyBop, DIPEA, EtNH2 HCl, DMF; (d) t-butyl nitrite, 2-PrOH/H2O (1:1); (e) iodide 47, Cs2CO3, DMF, room temperature; (f) saturated NH3, EtOH, 120°C; (g) 80% AcOH, 80°C; (h) KOH, MeOH, 70°C.
The various arylalkyl iodides used in this study, when not commercially available, were synthesized by the routes shown in Schemes 1 and 2. Tryptophol (3-(2-hydroxyethyl)indole) 41, 5-methoxytryptophol 42 and 5-hydroxy-tryptophol 43 were converted to the corresponding tosylates 44 – 46, respectively, followed by iodination with NaI to give the iodides 47 – 49. Indole-3-acetic acid analogues 50 – 52 having a functional group at the 2 and/or 5 position were transformed to the corresponding esters, which were reduced with lithium aluminum hydride to give the alcohols 53 – 55. Tosylation of the alcohols 53 – 55 followed by iodination with sodium iodide gave the corresponding iodides 59 – 61. Furthermore, other 5 or 6-substituted tryptophols 67 – 70 were prepared by refluxing the corresponding phenylhydrazine hydrochloride salts 63 – 66 and 2-ethoxytetrahydrofuran 62 to effect a Fischer indole ring cyclization.34 Compounds 67 – 70 were converted to the corresponding iodides 74 – 77, respectively, by using the conventional method mentioned above. Compounds 78 and 79 were transformed to the ethyl glyoxylate derivatives 80 and 81, which were reduced with lithium aluminum hydride to give the corresponding tryptophol derivatives 82 and 83, respectively. Compounds 82 and 83 were converted to the corresponding iodides 85 and 86 by using conventional methods.
A tosylated 2-tryptophol 88 was prepared by the palladium-mediated heteroannulation of 2-iodoaniline with 3-butyn-1-ol (Scheme 2). The amino group of 2-iodoaniline 87 was activated by the strong electron-withdrawing sulfonyl group, and the indole cyclization occurred in one pot to give the tosylated 2-tryptophol 8835. Compound 88 was converted to the corresponding iodide 89 with iodine, triphenylphosphine, and imidazole.
Reduction of benzoimidazol-1-yl-acetic acid 90 and benzotriazol-1-yl-acetic acid 91 with lithium aluminum hydride followed by iodination gave the corresponding iodides 94 and 95, respectively.
3-Pyrrolylacetic acid methyl ester 97 was synthesized from pyrrole by the known method36 involving Friedel-Crafts acylation followed by a Willgerodt-Kindler reaction. Reduction of 97 with lithium aluminum hydride produced the alcohol 98, which was converted to the corresponding iodide 99 by using iodine, triphenylphosphine, and imidazole.
The synthesis of the 5′-CH2OH analogues (Scheme 3) started from 9-(β-D-ribofuranosyl)-2-amino-6-chloropurine 100, which was converted to 9-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-6-chloro-2-hydroxypurine 101 as reported.33 Reaction of the hydroxyl group at the 2 position of 101 with various iodides 47 – 49, 59 – 61, 74 – 77, 85, 86, 1-iodo-3-phenylpropane, 89, 94 and 95, respectively, was carried out in the presence of cesium carbonate to give compounds 102 – 118. Simultaneous removal of the acetyl group and amination at the 6 position of 102 – 118 by using saturated ammonia in ethanol solution afforded compounds 18, 119 – 121, 26, 32, 125 – 126, 30, 8, 21, 35, 36, 127, respectively. Deprotection of the N-tosyl group of the indole or pyrrole ring of 18, 119–121, 26, 32, 122 – 126, 21 and 127 was conducted using potassium hydroxide in methanol to give compounds 17, 33, 34, 22, 24, 31, 23, 25, 27 – 29, 20 and 19, respectively.
Synthesis of N6-guanidino derivatives 37 and 38 began with compound 102 (Scheme 4). The guanidinolysis of 102 in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) afforded 37 and the tosylate 38. Treatment of 102 with ethyl amine and N,N-diisopropylethylamine in DMF at 140 °C followed by removal of the tosyl group with potassium hydroxide gave the N6-ethyl derivative 39.
Scheme 4.

Reagents and conditions: (a) guanidine solution,32 DABCO, EtOH, 110°C; (b) i. EtNH2 HCl, DIPEA, DMF, 140°C; ii. KOH, MeOH, 80°C.
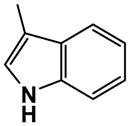
Synthesis of the 5′-N-ethylcarboxamido derivative 40 began with 100 (Scheme 5). Protection of the 1,2-diol of 100 afforded the acetonide 128. Oxidation of 128 with potassium permanganate gave the corresponding carboxylic acid derivative 129, which was converted to the 5′-N-ethylcarboxamide derivative 130 using benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP). Diazotization of 130 with t-butyl nitrite followed by hydrolysis gave the 2-hydroxy derivative 131 in 47% yield. Coupling of 131 with the iodide 47 in the presence of cesium carbonate in DMF gave the 2-O-alkylated derivative 132, which was followed by displacement of the 6-chloro moiety with ammonia to give 133. Removal of the 2′,3′-O-isopropylidene group of 133 with 80% acetic acid aqueous solution afforded compound 134. Treatment of 134 with potassium hydroxide in methanol resulted in the desired 5′-N-ethyluronamide derivative 40.
Biological Evaluation
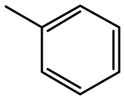
The AR binding affinities of novel compounds 8 and 17 – 40 were investigated in comparison to known (1 – 4, 5 – 7, and 9 – 16) nucleosides (Table 1). The SAR proceeded from known agonists with high potency at the A2BAR, i.e. 1 – 4. A previous difficulty has been targeting the A2BAR potency distinctly from the usually more potent activity at the A2AAR. Compound 4 was reported as a potent A2BAR agonist; nevertheless, it remained two orders of magnitude selective for the A2AAR in comparison to the A2BAR.31 Among 2 substituted derivatives, 2-ethers were more potent than the corresponding amines or thioethers.26 For example, a 2-phenylethyl ether 7 was only two-fold less potent than R-PIA 1 at the A2BAR. Nevertheless, the affinity in binding to the A2AAR was nearly 400-fold greater than the A2BAR functional potency. Therefore, great improvement was necessary in order to approach A2BAR-selectivity.
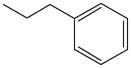
Elongation of the spacer alkyl chain beyond ethyl weakened the affinity against all ARs as shown in 7 – 9. Compound 10 is a variation of 8 in which a methyl group is branched in the alkyl chain, and its affinity was reduced in comparison to 8.
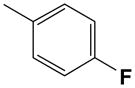
The effect of fluoro substitution of the phenyl ring was also probed. The 2-F 11, 4-F 12, and 3-F 13 analogues were invariant in affinity at A1, A2A, and A3 ARs, with Ki values at these subtypes ranging from 60 to 500 nM. These three analogues were also nearly inactive at the A2BAR.
Three other aromatic moieties in 2-(2-arylethyloxy) ethers 14 – 16 were reported in a previous study.26 2-Naphthyl and 2-thienyl moieties were tolerated at the A2BAR, while a 3-thienyl group resulted in inactivity at that subtype. Those modifications produced relatively minor changes at A1, A2A, and A3ARs in comparison to the phenyl analogue 7.
Interestingly, the 3-indolyl analogue 17 (a tryptophol ether) was 12-fold more potent than 7 at the A2BAR and 4 – 5 fold less potent than 7 in binding to the A2A and A3ARs. Also, 17 was only 2-fold less potent than 2 at the A2BAR. This considerable enhancement was exploited in subsequent SAR exploration. The corresponding N-tosyl derivative 18, as for similar N-tosyl derivatives, was considerably less potent at all ARs.
The 3-pyrrolyl derivative 19 was the most simple analogue in this study, whose pyrrole ring moiety is a critical component of an indole ring. Compound 19 was tolerated at the A2BAR, with a potency close to that of the 2-thienyl derivative 15, leading to 1.4-fold and 8-fold decreased potency at the A1 and A2AARs, respectively, and 2-fold similar potency at A3AR.
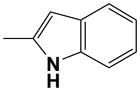
On the other hand the corresponding 2-indolyl derivative 20 decreased markedly A2BAR potency compared to 17. Thus, the 3 position was clearly the favored connection point and was utilized in subsequent synthesis. Its N-tosyl derivative 21 was less potent at all ARs, similar to results with 18.
It is known from SAR studies of A2B agonists that substitution of the 4′-hydroxymethyl group of an adenosine analogue with a 5′-N-ethylcarboxamido group often yields compounds endowed with higher affinity than the parent compound.31,37 Based on these findings we have prepared the 5′-N-ethyluronamido analogue 40 of 17. Unexpectedly, 40 was 3-fold less potent than the parent 5′-CH2OH compound 17 at the A2BAR and showed a 2-fold increased potency at only the A3AR.
Cristalli and colleagues have established that the N6-ethyl analogue of (S)-PHP-adenosine was more potent than the N6-methyl and N6-isopropyl derivatives at the A2B subtype.31 These findings were applied to 3-indolyl analogue 17, leading to the N6-ethyl analogue 39. Compound 39 was somewhat tolerated at A2BAR and 7-fold more potent than the parent compound 17 at the A3AR.
Another N6-functionalization for 17 was also investigated at ARs. Recently the N6-guanidino derivative 3 was reported to display a 3-fold potency enhancement over the parent 5′-N-ethyluronamide 2 at the A2BAR.32 However, introduction of a N6-guanidino group to 17 led to derivative 37 and its tosyl derivative 38 with decreased A2B potency compared to the parent compounds. Compound 37 showed a 2-fold increased potency at A1 and A3 ARs. Thus, the effect of N6-guanidinylation to enhance A2BAR selectivity is not always compatible with other beneficial structures.
Benzoimidazole and benzotriazole analogues 35 and 36 are simple congeners of 17, in which indole ring was substituted with an imidazole or triazole ring, respectively. Compounds 35 and 36 showed disappointingly low potency at all AR subtypes.
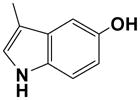
The effect of substitution of the indole ring moiety was tested. A 2″-methyl-5″-methoxy indolyl derivative 31 showed reduced potency compare to 17, especially at the A2BAR. Its N-tosyl derivative 32 lost potency at all ARs. A 5″-methoxy derivative 33 was tolerated at A2BAR, but the affinities against A1, A2A, and A3ARs were significantly reduced. 5″-Hydroxy analogue 34 was more potent than the 5″-methoxy analogue 33 at all ARs. Bulkiness of the substituent at the 5″ position might be related to the reduced affinity.
The effect on the 5″-halo-substitution of the indole moiety was also investigated. The corresponding 5″-halo analogues 22 – 25 were well tolerated by the A2BAR. Of these analogues, the 5″-bromo analogue 24 was equipotent to 17 at the A2BAR, but was 11-fold less potent than 17 in binding to the A2AAR. Compound 24 was roughly equipotent at all four ARs, however its selectivity was improved compared to the parent compound 17. This series of 5″-halo derivatives showed a tendency toward increased Ki values at A1 and A2AARs depending on the bulkiness of halogen atom.
The importance of bromo-substitution of 26 for A2BAR potency prompted us to design the positional isomers of bromo-substituted indole, leading to the 4″, 6″, or 7″-bromo derivatives 28 – 30. Of these bromo analogues, surprisingly, compound 28 surpassed the A2BAR potency of the 5″-bromo analogue 24, the parent compound 17, and even 2. Also, 28 displayed improved selectivity compared to 17 and 2. On the other hand the 5″-chloro analogue 27 showed a decreased potency at A2BAR compared to 28.
Activation curves were determined for 28 in comparison to 2 at the A1, A2A, A2BARs and A3AR (Figure 1). Compound 28 was a partial agonist at A1 and A3ARs and a full agonist at A2A and A2BARs.
Figure 1.

Functional effects of compound 28 on adenylate cyclase in CHO cells stably expressing the human ARs. Compound 28 was a full agonist at the A2A and A2BARs. In the curves shown the EC50 values for 2 were 21.9 (A2A) and 110 (A2B) nM, and for 28 EC50 values of were 39.7 (A2A) and 109 (A2B) nM were obtained. The relative maximal efficacy of 28 at the A1 and A3ARs was 31.8% and 20.2±1.0% of the full agonist 2, respectively.
Molecular Modeling
A recently published rhodopsin-based molecular model of the human A2BAR38 was adapted to study the binding mode of compound 28 after docking and energy optimization using Monte Carlo Multiple Minimum (MCMM) calculations.39 The position of the adenosine moiety of 28 in the A2BAR obtained after MCMM calculations was found to be similar to its initial positions. Furthermore, it was observed that the 2-(6-bromoindol-3-yl)-ethyloxy substituent fits the binding site well (Figure 2). In the resulting model, the oxygen atom of this moiety was found in proximity to the side chain amino group of Asn254 (6.55) and seemed to be involved in H-bonding with this residue. The indole ring occupied a pocket formed by several residues located in TM3 and EL2. In particular, the NH-group of the indole ring was found near the OH-group of Ser165 (EL2). Although, a H-bond between the NH-group of the indole ring and Ser165 was not observed in the model, a formation of this bond seems to be possible due to the rotation of the side chain of Ser165.
Figure 2.

Docking model of compound 28 in the binding site of the human A2BAR, showing residues in proximity (A) and the Van der Waals surface of the receptor (B).
DISCUSSION
The goal was to prepare novel A2BAR agonists having high potency and selectivity. Although the most potent agonist 28 is not truly selective for the A2BAR, it is effectively a mixed A2AAR/A2BAR agonist with minimal ability to activate A1 and A3ARs.
Initially we found a novel lead compound 17, in which adenosine is substituted with a 3-indolylethyloxy functional group at the 2 position as an A2BAR agonist having favorable pharmacological properties. The A2BAR potency (299 nM) of compound was similar to 2 (140 nM). These promising findings encouraged us to optimize the A2BAR activity and selectivity of 17 by derivatization at the indole 2 position and by modification at the ribose 5′ position or the purine 6 position. Generally, substitution of the 2 position of adenosine is not well tolerated by the A2BAR, however, (S)-PHP-Ado 13 and (S)-PHP-NECA 4 were known to show higher A2BAR potencies compared to 2.31 Distinct from the 2-ethynyl substituent of 4, exploration of the SAR of a 2-(3-indolylethyloxy) substituent could provide novel insights to molecular recognition at the A2BAR. We can speculate that the lack of an additive effect on A2B potency of combining the 3-(indolyl)ethyloxy- and 5′-N-ethyluronamido fragments (i.e., 40 in comparison to 2 and 17) may be due to an unfavorable change in the conformation or position of the ribose ring inside the ligand binding site.
First, we focused on the simplification, altered connectivity and mimicking of the indole ring of 17 as shown in the case of compounds 19, 8, 20, 35 and 36. Unfortunately, these approaches failed to maintain the A2BAR potency. Next, we tried to transform the 4′-hydroxymethyl moiety to an ethylcarboxamide, which was expected to favorably increase A2BAR potency. However, the 5′-N-ethyluronamide analogue 40 showed only a 2-fold increased potency at the A3AR compared to 17. In this respect 40 has quite different pharmacological characteristics from (S)-PHPNECA 4. Also, the 6′ modification of 17, as shown in the case of compound 37 and 38, did not improve potency at the A2BAR. Finally, we focused on functionalization of the indole moiety. Even minor modifications were examined because of the lack of a prior pharmacological precedent for the indole ring moiety at this position.
