

Abstract
Sedoheptulose 7-phosphate cyclases are enzymes that utilize the pentose phosphate pathway intermediate, sedoheptulose 7-phosphate, to generate cyclic precursors of many bioactive natural products, such as the antidiabetic drug acarbose, the crop protectant validamycin, and the natural sunscreens mycosporine-like amino acids. These proteins are phylogenetically related to the dehydroquinate (DHQ) synthases from the shikimate pathway, and are part of the more recently recognized superfamily of sugar phosphate cyclases, which includes DHQ synthases, aminoDHQ synthases and 2-deoxy-scyllo-inosose synthases. Through genome mining and biochemical studies, we identified yet another subset of DHQS-like proteins in the actinomycete Actinosynnema mirum and the myxobacterium Stigmatella aurantiaca DW4/3–1. These enzymes catalyze the conversion of sedoheptulose 7-phosphate to 2-epi-valiolone, which is predicted to be an alternative precursor for aminocyclitol biosynthesis. Comparative bioinformatics and biochemical analyses of these proteins with 2-epi-5-epi-valiolone synthases (EEVS) and desmethyl-4-deoxygadusol synthases (DDGS) provided further insights into their genetic diversity, conserved amino acid sequences, and plausible catalytic mechanisms. The results further highlight the uniquely diverse DHQS-like sugar phosphate cyclases, which may provide new tools for chemoenzymatic, stereospecific synthesis of various cyclic molecules.
INTRODUCTION
Sedoheptulose 7-phosphate, a phosphorylated monosaccharide with seven carbon atoms and a ketone functional group, is an intermediate in the pentose phosphate pathway (phosphogluconate pathway). This pathway is critical in living organisms, as its primary products include NADPH (used in reductive reactions within cells), ribose 5-phosphate (used in the biosynthesis of nucleotides), and erythrose 4-phosphate (one of the precursors in the formation of aromatic amino acids). In some organisms, sedoheptulose 7-phosphate is also involved in secondary metabolism. It serves as the substrate of 2-epi-5-epivaliolone synthases (EEVS), enzymes that are involved in the biosynthesis of C7N-aminocyclitol natural products, e.g., the antidiabetic drug acarbose and the crop protectant validamycin.1–3 EEVS are genetically related to the dehydroquinate (DHQ) synthases from the shikimate pathway. The latter enzymes catalyze the cyclization of 3-deoxy-d-arabinoheptulosonate 7-phosphate (DAHP), another C7-sugar phosphate, to 3-dehydroquinic acid. Other related proteins include aminodehydroquinate synthases (aminoDHQ), which cyclize aminoDAHP to aminoDHQ in the biosynthesis of 3-amino-5-hydroxybenzoic acid (3,5-AHBA),4 and 2-deoxy-scylloinosose (DOI) synthases,5 which are involved in the biosynthesis of many aminoglycoside antibiotics, e.g., kanamycin, neomycin, butirosin, and spectinomycin. Based on their substrates and catalytic functions, collectively these classes of enzymes form the sugar phosphate cyclase (SPC) superfamily.3
Through a comparative bioinformatics analysis of sugar phosphate cyclases we reported previously the identification of a new clade of proteins that are similar to EEVS.3 These proteins are widely distributed in cyanobacteria and fungi, such as Nos2 (Npun_5600) from Nostoc punctiforme and Anb2 (Ava_3858) from Anabaena variabilis.3 More recently, through seminal work of Balskus and Walsh, Npun_5600 has been demonstrated to be responsible for the formation of desmethyl-4-deoxygadusol (DDG), the precursor of the natural sunscreens mycosporine-like amino acids.6 Interestingly, similar to EEVS, Npun_5600 also utilizes sedoheptulose 7-phosphate as substrate, although the formation of DDG from sedoheptulose 7-phosphate most likely occurs through a somewhat divergent reaction mechanism.6
As part of our ongoing studies on the biosynthesis of aminocyclitol-derived natural products, we recently identified and characterized a pseudoglycosyltransferase enzyme (pseudo-GT, VldE) that catalyzes the coupling between GDP-valienol and validamine 7-phosphate in validamycin biosynthesis.7 To assess the distribution of pseudo-GTs in nature, we carried out bioinformatics analyses using the VldE sequence as a query and found two cryptic gene clusters that contain VldE-homologues (pseudo-GT20) in the genomes of Actinosynnema mirum DSM 43827 and Stigmatella aurantiaca DW 4/3–1 (Figure 1).8, 9 Based on the putative substrates for pseudo-GT enzymes,1, 2, 10–12 it is expected that the two clusters should also contain pseudo-sugar biosynthetic genes, e.g., the 2-epi-5-epivaliolone synthase (EEVS) gene. However, initial inspections of the clusters suggested that they do not contain the EEVS gene, but rather a putative dehydroquinate synthase (DHQS) gene, which raised questions as to whether DHQS are also involved in pseudosugar (cyclitol) formation.
Figure 1.
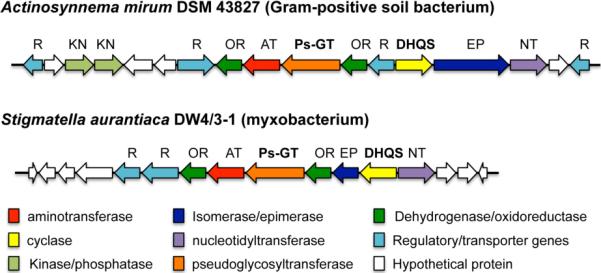
Genome mining leading to the discovery of new genes that encode DHQ synthase-like proteins. The presence of DHQS genes (yellow arrows) together with the putative pseudoglycosyltransferase (Ps-GT) genes (orange arrows) in the clusters raised questions as to whether DHQS are involved in pseudosugar (cyclitol) formation.
Within this study, the gene products (Amir_2000 and Staur_3140) were recombinantly prepared and subsequent enzymatic analysis revealed that these putative DHQS are neither DHQS nor EEVS. Instead, they utilize sedoheptulose 7-phosphate as substrate to form 2-epi-valiolone. The latter compound has never been identified in nature before, but it may well serve as an alternative precursor for aminocyclitol biosynthesis. Herein we report the identification and characterization of these new proteins, which are designated as 2-epi-valiolone synthases (EVS). We have also carried out comparative genetic, biochemical, kinetic, and in situ NMR analyses of the three different subsets of sedoheptulose 7-phophate cyclases (EEVS, EVS, and DDGS). The results provided further insights into their genetic diversity, conserved amino acid sequences, and plausible catalytic mechanisms.
RESULTS
Bioinformatic studies of Amir_2000 and Staur_3140
As misannotations within the DHQS-like proteins are rather common, we carried out detailed bioinformatics studies of the genes, amir_2000 (from A. mirum) and staur_3140 (from S. aurantiaca). DHQS and DHQS-like proteins from both primary and secondary metabolic pathways show relatively high sequence similarity (25–70% identity), and maximum likelihood analysis has revealed that protein similarity is highly correlated with the predicted enzyme function.3 To confirm that amir_2000 and staur_3140 are indeed more closely related to DHQS than to EEVS, we employed MEGA5 software with a WAG amino acid substitution model to generate a maximum likelihood phylogenetic tree of the various DHQS-like proteins (Figure 2).13, 14 Input sequences were selected from GenBank from each family of sugar phosphate cyclases including several putative DHQ synthase sequences such as Amir_2000 and Staur_3140. E. coli glycerol dehydrogenase was used as an out-group. The results revealed that Amir_2000 and Staur_3140 are phylogenetically indeed more closely related to DHQS than to EEVS.
