

Abstract
The synthetic caged Garcinia xanthone, cluvenone, has potent and selective cytotoxicity against numerous cancer cell lines including those that are multi-drug resistant. The direct target of this structurally and functionally unique agent is unknown and that of the parent natural product, gambogic acid (GA), presently in clinical trials, is not yet entirely clear. For the first time, using fluorescently labeled GA (GA-Bodipy), we determined that GA-Bodipy localized in mitochondria and was effectively displaced by cluvenone in competition experiments indicating that the direct target of cluvenone resided in mitochondria and was shared by GA. In agreement with these findings, treatment of HeLa cells with cluvenone or GA resulted in disruption of mitochondrial morphology within 4 h. Furthermore, experiments using the potential sensitive JC-1 dye demonstrated that cells treated with 1 μM cluvenone for 1 h had significant loss of MMP compared to control cells. Examination of Cyt c levels in leukemia cells treated with 1 μM cluvenone resulted in a 4-fold increase in levels of both cytosolic and mitochondrial Cyt c. In agreement with Cyt c release, caspase 9 activity was increased 2.6-fold after treatment of cells for 5 h with 1 μM cluvenone. Remarkably, the caspase-9 inhibitor, Z-LEHD-FMK, blocked cluvenone-induced apoptosis in a dose-dependent manner with apoptosis being completely blocked by 10 μM of the inhibitor. In conclusion, cluvenone, an agent with potent cytotoxicity against multi-drug resistant tumor cells, has direct targets in mitochondria thus setting precedence for drug discovery efforts against these targets in the treatment of refractory cancers.
Keywords: Cluvenone, mitocan, mitochondria, apoptosis, Garcinia, xanthone
Introduction
Despite the recent advances in molecular medicine, neoplastic disease remains a significant challenge and is the second leading cause of death in developed countries [1]. The main obstacle to successful treatment is the development of resistance to current chemotherapeutic agents resulting from the occurrence of cellular alterations allowing tumor cells to adapt and escape the toxicity of current chemotherapeutic agents. Hence, it is essential to investigate novel agents with unique mechanism(s) of action that are selective for tumor cells.
Mitochondria are the cells main energy producer and thus are essential for maintaining cell survival. However, mitochondria also play a major role in cell death and contain many of the important mediators of apoptosis. Mitochondrial membrane permeabilization resulting in the dissipation of the mitochondrial membrane potential, required for many of the functions of mitochondria, is the key event leading to apoptosis and is a point of no return [2]. Because of the important role of mitochondria in cell death as well as the bio-energetic differences between normal and cancer cell mitochondria, the mitochondrion has recently emerged as a promising target in the treatment of cancer [3]. Importantly, directly targeting mitochondria offers the advantage to initiate apoptosis independently of upstream signal transduction elements that are frequently impaired in human cancers, thus bypassing some mechanisms of chemo-resistance. Lastly, mitochondria being an invariant target common to all cells and in which mutations are very rare, suggests a low probability for acquired resistance to agents directed at mitochondria [4].
Agents that target mitochondria to exert their anti-cancer effects are collectively termed mitocans (4). Such agents often act by interfering with the energy-generating processes in the mitochondria of malignant cells leading to accumulation of elevated levels of ROS that in turn destabilize mitochondria to induce the intrinsic pathway of apoptosis [4, 5]. Many cancer cells have reduced levels of antioxidant enzymes such as the manganese superoxide dismutase (MnSOD) and are subjected to higher levels of oxidative stress than normal cells [6]. This characteristic renders cancer cells a lower tolerance for ROS and an increased susceptibility to apoptosis by mitocans that induce ROS formation, making such agents attractive for development as chemotherapeutics.
Natural products have been used to combat disease for thousands of years and have demonstrated potent anti-tumor properties [7]. The Garcinia genus of tropical plants has yielded a structurally intriguing family of caged xanthone-derived natural products that have antimicrobial and anticancer properties and a documented value in traditional eastern medicine [8]. We have recently demonstrated that a synthetic analogue of the caged Garcinia xanthone family of natural products, cluvenone, was an effective inducer of apoptosis in numerous tumor cell types [9, 10] and was equally cytotoxic to multi-drug resistant cells as the parental cell lines. The direct cellular target of this agent is not known and that of the parent natural product, GA, is not entirely clear. We present the results of studies examining the direct cellular target and pathway of apoptosis induced by cluvenone. Importantly, we provide evidence that mitochondria, when targeted directly, may bypass mechanisms of chemo-resistance. Secondly. We demonstrate, for the first time, that the direct targets of representatives of the caged Garcinia xanthones reside in mitochondria challenging the belief of the transferrin receptor being a direct target.