Eventually, through probing a relatively restrictive SAR, we achieved the new A2B agonist 28 that gained an advantage over the parent compounds 17 and 2 for the A2BAR in both potency and selectivity. In addition, compound 28 produced quite a different selectivity from (S)-PHPNECA 4 and 6-guanidino-NECA 3. Compound 4 showed a high potency/affinity at each AR (Table 1).31 Compound 3 displayed a 14 selectivity at A1 and A3ARs (A1: 7.0 nM, A2A: 628 nM, A2B: 54.5 nM, A3: 5.1 nM).32 Compound 28 showed the improved selectivity compared to compounds 2 – 4, providing a novel type of potent A2BAR agonist. Molecular modeling results with 28 docked in the human A2BAR demonstrated that in addition to all interactions proposed for adenosine, the 2-(6-bromoindol-3-yl)-ethyloxy fragment can provide additional favorable interactions of the ligand with a distal region of the putative agonist binding site of the receptor.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
Materials and instrumentation
2-Amino-6-chloropurine-9-riboside, tryptophol, 1-iodo-3-phenylpropane, 5-bromoindole-3-acetic acid, 5-methoxyindole-3-acetic acid and other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO), and 5-fluoroindole-3-acetic acid was purchased from Wako Chemicals USA, Inc. (Richmond, VA). Compound 69 was prepared as reported.34
1H NMR spectra were obtained with a Varian Gemini 300 spectrometer using CDCl3, CD3OD as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ0.00) for CDCl3 and (δ3.30) for CD3OD.
Purity of the nucleosides submitted for biological testing was checked using a Hewlett–Packard 1100 HPLC equipped with a Luna 5μ RP-C18(2) analytical column (250 X 4.6 mm; Agilent Technologies, Santa Clara, CA). System A: linear gradient solvent system: CH3CN/H2O from 20/80 to 40/60 in 20 min; the flow rate was 1 mL/min., System B: linear gradient solvent system: CH3CN/H2O from 20/80 to 60/40 in 20 min; the flow rate was 1 mL/min., System C: linear gradient solvent system: CH3CN/5mM TBAP from 20/80 to 60/40 in 20 min., the flow rate was 1 mL/min., System D: linear gradient solvent system: CH3CN/5mM TBAP from 5/95 to 80/20 in 20 min., the flow rate was 1 mL/min. Peaks were detected by UV absorption with a diode array detector. All derivatives tested for biological activity showed >98% purity in the HPLC systems.
TLC analysis was carried out on aluminum sheets precoated with silica gel F254 (0.2 mm) from Aldrich. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration using polyalanine. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
General tosylation procedure for the synthesis of 3-iodoethylindole derivative (44)~(46), (56)~(58), (71)~(73) and (84)
To a solution of the alcohol in THF (tetrahydrofuran) was added sodium hydride (60 %, 3 eq) at 0 °C, and the reaction mixture was stirred at 0 °C for 1 h. To the suspension was added tosyl chloride (3 eq) at 0 °C, the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate and washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to column chromatography on silica gel. Elution with a mixture of toluene and acetone (40:1) gave the tosylated derivative.
General iodination procedure for the synthesis of compounds 47 – 49, 59 – 61, 74 – 77, and 85
A solution of the tosylate and sodium iodide (3.5 eq) in N, N-dimethylformamide was stirred overnight at 60 °C. The reaction mixture was diluted with ethyl acetate and washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to column chromatography on silica gel. Elution with a mixture of hexanes and ethyl acetate (4:1) gave the iodide.
3-(p-Toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (44)
The yield was 62%: 1H NMR (CDCl3) δ 7.93 (1H, d with small coupling, J = 8.0 Hz), 7.74 (2H, d, J = 8.2 Hz), 7.56 (2H, d, J = 8.5 Hz), 7.12 – 7.34 (7H, m), 4.24 (2H, t, J = 6.6 Hz), 3.01 (2H, t, J = 6.6 Hz), 2.38 (3H, s), 2.32 (3H, s); HRMS (ESI-MS m/z) calcd for C24H23NO5S2Na (M+Na)+ 492.0915, found 492.0914.
5-Methoxy-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (45)
The yield was 56 %. 1H NMR (CDCl3) δ 7.82 (1H, d, J = 9.1 Hz), 7.71 (2H, d with small coupling, J = 8.5 Hz), 7.53 (2H, d with small coupling, J = 8.2 Hz), 7.27 (1H, s), 7.21 (2H, dd, J = 0.6 and 8.8 Hz), 7.15 (2H, dd, J = 0.6 and 8.5 Hz), 6.89 (1H, dd, J = 2.5 and 9.1 Hz), 6.71 (1H, d, J = 2.2 Hz), 4.23 (2H, t, J = 6.6 Hz), 3.77 (3H, s), 2.97 (2H, dt, J = 0.8 and 6.6 Hz), 2.39 (3H, s), 2.32 (3H, s); HRMS (ESI-MS m/z) calcd for C25H26NO6S2 500.1202 (M+H)+, found 500.1207.
1-(p-Toluenesulfonyl)-3-(p-toluenesulfonyloxyethyl)-5-(p-toluenesulfonyloxy) indole (46)
The yield was 57%: 1H NMR (CDCl3) δ 7.80 (1H, d, J = 9.1 Hz), 7.71 (2H, d with small coupling, J = 8.5 Hz), 7.82 (2H, d with small coupling, J = 8.5 Hz), 7.58 (2H, d, with small coupling, J = 8.2 Hz), 7.36 (1H, s), 7.31 (2H, dd, J = 0.6 and 8.5 Hz), 7.25 (2H, d, J = 8.4 Hz), 7.19 (2H, d, J = 8.2 Hz), 6.97 (1H, d, J = 2.2 Hz), 6.84 (1H, dd, J = 2.3 and 8.9 Hz), 4.18 (2H, t, J = 6.5 Hz), 2.90 (2H, t, J = 6.5 Hz), 2.46 (3H, s), 2.40 (3H, s), 2.35 (3H, s); HRMS (ESI-MS m/z) calcd for C31H30NO8S3 (M+H)+ 640.1134 found 640.1099.
3-Iodoethyl-1-(p-toluenesufonyl)indole (47)
The yield was 70%: 1H NMR (CDCl3) δ 7.98 (1H, d with small coupling, J = 8.2 Hz), 7.76 (2H, dt, J = 1.9 and 8.5 Hz), 7.45 (2H, m), 7.32 (1H, ddd, J = 1.3, 7.1, and 8.4 Hz), 7.23 – 7.28 (2H, m), 7.20 (2H, d with small coupling, J = 8.0 Hz), 3.41 (2H, t with small coupling, J = 7.1 Hz), 3.24 (2H, t with small coupling, J = 7.3 Hz), 2.33 (3H, s); HRMS (ESI-MS m/z) calcd for C17H17NO2SI (M+H)+ 426.0025, found 426.0016.
3-Iodoethyl-5-methoxy-1-(p-toluenesulfonyl)indole (48)
The yield was 74 %. 1H NMR (CDCl3) δ 7.74 (1H, d with small coupling, J = 9.1 Hz), 7.73 (2H, d with small coupling, J = 8.2 Hz), 7.40 (1H, s), 7.20 (2H, dd, J = 0.7 and 8.7 Hz), 6.92 (1H, dd, J = 2.5 and 9.2 Hz), 6.85 (1H, d, J = 2.5 Hz), 3.82 (3H, s), 3.40 (2H, dt, J = 0.7 and 7.6 Hz), 3.20 (2H, t, J = 7.3 Hz), 2.33 (3H, s); HRMS (ESI-MS m/z) calcd for C18H19NO3SI (M+H)+ 456.0130, found 456.0135.
3-Iodoethyl-1-(p-toluenesulfonyl)-5-(p-toluenesulfonyloxy) indole (49)
The yield was 83%. 1H NMR (CDCl3) δ 7.85 (1H, d, J = 9.1 Hz), 7.73 (2H, d with small coupling, J = 8.5 Hz), 7.68 (2H, d with small coupling, J = 8.2 Hz), 7.47 (1H, s), 7.30 (2H, d, J = 8.3 Hz), 7.23 (2H, d, J = 8.3 Hz), 7.02 (1H, d, J = 2.5 Hz), 6.91 (1H, dd, J = 2.3 and 8.9 Hz), 3.26 (2H, t with small coupling, J =7.4 Hz), 3.12 (2H, t with small coupling, J = 7.1 Hz), 2.46 (3H, s), 2.36 (3H, s); APCI-MS (m/z) 596.0 (M+H)+.
General procedure for the synthesis of 3-hydroxyethylindole derivatives 53 – 55
Esterification and reduction: To a solution of a 2- and/or 5-substituted-indole-3-acetic acid in methanol was added p-toluenesulfonic acid monohydrate (3 eq), and the reaction mixture was stirred at 60 °C overnight. After neutralization with 1N aqueous NaOH the solvent was evaporated leaving an oily residue, which was dissolved in ethyl acetate. The solution was washed with water, dried over MgSO4 and filtered. The filtrate was evaporated leaving an oily residue, which was subjected to column flush chromatography on silica gel. Elution with a mixture of toluene and acetone (5:1) gave the corresponding ester.
To a solution of the ester in THF was added lithium aluminum hydride (2.8 eq) at 0 °C, and the reaction mixture was stirred at 0 °C for 1 h and at room temperature for 1 h. After addition of ethyl acetate the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was diluted with ethyl acetate and washed with water, dried over MgSO4 and filtered. The filtrate was evaporated leaving an oily residue, which was subjected to column chromatography on silica gel. Elution with a mixture of toluene and acetone (3:1) gave the pure alcohol.
5-Fluoro-tryptophol (53)
Compound 53 was identical to the known compound reported by Mewshaw et al.40
5-Bromo-tryptophol (54)
Compound 54 was identical to the commercially available compound.
3-Hydroxyethyl-5-methoxy-2-methylindole (55)
The yield was 81%: 1H NMR (CDCl3) δ 7.27 (1H, br s), 7.16 (1H, d, J = 8.8 Hz), 6.97 (1H, d, J = 2.2 Hz), 6.78 (1H, dd, J = 2.5 and 8.5 Hz), 3.78 – 3.88 (2H, m overlapped with OCH3), 3.85 (3H, s), 2.94 (2H, t, J = 6.5 Hz), 2.39 (3H, s); HRMS (ESI-MS m/z) calcd for C12H16NO2 206.1181, found 206.1190.
5-Fluolo-1-(p-toluenesulfonyl)-3-(p-toluenesulfonyloxyethyl)indole (56)
The yield was 58 %. 1H NMR (CDCl3) δ 7.87 (1H, dd, J = 4.1 and 9.1 Hz), 7.72 (2H, d with small couplings, J = 8.5 Hz), 7.55 (2H, d with small coupling, J = 8.2 Hz), 7.36 (1H, S), 7.24 (2H, d with small coupling, J = 8.0 Hz), 7.16 (2H, dd, J = 0.7 and 8.7 Hz), 7.00 (1H, dt, J = 2.4 and 9.0 Hz), 6.89 (1H, dd, J = 2.2 and 8.5 Hz), 4.22 (3H, t, J = 6.5 Hz), 2.95 (3H, t, J = 6.5 Hz), 2.39 (3H, s), 2.34 (3H, s); HRMS (ESI-MS m/z) calcd for C24H23NO5S2F (M+H)+ 488.1002, found 488.0995.
5-Bromo-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (57)
The yield was 63%: 1H NMR (CDCl3) δ 7.80 (1H, d with small coupling, J = 9.6 Hz), 7.73 (2H, d with small coupling, J = 8.2 Hz), 7.52 (2H, d with small coupling, J = 8.5 Hz), 7.32–7.39 (3H, m), 7.25 (2H, d, J = 8.0 Hz), 7.13 (2H, d, J = 8.0 Hz), 4.23 (2H, t, J = 6.3 Hz), 2.95 (2H, dt, J = 0.8 and 6.5 Hz), 2.39 (3H, s), 2.34 (3H, s); HRMS (ESI-MS m/z) calcd for C24H22NO5S2BrLi (M+Li)+ 554.0283, found 554.0292.
5-Methoxy-2-methyl-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (58)
The yield was 30%: 1H NMR (CDCl3) δ 8.02 (1H, d, J = 9.1 Hz), 7.58 (2H, d with small coupling, J = 8.2Hz), 7.48 (2H, d with small coupling, J = 8.2 Hz), 7.18 (2H, dd, J = 0.7 and 8.7 Hz), 7.13 (2H, dd, J = 0.6 and 8.5 Hz), 6.84 (1H, dd, J = 2.8 and 9.1 Hz), 6.64 (1H, d, J = 2.5 Hz), 4.11 (2H, t, J = 6.7 Hz), 3.79 (3H, s), 2.90 (2H, t, J = 6.7 Hz), 2.43 (3H, s), 2.39 (3H, s), 2.33 (3H, s); HRMS (ESI-MS m/z) calcd for C26H27NO6S2Na (M+Na)+ 536.1178, found 536.1186.
3-Iodoethyl-5-fluoro-1-(p-toluenesulfonyl)indole (59)
Yield 70 %. 1H NMR (CDCl3) δ 7.92 (1H, ddd, J = 0.6, 4.3 and 9.1 Hz), 7.74 (2H, dt, J = 1.9 and 8.5 Hz), 7.48 (1H, s), 7.22 (2H, d, J = 8.0 Hz), 7.09 (1H, dd, J = 2.5 and 8.5 Hz), 7.04 (1H, dt, J = 2.5 and 9.1 Hz), 3.39 (2H, dt, J = 0.7 and 6.8 Hz), 3.19 (2H, t, J = 7.3 Hz), 2.35 (3H, s); HRMS (ESI-MS m/z) calcd for C17H15NO2FS 316.0808, found 316.0810.
5-Bromo-3-Iodoethyl-1-(p-toluenesufonyl)indole (60)
The yield was 65 %. 1H NMR (CDCl3) δ 7.85 (1H, d, J = 8.8 Hz), 7.74 (2H, d with small coupling, J = 8.2 Hz), 7.57 (1H, d, J = 1.9 Hz), 7.45 (1H, s), 7.41 (1H, dd, J = 1.9 and 8.8 Hz), 7.22 (2H, d, J = 8.2 Hz), 3.88 (2H, t, J = 7.0 Hz), 3.19 (2H, t, J = 7.3 Hz), 2.35 (3H, s); HRMS (ESI-MS m/z) calcd for C17H15NO2SBrI (M+H)+ 502.9025, found 502.9036.
3-Iodoethyl-5-methoxy-2-methyl-1-(p-toluenesufonyl)indole (61)
The yield was 85%: 1H NMR (CDCl3) δ 8.07 (1H, d, J = 9.1 Hz), 7.58 (2H, dt, J = 1.8 and 8.5 Hz), 7.17 (2H, d, J = 8.2 Hz), 6.87 (1H, dd, J = 2.5 and 9.1 Hz), 6.79 (1H, d, J = 2.5 Hz), 3.84 (3H, s), 3.26 (2H, m), 3.14 (2H, m), 2.52 (3H, s), 2.33 (3H, s); HRMS (ESIMS m/z) calcd for C19H21NO3IS (M+H)+ 470.0287, found 470.0294.
General synthetic procedure for 3-hydroxyethylindole derivatives (67) – (70) via Fischer indole ring preparation
A solution of substituted phenylhydrazine hydrochloride and ethoxytetrahydrofuran (1.5 eq) in 95 % ethanol was refluxed overnight. The reaction mixture was filtered through celite. The filtrate was evaporated to give a crude solid. The solid was dissolved in ethyl acetate, the solution was washed with water, dried over MgSO4, and filtered. The filtrate was evaporated to give a crude oil, which was subjected to column chromatography on silica gel. Elution with a mixture of toluene and acetone (2:1) gave the alcohol.
6-Chloro-tryptophol (67)41, 6-Bromo-tryptophol (68)42 and 5-Chloro-tryptophol (69)34 are identical to the known compounds.