Figure 2.
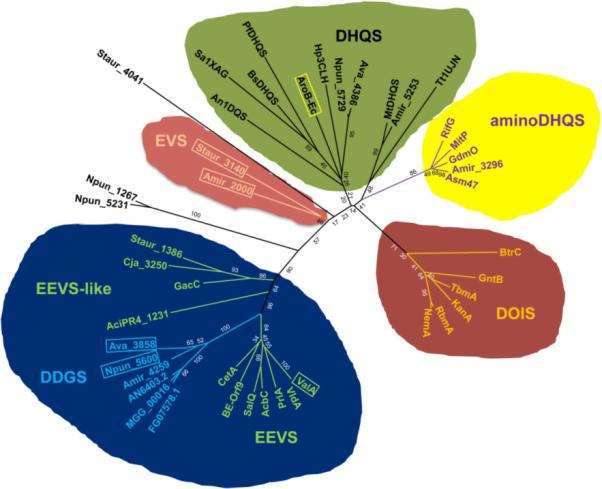
Phylogenetic analysis of the superfamily of sugar phosphate cyclases. Boxed proteins are representatives of families of sugar phosphate cyclases being studied in this paper. Maximum likelihood analysis was carried out using MEGA5 software with a WAG amino acid substitution model and a discrete gamma distribution was used to model evolutionary rate differences among sites. The robustness of the trees was assessed by bootstrap analysis (100 replicates). The source and accession number of the proteins are listed in Table S1.
Amir_2000 and Staur_3140 are not DHQ synthases
DHQS are enzymes that catalyze the cyclization of DAHP to DHQ, the first cyclic intermediate in the shikimate pathway. This pathway is commonly present in fungi, bacteria, protozoa, and plants for the synthesis of aromatic amino acids (e.g., phenylalanine, tyrosine, tryptophan), co-factors (e.g., pyrroloquinoline quinone (PQQ), ubiquinone, folate), and natural products (e.g., flavonoids, coumarins, tropanes, indole alkaloids, lignans and lignin). To investigate if Amir_2000 and Staur_3140 are indeed DHQS, the genes were cloned in the E. coli expression vector pRSET B (Invitrogen) and transferred into E. coli BL21(DE3)pLysS. Expression of amir_2000 and staur_3140 was induced by isopropyl-β-d-thiogalactopyranoside (IPTG) and the recombinant proteins (45.1 kDa and 47.7 kDa soluble polyhistidinetagged proteins, respectively) were purified using Ni-NTA columns (Figure 3A). Incubation of these two proteins with DAHP, in the presence of NAD+ and Co2+, however, did not give any product. On the other hand, a parallel experiment with DHQS from E. coli (AroBEc) resulted in the expected 3-dehydroquinate product (Figure 3B), eliminating the possibility of technical inadequacies, e.g., inappropriate assay condition or problems with product analysis; thus, suggesting that Amir_2000 and Staur_3140 may not be DHQS.
Figure 3.
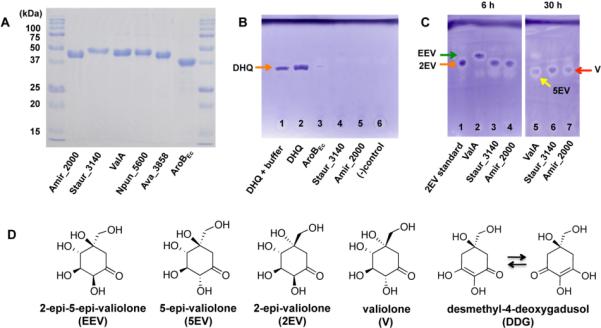
Characterization of Amir_2000, Staur_3140, and related enzymes. A, SDS-PAGE of Ni-NTA purified cyclases. B, TLC analysis of the cylases' products with DAHP as substrate. C, TLC analysis of the cyclases' products with sedoheptulose 7-phosphate as substrate. D, Chemical structures of 2-epi-5-epi-valiolone, 5-epi-valiolone, 2-epi-valiolone, valiolone, and desmethyl-4-deoxygadusol.
Characterization of Amir_2000 and Staur_3140 as sedoheptulose 7-phosphate cyclases
Whereas BLAST analysis revealed that the amino acid sequences of Amir_2000 and Staur_3140 are more similar to DHQS (Amir_2000 and Staur_3140 have 42% identity to DHQS from Desulfotomaculum gibsoniae DSM 7213 and Dialister succinatiphilus YIT 11850, respectively) than EEVS (Amir_2000 and Staur_3140 have 35% and 31% identity to ValA, respectively), the fact that the genes are clustering with the putative pseudoglycosyltransferase genes suggests that these enzymes may be involved in the formation of a cyclitol product. To explore this possibility, we incubated the two proteins with sedoheptulose 7-phosphate, NAD+ and Co2+ or Zn2+. Sedoheptulose 7-phosphate is the substrate for EEVS, whose product (2-epi-5-epi-valiolone, EEV) is the common cyclic intermediate in many aminocyclitol biosyntheses.1–3, 15 Careful monitoring of the catalytic reactions using TLC and GC-MS revealed that both Amir_2000 and Staur_3140 indeed convert sedoheptulose 7-phosphate to a product with Co2+ being the preferred cofactor (Figure S1).
However, direct comparison of the products with that of the well-characterized EEVS (ValA) from the validamycin pathway showed that on TLC the compound has an Rf value that is slightly lower than that of EEV, suggesting that the product is not EEV (Figure 3C).
Synthesis of 2-epi-valiolone and characterization of Amir_2000 and Staur_3140 products
Based on X-ray crystallographic studies, DHQS and DOIS have been proposed to catalyze the cyclization of their respective sugar phosphate substrates via aldol reactions. Because Amir_2000 and Staur_3140 are highly similar to DHQS, we suspected that they also adopt the same aldol mechanism. Consequently, the most likely product for such a cyclization reaction with sedoheptulose 7-phosphate as substrate, besides EEV, is 2-epi-valiolone (2EV). This is mechanistically plausible, as the aldol cyclization can take place from either face of the ketone, depending on the orientation of the intermediate within the active site of the enzyme. To determine if this is the case, we synthesized 2EV from (2S,3S,4S,5S)-2,3,4-tri-O-benzyl-5-C-[(benzyloxy)methyl]-6,6-dichloro-1-oxo-2,3,4,5-cyclohexanetetrol (2) (Scheme 1) and compared its physicochemical data with those of the enzyme products. 2 was prepared from commercially available 2,3,4,6-tetra-O-benzyl-d-mannopyranose (1) in five steps according to the method reported previously.10 The compound was then converted, via a free-radical reduction using Bu3SnH/AIBN, to 2,3,4,7-tetra-O-benzyl-2-epi-valiolone (4). Finally, 4 was deprotected by catalytic hydrogenation using wet 10% Pd/C as the catalyst to give 2EV in a quantitative yield.
Scheme 1.