Material and Methods
Cell Lines
T-cell acute lymphoblastic leukemia cells, CEM, were purchased from ATTC in 2008. The multi-drug resistant promyelocytic leukemia cells, HL60/ADR, were a gift from Dr. Michael J. Kelner. These leukemia cell lines were authenticated by observation of morphology and/or by measuring sensitivity to known agents and then comparing the IC50 for cytotoxicity to that reported in the literature. This testing is performed routinely in our laboratory. The prostate cancer cell line, PC3, was obtained from ATCC in 2000. These cells were authenticated by observation of their morphology. HeLa cells were obtained from Dr. Olivier Schwartz (Pasteur Institute) and maintained in DMEM supplemented with 10% FBS and 100 units/ml penicillin/streptomycin (complete DMEM). The HeLa cells were not authenticated in our laboratory. CEM, HL60/ADR, and PC3 cells were maintained in RPMI medium supplemented with 10% FBS, 2mM glutamine, and 100 units/ml penicillin/streptomycin (complete RPMI).
Examination of anti-proliferative activity of GA-Bodipy
The synthesis of GA-Bodipy was as described by us previously [9]. The anti-proliferative effect of GA-Bodipy was tested in a 3H-thymidine incorporation assay using HL60/ADR cells as described previously [9, 11].
Fluorescence microscopy
Intracellular localization studies
Hela cells (1 × 106) were placed on glass cover slips and were incubated with Mitotracker Red CMX Ros (Invitrogen, Carlsbad, CA) at a final concentration of 50 nM for 15 min. Cells were then fixed with 2% Formaldehyde for 2 min and treated with 1 μM GA-Bodipy for 30 min, followed by extensive washing in PBS. Stained cells were examined by fluorescence microscopy with excitation at 579 nm and emission at 599 nm using a Zeiss Observer Z1 inverted microscope with 63× objective controlled by AxioVision software (Zeiss, Thornwood, NY). For competition studies with cluvenone, HeLa cells were treated as above and further incubated in 0.1% DMSO or 20μM cluvenone for 1h then washed and visualized as above.
Determination of mitochondrial depolarization and loss of structural integrity
HeLa cells placed on cover slips were treated with 0.1% DMSO or 1 μM cluvenone for 1h followed by incubation with 5 μM JC-1 (a gift from Dr. Fabrizia Stavru) for 15min. The potential sensitive dye, JC-1, exists as aggregates which emit at 590 nm (red fluorescence) when concentrated in mitochondria having intact membrane potential. In depolarized mitochondria JC-1 exists as a monomer and yields green fluorescence (530 nm). Fluorescence was measured using a Zeiss Observer Z1 inverted microscope (excitation at 490 nm).
In studies examining mitochondrial structural integrity, HeLa cells were treated with 1 μM of each of cluvenone or GA (positive control), or with 0.1% DMSO (negative control) for 4 h. The cells were then fixed with 4% paraformaldehyde for 10 min followed by permeabilization with 0.1% Triton X-100 in PBS. Non-specific binding of antibodies was blocked by incubation with PBS containing 5% fetal goat serum (blocking buffer). Primary antibody, anti-Tom 20 (BD Bioscience, San Diego, CA), was resuspended in blocking buffer (1:500 dilution) and added to the cells for 1h at room temperature. Following three washes in PBS, the secondary antibody, Alexa Fluor 488 (excitation 495 nm/emission 519 nm) conjugated goat anti-mouse IgG (Invitrogen, Carlsbad, CA), was added to cells for 1h at a dilution of 1:500 in blocking buffer. The cells were then washed in PBS and stained with Hoescht (Invitogen, Carlsbad, CA) (1:100,000 in PBS) to stain DNA. Coverslips were mounted onto glass slides with Aqua-Poly/Mount (Polysciences Inc., Warrington, PA) and cells were visualized using a Zeiss Observer Z1 inverted microscope with 63× objective controlled by AxioVision software (Zeiss, Thornwood, NY).