5-Iodo-tryptophol (70)
The yield was 32 %. 1H NMR (CDCl3) δ 8.07 (1H, br s), 7.95 (1H, m), 7.45 (1H, dd, J = 1.7 and 8.5 Hz), 7.16 (1H, d, J = 8.5 Hz), 7.06 (1H, d, J = 2.2 Hz), 3.89 (2H, br s), 2.98 (2H, dt, J = 0.8 and 6.3 Hz), 1.45 (1H, br s); APCI-MS (m/z) 288.0 (M+H)+.
6-Chloro-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (71)
Yield 32%. 1H NMR (CDCl3) δ 7.94 (1H, d, J = 1.7 Hz), 7.74 (2H, d with small coupling, J = 8.5 Hz), 7.51 (2H, d with small coupling, J = 8.2 Hz), 7.29 (2H, d, J = 8.5 Hz), 7.25 (1H, s), 7.19 (1H, d, J = 8.2 Hz), 7.10–7.16 (3H, m), 4.27 (2H, t, J = 6.3 Hz), 2.97 (2H, t, J = 6.3 Hz), 2.40 (3H, s), 2.35 (3H, s); APCI-MS (m/z) 504.1 (M+H)+.
6-Bromo-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (72)
The yield was 30 %. 1H NMR (CDCl3) δ 8.10 (1H, d, J = 1.4Hz), 7.74 (2H, d with small coupling, J = 8.2 Hz), 7.51 (2H, d with small coupling, J = 8.2 Hz), 7.23–7.30 (5H, m), 7.13 (2H, dd, J = 1.7 and 8.2 Hz), 4.23 (2H, t, J = 6.3 Hz), 2.97 (2H, t, J = 6.3 Hz), 2.40 (3H, s), 2.35 (3H, s); APCI-MS (m/z) found 548.0 (M+H)+.
5-Chloro-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (73)
The yield was 30 %. 1H NMR (CDCl3) δ 7.85 (1H, dd, J = 0.6 and 8.8 Hz), 7.73 (2H, dt, J = 2.1 and 8.7 Hz), 7.53 (2H, dt, J = 1.8 and 8.5 Hz), 7.36 (1H, s), 7.21 – 7.26 (3H, m), 7.19 (1H, dd, J = 0.6 and 1.9 Hz), 7.14 (2H, dd, J = 0.5 and 8.5 Hz), 4.23 (2H, t, J = 6.5 Hz), 2.95 (2H, dt, J = 0.8 and 6.5 Hz), 2.39 (3H, s), 2.34 (3H, s); HRMS (ESI-MS m/z) calcd for C24H22NO5NaS2Cl 526.0526, found 526.0535.
6-Chloro-3-iodoethyl-1-(p-toluenesulfonyl)indole (74)
The yield was 67 %. 1H NMR (CDCl3) d 8.00 (1H, d, J = 1.7 Hz), 7.76 (2H, d with small coupling, J = 8.5 Hz), 7.43 (1H, s), 7.36 (1H, d, J = 8.5 Hz), 7.25 ~ 7.28 (2H overlapped with CHCl3), 7.22 (1H, dd, J = 1.8 and 8.4 Hz), 3.39 (2H, t with small coupling, J = 7.3 Hz), 3.21 (2H, t, J with small coupling, J = 7.3 Hz), APCI-MS (m/z) 460.0 (M+H)+.
6-Bromo-3-iodoethyl-1-(p-toluenesulfonyl)indole (75)
The yield was 67 %. 1H NMR (CDCl3) δ 8.16 (1H, d, J = 1.7 Hz), 7.76 (2H, dd, J = 1.9 and 8.5 Hz), 7.42 (1H, br s), 7.36 (1H, dd, J = 1.7 and 8.5 Hz), 7.30 (1H, d, J = 8.2 Hz), 7.25 (2H, d, J = 8.5 Hz), 3.38 (2H, t with small coupling, J = 7.4 Hz), 3.21 (2H, t with small coupling, J = 7.3 Hz), 2.36 (3H, s); HRMS (ESI-MS m/z) calcd for C17H15BrINO2S (M)+ 502.9052, found 502.9066.
5-Chloro-3-iodoethyl-1-(p-toluenesulfonyl)indole (76)
The yield was 60 %. 1H NMR (CDCl3) δ 7.90 (1H, dd, J = 0.6 and 8.8 Hz), 7.74 (2H, d with small coupling, J = 8.2 Hz), 7.47 (1H, s), 7.41 (1H, dd, J = 0.6 and 1.9 Hz), 7.24–7.29 (1H overlaped with CHCl3), 7.22 (2H, d, J = 8.3 Hz), 3.39 (2H, dt, J = 0.8 and 7.3 Hz), 3.20 (2H, t, J = 7.3 Hz), 2.35 (3H, s); HRMS (ESI-MS m/z) calcd for C17H16NO2IClS (M+H)+ 459.9635, found 459.9625.
5-Iodo-3-iodoethyl-1-(p-toluenesulfonyl)indole (77)
The yield was 68 %. 1H NMR (CDCl3) δ 7.77 (1H, d, J = 1.6 Hz), 7.74 (1H, d, J = 8.8 Hz), 7.73 (2H, d with small coupling, J = 8.5 Hz), 7.58 (1H, dd, J = 1.7 and 8.5 Hz), 7.41 (1H, s), 7.22 (2H, d, J = 8.2 Hz), 3.83 (2H, t, J = 7.2 Hz), 3.19 (2H, t, J = 7.3 Hz), 2.35 (3H, s); APCI-MS (m/z) 551.9 (M+H)+.
Ethyl 4-bromo-3-indolylglyoxylate (80)
To a solution of 78 (2.94 g, 15.0 mmol) in diethyl ether (60 mL) was added oxalyl chloride (3.01 ml, 34.5 mmol) at 0 °C, and the reaction mixture was stirred at room temperature for 10 h. After evaporation, ethanol (30 mL) was added to the solids, and the solution was stirred at room temperarure overnight. The solvent was evaporated to give a solid. This residue was dissolved in ethyl acetate, washed with water, dried over MgSO4, and filtered. The filtrate was evaporated to give a crude solid, which was subjected to column chromatography on silica gel. Elution with a mixture of hexanes and ethyl acetate (1:1) gave 80 (2.3g, 52 %).
1H NMR (CDCl3) δ 9.55 (1H, br s), 8.24 (1H, d, J = 3.3 Hz), 7.50 (1H, dd, J = 0.8 and 7.7 Hz), 7.42 (1H, dd, J = 0.8 and 8.3 Hz), 7.13 (1H, t, J = 8.0 Hz), 4.41 (2H, q, J = 7.1 Hz), 1.40 (3H, t, J = 7.1 Hz); APCI-MS (m/z) 296.0 (M+H)+.
Ethyl 7-Bromo-3-indolylglyoxylate (81)
Compound 81 was obtained from 79 by the similar procedure for the preparation of 80 (yield 70 %).
1H NMR (CDCl3) δ 8.96 (1H, br s), 8.55 (1H, d, J = 3.3 Hz), 8.39 (1H, dd, J = 0.6 and 8.0 Hz), 7.48 (1H, dd, J = 0.8 and 8.0 Hz), 7.23 (1H, t, J = 7.8 Hz), 4.43 (2H, q, J = 7.1 Hz), 1.44 (3H, t, J = 7.1 Hz), APCI-MS (m/z) 296.0 (M+H)+.
4-Bromo-tryptophol (82)
To a solution of 80 (20 mg, 0.0675 mmol) in THF (1.4 mL) was added lithium aluminum hydride (17.4 mg, 0.459 mmol), and the reaction mixture was refluxed for 2 h. The mixture was diluted with ethyl acetate and washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of hexanes and ethyl acetate (1:1) to give 82 (10 mg, 63 % yield). 1H NMR (CDCl3) δ 8.12 (1H, br s), 7.32 (1H, dd, J = 0.8 and 7.7 Hz), 7.28 (1H, dd, J = 0.8 and 7.7 Hz), 7.14 (1H, d, J = 2.5 Hz), 7.02 (1H, t, J = 7.8 Hz), 3.97 (2H, q, J = 6.1 Hz), 3.29 (2H, dt, J = 0.6 and 6.5 Hz), 1.46 (1H, t, J = 5.6 Hz); HRMS (ESI-MS m/z) calcd for C10H11BrNO (M+H)+ 240.0024, found 240.0028.
7-Bromo-tryptophol (83)
Compound 83 was obtained from 81 by the similar procedure for the preparation of 82 (yield 52 %).
1H NMR (CDCl3) δ 8.82 (1H, br s), 7.57 (1H, d, J = 8.0 Hz), 7.36 (1H, d, J = 8.0 Hz), 7.16 (1H, d, J = 2.2 Hz), 7.02 (1H, t, J = 7.8 Hz), 3.91 (2H, q, J = 6.2 Hz), 3.02 (2H, dt, J = 0.5 and 6.3 Hz), 1.46 (1H, t, J = 6.0 Hz); HRMS (ESI-MS m/z) calcd for C10H11NOBr (M+H)+ 240.0024, found 240.0031.
4-Bromo-3-(p-toluenesulfonyloxyethyl)-1-(p-toluenesulfonyl)indole (84)
The yield was 41 %. 1H NMR (CDCl3) δ 7.91 (1H, dd, J = 0.8 and 8.2 Hz), 7.75 (2H, d with small coupling, J = 8.5 Hz), 7.55 (2H, d with small coupling, J = 8.2 Hz), 7.41 (1H, s), 7.28 (1H, dd, J = 1.0 and 7.8 Hz), 7.25 (2H, d, J = 8.2 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.09 (1H, t, J = 8.0 Hz), 4.32 (2H, t, J = 6.5 Hz), 3.25 (2H, t, J = 6.3 Hz), 2.34 (6H, s); APCI-MS (m/z) 548.0 (M+H)+.
4-Bromo-3-iodoethyl-1-(p-toluenesulfonyl)-indole (85)
The yield was 67 %. 1H NMR (CDCl3) δ 7.96 (1H, dd, J = 0.8 and 8.2 Hz), 7.76 (2H, d with small coupling, J = 8.5 Hz), 7.52 (1H, s), 7.38 (1H, dd, J = 0.8 and 8.0 Hz), 7.23 (2H, dd, J = 0.6 and 8.5 Hz), 7.13 (1H, t, J = 8.1 Hz), 3.45 (4H, s), 2.35 (2H, s); HRMS (ESI-MS m/z) calcd for C17H16NO2IBrS (M+H)+ 509.9130, found 509.9114.
7-Bromo-3-iodoethylindole (86)
The yield was 88 %. 1H NMR (CDCl3) δ 8.21 (1H, br s), 7.52 (1H, dd, J = 0.8 and 8.0 Hz), 7.36 (1H, d, J = 7.7 Hz), 7.16 (1H, d, J = 2.2 Hz), 7.02 (1H, t, J = 7.7 Hz), 3.39–3.46 (2H, m), 3.29–3.37 (2H, m); HRMS (ESI-MS m/z) calcd for C10H10NBrI (M+H)+ 349.9041, found 349.9036.
2-Hydroxyethyl-1-(p-toluenesulfonyl)-indole (88)
To a solution of N-tosyl-2-iodoanilide (1.172g. 3.14 mmol) in DMF were added 3-butyn-1-ol (1.42 ml, 18.8 mmol), copper iodide (119 mg, 0.628 mmol), triethyl amine (13.56 mL, 97.3 mmol) and Pd(PPh3)2Cl2 (220.3 mg, 0.314 mmol), and the reaction mixture was stirred at 70 °C overnight. The reaction mixture was diluted with ethyl acetate. The solution was washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give an oil, which was subjected to column chromatography on silica gel. Elution with a mixture of hexanes and ethyl acetate (1:1) gave 88 (820 mg, 83%). 1H NMR (CDCl3) δ 8.16 (1H, d, J = 8.2 Hz), 7.61 (2H, d with small coupling, J = 8.2 Hz), 7.42 (1H, dd, J = 1.7 and 7.4 Hz), 7.28 (1H, dt, J = 2.2 and 7.6 Hz), 7.23 (1H overlapped with Ph), 7.18 (2H, d, J = 8.5 Hz), 6.50 (1H, s), 4.01 (2H, t, J = 6.1 Hz), 3.29 (2H, t, J = 6.2 Hz), 2.33 (3H, s); APCI-MS (m/z) 316.0 (M+H)+.
2-Iodoethyl-1-(p-toluenesulfonyl)-indole (89)
To a solution of 88 (638 mg, 2.02 mmol) in a mixture of diethylether (24 mL) and acetnitrile (8 mL) were added triphenylphosphine (1.589 g, 6.06 mmol), imidazole (439 mg, 6.46 mmol) and iodine (1.639 g, 6.46 mmol), the reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was diluted with ethyl acetate. The solution was washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give an oil, which was subjected to column chromatography on silica gel. Elution with a mixture of hexanes and ethyl acetate (4:1) gave 89 (847 mg, 98 %).
1H NMR (CDCl3) δ 8.14 (1H, d with small coupling, J = 8.2 Hz), 7.60 (2H, dt, J = 2.0 and 8.6 Hz), 7.45 (1H, dd, J = 1.0 and 6.7 Hz), 7.30 (1H, dt, J = 1.5 and 8.0 Hz), 7.23 (1H, dd, J = 1.2 and 7.6 Hz), 7.18 (2H, d with small coupling, J = 8.0 Hz), 6.50 (1H, s), 3.46 – 3.60 (4H, m), 2.33 (3H, s); HRMS (ESI-MS m/z) calcd for C17H17NO2IS (M+H)+ 426,0025 found 426.0035.
Benzoimidazol-1-yl-ethanol (92)
To a solution of benzoimidazol-1-yl-acetic acid (893 mg, 5.06 mmol) in THF (20 mL) was added lithium aluminum hydride (672 mg, 17.7 mmol) at 0 °C, and the reaction mixture was stirred at room temperature for 5 h. After dilution with ethyl acetate the solution was washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to column chromatography on silica gel. Elution was with a mixture of chloroform and methanol (8:1) to give 92 (620 mg, 76 %).
1H NMR (CDCl3) δ 7.63 (1H, s), 7.38 (1H, dt, J = 1.0 and 8.2 Hz), 7.31 (1H, dt, J = 1.0 and 8.0 Hz), 7.17 (1H, ddd, J = 1.0, 7.1, and 8.1 Hz), 7.06 (1H, ddd, J = 1.1, 7.1, and 8.1 Hz), 4.22 (2H, t, J = 4.9 Hz), 3.99 (2H, t, J = 4.8 Hz); HRMS (ESI-MS m/z) calcd for C9H11N2O (M+H)+ 163.0871, found 163.0880.
Benzotriazol-1-yl-ethanol (93)
Procedure used for the preparation of 93 from 91 was similar to those used for the preparation of 92 from 90; amorphous solid, the yield was 59 %.
1H NMR (CDCl3) δ (1H, d with small coupling, J = 8.5 Hz), 7.61 (1H, d with small coupling, J = 8.2 Hz), 7.50 (1H, ddd, J = 1.1, 7.0 and 8.1 Hz), 7.36 (1H, ddd, J = 1.2, 6.9 and 8.1 Hz), 4.75 (2H, t, J = 5.1 Hz), 4.25 (2H, dd, J = 5.9 and 10.3 Hz), 2.55 (1H, t, J = 6.0 Hz); HRMS (ESI-MS m/z) calcd for C8H10N3O 164.0824 found 164.0812.
Benzoimidazol-1-yl-ethyliodide (94)
To a solution of 92 (22 mg, 0.134 mmol) in a mixture of acetonitrile (0.3 mL) and diethylether (0.9 mL) were added triphenylphosphine (105 mg, 0.402 mmol), imidazole (29 mg, 0.428 mmol), and iodine (108 mg, 0.428 mmol) at 0 °C, the reaction mixture was stirred for 2 h. The mixture was diluted with ethyl acetate, washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of toluene and acetone (2:1) to give 94 (32.3 mg, 88 %).