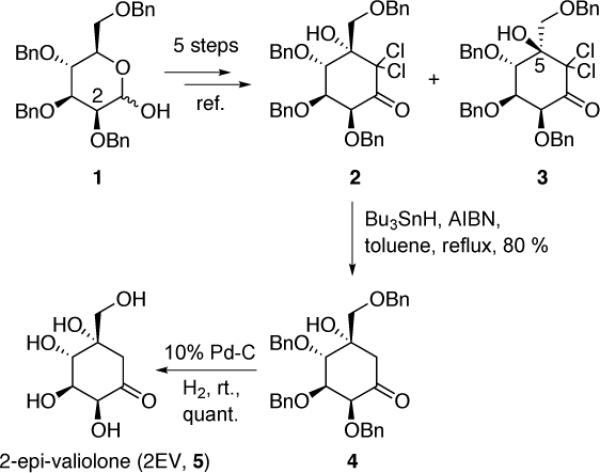
Synthesis of 2-epi-valiolone
The synthetic 2EV was then used as an authentic sample for the characterization of Amir_2000 and Staur_3140 products. Preliminary comparative analysis using TLC revealed that the enzyme products displayed a similar Rf value to that of the synthetic compound. Further analysis of the products, after trimethylsilyl derivatization, using GC-MS showed identical retention time and fragmentation pattern as those of 2EV (Figure S2), suggesting that the enzyme products are indeed 2EV. This was further confirmed by comparison of the 1H NMR spectra of Amir_2000 and Staur_3140 products with that of the synthetic 2EV (Figure S3). All together, the data suggest that Amir_2000 and Staur_3140 are neither DHQS nor EEVS, but yet another subset of DHQS-like proteins, which we identify as 2-epi-valiolone synthases (EVS).
Interestingly, in a prolonged incubation time at pH 7.4, the product of Amir_2000 and Staur_3140 (2EV) is prone to non-enzymatic epimerization. A similar conversion was also observed with synthetically prepared 2EV when stored in an aqueous solution. Comparative analyses by TLC and GC-MS with synthetically prepared compounds suggest that the epimerization product is valiolone (V) (Figure 3C). Epimerization at C-2 from a β-configuration (mannose-like) to an α-configuration (glucose-like) appears to occur more readily in C7-cyclitols. Although in the validamycin pathway a dedicated epimerase enzyme (ValD) is involved in the conversion of EEV to 5-epi-valiolone (5EV), the conversion may also occur non-enzymatically, albeit at a much lower rate.16
In situ NMR analysis of ValA, Amir_2000, Staur_3140, Npun_5600, and Ava_3858
The discovery of Amir_2000 and Staur_3140 as 2-epi-valiolone synthases (EVS) expanded the diverse groups of DHQS-like proteins. Together with the EEVS and the DDGS, they represent the subgroup of sedoheptulose 7-phosphate cyclases, which fascinatingly furnish three distinct products, 2EV, EEV, and DDG, respectively. Therefore, it is intriguing to explore whether any of these enzymes share a diffusible common intermediate(s) during or after catalysis. To investigate this possibility, we set up in situ NMR analyses of the enzymatic assays with EEVS (ValA), EVS (Amir_2000 and Staur_3140), and DDGS (Npun_5600 and Ava_3858) proteins. The catalytic function of Ava_3858 has not previously been characterized, however, its high sequence identity to Npun_5600 suggested that it is a DDGS. This activity was confirmed in the present study (see below). ValA was recombinantly prepared in E. coli and purified according to the method we reported previously.2 The Npun_5600 and Ava_3858 genes, amplified from the chromosomes of the cyanobacteria N. punctiforme and A. variabilis, respectively, were cloned in pRSET B and expressed in E. coli BL21(DE3)pLysS. The recombinant proteins were purified by Ni-NTA columns.
To monitor the catalytic reactions of these five enzymes, we carried out time-course 1H NMR experiments in D2O/H2O. The substrate (2 mg, as a barium salt) was dissolved in D2O (152.5 μL) and potassium phosphate buffer (12.5 μL of 0.4 M solution, pH 7.4), NAD+ (2.5 μL of 100 mM solution) and Co2+ (2.5 μL of 20 mM solution) (all in H2O) were added. The mixture was transferred into a Shigemi NMR tube and the reaction was started by adding the purified enzyme solution (80 μL). The reactions were carried out at 25 °C and 1H NMR measurements were performed under water suppression conditions at 0 min (no enzyme), 30 min, 1 h, 3h, and 5h (Figures 4 and S3). A decreased intensity of the proton signals of the substrate sedoheptulose 7-phosphate and an increased intensity of those of the products were observed in the NMR spectra, confirming the distinct function of each of those enzymes.
Figure 4.

In situ 1H NMR analyses of ValA (A), Amir_2000 (B), and Npun_5600 (C) reactions.
1H NMR spectrum of the ValA product showed a set of geminal proton signals at 2.45 (d, J = 14.0 Hz, 6-Ha) and 2.91 (d, J = 14.0 Hz, 6-Hb) (Figure 4A), consistent with those expected for EEV. On the other hand, 1H NMR spectra of DDG (the product of Npun_5600 and Ava_3858) showed geminal proton signals at 2.41 (2H, d, J = 16.9 Hz, 4-Ha and 6-Ha), 2.68 (2H, d, J = 16.9 Hz, 4-Hb and 6-Hb) and 3.50 (2H, s, 7-H2) (Figures 4C and S3C). The overlapping C-4 and C-6 methylene proton signals in DDG suggest that the compound exists in an equilibrium of two reversible enol forms. However, no indication of the presence of diffusible common intermediate(s) was observed in the 1H NMR spectra of these reactions, thus, eliminating the possibility of non-enzymatic conversions of any of these products.
Interestingly, cyclization of sedoheptulose 7-phosphate by Amir_2000 and Staur_3140 gave a product(s) with two sets of geminal proton signals between 1.5 and 3.0 ppm [set A, 2.65 (dd, J = 1, 14.4 Hz, 6-Ha) and 2.89 (d, J = 14.4 Hz, 6-Hb); set B, 1.83 (dd, J = 2.0, 14.6 Hz, 6-Ha) and 2.08 (d, J = 14.6 Hz, 6-Hb)] (Figures 4B and S3B). None of these signals belong to EEV or DDG, excluding the possibility that Amir_2000 and Staur_3140 also produce those compounds. The signals are also different from those of V [2.39 (d, J = 14.6 Hz, 6-Ha)] and 2.79 (d, J = 14.6 Hz, 6-Hb)].17 More importantly, identical two sets of geminal proton signals were also observed in the 1H NMR spectrum of the synthetic compound (Figure S3A), confirming that the signals indeed belong to 2EV. Therefore, we predicted that in an aqueous solution 2EV exists in two stable conformational forms.
Computational studies of the valiolones
To confirm the presence of two stable conformers of 2EV in an aqueous solution, we performed computational modeling of 2EV together with EEV, 5EV and V using MacroModel Force Field-based Molecular Modeling and the Jaguar ab initio Quantum Mechanics Programs (Schrödinger). Conformational searches were performed using AMBER* (water). Low-energy structures found for each compound were selected and subjected to unrestrained quantum mechanical geometry minimization using Hybrid B3LYP/6–31G**-SCF [PBF(water)]. Optimizations were followed by single point energy calculations to obtain more accurate energies for each structure. Superimposition of twenty low-energy structures of EEV, 5EV, and V revealed that the majority adopts a conserved chair conformation (Figure 5A, 5B, and 5D). On the other hand, 2EV appears to adopt at least two different conformations in water (Figure 5C). The quantum calculations found that the two conformations were highly similar in enthalpy (within 0.2 kcal/mol), suggesting both conformers may coexist in solution (Figure 5E and 5F). This is in complete agreement with the observed two sets of geminal proton signals in the 1H NMR spectrum. The quantum calculations also revealed that V (31.8 kcal/mol) is thermodynamically more stable than 2EV (33.7 kcal/mol), which explains the tendency for non-enzymatic epimerization of this compound. In contrast to the computed structures of 2EV, which are highly variable (Figure 5C), all twenty low-energy structures for V consistently adopt chair conformations with the C-2, C-3, and C-4 hydroxy groups, as well as the C-5 hydroxymethylene group, oriented in equatorial positions (Figure 5D).