Measurement of Cytochrome c
CEM cells were plated at 10–15 × 106 cells/T-75 flask and treated with 1μM cluvenone or 0.1% DMSO for 7 h before harvesting. Cytosolic and mitochondrial extracts were generated using a digitonin-based subcellular fractionation kit (Enzo Life Sciences, Plymouth Meeting, PA) as recommended by manufacturer. Briefly, cell pellets were resuspended in 300μl of digitonin cell permeabilization buffer and placed on ice for 5 min. Suspensions were then centrifuged at 1000 g for 5 min at 4°C. Supernatant containing the cytosolic fraction was removed and stored at −80°C until further use. The remaining pellet was resuspended in 250μl RIPA cell lysis buffer and placed on ice for 5 min before centrifuging at 10,000g for 10 min at 4°C to obtain the supernatant containing the mitochondrial fraction. Cytochrome c (Cyt c) in cell extracts was measured by an ELISA assay using the cytochrome c EIA kit (Enzo Life Sciences) according to manufacturer’s recommendations. All conditions were performed in duplicate and absorbance was measured in a Synergy Mx microplate reader (BioTek Instruments Inc., Winooski, VT) at 405 nm with correction at 580 nm. Levels of Cyt c in samples were determined by comparison to standards provided in the kit and all values were reported per 10 × 106 cells. Background was determined in the absence of extract.
Measurement of caspase-9 activity
Caspase-9 activity was measured using a colorimetric assay kit from BioVision (Mountain View, CA) according to the manufacturer’s recommendations. Briefly, CEM cells were plated at 5 × 106 cells/well in a 6-well plate in complete RPMI. Cells were then treated with 0.1% DMSO or 1 μM cluvenone for 5 h before harvesting and extraction of cell protein. Cell lysates were incubated with caspase-9 substrate for 2 h at 37°C and resulting color was measured in a Synergy Mx microplate reader (BioTek Instruments Inc., Winooski, VT) at 405 nm. Background was determined in the absence of substrate and subtracted from values obtained from untreated and treated cells. All conditions were performed in triplicate.
Determination of the effect of a Caspase-9 inhibitor on apoptosis induced by cluvenone
PC3 cells were plated at 10,000 cells\well (96-well plate) in complete RPMI and allowed to attach overnight. Following day, cells were pre-treated with increasing concentrations of the caspase 9 inhibitor, Z-LEHD-FMK (BD Biosciences, Bedford, MA) for 30 min prior to treatment with 1.5μM cluvenone. Control cells received 0.2% DMSO, cluvenone alone, or Z-LEHD-FMK alone. After a 7 h incubation with the additions, cells were lysed and cytoplasmic fractions were obtained after centrifugation at 200 × g for 10 min. Apoptosis was measured by a photometric enzyme-immunoassay which measures cytoplasmic histone-associated-DNA fragments with the Cell Death Detection ELISAplus kit (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s recommendations.
Results
In order to gain information on the possible cellular targets of cluvenone, a fluorescent analog (GA-Bodipy) of the parent molecule, GA, known to induce the intrinsic pathway of apoptosis, was synthesized since cluvenone was not amenable to functionalization. In this way, it was possible to visualize not only the sub-cellular component(s) to which GA localizes but, more importantly, to determine potential sub-cellular targets of cluvenone that are shared with GA via competition experiments. GA-Bodipy was first tested for bioactivity in a proliferation assay to ensure that it had not lost activity due to labeling. In a 3H-thymidine incorporation assay, GA-Bodipy was similarly cytotoxic to GA and cluvenone with an IC50 of 0.6 μM in HL60/ADR cells indicating that GA-Bodipy retained biological activity [9]. To determine the intracellular localization of GA-Bodipy, HeLa cells were stained with the mitochondrial dye MitoTracker Red and then fixed prior to treatment with 1 μM GA-Bodipy. As shown in Figure 1, the green fluorescence due to GA-Bodipy clearly co-localized with MitoTracker Red indicating that GA-Bodipy localized in the mitochondria. In addition, incubation of cells with GA-Bodipy resulted in a diffuse, bright, peri-nuclear signal which could not be ascribed to any specific intra-cellular structure. At this point, it is not clear whether this signal represents a nonspecific cytosolic accumulation or indicates the presence of additional GA targets.