1H NMR (CDCl3) δ 7.97 (1H, s), 7.80–7.87 (1H, m), 7.28–7.42 (3H, m), 4.58 (2H, t, J = 7.0 Hz), 3.51 (2H, t, J = 7.1 Hz); APCI-MS (m/z) 273.0 (M+H)+.
Benzotriazol-1-yl-ethyliodide (95)
Procedure used for the preparation of 95 from 93 was similar to those used for the preparation of 94 from 92; amorphous solid, the yield was 88 %. 1H NMR (CDCl3) δ 8.09 (1H, dt, J = 1.0 and 8.3 Hz), 7.50 – 7.60 (2H, m), 7.40 (1H, ddd, J = 1.8, 6.3, and 8.1 Hz), 5.02 (2H, t, J = 7.3 Hz), 3.67 (2H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C8H9N3I (M+H)+ 273.9841, found 273.9833.
3-Hydroxyethyl-1-(p-toluenesulfonyl)pyrrole (98)
To a solution of 97 (1.24 g, 4.21 mmol) in THF (20 mL) was added lithium aluminum hydride (340 mg, 6.32 mmol) at 0 °C. The reaction mixture was stirred for 2 h. After addition of ethyl acetate the mixture was stirred for 30 min. The mixture was diluted with ethyl acetate, washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to column chromatography on silica gel. Elution with a mixture of chloroform and methanol (20:1) gave 98 (692 mg, 62 %).
1H NMR δ 7.74 (2H, d, J = 8.4 Hz), 7.28 (2H, d, J = 8.7 Hz), 7.10 (1H, t, J = 2.7 Hz), 6.99 (1H, m), 6.19 (1H, dd, J = 1.5 and 3.3 Hz), 3.74 (2H, t, J = 6.5 Hz), 2.64 (2H, t, J = 6.5 Hz), 2.40 (3H, s); HRMS (ESI-MS m/z) calcd for C13H16NO3S (M+H)+ 266.0851, found 266.0837.
3-Iodoethyl-1-(p-toluenesulfonyl)pyrrole (99)
The procedure used for preparation of 99 from 98 is similar to those used for the preparation of 89 from 88; the yield was 62 %.
1H NMR (CDCl3) δ 7.73 (2H, d, J = 8.2 Hz), 7.29 (2H, d, J = 8.5 Hz), 7.08 (1H, t, J = 2.8 Hz), 6.99 (1H, m), 6.16 (1H, dd, J = 1.6 and 3.3 Hz), 3.24 (2H, t, J = 7.3 Hz), 2.96 (2H, t, J =7.6 Hz), 2.40 (3H, s); HRMS (ESI-MS m/z) calcd for C13H15NO2SI (M+H)+ 375.9868, found 375.9857.
General synthetic procedure for 2-substituted adenosine derivatives
To a solution of 2′, 3′, 5′-triacetyl-6-chloroguanosine in N,N-dimethylformamide were added iodide (1.8 eq) and Cs2CO3 (2.7 eq) at room temperature, and the reaction mixture was stirred overnight. After dilution with ethyl acetate the solution was washed with water twice, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil which was purified by column chromatography or preparative TLC on silica gel. Elution or developing with a mixture of toluene and acetone (4:1) gave the 2-substituted 2′, 3′, 5′-triacetyl-6-chloroadenosine derivative.
A solution of 2-substituted 2′, 3′, 5′-triacetyl-6-chloroadenosine derivative in saturated ammonia ethanol solution was stirred in sealed tube overnight at 110 – 120 °C. The solvent was evaporated to give an oil, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (8:1) to give the 2-substituted adenosine derivative.
In case of deprotection of tosyl group of 2-substituent, tosylated adenosine derivative was treated with KOH (20 eq) in methanol overnight at 70 °C in sealed tube. The reaction mixture was concentrated to a small amount of solution, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (5:1) to give the final product.
6-Chloro-2-(3″-(1″-(p-toluenesulfonyl)indolyl)ethyloxy)-3′, 4′, 5′-triacetyladenosine (102)
The yield was 86 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.98 (1H, d with small coupling, J = 6.6 Hz), 7.76 (2H, d with small coupling, J = 8.2 Hz), 7.61 (1H, d with small coupling, J = 7.1 Hz), 7.53 (1H, s), 7.14 – 7.36 (6H, m), 6.13 (1H, d, J = 4.7 Hz), 5.93 (1H, t, J = 5.1 Hz), 5.65 (1H, t, J = 5.5 Hz), 4.71 (2H, m), 4.39 – 4.49 (2H, m), 4.32 (1H, dd, J = 4.7 and 12.6 Hz), 3.24 (2H, t, J = 7.0 Hz), 2.32 (3H, s), 2.14 (3H, s), 2.09 (3H, s), 2.05 (3H, s); HRMS (ESI-MS m/z) calcd for C33H32N5O10ClSNa (M+Na)+ 748.1456, found 748.1455.
6-Chloro-2-(3″-(5″-methoxy-1″-(p-toluenesulfonyl)indolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (103)
The yield was 72 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.86 (1H, dd, J = 9.1 Hz), 7.73 (2H, d, J = 8.5 Hz), 7.48 (1H, s), 7.20 (2H, d, J = 8.2 Hz), 7.03 (1H, d, J = 2.5 Hz), 6.92 (1H, dd, J = 2.3 and 8.9 Hz), 6.12 (1H, d, J = 4.7 Hz), 5.93 (1H, t, J = 5.1 Hz), 5.66 (1H, t, J = 5.6 Hz), 4.69 (2H, ddd, J = 3.8, 7.4, and 14.3 Hz), 4.40 – 4.49 (2H, m), 4.31 (1H, dd, J = 4.4 and 12.6 Hz), 3.85 (3H, s), 3.19 (2H, t, J = 6.7 Hz), 2.32 (3H, s), 2.14 (3H, s), 2.09 (3H, s), 2.05 (3H, s); HRMS (ESI-MS m/z) calcd for C34H35N5O11SCl (M+H)+ 756.1742, found 756.1735.
6-Chloro-2-(3″-(5″-(p-toluenesulfonyloxy) -1″-(p-toluenesulfonyl)indolyl)ethyloxy)-3′, 4′, 5′-triacetyladenosine (104)
The yield was 51 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.83 (1H, d, J = 8.5 Hz), 7.72 (2H, d with small coupling, J = 8.5 Hz), 7.68 (2H, d with small coupling, J = 8.2 Hz), 7.57 (1H, s), 7.25–7.31 (3H, m), 7.22 (2H, d, J = 8.0 Hz), 6.83 (1H, dd, J = 2.3 and 8.9 Hz), 6.12 (1H, d, J = 4.4 Hz), 5.94 (1H, dd, J = 4.7 and 5.5 Hz), 5.67 (1H, t, J = 5.4 Hz), 4.64 (2H, m), 4.40–4.50 (2H, m), 4.31 (1H, dd, J = 5.1 and 12.8 Hz), 3.13 (2H, t, J = 6.7 Hz), 2.42 (3H, s), 2.34 (3H, s), 2.15 (3H, s), 2.08 (3H, s), 2.03 (3H, s); HRMS (ESI-MS m/z) calcd for C40H39ClN5O13S2 (M+H)+ 896.1674 found 896.1638.
6-Chloro-2-(3″-(5″-fluoro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)-3′, 4′, 5′-triacetyladenosine (105)
The yield was 59 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.91 (1H, dd, J = 4.4 and 9.1 Hz), 7.74 (2H, d with small coupling, J = 8.5 Hz), 7.57 (1H, s), 7.25–7.31 (1H overlapped with CHCl3), 7.22 (2H, d, J = 8.0 Hz), 7.04 (1H, dt, J = 2.6 and 9.0 Hz), 6.11 (1H, d, J = 4.7 Hz), 5.94 (1H, t, J = 4.9 Hz), 5.67 (1H, t, J = 5.5 Hz), 4.69 (2H, m), 4.40–4.50 (2H, m), 4.31 (1H, dd, J = 4.9 and 12.9 Hz), 3.18 (2H, t, J = 6.9 Hz), 2.33 (3H, s), 2.15 (3H, s), 2.09 (3H, s), 2.04 (3H, s); HRMS (ESI-MS m/z) calcd for C32H23N5O10SFCl (M+H)+ 744.1542, found 744.1522.
2-(3″-(5″-Bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)-6-chloro-3′, 4′, 5′-triacetyladenosine (106)
The yield was 47 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.84 (1H, d, J = 8.8 Hz), 7.75 (1H, s), 7.73 (2H, d with small coupling, J = 6.6 Hz), 7.54 (1H, s), 7.40 (1H, dd, J = 1.8 and 8.9 Hz), 7.22 (2H, d, J = 8.5 Hz), 6.13 (1H, d, J = 4.7 Hz), 5.94 (1H, t, J = 4.9 Hz), 5.67 (1H, t, J = 5.4 Hz), 4.69 (2H, t, J = 6.6 Hz), 4.40–4.50 (2H, m), 4.31 (1H, dd, J = 5.2 Hz), 3.19 (2H, t, J = 6.7 Hz), 2.33 (3H,s), 2.15 (3H, s), 2.09 (3H, s), 2.03 (3H, s); HRMS (ESIMS m/z) calcd for C33H32N5O10BSClBr (M+H)+ 804.0742, found 804.0752.
6-Chloro-2-(3″-(5″-methoxy-2″-methyl-1″-(p-toluenesulfonyl)indolyl)ethyloxy)-3′,4′,5′-triacetyladenosine (107)
The yield was 72 %. 1H NMR (CDCl3) δ 8.08 (1H, s), 8.07 (1H, d, J = 9.6 Hz), 7.60 (2H, d with small coupling, J = 8.5 Hz), 7.17 (2H, d, J = 8.0 Hz), 6.98 (1H, d, J = 2.5 Hz), 6.87 (1H, dd, J = 2.8 and 9.1 Hz), 6.12 (1H, d, J = 5.0 Hz), 5.83 (1H, t, J = 5.2 Hz), 4.36 – 4.54 (5H, m), 4.31 (1H, dd, J = 4.1 and 12.4 Hz), 3.86 (3H, s), 3.13 (2H, t, J = 7.8 Hz), 2.61 (3H, s), 2.32 (3H, s), 2.13 (3H,s), 2.06 (3H,s), 2.05 (3H, s); HRMS (ESI-MS m/z) calcd for C35H37N5O11SCl (M+H)+ 770.1899, found 770.1895.
6-Chloro-2-(3″-(6″-chloro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)-3′, 4′, 5′-triacetyladenosine (108)
The yield was 70 %. 1H NMR (CDCl3) d 8.09 (1H, s), 7.99 (1H, d, J = 1.9 Hz), 7.76 (2H, d with small coupling, J = 8.5 Hz), 7.53 (1H, d, J = 8.5 Hz), 7.52 (1H, s), 7.20 – 7.28 (3H, m), 6.11 (1H, d, J = 4.7 Hz), 5.95 (1H, t, J = 4.9 Hz), 5.66 (1H, t, J = 5.5 Hz), 4.68 (2H, m), 4.38–4.50 (2H, m), 4.31 (1H, dd, J = 4.5 and 12.2 Hz), 3.20 (2H, t, J = 6.9 Hz), 2.35 (3H, s), 2.14 (3H, s), 2.09 (3H, s), 2.06 (3H, s); APCI-MS (m/z) 760.1 (M+H)+.
2-(3″-(6″-Bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)-6-chloro-3′, 4′, 5′-triacetyladenosine (109)
The yield was 45 %. 1H NMR (CDCl3) δ 8.15 (1H, d, J = 1.6 Hz), 8.09 (1H, s), 7.76 (2H, d with small coupling, J = 8.2 Hz), 7.50 (1H, s), 7.49 (1H, d, J = 9.1 Hz), 7.38 (1H, dd, J = 1.7 and 8.5 Hz), 7.25 (2H, d, J = 8.2 Hz), 6.11 (1H, d, J = 4.7 Hz), 5.95 (1H, t, J = 4.9 Hz), 5.66 (1H, t, J = 5.5 Hz), 4.68 (2H, m), 4.18–4.50 (2H, m), 4.31 (1H, dd, J = 4.5 and 12.5 Hz), 3.20 (2H, t, J = 7.0 Hz), 2.35 (3H, s), 2.14 (3H, s), 2.09 (3H, s), 2.06 (3H, s); HRMS (ESI-MS m/z) calcd for C33H32N5O10SCl Br(M+H)+ 804.0742, found 804.0760.
6-Chloro-2-(3″-(5″-chloro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (110)
The yield was 46 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.89 (1H, d, J = 9.1 Hz), 7.73 (2H, d with small coupling, J = 8.2 Hz), 7.59 (1H, d, J = 2.2 Hz), 7.24–7.30 (1H, m), 7.22 (2H, d, J = 8.0 Hz), 6.12 (1H, d, J = 4.4 Hz), 5.94 (1H, t, J = 5.1 Hz), 5.67 (1H, t, J = 5.4 Hz), 4.69 (2H, m), 4.40–4.49 (2H, m), 4.31 (1H, dd, J = 4.9 and 13.2 Hz), 3.18 (2H, t, J = 6.7 Hz), 2.33 (3H, s), 2.15 (3H, s), 2.09 (3H, s), 2.04 (3H, s); HRMS (ESI-MS m/z) calcd for C33H31N5O10SCl2Na (M+Na)+ 782.1066, found 782.1071.
6-Chloro-2-(3″-(5″-iodo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (111)
The yield was 45 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.93 (1H, d, J = 1.7 Hz), 7.73 (3H, m), 7.57 (1H, dd, J = 1.8 and 8.7 Hz), 7.50 (1H, s), 7.22 (2H, d, J = 8.0 Hz), 6.13 (1H, d, J = 4.7 Hz), 5.94 (1H, t, J = 5.1 Hz), 5.67 (1H, t, J = 5.4 Hz), 4.68 (2H, t, J = 7.0 Hz), 4.40–4.48 (2H, m), 4.31 (1H, dd, J = 5.1 and 13.3 Hz), 3.18 (2H, t, J = 6.9 Hz), 2.33 (3H, s), 2.15 (3H, s), 2.09 (3H, s), 2.04 (3H, s); HRMS (ESI-MS m/z) calcd for C33H32N5O10SClI (M+H)+ 852.0603, found 852.0566.
6-Chloro-2-(3″-(4″-bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (112)
The yield was 40 %. 1H NMR (CDCl3) δ 8.09 (1H, s), 7.95 (1H, dd, J = 0.8 and 8.2 Hz), 7.75 (2H, d with small coupling, J = 8.5 Hz), 7.59 (1H, s), 7.39 (1H, dd, J = 0.8 and 8.0 Hz), 7.23 (2H, d, J = 8.5 Hz), 7.12 (1H, t, J = 8.1 Hz), 6.13 (1H, d, J = 5.0 Hz), 5.92 (1H, t, J = 5.1 Hz), 5.64 (1H, t, J = 5.4 Hz), 4.75 (2H, m), 4.38–4.50 (2H, m), 4.33 (1H, dd, J = 4.7 and 12.9 Hz), 3.53 (2H, t, J = 7.0 Hz), 2.34 (3H, s), 2.13 (3H, s), 2.09 (3H, s), 2.05 (3H, s); APCI-MS (m/z) 806.1 (M+H)+.
6-Chloro-2-(3″-(7″-bromoindolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (113)
The yield was 10 %. 1H NMR (CDCl3) δ 8.06 (1H, s), 7.66 (1H, d, J = 8.0 Hz), 7.35 (1H, d, J = 7.7 Hz), 7.27 (1H overlapped with CHCl3), 7.03 (1H, t, J = 7.7 Hz), 6.11 (1H, d, J = 5.0 Hz), 5.91 (1H, t, J = 5.2 Hz), 5.64 (1H, t, J = 5.2 Hz), 4.70 (2H, m), 4.37–4.46 (2H, m), 4.32 (1H, dd, J = 5.4 and 13.1 Hz), 3.30 (2H, t, J = 7.1 Hz), 2.13 (3H, s), 2.07 (3H, s), 2.07 (3H, s); APCI-MS (m/z) 672.1 (M+Na)+.