Figure 5.

Superimposition of twenty low-energy structures of EEV (A), 5EV (B), 2EV (C), and V (D). The first two low-energy conformations of 2EV are shown in the ball-and-stick representation (E and F).
Kinetic studies of ValA, Amir_2000, Staur_3140, Npun_5600, and Ava_3858
To compare the affinity of each of these enzymes toward the substrate sedoheptulose 7-phosphate and determine their catalytic efficiency, we carried out kinetic studies of ValA, Amir_2000, Staur_3140, Npun_5600, and Ava_3858. The kinetic properties were determined using the EnzChek Pyrophosphate Assay Kit18 (Molecular Probes) (Scheme S1). Each enzyme, in Tris-HCl buffer, was pre-incubated with NAD+, CoCl2, 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG), and purine nucleoside phosphorylase (PNP) for 10 min at 28 °C. Reactions were initiated by addition of sedoheptulose 7-phosphate and monitored at A360 every 8 sec. The apparent kinetic values, obtained from Hanes-Woolf plots, are shown in Table 1. The results suggest that despite their different catalytic mechanisms and products, all tested proteins have relatively similar substrate affinity and catalytic efficiency.
Table 1.
Apparent kinetic values for ValA, Amir_2000, Staur_3140, Npun_5600, and Ava_3858.
| Protein | Km (μM) | kctA (min−1) | kcat/Km (μM−1•min−1) |
|---|---|---|---|
| ValA | 25.6 ± 5.2 | 2.7 ± 0.1 | 0.105 ± 0.01 |
| Amir_2000 | 59.9 ± 13.4 | 2.0 ± 0.1 | 0.033 ± 0.004 |
| Staur_3140 | 38.6 ± 5.2 | 3.7 ± 0.2 | 0.096 ± 0.007 |
| Npun_5600 | 65.1 ± 12.4 | 5.0 ± 0.5 | 0.077 ± 0.006 |
| Ava_3858 | 21.6 ± 2.8 | 3.2 ± 0.3 | 0.148 ± 0.005 |
Identification of sequences characteristic to EVS
Finally, to be able to recognize EVS proteins at the genetic level, it is important to establish their sequence characteristics that will enable us to distinguish them from DHQS and their other close relatives. Previously, we established unique signature sequences for each of the known sugar phosphate cyclases (SPCs) that can be used to accurately annotate sequences according to their function.3 Using a similar approach we investigated the sequences of EVS to identify characteristic residues that can differentiate EVS from DHQS and other SPCs. Thus, amino acid sequences of representative members of each family of SPCs (i.e., DHQS, aminoDHQS, DOIS, EEVS, EVS, and DDGS) were compared using the multiple alignment program ClustalW. The active site of DHQS is formed within a cleft between the N-and C-terminal domains and is lined by 14 amino acid residues. These active site amino acid residues (Table 2) are identified based on the x-ray crystal structure of the DHQS from Aspergillus nidulans.19 The family more related to DHQS is the aminoDHQS from the rifamycin (RifG),20 ansamitocin (Asm47),21 mitomycin (MitP),22 and geldanamycin (GdmO)23 pathways. Sequence alignment of the binding pocket residues revealed that DHQS and aminoDHQS retain high sequence conservation, only differing at positions K197 and E260. Similarly, the DOIS family is also closely related to DHQS, as both have conserved active site residues, except for those at positions R264 and N268. In DOIS from the tobramycin (TbmA), kanamycin (KanA), ribostamycin (RbmA), gentamycin (GntB), and butirosin (BtrC) pathways,24, 25 the arginine and asparagine residues corresponding to position 264 and 268 in DHQS have been altered to conserved glycine and glutamate residues, respectively.
Table 2.
Sequence alignment of the proposed active site residues of currently known DHQS-like sugar phosphate cyclases
| Protein | 130 | 146 | 152 | 162 | 194 | 197 | 250 | 260 | 264 | 268 | 271 | 275 | 287 | 356 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DHQS | R | D | K | N | E | K | K | E | R | N | H | H | H | K |
| aDHQS | R | D | K | N | E | R | K | D | R | N | H | H | H | K |
| DOIS | R | D | K | N | E | K | K | E | G | E | H | H | H | K |
| EEVS | R | D | K | N | E | K | M | E | R | D | H | P | H | P |
| EVS | R | D | K | N | E | K | K | E | R | N | H | H | H | R |
| DDGS | R | D | K | N | E | K | M | E | R | A | H | P | H | P |
Multiple amino acid sequence alignment was conducted by ClustalW. Full-length amino acid sequences were aligned using the following parameters: protein weight matrix=Blosum, gap open penalty=15, gap extension penalty=0.2. Numbering is based on DHQS domain of AROM from A. nidulans. Proteins used for multiple sequence alignment are listed in Supplemental Table S1.
In EEVS (e.g., ValA, CetA, SalQ, PrlA) and DDGS (e.g., Npun_5600, Ava_3858), one third of the active site residues are consistently altered from those of DHQS including H275, which is believed to play a critical role in enzyme catalysis. Amino acid residues corresponding to DHQS K250, H275 and K356 are highly conserved methionine, proline, and proline residues, respectively, in EEVS and DDGS (Table 2). On the other hand, the basic N268 in DHQS is a conserved aspartate residue in EEVS and a conserved alanine residue in DDGS. Interestingly, despite the fact that EVS are catalytically more similar to EEVS and DDGS, genetically they demonstrate higher similarity with DHQS. EVS retain high active site residue conservation with DHQS, only differing at position K356. In both Amir_2000 and Staur_3140, the lysine corresponding to position 356 in DHQS has been altered to a conserved arginine residue (Table 2).
DISCUSSION
Since the discovery of the AROM protein in Aspergillus nidulans and Saccharomyces cerevisiae, a substantial list of DHQS have been reported from other fungi, bacteria, apicomplexans, and plants, suggesting that the shikimate path way is a common metabolic pathway in these organisms. However, in certain host-associated bacteria, some of the shikimate pathway genes appeared to be missing.26 Subsequently, through seminal work by Floss and others,4, 27 aminoDHQS, a closely related family of DHQS, were found to be involved in the biosynthesis of rifamycin, ansamitocin, geldanamycin, and mitomycin C. The next well-studied DHQS-like proteins are DOIS,5, 28, 29 required for the biosynthesis of aminoglycoside antibiotics, followed by the EEVS that are involved in C7N-aminocyclitol-biosynthesis.1–3, 12, 15
The recent characterization of DDGS involved in the biosynthesis of sunscreen compounds in cyanobacteria and the characterization of EVS reported in this study further underscore the uniquely diverse DHQS-like sugar phosphate cyclases.3, 6, 30 Interestingly, except for aminoDHQS, whose substrate is derived from a discrete pathway involving kanosamine 6-phosphate and 3-amino-3-deoxy-D-fructose 6-phosphate,4, 31, 32 the substrates for DHQS, EEVS, EVS, DDGS, and DOIS are all directly derived from the pentose phosphate pathway (Scheme 2). Particularly intriguing are the EEVS, EVS, and DDGS that all utilize sedoheptulose 7-phosphate as substrate but yield three different cyclic products. This raises an important mechanistic question as to how three very closely related enzymes utilize a common substrate but produce three different products.
Scheme 2.