6-Chloro-2-phenypropoxy-3′, 4′, 5′-triacetyladenosine (114)
The yield was 63 %. 1H NMR (CDCl3) δ 8.08 (1H, s), 7.16–7.34 (5H, m), 6.14 (1H, d, J = 4.9 Hz), 5.91 (1H, t, J = 5.4 Hz), 5.63 (1H, t, J = 5.2 Hz), 4.38 – 4.52 (5H, m), 4.31 (1H, dd, J = 3.8 and 11.8 Hz), 2.85 (2H, dd, J = 7.4 and 8.0 Hz), 2.12 – 2.24 (2H, m), 2.15 (3H, s), 2.10 (3H, s), 2.09 (3H, s); HRMS (ESI-MS m/z) calcd for C25H27N4O8ClLi (M+Li)+ 553.1677, found 553.1661.
6-Chloro-2-(2″-(1″-(p-toluenesulfonyl)indolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (115)
The yield was 40 %. 1H NMR (CDCl3) δ 8.16 (1H, d, J = 8.2 Hz), 8.09 (1H, s), 7.63 (2H, d, J = 8.5 Hz), 7.42 (2H, d, J = 7.1 Hz), 7.15 – 7.32 (5H, m), 6.59 (1H, s), 6.16 (1H, d, J = 5.0 Hz), 5.87 (1H, t, J = 5.2 Hz), 5.61 (1H, t, J = 5.2 Hz), 4.83 (2H, m), 4.40 – 4.48 (2H, m), 4.34 (1H, dd, J = 4.9 and 13.2 Hz), 3.58 (2H, t, J = 6.6 Hz), 2.33 (3H, s), 2.12 (3H, s), 2.08 (3H,s), 2.07 (3H,s); HRMS (ESI-MS m/z) calcd for C33H33N5O10ClS (M+H)+ 726.1637, found 726.1640.
6-Chloro-2-(3″-(benzoimidazol-1″-yl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (116)
The yield was 51 %. 1H NMR (CD3OD) δ 8.09 (2H, d, J = 8.0 Hz), 7.79 (1H, dd, J = 1.4 and 7.1 Hz), 7.55 (1H, dd, J = 1.1 and 7.1 Hz), 7.34 (1H, dt, J = 1.4 and 7.4 Hz), 7.28 (1H, dt, J = 1.4 and 7.5 Hz), 6.05 (1H, d, J = 4.7 Hz), 5.92 (1H, t, J = 4.9 Hz), 5.67 (1H, t, J = 5.4 Hz), 4.82 (2H, m), 4.66 (2H, t, J = 5.4 Hz), 4.39 – 4.48 (2H, m), 4.27 (1H, dd, J = 5.2 and 13.2 Hz), 2.16 (3H, s), 2.09 (3H, s), 2.02 (3H, s); HRMS (ESI-MS m/z) calcd for C25H26N6O8Cl (M+H)+ 573.1501, found 573.1503.
6-Chloro-2-(3″-(benzotriazol-1″-yl)ethoxy)- 3′, 4′, 5′-triacetyladenosine (117)
The yield was 53 %. 1H NMR (CDCl3) δ 8.08 (1H, s), 8.03 (1H, dt, J = 0.8 and 8.5 Hz), 7.72 (1H, dt, J = 0.8 and 8.5 Hz), 7.52 (1H, ddd, J = 1.0, 7.1 and 8.1 Hz), 7.36 (1H, ddd, J = 1.1, 7.1 and 8.2 Hz), 6.07 (1H, d, J = 4.4 Hz), 5.89 (1H, dd, J = 4.7 and 5.5 Hz), 5.62 (1H, t, J = 5.4 Hz), 5.11 (2H, m), 5.00 (2H, m), 4.40–4.48 (2H, m), 4.26–4.34 (1H, m), 2.15, 2.10 and 2.05 (each 3H, s); HRMS (ESI-MS m/z) calcd for C24H25N7O8Cl (M+H)+ 574.1453, found 574.1456.
6-Chloro-2-(3″-(1″-p-toluenesulfonyl)pyrrolyl)ethyloxy)- 3′, 4′, 5′-triacetyladenosine (118)
The yield was 61 %. 1H NMR (CDCl3) δ 8.08 (1H, s), 7.73 (1H, d, J = 8.5 Hz), 7.27 (2H, d overlapped with CHCl3), 7.08 (2H, m), 6.28 (1H, dd, J = 1.9 and 3.0 Hz), 6.13 (1H, d, J = 5.0 Hz), 5.91 (1H, t, J = 5.1 Hz), 5.63 (1H, t, J = 5.4 Hz), 4.57 (1H, dd, J = 2.8 and 7.4 Hz), 4.53 (1H, dd, J = 3.0 and 7.7 Hz), 4.38 – 4.45 (2H, m), 4.32 (1H, dd, J = 4.3 and 12.2 Hz), 2.95 (2H, t, J = 7.0 Hz), 2.40(3H, s), 2.15(3H, s), 2.09 (3H, s), 2.07 (3H, s); HRMS (ESI-MS m/z) calcd for C29H31N5O10SCl (M+H)+ 676.1480, found 676.1450.
2-(3″-(5″-Methoxy-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (119)
The yield was 56 %. 1H NMR (CD3OD) δ 8.14 (1H, s), 7.81 (1H, d, J = 9.3 Hz), 7.67 (2H, dt, J =1.9 and 8.5 Hz), 7.52 (1H, s), 7.15 (2H, dd, J = 0.6 and 8.5 Hz), 7.02 (1H, d, J =2.5 Hz), 6.89 (1H, dd, J = 2.8 and 8.8 Hz), 5.89 (1H, d, J = 6.0 Hz), 4.72 (1H, t, J = 5.6 Hz), 4.55 (2H, dt, J = 1.1 and 6.3 Hz), 4.33 (1H, dd, J = 3.3 and 5.0 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.88 (1H, dd, J = 2.7 and 12.4 Hz), 3.78 (3H, s), 3.74 (1H, dd, J =3.2 and 12.5 Hz), 3.11 (2H, t, J = 6.3 Hz), 2.25 (3H, s); HRMS (ESI-MS m/z) calcd for C28H31N6O8S (M+H)+ 611.1924, found 611.1899.
2-(3″-(5″-(p-toluenesulfonyloxy) -1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (120)
The yield was 63 %. 1H NMR (CD3OD) δ 8.15 (1H, s), 7.86 (1H, d, J = 8.8 Hz), 7.71 (2H, d with small coupling, J = 8.5 Hz), 7.64 (1H, s), 7.60 (2H, d with small coupling, J = 8.2 Hz), 7.28 (2H, d, J = 8.5 Hz), 7.21 (2H, d, J = 8.0 Hz), 7.08 (1H, d, J = 2.5 Hz), 6.93 (1H, dd, J = 2.2 and 9.1 Hz), 5.90 (1H, d, J = 6.1 Hz), 4.71 (1H, t, J = 5.6 Hz), 4.42 (2H, t, J = 6.6 Hz), 4.33 (2H, t, J = 6.6 Hz), 4.33 (1H, dd, J = 3.2 and 5.1 Hz), 4.14 (1H, q, J = 3.0 Hz), 3.90 (1H, dd, J = 2.8 and 12.6 Hz), 3.74 (1H, dd, J = 3.2 and 12.5 Hz), 3.00 (2H, t, J = 6.6 Hz), 2.34 (3H, s), 2.29 (3H, s); HRMS (ESI-MS m/z) calcd for C34H35N6O10S2 (M+H)+ 751.1856, found 751.1819.
2-(3″-(5″-Fluoro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (121)
The yield was 55 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 7.91 (1H, dd, J = 4.4 and 9.1 Hz), 7.70 (2H, d with small coupling, J = 8.5 Hz), 7.64 (1H, s), 7.29 (1H, dd, J = 2.5 and 8.8 Hz), 7.16 (2H, d, J = 8.0 Hz), 7.05 (1H, dt, J = 2.5 and 9.1 Hz), 5.89 (1H, d, J = 6.0 Hz), 4.73 (1H, t, J = 5.5 Hz), 4.54 (2H, t, J = 6.2 Hz), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.89 (1H, dd, J = 2.7 and 12.4 Hz), 3.74 (1H, dd, J = 3.2 and 12.5 Hz), 3.10 (2H, t, J = 6.3 Hz), 2.26 (3H, s); HRMS (ESI-MS m/z) calcd for C27H28N6O7SF (M+H)+ 599.1724, found 599.1714.
2-(3″-(6″-Chloro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (122)
The yield was 51 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 7.92 (1H, d, J = 1.7 Hz), 7.72 (2H, d with small coupling, J = 8.2 Hz), 7.61 (1H, s), 7.58 (1H, d, J = 8.5 Hz), 7.25 (1H, dd, J = 1.9 and 8.5 Hz), 7.21 (2H, dd, J = 0.7 and 8.7 Hz), 5.89 (1H, d, J = 6.1 Hz), 4.72 (1H, t, J = 5.5 Hz), 4.55 (2H, m), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 2.9 Hz), 3.88 (1H, dd, J = 2.8 and 12.4 Hz), 3.74 (1H, dd, J = 3.3 and 12.4 Hz), 3.12 (2H, t, J = 6.5 Hz), 2.28 (3H, s); HRMS (ESI-MS m/z) calcd for C27H28N6O7SCl (M+H)+ 615.1429, found 615.1413.
2-(3″-(6″-Bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (123)
The yield was 50 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 8.08 (1H, d, J = 1.7 Hz), 7.71 (2H, dt, J = 2.1 and 8.5 Hz), 7.60 (1H, s), 7.53 (1H, d, J = 8.2 Hz), 7.38 (1H, dd, J = 1.7 and 8.2 Hz), 7.21 (2H, d with small coupling, J = 8.2 Hz), 5.89 (1H, d, J = 6.1 Hz), 4.72 (1H, t, J = 5.5 Hz), 4.54 (2H, m), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.88 (1H, dd, J = 2.7 and 12.4 Hz), 3.74 (1H, dd, J = 3.3 and 12.4 Hz), 3.12 (2H, t, J = 6.5 Hz), 2.28 (3H, s); HRMS (ESI-MS m/z) calcd for C27H28N6O7BrS (M+H)+ 659.0924, found 659.0910.
2-(3″-(5″-Chloro-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (124)
The yield was 62 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 7.90 (1H, d, J = 8.8), 7.71 (2H, d with small coupling, J = 8.8 Hz), 7.64 (1H, s), 7.57 (1H, d, J = 1.9 Hz), 7.27 (1H, dd, J = 1.9 and 8.8 Hz), 7.18 (2H, d, J = 8.2 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.73 (1H, t, J = 5.6 Hz), 4.55 (2H, t, J = 6.5 Hz), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.1 Hz), 3.89 (1H, dd, J = 2.7 and 12.6 Hz), 3.74 (1H, dd, J = 3.2 and 12.5 Hz), 3.11 (2H, t, J = 6.3 Hz), HRMS (ESI-MS m/z) calcd for C27H28N6O7SCl (M+H)+ 615.1429, found 615.1401.
2-(3″-(5″-Iodo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (125)
The yield was 71 %. 1H NMR (CD3OD) δ 8.14 (1H, s), 7.89 (1H, d, J = 1.7 Hz), 7.73 (1H, d, J = 9.1 Hz), 7.71 (2H, d with small coupling, J = 8.5 Hz), 7.58 (1H, s), 7.57 (1H, dd, J = 1.8 and 8.7 Hz), 7.18 (2H, d, J = 8.2 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.73 (1H, t, J = 5.5 Hz), 4.54 (2H, t, J = 6.3 Hz), 4.34 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.89 (1H, dd, J = 2.9 and 12.5 Hz), 3.75 (1H, dd, J = 3.3 and 12.4 Hz), 3.10 (2H, t, J = 6.3 Hz), 3.27 (3H, s); APCI-MS (m/z) 707.0 (M+H)+.
2-(3″-(4″-Bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (126)
The yield was 43 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 7.95 (1H, dd, J = 0.7 and 8.4 Hz), 7.73 (2H, d with small coupling, J = 8.5 Hz), 7.69 (1H, s), 7.39 (1H, dd, J = 0.8 and 7.7 Hz), 7.19 (2H, dd, J = 0.6 and 8.5 Hz), 7.15 (1H, t, J = 8.1 Hz), 5.89 (1H, d, J = 6.1 Hz), 4.74 (1H, t, J = 5.5 Hz), 4.61 (2H, m), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.1 Hz), 3.89 (1H, dd, J = 2.8 and 12.4 Hz), 3.75 (1H, dd, J = 3.3 and 12.4 Hz), 3.44 (2H, m), 2.27 (3H, s); APCI-MS (m/z) 659.1 (M+H)+.
2-(3″-(1″-(p-toluenesulfonyl)pyrrolyl)ethyloxy)-adenosine (127)
The yield was 69 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.72 (2H, d with small coupling, J = 8.5 H), 7.29 (2H, d, J = 8.5Hz), 7.08–7.14 (2H, m), 6.30 (1H, dd, J = 1.7 and 3.2 Hz), 5.88 (1H, d, J = 6.0 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.41 (2H, t, J = 6.7 Hz), 4.32 (1H, dd, J = 3.4 and 5.1 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.86 (1H, dd, J = 2.6 and 12.5 Hz), 3.73 (1H, dd, J = 3.3 and 12.7 Hz), 2.85 (2H, t, J = 6.5 Hz), 2.36 (3H, s); HRMS (ESI-MS m/z) calcd for C23H27N6O7S (M+H)+ 531.1662, found 531.1667.
(2R, 3S, 4S, 5R)-2-(2′-Amino-6′-chloropurin-9′-yl)-5-hydroxymethyl-3, 4-O-isopropylidene-tetrahydrofuran (128)
To a solution of 2-amino-6-chloropurine-9-riboside (100 mg, 0.331 mmol) in N,N-dimethylformamide (2 mL) were added 2,2-dimethoxypropane (0.242 ml, 1.97 mmol) and p-toluenesulfonic acid monohydrate (188 mg, 0.993 mmol), and the reaction mixture was stirred overnight at room temperature. The reaction was diluted with ethyl acetate, washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give an oil, which was subjected to preparative TLC developed with a mixture of toluene and acetone (1:1) to give 128 (56 mg, 50 %):
1H NMR (CDCl3) δ 7.81 (1H, s), 5.79 (1H, d, J = 4.9 Hz), 5.68 (1H, dd, J = 1.4 and 11.3 Hz), 5.14 – 5.24 (3H, m), 5.08 (1H, dd, J = 1.4 and 6.0 Hz), 4.51 (1H, d, J = 1.7 Hz), 3.97 (1H, d with small coupling, J = 12.6 Hz), 3.78 (1H, ddd, J = 1.9, 11.3 and 13.2 Hz), 1.64 (3H, s), 1.38 (3H, s); HRMS (ESI-MS m/z) calcd for C13H17N5O4Cl (M+H)+ 342.0969, found 342.0979.
(2R, 3S, 4S, 5R)- 2-(2′-Amino-6′-chloropurin-9′-yl)-5-carboxy-3, 4-O-isopropylidene-tetrahydrofuran (129)
To a solution of 128 (16.9 mg, 0.0494 mmol) in water (4.5 mL) were added potassium permanganate (70.3 mg, 0.445 mmol) and potassium hydroxide (25 mg, 0.444 mmol), and the reaction mixture was stirred for 1 h. After addition of isopropanol the reaction mixture was filtered. The filtrate was neutralized with 0.1N hydrochloric acid aqueous solution and evaporated to give a crude solid, which was subjected to preparative TLC developed with a mixture of chloroform, methanol and saturated aqueous ammonia (2: 1: 0.3) to give 129 (9 mg, 51 %).
1H NMR (CD3OD) δ 8.29 (1H, s), 6.18 (1H, d, J = 1.2 Hz), 5.52 (1H, dd, J = 1.7 and 6.2 Hz), 5.37 (1H, d, J = 6.0 Hz), 4.59 (1H, d, J = 1.7 Hz), 1.55 (3H, s), 1.39 (3H, S); HRMS (ESI-MS m/z) calcd for C13H13N5O5Cl (M-H)− 354.0605, found 354.0622.
(2R, 3S, 4S, 5R)- 2-(2′-Amino-6′-chloropurin-9′-yl)-5-ethoxycarboxyamide-3, 4-isopropylidene-tetrahydrofuran (130)
To a solution of 129 (11.9 mg, 0.0334 mmol) in N, N-dimethylformamide (0.8 mL) were added ethylamine hydrochloride (8.1 mg, 0.100 mmol), N, N-diisopropylethylamine (0.035 ml, 0.200 mmol), and (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (22.5 mg, 0.0434 mmol), and the reaction mixture was stirred overnight. The mixture was diluted with ethyl acetate, washed with water, dried over MgSO4, and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (10:1) to give 130 (10 mg, 78 %)
1H NMR (CD3OD) δ 8.16(1H, s), 6.27 (1H, s), 5.73 (1H, dd, J = 1.9 and 6.3 Hz), 5.43 (1H, d, J = 6.3 Hz), 4.62 (1H, d, J = 1.7 Hz), 2.91 (1H, dt, J = 6.0 and 13.3 Hz), 2.80 (1H, dt, J = 6.0 and 13.3 Hz), 1.55 (3H, s), 1.40 (3H,s), 0.61 (3H, t, J = 7.3 Hz); HRMS (ESIMS m/z) calcd for C15H20N6O4Cl (M+H)+383.1235, found 383.1229.
(2R, 3S, 4S, 5R)-5-Ethoxycarboxyamide-2-(2′-hydroxy-6′-chloropurin-9′-yl)-3, 4-O-isopropylidene-tetrahydrofuran (131)
To a solution of 130 (10 mg, 0.026 mmol) in a mixture of 2-propanol (0.4 mL) and water (0.4 mL) was added t-butylnitrite (13.3 μl, 0.115 mmol) at 4 °C, and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate, washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (10:1) to give 131 (5 mg, 50 %).
1H NMR (CDCl3) δ 7.99 (1H, s), 6.37 (1H, br t, J = 6.1Hz), 6.11 (1H, d, J = 1.6 Hz), 5.72 (1H, dd, J = 1.7 and 6.1 Hz), 5.36 (1H, dd, J = 1.7 and 6.0 Hz), 4.75 (1H, s), 3.05 (2H, m), 1.61 (3H, s), 1.41 (3H, s), 0.77 (3H, t, J = 7.3 Hz); APCI-MS (m/z) 384.1 (M+H)+.
(2R, 3S, 4S, 5R)-5-Ethylcarboxyamide-2–2′-(3″-(1″-(p-toluenesufonyl) indolyl)ethyloxy)-6′-chloropurin-9′-yl)-3, 4-isopropylidene-tetrahydrofuran (132)
To a solution of 131 (19.4 mg, 0.0505 mmol) in N, N-dimethylformamide (0.8 mL) was added iodide 47 (43 mg, 0.101 mmol) and cesium carbonate (49.3 mg, 0.151 mmol), and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate, washed with water, dried over MgSO4 and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of toluene and acetone (4:1) to give 132 (24 mg, 70 %).
1H NMR (CDCl3) δ 7.99 (1H, s), 7.98 (1H, d, J = 8.2 Hz), 7.77 (2H, d with small coupling, J = 8.5 Hz), 7.61 (1H, d with small coupling, J = 7.7 Hz), 7.56 (1H, s), 7.24- 7.35 (2H, m), 7.21 (2H, d, J = 8.5 Hz), 6.27 (1H, t, J = 6.0 Hz), 6.14 (1H, d, J = 2.2 Hz), 5.52 (1H, dd, J = 1.9 and 6.1 Hz), 5.39 (1H, dd, J = 2.1 and 6.2 Hz), 4.60–4.80 (3H, m), 3.27 (2H, t, J = 6.7 Hz), 2.97 (2H, m), 2.32 (3H, s), 1.61 (3H, s), 1.35 (3H, s), 0.69 (3H, t, J = 7.3Hz); HRMS (ESI-MS m/z) calcd for C32H33N6O7SCl Na (M+Na)+ 703.1718, found 703.1732.
(2R, 3S, 4S, 5R)-5-Ethylcarboxyamide-2-(6′-amino -2′-(3″-(1″-(p-toluenesufonyl) indolyl)ethyloxy)-purin-9′-yl)-3, 4-O-isopropylidene-tetrahydrofuran (133)
A solution of 132 in saturated ammonia ethanol solution was stirred at 120 °C overnight. The solvent was evaporated to give an oil, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (10:1) to give 133 (17 mg, 88 % yield).
1H NMR (CDCl3) δ 7.96 (1H, d, J = 7.7 Hz), 7.77 (2H, d, J = 8.2 Hz), 7.68 (1H, s), 7.54–7.60 (2H, m), 7.30 (1H, dt, J = 1.6 and 7.7 Hz), 7.24 (1H, overlapped with CHCl3), 7.19 (2H, d, J = 8.5 Hz), 6.44 (1H, t, J = 5.9 Hz), 6.07 (1H, d, J = 1.9 Hz), 5.59 (1H, br s), 5.51 (1H, dd, J = 1.9 and 6.0 Hz), 5.43 (1H, dd, J = 1.8 and 6.2 Hz), 4.70 (1H, d, J = 1.9 Hz), 4.61 (2H, m), 3.18 (2H, t, J = 6.7 Hz), 2.96 (2H, m), 2.31 (3H, s), 1.64 (3H, s), 1.31 (3H, s), 0.69 (3H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C32H36N7O7S (M+H)+ 662.2397, found 662.2374.
(2R, 3S, 4S, 5R)-5-Ethylcarboxyamide-2-(6′-amino -2′-(3″-(1″-(p-toluenesufonyl) indolyl)ethyloxy)-purin-9′-yl)-tetrahydrofuran (134)
A solution of 133 (13.4 mg, 0.0202 mmol) in 80 % acetic acid aqueous solution was stirred at 80 °C for 63 h and evaporated to give an oil, which was subjected to preparative TLC developed with a mixture of chloroform and methanol (8:1) to give 134 (9 mg, recovery yield 85 %).
1H NMR (CD3OD) δ 8.09 (1H, s), 7.94 (1H, d, J = 7.7 Hz), 7.69 (2H, d with small coupling, J = 8.5 Hz), 7.56 (1H, d with small coupling, J = 7.7 Hz), 7.52 (1H, s), 7.30 (1H, dt, J = 1.3 and 7.7 Hz), 7.16–7.26 (3H, m), 5.93 (1H, d, J = 7.4 Hz), 4.54–4.76 (2H, m), 4.42 (1H, d, J = 1.9 Hz), 4.31 (1H, dd, J = 1.8 and 4.8 Hz), 3.00–3.18 (4H, m), 2.28 (3H, s), 0.83 (3H, t, J = 7.1 Hz); HRMS (ESI-MS m/z) calcd for C29H32N7O7S (M+H)+ 622.2084, found 622.2095.
2-Phenylpropoxyadenosine (8)
The yield was 66 %. 1H NMR (CD3OD) δ 8.11 (1H, s), 7.10 – 7.28 (5H, m), 5.88 (1H, d, J = 6.1 Hz), 4.72 (1H, t, J = 5.5 Hz), 4.30 – 4.34 (1H overlaped with CH2), 4.29 (2H, t, J = 6.6 Hz), 4.10 (1H, q, J = 3.2 Hz), 3.85 (1H, dd, J = 2.9 and 12.5 Hz), 3.72 (1H, dd, J = 3.3 and 12.4 Hz), 2.78 (2H, t, J = 7.7 Hz), 2.06 (2H, dt, J = 6.4 and 15.3 Hz); HRMS (ESI-MS m/z) calcd for C19H24N5O5(M+H)+ 402.1777, found 402.1771; HPLC (system A) 14.1 min (99%), (system C) 10.7 min (99%).
2-(3″-Indolylethyloxy)adenosine (17)
The yield was 72 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.60 (1H, d with small coupling, J = 7.7 Hz), 7.32 (1H, d with small coupling, J = 7.7 Hz), 7.15 (1H, s), 7.07 (1H, dt, J = 1.2 and 7.5 Hz), 7.01 (1H, dt, J = 1.2 and 7.3 Hz), 5.90 (1H, d, J = 5.5 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.58 (2H, m), 4.31 (1H, dd, J = 3.6 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.85 (1H, dd, J = 2.8 and 12.4 Hz), 3.73 (1H, dd, J = 3.4 and 12.2 Hz), 3.24 (2H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C20H23N6O5 (M+H)+ 427.1730, found 427.1711; HPLC (system A) 11.4 min (99 %), (system C) 9.3 min (99 %).
2-(3″-(1″-(p-Toluenesulfonyl)indolyl)ethyloxy)adenosine (18)
The yield was 60 %. 1H NMR (CD3OD) δ 8.14 (1H, s), 7.93 (1H, d with small coupling, J = 7.4 Hz), 7.70 (2H, d with small coupling, J = 8.2 Hz), 7.59 (2H, m), 7.29 (1H, dt, J = 1.7 and 8.1 Hz), 7.23 (1H, dt, J = 1.5 and 8.1 Hz), 7.17 (2H, d, J = 8.0 Hz), 5.90 (1H, d, J = 6.0 Hz), 4.73 (1H, t, J = 5.6 Hz), 4.57 (2H, m), 4.33 (1H, dd, J = 3.2 and 5.1 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.88 (1H, dd, J = 2.8 and 12.4 Hz), 3.74 (1H, dd, J = 3.3 and 12.4 Hz), 3.14 (2H, t, J = 6.3 Hz); HRMS (ESI-MS m/z) calcd for C27H29N6O7S (M+H)+ 581.1818, found 581.1797; HPLC (system B) 16.5 min (99%), (system C) 16.7 min(99%).
2-(3″-Pyrrolylethyloxy)adenosine (19)
The yield was 57 %. 1H NMR (CD3OD) δ 8.11(1H, s), 6.63(2H, d, J = 2.2 Hz), 6.04 (1H, t, J = 2.1 Hz), 5.88 (1H, d, J = 5.8 Hz), 4.72 (1H, t, J = 5.5 Hz), 4.42 (2H, t, J = 7.4 Hz), 4.31 (1H, dd, J = 3.4 and 5.1 Hz), 4.10 (1H, q, J =3.2 Hz). 3.85 (1H, dd, J = 3.0 and 12.4 Hz), 3.73 (1H, dd, J = 3.6 and 12.4 Hz), 2.91 (2H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C16H21N6O5 (M+H)+ 377.1573, found 377.1577; HPLC (system A) 4.5 min (99%), (system C) 4.7 min (99%).
2-(2″-Indolylethyloxy)adenosine (20)
The yield was 32 %. 1H NMR (CD3OD) δ 8.08 (1H, s), 7.37 (1H, d with small couplings, J = 7.4 Hz), 7.25 (1H, d with small coupling, J = 8.0 Hz), 6.97 (1H, dt, J = 1.4 and 7.1 Hz), 6.88 (1H, dt, J = 1.1 and 7.2 Hz), 6.21 (1H, d, J = 0.8 Hz), 5.86 (1H, d, J = 5.8 Hz), 4.69 (1H, t, J = 5.5 Hz), 4.57 (2H, t, J = 6.6 Hz), 4.30 (1H, dd, J = 3.3 and 5.2 Hz), 4.08 (1H, q, J = 3.3 Hz), 3.83 (1H, dd, J = 2.9 and 12.2 Hz), 3.72 (1H, dd, J = 3.3 and 12.4 Hz), 3.18 (2H, t, J = 6.7 Hz); HRMS (ESI-MS m/z) calcd for C20H23N6O5 (M+H)+ 427.1730, found 427.1735; HPLC (system A) 14.6 min (99 %), (system C) 10.9 min (99 %).
2-(2″-(1″-(p-Toluenesulfonyl)indolyl)ethyloxy)adenosine (21)
The yield was 55 %. 1H NMR (CDCl3) δ 8.10 (1H, d, J = 8.2 Hz), 7.57–7.64 (3H, m), 7.37 (1H, d, J = 7.7 Hz), 7.11–7.26 (4H, m), 6.52 (1H, s), 5.71 (1H, d, J = 6.9 Hz), 5.66 (2H, br s), 5.03 (1H, t, J = 6.9 Hz), 4.62 (1H, m), 4.62 (1H, m), 4.46 (2H, m), 4.27 (1H, s), 3.89 (1H, d, J = 11.5 Hz), 3.74 (1H, d, J = 11.5 Hz), 3.43 (2H, m), 3.19 (1H, br s), 2.30 (3H, s); HRMS (ESI-MS m/z) calcd for C27H29N6O7S (M+H)+ 581.1818, found 581.1802; HPLC (system A) 23.0 min (99%), (system C) 16.7 min (99%).
2-(3″-(5″-Fluoro-indolyl)ethyloxy)adenosine (22)
The yield was 80 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.20 ~ 7.30 (3H, m), 6.83 (1H, dt, J = 2.5 and 9.1 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.72 (1H, t, J = 5.5 Hz), 4.55 (2H, t, J = 7.0 Hz), 4.31 (1H, dd, J = 3.3 and 5.2 Hz), 4.11 (1H, q, J = 3.3 Hz), 3.86 (1H, dd, J = 2.8 and 12.4 Hz), 3.73 (1H, dd, J = 3.6 and 12.4 Hz), 3.16 (2H, t, J = 7.1 Hz); HRMS (ESI-MS m/z) calcd for C20H21N6O5FNa (M+Na)+ 467.1455, found 467.1479; HPLC (system A) 13.1 min (99%), (system C) 10.2 min (99 %).
2-(3″-(6″-Chloro-indolyl)ethyloxy)adenosine (23)
The yield was 54 %. 1H NMR (CD3OD) δ 8.08 (1H, s), 7.52 (1H, d, J = 8.5 Hz), 7.27 (1H, d, J = 1.7 Hz), 7.13 (1H, s), 6.95 (1H, dd, J = 1.9 and 8.5 Hz), 5.86 (1H, d, J = 6.0 Hz), 4.67 (1H, t, J = 5.5 Hz), 4.51 (2H, m), 4.28 (1H, dd, J = 3.4 and 5.1 Hz), 4.07 (1H, q, J = 3.2 Hz), 3.81 (1H, dd, J = 2.8 and 12.4 Hz), 3.69 (1H, dd, J = 3.3 and 12.4 Hz), 3.14 (2H, t, J = 7.0 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Cl (M+H)+ 461.1340, found 461.1339; HPLC (system A) 15.7 min (99 %), (system C) 11.9 min (99 %).
2-(3″-(5″-Bromo-indolyl)ethyloxy)adenosine (24)
The yield was 70 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.70 (1H, d, J = 1.9 Hz), 7.24 (1H, d, J = 8.8 Hz), 7.20 (1H, s), 7.15 (1H, dd, J = 1.8 and 8.7 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.56 (2H, t, J = 7.0 Hz), 4.32 (1H, dd, J = 3.6 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.86 (1H, dd, J = 2.8 and 12.4 Hz), 3.73 (1H, dd, J = 3.6 and 12.4 Hz), 3.17 (2H, t, J = 6.9 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Br (M+H)+ 505.0835, found 505.0822; HPLC (system A) 15.2 min(98 %), (system C) 12.2 min (98%).