Most substrates for DHQ synthase-like cyclases are directly derived from the pentose phosphate pathway. EEVS, EVS, and DDGS all utilize sedoheptulose 7-phosphate as substrate but yield three different cyclic products.
The catalytic mechanism of DHQS has been proposed to proceed through a multi-step process including alcohol oxidation, phosphate β-elimination, carbonyl reduction, ring opening and intramolecular aldol condensation.19 Similar mechanisms were also proposed for DOIS, EEVS, and DDGS.1, 5, 6 Therefore, it is tempting to assume that EVS also adopt a similar mechanism, albeit the substrate may be oriented differently within the enzyme active site pocket (Scheme 3). In EEVS and EVS, the reaction is assumed to be initiated by transient dehydrogenation of C-5 to a ketone, which sets the stage for the elimination of phosphate, followed by reduction of the C-5 ketone and ring opening to produce the enol of a 6,7-methyl ketone. The latter then undergoes intramolecular aldol condensation to give either 2-epi-5-epi-valiolone or 2-epi-valiolone, depending on the orientation of the aldol acceptor. In DDGS,6 the reaction is also assumed to be initiated by dehydrogenation of C-5 to a ketone, followed by the elimination of phosphate. However, ring opening and aldol condensation should occur prior to enolization, dehydration, reduction, and tautomerization to give desmethyl 4-deoxygadusol (Scheme 3). However, how exactly these different processes take place in EEVS, EVS, and DDGS remain obscure. Further investigations of these enzymes at the molecular and structural levels may help dissect the unique catalytic evolution of this fascinating family of proteins.
Scheme 3.

Proposed catalytic mechanisms for EEVS, EVS, and DDGS.
Since our first report on the identification of EEVS in the late 1990s,1 dozens of EEVS and EEVS-like proteins have been reported in the NCBI database. Mostly, they are of bacterial origin, including marine actinomycetes and myxobacteria, but genes encoding EEVS-like proteins are also found in fungal, fish, and frog genomes. This is rather surprising, as only a relatively limited number of C7N-aminocyclitol-containing natural products have been reported in the literature and most of them were from bacteria. Genome mining, however, suggests the likelihood that aminocyclitol-containing compounds are more common in nature than currently appreciated. However, due to their physicochemical properties, e.g., high polarity, they may be overlooked during the isolation of natural products. On the other hand, EEVS or EEVS-like proteins may also be involved in primary metabolism, providing biosynthetic intermediates that will ultimately contribute to the structural or physiological functions within the organisms.
Unlike EEVS, EVS appear to be less common in nature. A preliminary inspection of the database resulted in the discovery of only a few proteins that are homologous to EVS. This could be due to the fact that the identification of EVS based on sequence similarity is more challenging. EVS are highly similar to DHQS, which by April 2012 have topped 6,875 entries in the NCBI database. Our comparative analysis based on the predicted active site residues of Aspergillus nidulans DHQS showed that only a single putative active site residue of EVS differs from those in DHQS (Table 2). It remains to be determined if this single active site difference is enough to cause the change in substrate affinity between DHQS and EVS enzymes. Other modifications near the identified active site residues may play a more fundamental role in substrate specificity within these two classes of enzymes, making annotation of enzymatic function based on protein sequence alone more difficult. In contrast, almost one third of the active site residues in EEVS and DDGS are different from those in DHQS, making them readily distinguishable at the protein sequence level. Thus, the characterization of Amir_2000 and Staur_3140 reported here provide the foundation for further understanding of this unique family of sedoheptulose 7-phosphate cyclases.
CONCLUSION
Using genome mining and biochemical studies we identified a new subset of DHQS-like proteins in the actinomycete A. mirum and the myxobacterium S. aurantiaca DW4/3–1 that catalyze the conversion of sedoheptulose 7-phosphate to 2EV. Comparative bioinformatic analyses revealed that at the amino acid sequence level EVS is more similar to DHQS than to EEVS and DDGS. The latter enzymes share the same substrate with EVS, but produce two other distinct products. Further analysis of the products using in situ NMR and computational calculations revealed that in an aqueous solution 2EV exists in two stable conformational forms. Kinetic studies of EEVS, EVS, and DDGS suggest that despite their different catalytic mechanisms and products, they have relatively similar substrate affinity and catalytic efficiency. The results not only highlight the uniquely diverse DHQS-like sugar phosphate cyclases, which may provide new tools for chemoenzymatic, stereospecific synthesis of various cyclic molecules, but it will also enable the identification of cryptic metabolic pathways involved in the biosynthesis of biologically active secondary metabolites with potential applications in medicine, agriculture, and other industrial processes.
EXPERIMENTAL SECTION
General
All chemical reactions were performed under an argon or nitrogen atmosphere employing oven-dried glassware. Analytical thin-layer chromatography (TLC) was performed using silica plates (60 Å) with a fluorescent indicator (254 nm), which were visualized with a UV lamp and ceric ammonium molybdate (CAM) solution. Chromatographic purification of products was performed on silica gel (60 Å, 72–230 mesh). Proton NMR spectra were recorded on Bruker 300 or 400 MHz spectrometers. Proton chemical shifts are reported in ppm (δ) relative to the residual solvent signals as the internal standard (CDCl3: δH 7.26; D2O: δH 4.79). Multiplicities in the 1H NMR spectra are described as follows: s = singlet, bs = broad singlet, d = doublet, bd = broad doublet, t = triplet, bt = broad triplet, q = quartet, m = multiplet; coupling constants are reported in Hz. Carbon NMR spectra were recorded on a Bruker 300 (75 MHz) spectrometer with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to the residual solvent signal as the internal standard (CDCl3: δC 77.16), or with sodium 2,2-dimethylsilapentane-5-sulphonate (DSS) (δ 0.0) as an external standard. Low-resolution electrospray ionization (ESI) mass spectra were recorded on a ThermoFinnigan liquid chromatograph-ion trap mass spectrometer, and high-resolution electrospray mass spectra were recorded on a Waters/Micromass LCT spectrometer. Size exclusion chromatography was done on Sephadex LH-20 (Pharmacia).
Molecular phylogenetic analysis by maximum likelihood method
For phylogenetic analysis, full-length amino acid sequences were aligned using ClustalW with the following parameters: Protein Weight Matrix=Blosum; Gap Open Penalty=15; Gap Extension penalty=0.2. Maximum likelihood analysis was carried out using MEGA5 software with a WAG amino acid substitution model and a discrete gamma distribution was used to model evolutionary rate differences among sites. The robustness of the trees was assessed by bootstrap analysis (100 replicates).
Chemical synthesis of 2-epi-valiolone
2,3,4,7-tetra-O-benzyl-2-epi-valiolone (4)
To a solution of 2 (25 mg, 0.040 mmol) in toluene (0.25 mL), tributyltin hydride (0.043 mL, 0.161 mmol) and AIBN (2.6 mg, 0.016 mmol) were added and the mixture was refluxed for 2 h and then cooled to room temperature. The products were extracted with EtOAc (3 mL) and the organic solution was washed with 2N HCl, sat. aq. NaHCO3, and brine. The organic solvent was evaporated under reduced pressure and the extract was purified over a silica gel column (nhexane/EtOAc 25:1 - 5:1) to give 4 (11 mg, 49 %); colorless syrup, 1H-NMR (300 MHz, CDCl3) δ: 2.37 (d, J = 14.4 Hz, 6-Ha), 2.61 (s, 5-OH), 3.15 (d, J = 14.4 Hz, 6-Hb), 3.23 (d, J = 8.9 Hz, 7-Ha), 3.49 (d, J = 8.9 Hz, 7-Hb), 3.93 (dd, J = 7.6, 3 Hz, 3-H), 4.10 (d, J = 3 Hz, 4-H), 4.30 (d, J = 7.6 Hz, 2-H), 4.40–4.87 (PhCH2- x 4), 7.15–7.40 (C6H5 x 4). 13C-NMR (75 MHz, CDCl3) δC: 44.9 (t, C-7), 72.1, 72.6, 73.3, 73.4 (all t, PhCH2 x 4), 74.0 (d, C-4), 77.4 (s, C-5), 79.5 (d, C-3), 81.0 (d, C-2), 127.7–128.4 (all d) and 137.2–137.9 (all s, C6H5 x 4), 206.6 (s, C-1). LRMS (ESI-TOF) m/z 575 [M+Na]+. HRMS (ESI-TOF) m/z 575.2391 (calcd for C35H36O6Na [M+Na]+: 575.2410).