2-(3″-(5″-Iodo-indolyl)ethyloxy)adenosine (25)
The yield was 56 %. 1H NMR (CD3OD) δ 8.12 (1H, s) 7.89 (1H, dd, J = 0.6 and 1.7 Hz), 7.32 (1H, dd, J = 1.7 and 8.5 Hz), 7.16 (1H, s), 7.15 (1H, dd, J = 0.6 and 8.5 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.55 (2H, t, J = 7.0 Hz), 4.32 (1H, dd, J = 3.6 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.86 (1H, dd, J = 2.8 and 12.4 Hz), 3.73 (1H, dd, J = 3.3 and 12.4 Hz), 3.16 (2H, t, J = 6.9 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5I (M+H)+ 553.0696, found 553.0681; HPLC (system A) 19.0 min (98 %), (system C) 13.1 min (98 %).
2-(3″-(5″-Bromo-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (26)
The yield was 68 %. 1H NMR (CD3OD) δ 8.14 (1H, s), 7.86 (1H, dd, J = 0.6 and 8.8 Hz), 7.68 – 7.74 (3H, m), 7.63 (1H, s), 7.40 (1H, dd, J = 1.8 and 8.8 Hz), 7.18 (2H, d with small coupling, J = 8.5 Hz), 5.89 (1H, d, J = 6.0 Hz), 4.73 (1H, t, J = 5.6 Hz), 4.55 (2H, t, J = 6.3 Hz), 4.33 (1H, dd, J = 3.3 and 5.2 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.89 (1H, dd, J = 2.9 and 12.5 Hz), 3.74 (1H, dd, J = 3.3 and 12.4 Hz), 3.11 (2H, t, J = 6.2 Hz), 2.27 (3H, s); HRMS (ESI MS m/z) calcd for C27H28N6O7BrS (M+H)+ 659.0924, found 659.0921.
2-(3″-(5″-Chloro-indolyl)ethyloxy)adenosine (27)
The yield was 54 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.55 (1H, d, J = 1.9 Hz), 7.28 (1H, dd, J = 0.5 and 8.5 Hz), 7.22 (1H, s), 7.03 (1H, dd, J = 1.9 and 8.5 Hz), 5.89 (1H, d, J = 6.0 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.56 (2H, t, J = 7.0 Hz), 4.31 (1H, dd, J = 3.6 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.86 (1H, dd, J = 2.9 and 12.5 Hz), 3.73 (1H, dd, J = 3.3 and 12.4 Hz), 3.17 (2H, t, J = 7.0 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Cl (M+H)+ 461.1340, found 461.1332; HPLC (system A) 16.6 min (99 %), (system C) 11.8 min (99 %).
2-(3″-(6″-Bromo-indolyl)ethyloxy)adenosine (28)
The yield was 71 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.52 (1H, d, J = 8.5 Hz), 7.48 (1H, d, J = 1.7 Hz), 7.17 (1H, s), 7.12 (1H, dd, J = 1.7 and 8.5 Hz), 5.90 (1H, d, J = 5.8 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.56 (2H, m), 4.31 (1H, dd, J = 3.3 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.85 (1H, dd, J = 3.4 and 12.2 Hz), 3.73 (1H, dd, J =3.4 and 12.2 Hz), 3.21 (2H, t, J = 7.2 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Br (M+H)+ 505.0835 found 505.0840; HPLC (system A) 18.1 min (98 %), (system C) 12.6 min (98 %).
2-(3″-(4″-Bromo-indolyl)ethyloxy)adenosine (29)
The yield was 44 %. 1H NMR (CD3OD) δ 8.11 (1H, s), 7.31 (1H, dd, J = 0.8 and 8.0 Hz), 7.24 (1H, s), 7.16 (1H, dd, J = 0.8 and 7.4 Hz), 6.93 (1H, t, J = 7.8 Hz), 5.89 (1H, d, J = 6.0 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.60 (2H, m), 4.31 (1H, dd, J = 3.6 and 5.0 Hz), 4.10 (1H, q, J = 3.2 Hz), 3.85 (1H, dd, J = 2.9 and 12.5 Hz), 3.73 (1H, dd, J = 3.4 and 12.5 Hz), 3.47 (1H, t, J = 7.1 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Br (M+H)+ 505.0835, found 505.0843; HPLC (system A) 15.3 min (99 %), (system C) 11.7 min (99 %).
2-(3″-(7″-Bromo-indolyl)ethyloxy)adenosine (30)
The yield was 27 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.60 (1H, d, J = 8.0 Hz), 7.25 (1H, d, J = 7.4 Hz), 7.24 (1H, s), 6.95 (1H, t, J = 7.8 Hz), 5.90 (1H, d, J = 5.8 Hz), 4.71 (1H, t, J = 5.6 Hz), 4.58 (2H, m), 4.31 (1H, dd, J = 3.6 and 4.9 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.85 (1H, dd, J = 2.9 and 12.2 Hz), 3.73 (1H, dd, J = 3.3 and 12.4 Hz), 3.20 (2H, t, J = 6.9 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O5Br (M+H)+ 505.0835, found 505.0837; HPLC (system A) 15.9 min (99 %), (system C) 12.1 min (99 %).
2-(3″-(5″-Methoxy-2″-methylindolyl)ethyloxy)adenosine (31)
The yield was 29 %. 1H NMR (CD3OD) δ 8.13 (1H, s), 7.10 (1H, dd, J = 0.6 and 8.8 Hz), 6.96 (1H, d, J = 2.2 Hz), 6.65 (1H, dd, J = 2.3 and 8.7 Hz), 5.90 (1H, d, J = 5.8 Hz), 4.68 (1H, t, J = 5.5 Hz), 4.47 (2H, m), 4.30 (1H, dd, J = 3.6 and 5.2 Hz), 4.10 (1H, q, J = 3.2 and 6.5 Hz), 3.84 (1H, dd, J = 2.9 and 12.2 Hz), 3.72 (1H, dd, J = 3.3 and 12.4 Hz), 3.13 (2H, t, J = 7.3 Hz), 2.37 (3H, s); HRMS (ESI-MS m/z) calcd for C22H26N6O8Na (M+Na)+ 493.1812, found 493.1792; HPLC (system A) 11.8 min (98%), (system C) 9.4 min (98 %).
2-(3″-(5″–Methoxy-2″-methyl-1″-(p-toluenesulfonyl)indolyl)ethyloxy)adenosine (32)
The yield was 71 %. 1H NMR (CD3OD) δ 8.12 (1H,s), 7.96 (1H, d, J = 9.1 Hz), 7.52 (2H, d, J = 8.5 Hz), 7.11 (2H, d, J = 8.5 Hz), 6.92 (1H, d, J = 2.5 Hz), 6.82 (1H, dd, J = 2.5 and 9.1 Hz), 5.87 (1H, d, J = 6.0 Hz), 4.66 (1H, t, J = 5.6 Hz), 4.41 (2H, m), 4.29 (1H, dd, J = 3.2 and 5.1 Hz), 4.10 (1H, q, J =2.9 Hz), 3.82 (1H, dd, J = 2.8 and 12.4 Hz), 3.77 (3H, s), 3.70 (1H, dd, J = 3.2 and 12.5 Hz), 3.06 (2H, t, J = 6.6 Hz), 2.56 (3H, s), 2.21 (3H, s); HRMS (ESI-MS m/z) calcd for C29H33N6O8S (M+H)+ 625.2081, found 625.2079; HPLC (system B) 17.3 min (99 %), (system C) 17.6 min (99 %).
2-(3″-(5″-Methoxy-indolyl)ethyloxy)adenosine (33)
The yield was 54 %. 1H NMR (CD3OD) δ 8.12 (1H, s), 7.20 (1H, d, J = 8.8 Hz), 7.12 (1H, s), 7.05 (1H, d, J = 2.5 Hz), 6.73 (1H, dd, J = 2.5 and 8.8 Hz), 5.89 (1H, d, J = 5.8 Hz), 4.71 (1H, t, J = 5.5 Hz), 4.56 (2H, t, J =7.1 Hz), 4.31 (1H, dd, J =3.3 and 5.2 Hz), 4.10 (1H, q, J = 3.2 Hz), 3.84 (1H, dd, J = 2.8 and 12.4 Hz), 3.72 (1H, dd, J = 3.3 and 12.4 Hz), 3.18 (2H, t, J = 7.1 Hz), HRMS (ESI-MS m/z) calcd for C21H25N6O6 (M+H)+ 457.1836, found 457.1815; HPLC (system A) 10.7 min (98 %), (system C) 8.8 min (98%).
2-(3″-(5″-Hydroxyindolyl)ethyloxy)adenosine (34)
The yield was 31 %; 1H NMR (CD3OD) δ 8.12 (1H, s), 7.15 (1H, dd, J = 0.6 and 8.8 Hz), 7.09 (1H, s), 7.01 (1H, dd, J = 0.5 and 2.5 Hz), 6.65 (1H, dd, J = 2.3 and 8.4 Hz), 5.90 (1H, d, J = 5.8 Hz), 4.72 (1H, t, J = 5.6 Hz), 4.54 (2H, t, J = 7.4 Hz), 4.31 (1H, dd, J = 3.3 and 5.2 Hz), 4.11 (1H, q, J = 3.2 Hz), 3.86 (1H, dd, J = 2.7 and 12.4 Hz), 3.74 (1H, dd, J = 3.3 and 12.4 Hz), 3.13 (2H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C20H22N6O6Na (M+Na)+ 465.1499, found 465.1471; HPLC (system A) 5.5 min (98%), (system C) 5.3 min (98 %).
2-(3″-(Benzoimidazole-1″-yl)ethyloxy)adenosine (35)
The yield was 57 %. 1H NMR (CD3OD) δ 8.22 (1H, s), 8.10 (1H, s), 7.62 (2H, d with small coupling, J = 8.2 Hz), 7.31 (1H, dt, J = 1.2 and 7.6 Hz), 7.24 (1H, dt, J = 1.3 and 7.6 Hz), 5.84 (1H, d, J = 5.8 Hz), 4.63 – 4.74 (5H, m), 4.31 (1H, dd, J = 3.2 and 5.1 Hz), 4.11 (1H, q, J = 3.0 Hz), 3.86 (1H, dd, J = 2.8 and 12.4 Hz), 3.73 (1H, dd, J = 3.0 and 12.4 Hz); HRMS (ESI-MS m/z) calcd for C19H22N7O5 (M+H)+ 428.1682, found 428.1691; HPLC (system A) 4.9 min (99%), (system D) 8.3 min (99 %).
2-(3″-(Benzotriazole-1″-yl)ethyloxy)adenosine (36)
The yield was 40 %. 1H NMR (CD3OD) δ 8.10 (1H, s), 7.93(1H, dt, J =1.0 and 7.4 Hz), 7.80 (1H, dt, J =0.8 and 8.2 Hz), 7.52 (1H, ddd, J = 1.0, 7.1 and 8.1 Hz), 7.38 (1H, ddd, J =1.0, 7.1 and 8.1 Hz), 5.83 (1H, d, J = 5.8 Hz), 5.13 (2H, m), 4.82–4.92 (2H, m, overlapped with HDO), 4.64 (1H, t, J = 5.6 Hz), 4.31 (1H, dd, J = 3.3 and 5.2 Hz), 4.10 (1H, q, J = 3.1 Hz), 3.83 (1H, dd, J = 2.8 and 12.6 Hz), 3.72 (1H, dd, J = 3.3 and 12.4 Hz); HRMS (ESI- MS m/z) calcd for C18H21N8O5 (M+H)+ 429.1635, found 429.1642; HPLC (system A) 4.7 min (99 %), (system C) 4.8 min (99 %).
6-Guanidino-2-(3″-indolylethyloxy)adenosine (37) and 6-Guanidino-2-(3″-(1″-(p-Toluenesulfonyl)indolyl)ethyloxy)adenosine (38)
To a solution of guanidine hydrochloride (98 mg, 1.02 mmol) in acetonitrile (2.2 mL) and N, N-dimethylformamide (1.1 mL) was added sodium hydride (60 %) (41.2 mg, 1.02 mmol) at room temperature, the reaction mixture was stirred overnight. This guanidine solution was added to a mixture of compound 102 (46 mg, 0.0633 mmol) and 1,4- diazabicyclo[2.2.2]octane (14 mg, 0.0126 mmol), and the resulting mixture was stirred overnight at 110 °C and filtered. The filtrate was evaporated to give a crude oil, which was subjected to preparative TLC developed with a mixture of chloroform, methanol and saturated aqueous ammonia (2:1:0.3) to give 37 (2.3 mg, 8 %) and 38 (9 mg, 23 %) as an amorphous solid:
37
1H NMR (CD3OD) δ 8.09 (1H, s), 7.60 (1H, d with small coupling, J = 7.7 Hz), 7.32 (1H, d with small coupling, J = 7.2 Hz), 7.16 (1H, s), 7.08 (1H, dt, J = 1.3 and 7.6 Hz), 7.01 (1H, dt, J = 1.2 and 7.3 Hz), 5.90 (1H, d, J = 6.0 Hz), 4.73 (1H, t, J = 5.5 Hz), 4.54 (2H, t, J = 7.1 Hz), 4.32 (1H, dd, J = 3.3 and 4.9 Hz), 4.12 (1H, q, J = 3.0 Hz), 3.87 (1H, dd, J = 2.8 and 12.6 Hz), 3.73 (1H, dd, J = 3.3 and 12.4 Hz), 3.24 (2H, t, J = 7.4 Hz); HRMS (ESI-MS m/z) calcd for C21H25N8O5 (M+H)+ 469.1948, found 469.1952; HPLC (system B) 8.9 min (97 %), (system D) 7.5 min (97 %).
38
1H NMR (CD3OD) δ 8.25 (1H, s), 7.93 (1H, dd, J = 1.4 and 7.4 Hz), 7.71 (2H, d with small coupling, J = 8.5 Hz), 7.61 (1H, dd, J = 1.4 and 7.1 Hz), 7.59 (1H, s), 7.30 (1H, dt, J = 1.2 and 7.6 Hz), 7.24 (1H, dt, J = 1.2 and 7.6 Hz), 7.18 (2H, d with small coupling, J = 8.5 Hz), 5.96 (1H, d, J = 6.0 Hz), 4.74 (1H, t, J = 5.5 Hz), 4.59 (2H, t, J = 6.5 Hz), 4.35 (1H, dd, J = 3.4 and 5.1 Hz), 4.13 (1H, q, J = 3.2 Hz), 3.89 (1H, dd, J = 2.9 and 12.5 Hz), 3.76 (1H, dd, J = 3.6 and 12.4 Hz), 3.18 (2H, t, J = 6.5 Hz), 3.26 (3H, s); HRMS (ESIMS m/z) calcd for C28H31N8O7S (M+H)+ 623.2036, found 623.2045; HPLC (system A) 16.3 min (97 %), (system C) 8.7 min (97 %).
6-Ethylamino-2-(3″-indolyl)ethyloxy)adenosine (39)
To a solution of 102 (28.3 mg, 0.0389 mmol) in DMF (1.4 mL) in sealed tube were added ethylamine hydrochloride (63.5 mg, 0.779 mmol) and N, N-diisopropylethylamine (0.271 mL), and the reaction mixture was stirred at 140 °C overnight and evaporated to give an oil. The oil was dissolved in methanol (1 mL), KOH (14.6 mg, 0.261 mmol) was added, and the reaction mixture was stirred for 42 h at 80 °C. The solvent was evaporated to give a crude solid which was subjected to preparative TLC developed with a mixture of chloroform and methanol (5:1) to give 39 (2.2 mg, 28 % yield in two steps).