2-epi-valiolone (5)
To a solution of 4 (8 mg) in 95 % aqueous ethanol (0.80 mL) was added wet 10 % Pd/C (8 mg) and the mixture was stirred at room temperature under an H2 atmosphere for 2 h. The suspension was passed through a Celite column to remove the catalyst and then filtered through a membrane filter. The solvent was evaporated in vacuo to give pure 5 (2.7 mg); white solid, 1H-NMR (300 MHz, CD3OD) δ: 1.83 (dd, J = 14.1, 2.1 Hz, 6-Ha1) 2.03 (d, J = 14.1 Hz, 6-Hb1), 2.47 (d, J = 14.1, 1.2 Hz, 6-Ha2) 2.87 (d, J = 14.1 Hz, 6-Hb2), 3.41 (d, J = 11.1Hz, 7-Ha) 3.64 (d, J = 11.1, 7Hb) 3.98–4.02 (m, 3-H and 4-H), 4.31 (d, J = 3.3 Hz, 2-H). 13C-NMR (75 MHz, CD3OD) δC: 44.1 70.4 (t, C-6), 71.3 (d, C-4), 73.7(d, C-2), 75.1 (s, C-5), 75.6 (d, C-3), 208.4 (s, C-1). 1H-NMR (300 MHz, D2O) δ: 1.85 (d, J = 15 Hz, 6-Ha1, 1H), 2.11 (d, J = 15 Hz, 6-Hb1, 1H), 2.67 (d, J = 14 Hz, 6-Ha2, 1H), 2.91 (d, J = 14 Hz, 6-Hb2, 1H), 3.48–3.86 (m, 7H), 3.9–4.2 (m, 3H), 4.52 (d, J = 3.9 Hz, 2-H, 1H). LRMS (ESI-TOF) m/z 191 [M−H]−. HRMS (ESI-TOF) m/z 191.0554 (calcd for C7H11O6 [M−H]−: 191.0556).
Construction of ValA, Amir_2000, Staur_3140, Npun_5600, Ava_3858, and AroBEc expression plasmids
Construction of an expression plasmid for recombinant ValA from Streptomyces hygroscopicus subsp. jinggangensis was reported in our previous paper.2
Construction of an expression plasmid for recombinant Amir_2000
Actinosynemma mirum DSM 43827 was grown on YMG agar plate for 4 days and the grown cells were used to inoculate YMG liquid media. After incubation at 30 °C, 200 rpm for 2 days, cells were harvested and their genomic DNA isolated. The amir_2000 gene was amplified by PCR using two primers (5′-GAA GAT CTT ATG GAC AGT CCC GCT GGT TAC C-3′ and 5′-CGG AAT TCA GGC TCA TCG CAG CCT CAC C-3′, BglII and EcoRI are underlined). BglII and EcoRI digested PCR product was ligated into BamHI and EcoRI sites of pRSET B to generate pRSET B-amir_2000.
Construction of an expression plasmid for recombinant Staur_3140
Stigmatella aurantiaca DW4/3–1 was grown on agar plate media containing: 1% Tryptone, 0.2% MgSO4 × 7H2O, 1.2% HEPES, and 1.5% agar, pH adjusted to 7.2 with KOH, for 10 days at 30 °C and the grown cells were used to inoculate liquid media containing: 1% Tryp-tone, 0.2% MgSO4 × 7H2O, 0.4 % soluble starch, 1.2% HEPES, pH 7.2 with KOH). After culture at 30 °C, 200 rpm for 4 days, cells were harvested and their genomic DNA was isolated. A staur_3140 gene was amplified by PCR using two primers (5'-GAA GAT CTC GAC CCA ATG CCT TCC ACT G-3' and 5'-CGG AAT TCA TGT AGG CCG TGG ACG CGA G-3', BglII and EcoRI are underlined). BglII and EcoRI digested PCR product was ligated into BamHI and EcoRI sites of pRSET B to generate pRSET B-staur_3140.
Construction of an expression plasmid for recombinant Npun_5600 and Ava_3858
Frozen stocks of Nostoc punctiforme ATCC 29133 and Anabaena variabilis ATCC 29413 were used to inoculate Allen and Arnon liquid media and the cultures were statically grown with filtered air infused into the cultures using an air stone.33 After culture at 28 °C for 21 days, 50 mL of culture (~ 1 gram of cell mass) were harvested by centrifugation at 3,000 × g and the genomic DNA was isolated using a modified STE protocol. Briefly, the cell pellet was frozen and ground in liquid nitrogen using a mortar and pestle to fully disrupt the cell wall components. One gram of cell mass was resuspended in 5 mL of STE buffer containing 20 mg of lysozyme and incubated at 37 °C for 10 min. EDTA was added to a final concentration of 0.1 M containing 1% SDS, 0.5 mg of RNAse, and 0.15 mg Pronase and the sample was incubated for an additional 15 min at 37 °C. The sample was extracted with phenol:chloroform and precipitated with 0.3 M sodium acetate, pH 5.2 and 70% isopropanol. Spooled DNA was washed with 70% ethanol, briefly air-dried and resuspended in sterile water. The npun_5600 (DDGS) gene was amplified by PCR using two primers (5′-GAA GAT CTG CAT ATG AGT AAT GTT CAA GCA TCG T-3' and 5'-CGG GGT ACC TCA CAC TCC CAA TAG TTT GGA-3′, BglII and KpnI are under-lined). BglII and KpnI digested PCR product was ligated into BamHI and KpnI sites of pRSET B to give pRSET B-npun_5600. The ava_3858 (DDGS) gene was amplified by PCR using two primers (5′-GAA GAT CTG CAT ATG AGT ATC GTC CAA GCA AAG-3′ and 5′-CGG GGT ACC TTA TTT AAC ACT CCC GAT TAT T-3′, BglII and KpnI are under-lined). BglII and KpnI digested PCR product was ligated into BamHI and KpnI sites of pRSET B to give pRSET B-ava_3858.
Construction of an expression plasmid for recombinant AroB
The aroB gene from Escherichia coli (aroBEc) was amplified by PCR from pJB14 (a gift from John W. Frost) using two primers (5′-TGG ATG CTC GAG TAT GGA GAG G-3' and 5'-CCT TTC GAA TTC TCA CGC TGA-3′, XhoI and EcoRI are under-lined). XhoI and EcoRI digested PCR product was ligated into the corresponding sites of pRSET B to give pRSET B-aroBEc. DNA sequences of all PCR amplified clones were confirmed by the Oregon State University Center for Genome Research and Biocomputing (CGRB) core lab.