1H NMR (CD3OD) δ 8.05 (1H, s), 7.60 (1H, d with small coupling, J = 7.1 Hz), 7.32 (1H, d with small coupling, J = 7.7 Hz), 7.14 (1H, s), 7.07 (1H, dt, J = 1.3 and 7.6 Hz), 7.00 (1H, dt, J = 1.1 and 7.4 Hz), 5.87 (1H, d, J = 6.0 Hz), 4.71 (1H, t, J = 5.6 Hz), 4.60 (2H, t, J = 7.0 Hz), 4.30 (1H, dd, J = 3.2 and 5.1 Hz), 4.11 (1H, q, J = 3.0 Hz), 3.86 (1H, dd, J = 2.6 and 12.5 Hz), 3.73 (1H, dd, J = 3.2 and 12.5 Hz), 3.50–3.66 (2H, br m), 3.22 (2H, t, J = 7.1 Hz), 1.26 (3H, t, J = 7.3 Hz); HRMS (ESI-MS m/z) calcd for C22H27N6O5 (M+H)+ 455.2043, found 455.2063; HPLC (system A) 19.5 min (99 %), (system C) 13.5 min (99 %).
(2R, 3S, 4S, 5R)-6-Amino-5-Ethylcarboxyamide-2-(3″-indolyl)ethyloxy)-purin-9-yl)-tetrahydrofuran (40)
Potassium hydroxide (12.6 mg, 0.025 mmol) was added to a solution of 134 (7.0 mg, 0.0112 mmol) in methanol (1.5 mL), and the reaction mixture was stirred at 70 °C overnight. The mixture was evaporated to a small amount of solution which was subjected to preparative TLC developed with a mixture of chloroform and methanol (5:1) to give 40 (1.7 mg, 33% yield).
1H NMR (CD3OD) δ 8.06 (1H, s), 7.56 (1H, d with small coupling, J = 8.0 Hz), 7.32 (1H, d with small coupling, J = 8.0 Hz), 7.11 (1H, s), 7.08 (1H, dt, J = 1.2 and 8.1 Hz), 7.00 (1H, dt, J = 1.1 and 8.1 Hz), 5.91 (1H, d, J = 7.4 Hz), 4.74 (1H, dd, J = 4.8 and 7.3 Hz), 4.61 (2H, m), 4.41 (1H, d, J = 1.9 Hz), 4.30 (1H, dd, J = 1.7 and 4.9 Hz), 3.15–3.26 (4H, m), 0.99 (3H, t, J = 7.2 Hz); HRMS (ESI MS m/z) calcd for C22H26N7O5(M+H)+ 53 468.1995, found 468.2015; HPLC (system A) 15.7 min (98 %), (system C) 11.4 min (98 %).
Pharmacological Methods
[125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (I-AB-MECA; 2000 Ci/mmol), [3H]CCPA (2-chloro-N6-cyclopentyladenosine, 42.6 Ci/mmol), [3H]CGS21680 (2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamido-adenosine, 47 Ci/mmol) and [3H]cyclic AMP (40 Ci/mmol) were from Amersham Pharmacia Biotech (Buckinghamshire, UK).
Cell culture and membrane preparation
CHO (Chinese hamster ovary) cells expressing the recombinant human ARs26 were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 μmol/mL glutamine and 800 μg/mL geneticin. Cells were harvested by trypsinization. After homogenization and suspension, cells were centrifuged at 500 g for 10 min, and the pellet was re-suspended in 50 mM Tris·HCl buffer (pH 8.0) containing 10 mM MgCl2, 1 mM EDTA and 0.1 mg/mL CHAPS. The suspension was homogenized with an electric homogenizer for 10 sec, and was then re-centrifuged at 20,000 g for 20 min at 4°C. The resultant pellets were resuspended in buffer in the presence of 3 Units/ml adenosine deaminase, and the suspension was stored at −80°C until the binding experiments. The protein concentration was measured using the Bradford assay.43
Binding assay
Human A1 and A2A Receptors
For binding to human A1 receptors, [3H]CCPA (1 nM) was incubated with membranes (40 μg/tube) from CHO cells stably expressing human A1 receptors at 25°C for 60 min in 50 mM Tris·HCl buffer (pH 7.4; MgCl2, 10 mM) in a total assay volume of 200 μL. Nonspecific binding was determined using 10 μM of CPA. For human A2A receptor binding, membranes (20 μg/tube) from HEK-293 cells stably expressing human A2A receptors were incubated with 15 nM [3H]CGS21680 at 25°C for 60 min in 200 μl 50 mM Tris·HCl, pH 7.4, containing 10 mM MgCl2. NECA (10 μM) was used to define nonspecific binding. Reaction was terminated by filtration with GF/B filters.
Human A3 Receptor
For competitive binding assay, each tube contained 100 μL membrane suspension (20 μg protein), 50 μL of [125I]I-AB-MECA (0.5 nM), and 50 μL of increasing concentrations of the nucleoside derivative in Tris·HCl buffer (50 mM, pH 7.4) containing 10 mM MgCl2, 1 mM EDTA. Nonspecific binding was determined using 10 μM of Cl-IB-MECA in the buffer. The mixtures were incubated at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburgh, MD, USA). Filters were washed three times with 9 mL ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter.
Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.44, 45 CHO cells that expressed recombinant human A3ARs were harvested by trypsinization. After centrifugation and resuspension in medium, cells were plated in 24-well plates in 1.0 ml medium. After 24 hr, the medium was removed and cells were washed three times with 1 ml DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min forskolin (10 μM) was added to the medium, and incubation was continued an additional 15 min. The reaction was terminated upon removal of the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1 M HCl or 50 μL of cyclic AMP solution (0–16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
Statistical analysis
Binding and functional parameters were calculated using Prism 4.0 software (GraphPAD, San Diego, CA, USA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation.46 Data were expressed as mean ± standard error.
Molecular modeling
A published molecular model of the human A2BAR in which (S)-PHPNECA was docked38 was utilized to study the binding mode of 28. Toward this goal, the ribose and adenine moieties of 28 were superimposed upon the corresponding moieties of (S)-PHPNECA located inside the binding site. Then (S)-PHPNECA was removed from the receptor. The obtained complex of the A2BAR with 28 was subjected to Monte Carlo Multiple Minimum (MCMM) calculations using MacroModel software.38 The MCMM calculations were performed for 28 and all residues located within 5Å from the ligand, using a shell of residues located within 2Å. The following parameters were used: MMFFs force field, water was used as an implicit solvent, a maximum of 1000 iterations of the Polak-Ribier Conjugate Gradient (PRCG) minimization method was used with a convergence threshold of 0.05 kJ·mol−1·Å−1, the number of conformational search steps = 100, the energy window for saving structures = 1000 kJ·mol−1.
Acknowledgments
We thank Dr. John W. Daly, Dr. Peiran Chen, Dr. Hea Ok Kim, and Dan Margulies (NIDDK) for helpful discussions and Dr. John Lloyd (NIDDK) for mass spectral determinations. We are grateful to the Cystic Fibrosis Foundation and Microbial Chemistry Research Foundation for financial support provided to H. Adachi.
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YC, Ji X-d, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J Med Chem. 2000;43:1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Webb TR, Lvovskiy D, Kim SA, Ji X-d, Melman N, Linden J, Jacobson KA. Quinazolines as adenosine receptor antagonists: SAR and selectivity for A2B receptors. Bioorg Med Chem. 2003;11:77–85. doi: 10.1016/s0968-0896(02)00323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harada H, Asano O, Hoshino Y, Yoshikawa S, Matsukura M, Kabasawa Y, Niijima J, Kotake Y, Watanabe N, Kawata T, Inoue T, Horizoe T, Yasuda N, Minami H, Nagata K, Murakami M, Nagaoka J, Kobayashi S, Tanaka I, Abe S. 2-Alkynyl-8-aryl-9-methyladenines as novel adenosine receptor antagonists: Their synthesis and structure-activity relationships toward hepatic glucose production induced via agonism of the A2B receptor. J Med Chem. 2001;44:170–179. doi: 10.1021/jm990499b. [DOI] [PubMed] [Google Scholar]
- 6.Hayallah AM, Sandoval-Ramirez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE. 1,8-Disubstituted xanthine derivatives: Synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem. 2002;45:1500–1510. doi: 10.1021/jm011049y. [DOI] [PubMed] [Google Scholar]
- 7.Elzein E, Kalla R, Li X, Perry T, Parkhill E, Palle V, Varkhedkar V, Gimbel A, Zeng D, Lustig D, Leung K, Zablocki J. Novel 1,3-dipropyl-8-(1-heteroarylmethyl-1H-pyrazol-4-yl)-xanthine derivatives as high affinity and selective A2B receptor antagonists. Bioorg Med Chem Lett. 2006;16:302–306. doi: 10.1016/j.bmcl.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Varani K, Gessi S, Merighi S, Vincenzi F, Cattabriga E, Benini A, Klotz KN, Baraldi PG, Tabrizi AM, Lennan SM, Leung E, Borea PA. Pharmacological characterization of novel adenosine ligands in recombinant and native human A2B receptors. Biochem Pharmacol. 2005;70:1601–1612. doi: 10.1016/j.bcp.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Feoktistov I, Ryzhov S, Zhong H, Goldstein AE, Matafonov A, Zeng D, Biaggioni I. Hypoxia modulates adenosine receptors in human endothelial and smooth muscle cells toward an A2B angiogenic phenotype. Hypertension. 2004;44:649–654. doi: 10.1161/01.HYP.0000144800.21037.a5. [DOI] [PubMed] [Google Scholar]
- 10.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in post-hypoxic endothelium: role of ectonucleotidases and adenosine A2B-receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Németh ZH, Lutz CS, Csóka B, Deitch EA, Leibovich SJ, Gause WC, Tone M, Pacher P, Vizi ES, Haskó G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Kolachala V, Walia B, Balasubramanian S, Hall RA, Merlin D, Sitaraman SV. Agonist-induced polarized trafficking and surface expression of the adenosine 2b receptor in intestinal epithelial cells: role of SNARE proteins. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1100–1107. doi: 10.1152/ajpgi.00164.2004. [DOI] [PubMed] [Google Scholar]
- 13.Kolachala V, Asamoah V, Wang L, Srinivasan S, Merlin D, Sitaraman SV. Interferon-gamma down-regulates adenosine 2b receptor-mediated signaling and short circuit current in the intestinal epithelia by inhibiting the expression of adenylate cyclase. J Biol Chem. 2005;280:4048–4057. doi: 10.1074/jbc.M409577200. [DOI] [PubMed] [Google Scholar]
- 14.Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S, Boucher RC. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J Biol Chem. 2004;279:36855–36864. doi: 10.1074/jbc.M405367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auchampach JA, Jin J, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptors and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol. 1997;52:846–860. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 16.Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I. Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B and A3 adenosine receptors. Circ Res. 2003;92:485–492. doi: 10.1161/01.RES.0000061572.10929.2D. [DOI] [PubMed] [Google Scholar]
- 17.Yan L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerging Drug. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 18.Clancy JP, Ruiz FE, Sorscher EJ. Adenosine and its nucleotides activate wild-type and R117H CFTR through an A2B receptor-coupled pathway. Am J Physiol. 1999;276:C361–C369. doi: 10.1152/ajpcell.1999.276.2.C361. [DOI] [PubMed] [Google Scholar]
- 19.Dubey RK, Gillespie DG, Mi Z, Jackson EK. Adenosine inhibits PDGF-induced growth of human glomerular mesangial cells via A2B receptors. Hypertension. 2005;46:628–634. doi: 10.1161/01.HYP.0000178464.63393.88. [DOI] [PubMed] [Google Scholar]
- 20.Mohsenin A, Blackburn MR. Adenosine signaling in asthma and chronic obstructive pulmonary disease. Current Opin in Pulmonary Med. 2006;12:54–59. doi: 10.1097/01.mcp.0000199002.46038.cb. [DOI] [PubMed] [Google Scholar]
- 21.Chiang PH, Wu SN, Tsai EM, Wu CC, Shen MR, Huang CH, Chiang CP. Adenosine modulation of neurotransmission in penile erection. Br J Clin Pharmacol. 1994;38:357–362. doi: 10.1111/j.1365-2125.1994.tb04366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, Lamperti E, Schreiber BM, Gavras H, Wagner DD, Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2B receptor cascade. Cardiovasc Res. 2006;70:308– 314. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Daly JW, Butts-Lamb P, Padgett W. Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell Mol Neurobiol. 1983;3:69–80. doi: 10.1007/BF00734999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hide I, Padgett WL, Jacobson KA, Daly JW. A2a-Adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: Characterization with radioligand binding and by activation of adenylate cyclase. Mol Pharmacol. 1992;41:352–359. [PMC free article] [PubMed] [Google Scholar]
- 26.Gao ZG, Mamedova L, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes – characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 28.de Zwart M, Link R, von Frijtag Drabbe Künzel JK, Cristalli G, Jacobson KA, Townsend-Nicholson A, IJzerman AP. A screening of adenosine analogues on the human adenosine A2B receptor as part of a search for potent and selective agonists. Nucleos Nucleotid. 1998;17:969–986. doi: 10.1080/07328319808004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 30.Vittori S, Costanzi S, Lambertucci C, Portino FR, Taffi S, Volpini R, Klotz KN, Cristalli G. A2B adenosine receptor agonists: synthesis and biological evaluation of 2-phenylhydroxypropynyl adenosine and NECA derivatives. Nucleosides Nucleotides Nucleic Acids. 2004;23:471–481. doi: 10.1081/ncn-120028340. [DOI] [PubMed] [Google Scholar]
- 31.Volpini R, Costanzi S, Vittori S, Cristalli G, Klotz KN. Medicinal chemistry and pharmacology of A2B adenosine receptors. Curr Top Med Chem. 2003;3:427–443. doi: 10.2174/1568026033392264. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem Biol. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorman AR. Synthesis of 2-aralkoxy adenosines and 2-alkoxyadenosines. 2003035662 WO patent.
- 34.Chou SY. A novel substitution reaction of tetrahydropyrano[3,4-b]indole derivative chain extension and structural correlation study. Heterocycles. 2003;60:1095–1110. [Google Scholar]
- 35.Zhang H-C, Ye H, Moretto AF, Brumfielf KK, Maryanoff BE. Facile solid phase construction of indole derivatives based on a traceless, activating sulfonyl linker. Org Lett. 2000;2:89–92. doi: 10.1021/ol991255o. [DOI] [PubMed] [Google Scholar]
- 36.Ho-Hoang A, Fache F, Lemaire M. Synthesis and polymerization of 2-(3-pyrrolyl)acetic acid derivatives from pyrrole. Synth Commun. 1996;26:1289–1304. [Google Scholar]
- 37.Beukers MW, Meurs I, IJzerman AP. Structure-affinity relationships of adenosine A2B receptor ligands. Med Res Rev. 2006;26:667–698. doi: 10.1002/med.20069. [DOI] [PubMed] [Google Scholar]
- 38.Ivanov AA, Palyulin VA, Zefirov NS. Computer aided analysis of the binding modes of the adenosine receptor agonists for all known subtypes of adenosine receptors. J Mol Graph Mod. 2006 doi: 10.1016/j.jmgm.2006.06.004. in press. [DOI] [PubMed] [Google Scholar]
- 39.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. MacroModel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
- 40.Mewshaw RE, Zhou D, Zhou P, Shi X, Hornby G, Spanger T, Scerni R, Smith D, Schechter LE, Andree TH. Studies toward the discovery of the next generation of antidepressants. 3. Dual 5-HT1A and serotonin transfer affinity within a class of N-aryloxyethylindolylalkyl amines. J Med Chem. 2004;47:3823–3842. doi: 10.1021/jm0304010. [DOI] [PubMed] [Google Scholar]
- 41.Bang-Andersen B, Kehler J, Felding J. Preparation of phenylpiperazines as dopamine D4 and D3 receptors ligands. 2001049679 WO patent.
- 42.Fuchs JR, Funk RL. Total synthesis of (±)-Perophoramidine. J Am Chem Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 43.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 44.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 45.Post SR, Ostrom RS, Insel PA. Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol Biol. 2000;126:363–374. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 46.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]