Expression of valA, amir_2000, npun_5600, ava_3858, and aroBEc
Expression plasmids were used to transform E. coli BL21(DE3) pLysS. Transformants were grown overnight at 37 °C on LB agar plates containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol. A single colony was inoculated into 3 mL of LB medium and cultured at 37 °C for 6 h and then 1 mL of seed culture was transferred into 100 mL of LB medium in a 500 mL flask and grown at 28 °C until OD600 reached 0.3–0.5. Subsequently, the temperature was reduced to 18 °C and after 1 h adaptation 0.1 mM IPTG was added to induce production of the N-terminal hexahistidine-tagged recombinant proteins. After further growth for 16–20 h, the cells were harvested by centrifugation (5,000 rpm, 10 min, 4 °C) and stored at −80 °C until used.
Expression of staur_3140
The expression plasmid was used to transform E. coli BL21(DE3) pLysS. A transformant was grown overnight at 37 °C on an LB agar plate containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol. A single colony was inoculated into 3 mL of LB medium and cultured at 37 °C for 6 h and then 0.1 mL of seed culture was transferred into 100 mL of M9-glucose medium in a 500 mL flask and grown at 15 °C for 2 days until OD600 reached 0.3. Subsequently, 0.1 mM IPTG was added to induce production of the N-terminal hexahistidine-tagged Staur_3140 protein. After further grown for 24 h, the cells were harvested by centrifugation (5,000 rpm, 10 min, 4 °C) and stored at −80 °C until used.
Protein purification for enzyme assay
Cell pellets from 50 mL of culture were washed with binding (B) buffer (1 mL) (40 mM KPi, 300 mM NaCl, and 10 mM imidazole, pH 7.4) and centrifuged (8,000 rpm, 3 min, 4 °C). After removal of the supernatant, B buffer (1 mL) was added and the suspension sonicated (8 watts, 10 sec, 5 times). After centrifugation (14,500 rpm, 20 min, 4 °C), the supernatants (0.8 mL) were each mixed with B buffer-equilibrated Ni-NTA resin (QIAGEN) (0.2 mL) and incubated for 1 h at 4 °C. After incubation, the mixtures were centrifuged (4,000 rpm, 3 min, 4 °C) and the supernatants were discarded. One mL of washing (W) buffer [40 mM KPi, 300 mM NaCl, and 20 mM imidazole (50 mM imidazole in case of ValA and Staur_3140), pH 7.4] was added and the mixture was centrifuged (4,000 rpm, 3 min, 4 °C). This washing step was repeated three times. Subsequently, elution (E) buffer (0.5 mL) (40 mM KPi pH 7.4, 300 mM NaCl, and 500 mM imidazole, pH 7.4) was added and incubated at 4 °C for 20 min to eluted the desired proteins. This elution step was repeated twice. Eluted proteins were dialyzed against dialysis (D) buffer (1 L) (10 mM KPi, 0.1 mM DTT, and 1 mM NaF, pH 7.4) 3 times for 3 h each.
Protein purification for kinetic studies using Enzchek phosphate assays
Enzyme purification for kinetic determinations using the Enzchek phosphate assay kit (Invitrogen) was done similar to the procedure mentioned above except that B2 buffer (40 mM HEPES, 300 mM NaCl, 10% glycerol, and 10 mM imidazole, pH 7.4) was used for binding, W2 buffer [40 mM HEPES, 300 mM NaCl, 10% glycerol, and 20 mM imidazole (50 mM imidazole in case of ValA and Staur_3140), pH 7.4] for washing, E2 buffer (40 mM HEPES, 300 mM NaCl, 10% glycerol, and 500 mM imidazole, pH 7.4) for elution, and D2 buffer [10 mM Tris-HCl, 0.1 mM DTT, pH 7.0 (pH 7.5 in case of ValA)] for dialysis. The Ni-NTA purified proteins were analyzed by SDS-PAGE and concentrated by ultrafiltration using Amicon YM-10 (Millipore). Protein concentration was determined using the Bradford assay (BIO-RAD) with BSA as standard.
Enzyme Assays for TLC and GC-MS analyses
Each reaction mixture (20 μL, or 40 μL in case of reactions for GS-MS analysis) contains potassium phosphate buffer (20 mM, pH 7.4), NAD+ (1 mM), CoCl2 (0.2 mM), sedoheptu-lose 7-phosphate (5 mM), and enzyme (10–20 μM). The mixture was incubated at 30 °C for a total of 6 h. For TLC analysis, aliquots were taken at 2 h, 4 h, and 6 h. For GC-MS analysis, aliquots were taken at 3 h.
GC-MS sample preparation for ValA, Amir_2000, and Staur_3140 reaction products
Reaction mixtures (40 μL) were lyophilized and Sil-A (Sigma-Aldrich) (~80 μL) were added to the lyophilized samples and let stand for 20 min. The solvent was evaporated with argon gas and the products were extracted with hexanes (100 μL) and subjected to GC-MS analysis.
GC-MS condition
A HP GC-5890 series II and MS-5971 (Hewlett-Packard) system, HP-5ms (0.25 μm, Agilent technologies) and CycloSil-beta (0.25 μm, Agilent technologies) columns were used for analysis. Temperature gradient was set as 7 °C/min from 70 °C at 2 min until 300 °C for HP-5ms column and 5 °C/min from 50 °C at 2 min until 240 °C for CycloSil-beta column.
Reaction condition for monitoring product conversion by 1H NMR spectroscopy
Sedoheptulose 7-phosphate (Ba2+ salt, Carbosynth Ltd.) (2.0 mg) was dissolved in D2O (152.5 μL). Then, potassium phosphate buffer (12.5 μL of 0.4 M solution, pH 7.4), NAD+ (2.5 μL of 100 mM solution) and CoCl2 (2.5 μL of 20 mM solution), all in H2O, were added to the D2O solution mixture. The solution was loaded into Shigemi (BMS-003) tube and the enzyme solution (80 μL) was added to the mixture and mixed gently. The conversion of substrate to product was monitored by 1H NMR spectroscopy under water suppression condition (PL9 value 45.0) at 0 h, 0.5 h, 1 h, 3 h, 5 h. Temperature was set between 297.9 and 298.2 K and data acquisition was carried out with 64 scans for each time point.
Determination of the Km and kcat values of ValA, Amir_2000, Staur_3140, Npun_5600, and Ava_3858
A typical reaction mixture (100 μL) contained Tris-HCl (50 mM, pH 7.0 for Amir_2000, Staur_3140, Npun_5600, and Ava_3858, and pH 7.5 for ValA), NAD+ (200 μM), CoCl2 (50 μM), sedoheptulose 7-phosphate (0, 5, 10, 25, 50, 100, and 200 μM), enzyme (1 μM), 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG) (0.2 mM), purine nucleoside phosphorylase (PNP) (0.1 U). Optimal concentrations of the cofactors (NAD+ and CO2+) were first determined to obtain saturated amount that would give Vmax with no inhibition of the reaction. All components, except the substrate sedoheptulose 7-phosphate, were incubated at 28 °C for 10 min. Then, the reaction was initiated by addition of the substrate. The reaction was monitored at 360 nm every 8 sec. Kinetic constants were derived from a Hanes-Woolf plot.
High-resolution mass spectral analysis of Amir_2000 product
HRMS (ESI-TOF) m/z 191.0548 (calcd for C7H11O6 [M-H]− : 191.0556).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Heinz G. Floss for his comments and assistance in the preparation of this manuscript, Rolf Müller for providing Stigmatella aurantiaca DW4/3–1, Kerry McPhail and Justyna Sikorska for technical assistance. The project described was supported by grant R01AI061528 from the National Insti tute of Allergy and Infectious Diseases and by the Herman-Frasch Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health (NIH).
Footnotes
Supporting Information. Proteins and accession numbers used in alignment and phylogenetic studies (Table S1), TLC analysis of reaction products (Fig. S1), GC/MS analysis of tetrasilylated 2-epi-valiolone standard and Amir_2000, Staur_3140, and ValA reaction products (Figs S2–S3), 1H NMR spectrum of 2-epi-valiolone standard and in situ 1H NMR analyses of Staur_3140, and Ava_3858 reactions (Fig. S4), kinetic data (Fig. S5), 1H and 13C spectra of synthetic compounds generated in this study (Figs. S6 – S10). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Stratmann A, Mahmud T, Lee S, Distler J, Floss HG, Piepersberg W. J. Biol. Chem. 1999;274(16):10889–96. doi: 10.1074/jbc.274.16.10889. [DOI] [PubMed] [Google Scholar]
- (2).Yu Y, Bai L, Minagawa K, Jian X, Li L, Li J, Chen S, Cao E, Mahmud T, Floss HG, Zhou X, Deng Z. Appl. Environ. Microbiol. 2005;71(9):5066–76. doi: 10.1128/AEM.71.9.5066-5076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wu X, Flatt PM, Schlorke O, Zeeck A, Dairi T, Mahmud T. Chembiochem. 2007;8(2):239–48. doi: 10.1002/cbic.200600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Floss HG, Yu TW, Arakawa K. J. Antibiot. 2011;64(1):35–44. doi: 10.1038/ja.2010.139. [DOI] [PubMed] [Google Scholar]
- (5).Nango E, Kumasaka T, Hirayama T, Tanaka N, Eguchi T. Proteins. 2008;70(2):517–27. doi: 10.1002/prot.21526. [DOI] [PubMed] [Google Scholar]
- (6).Balskus EP, Walsh CT. Science. 2010;329(5999):1653–6. doi: 10.1126/science.1193637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Asamizu S, Yang J, Almabruk KH, Mahmud T. J. Am. Chem. Soc. 2011;133(31):12124–35. doi: 10.1021/ja203574u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Land M, Lapidus A, Mayilraj S, Chen F, Copeland A, Del Rio TG, Nolan M, Lucas S, Tice H, Cheng JF, Chertkov O, Bruce D, Goodwin L, Pitluck S, Rohde M, Goker M, Pati A, Ivanova N, Mavromatis K, Chen A, Palaniappan K, Hauser L, Chang YJ, Jeffries CC, Brettin T, Detter JC, Han C, Chain P, Tindall BJ, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP. Stand. Genomic Sci. 2009;1(1):46–53. doi: 10.4056/sigs.21137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huntley S, Hamann N, Wegener-Feldbrugge S, Treuner-Lange A, Kube M, Reinhardt R, Klages S, Müller R, Ronning CM, Nierman WC, Sogaard-Andersen L. Mol. Biol. Evol. 2011;28(2):1083–97. doi: 10.1093/molbev/msq292. [DOI] [PubMed] [Google Scholar]
- (10).Mahmud T, Tornus I, Egelkrout E, Wolf E, Uy C, Floss HG, Lee S. J. Am. Chem. Soc. 1999;121(30):6973–6983. [Google Scholar]
- (11).Bai L, Li L, Xu H, Minagawa K, Yu Y, Zhang Y, Zhou X, Floss HG, Mahmud T, Deng Z. Chem. Biol. 2006;13(4):387–97. doi: 10.1016/j.chembiol.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Choi WS, Wu X, Choeng YH, Mahmud T, Jeong BC, Lee SH, Chang YK, Kim CJ, Hong SK. Appl. Microbiol. Biotechnol. 2008;80(4):637–45. doi: 10.1007/s00253-008-1591-2. [DOI] [PubMed] [Google Scholar]
- (13).Whelan S, Goldman N. Mol. Biol. Evol. 2001;18(5):691–9. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- (14).Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mol. Biol. Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wu X, Flatt PM, Xu H, Mahmud T. Chembiochem. 2009;10(2):304–14. doi: 10.1002/cbic.200800527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xu H, Zhang Y, Yang J, Mahmud T, Bai L, Deng Z. Chem. Biol. 2009;16(5):567–76. doi: 10.1016/j.chembiol.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Floss HG, Lee S, Tornus I. U.S. Patent 6,150,568. 2000 Nov 21;
- (18).Upson RH, Haugland RP, Malekzadeh MN, Haugland RP. Anal. Biochem. 1996;243(1):41–5. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- (19).Carpenter EP, Hawkins AR, Frost JW, Brown KA. Nature. 1998;394(6690):299–302. doi: 10.1038/28431. [DOI] [PubMed] [Google Scholar]
- (20).August PR, Tang L, Yoon YJ, Ning S, Müller R, Yu TW, Taylor M, Hoffmann D, Kim CG, Zhang X, Hutchinson CR, Floss HG. Chem. Biol. 1998;5(2):69–79. doi: 10.1016/s1074-5521(98)90141-7. [DOI] [PubMed] [Google Scholar]
- (21).Yu TW, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG. Proc. Natl. Acad. Sci. U.S.A. 2002;99(12):7968–73. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mao Y, Varoglu M, Sherman DH. Chem. Biol. 1999;6(4):251–63. doi: 10.1016/S1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- (23).Rascher A, Hu Z, Buchanan GO, Reid R, Hutchinson CR. Appl. Environ. Microbiol. 2005;71(8):4862–71. doi: 10.1128/AEM.71.8.4862-4871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Flatt PM, Mahmud T. Nat. Prod. Rep. 2007;24(2):358–392. doi: 10.1039/b603816f. [DOI] [PubMed] [Google Scholar]
- (25).Mahmud T. Curr Opin Chem Biol. 2009;13(2):161–70. doi: 10.1016/j.cbpa.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zucko J, Dunlap WC, Shick JM, Cullum J, Cercelet F, Amin B, Hammen L, Lau T, Williams J, Hranueli D, Long PF. BMC Genomics. 2010;11:628. doi: 10.1186/1471-2164-11-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kim CG, Kirschning A, Bergon P, Zhou P, Su E, Sauerbrei B, Ning S, Ahn Y, Breuer M, Leistner E, Floss HG. J. Am. Chem. Soc. 1996;118:7486–7491. [Google Scholar]
- (28).Kudo F, Hosomi Y, Tamegai H, Kakinuma K. J. Antibiot. 1999;52(2):81–8. doi: 10.7164/antibiotics.52.81. [DOI] [PubMed] [Google Scholar]
- (29).Nango E, Eguchi T, Kakinuma K. J. Org. Chem. 2004;69(3):593–600. doi: 10.1021/jo034706y. [DOI] [PubMed] [Google Scholar]
- (30).Schmidt EW. Chembiochem. 2011;12(3):363–5. doi: 10.1002/cbic.201000709. [DOI] [PubMed] [Google Scholar]
- (31).Arakawa K, Müller R, Mahmud T, Yu TW, Floss HG. J. Am. Chem. Soc. 2002;124(36):10644–5. doi: 10.1021/ja0206339. [DOI] [PubMed] [Google Scholar]
- (32).Guo J, Frost JW. J. Am. Chem. Soc. 2002;124(4):528–9. doi: 10.1021/ja016963v. [DOI] [PubMed] [Google Scholar]
- (33).Allen MB, Arnon DI. Plant. Physiol. 1955;30(4):366–72. doi: 10.1104/pp.30.4.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.