Figure 1.

GA-Bodipy localizes to mitochondria. HeLa cells were incubated with MitoTracker Red at 50 nM for 15 min. Cells were then fixed and treated with GA-Bodipy at 1uM for 30 min, followed by washing with PBS. Stained cells were then examined by fluorescence microscopy with excitation at 579 nm and emission at 599 nm. Mitochondria (c) are shown in red and BODIPY (b) in green.
The localization of GA-Bodipy in mitochondria led to the question of whether cluvenone also localized to the mitochondria and shared targets with GA. To address this question, competition experiments were conducted in which cells stained with MitoTracker Red and GA-Bodipy, were then incubated with cluvenone or DMSO as control. In the presence of DMSO, GA-Bodipy maintained mitochondrial localization (Figure 2a, b, c). However, in the presence of cluvenone, GA-Bodipy was significantly displaced (Figure 2d, e, f) suggesting that GA and cluvenone share targets in the mitochondria. The finding that cluvenone localized in the mitochondria suggested that it may disrupt mitochondrial function and trigger the mitochondrial pathway of apoptosis.
Figure 2.

Cluvenone competes with GA-Bodipy for sites in mitochondria. HeLa cells were incubated with MitoTracker Red at 50 nM for 15 min and then fixed. Fixed cells were then treated with 1 μM GA-Bodipy for 30 min followed by washing. Cells were then further incubated with 0.1% DMSO (a, b, c) or 20μM cluvenone (d, e, f) for 1h followed by washing. Fluorescence microscopy was performed with excitation at 579 nm and emission at 599 nm. Mitochondria (red) are shown in c) and f). BODIPY fluorescence (green) is shown in b) and e). Merged images are in a) and d).
To determine whether cluvenone disrupted mitochondrial function, mitochondrial membrane potential was measured qualitatively in HeLa cells treated with cluvenone or DMSO using the mitochondrial membrane potential (MMP) sensitive dye JC-1 (Fig. 3, top panel). Regions of high mitochondrial polarization are indicated by red fluorescence due to aggregate formation by the JC-1 dye. Depolarized regions are indicated by the green fluorescence of the JC-1 monomers. The results of fluorescence microscopy experiments using JC-1 show that cells treated with cluvenone (Figure 3 Top panel d, e, f) display a greater level of green fluorescence and less red fluorescence compared to control cells (Figure 3 Top panel a, b, c) after 1h treatment. These observations indicate that mitochondria in cells treated with cluvenone have lost MMP and have become depolarized.
Figure 3.

Loss of mitochondrial membrane potential and structural integrity in cells treated with cluvenone. Mitochondrial membrane potential was examined in HeLa cells after treatment with 0.1% DMSO (a, b, c), or 1 μM cluvenone (d, e, f) for 1h, followed by staining with JC-1 for 15min. Cells with high membrane potential promote the formation of JC-1 aggregates which fluoresce red (a, d). In cells with low potential monomeric JC-1 fluoresces green (b, e). Yellow color in merged images (c, f) indicates depolarized mitochondria (Top Panel). Structural integrity of mitochondria was examined in HeLa cells treated for 4 h with 0.1% DMSO (a), or with 1 μM each of GA (b), or cluvenone (c). Fixed and permeabilized cells were then stained for the mitochondrial protein Tom 20 (green) as well as for nuclei, shown in blue (Bottom Panel).
Additional evidence suggesting that cluvenone disrupts mitochondrial function comes from examination of mitochondrial morphology by immuno-fluorescence staining of the outer mitochondrial membrane receptor protein, Tom20, in cells treated with cluvenone, GA, or DMSO. HeLa cells treated for 4 h with 1 μM cluvenone or GA revealed a dramatic change in mitochondrial morphology compared to control cells (Figure 3, bottom panel). The complex branching network of tubular shaped mitochondria was converted into vesicular shaped organelles. Longer incubations resulted in an even more dramatic phenotype with a decrease in size and a reduction in the number of mitochondria (data not shown). In summary, the fluorescent analog of GA was found to localize in mitochondria which could be displaced by cluvenone suggesting shared mitochondrial targets between GA and cluvenone. Furthermore, cluvenone disrupted mitochondrial morphology and resulted in mitochondrial depolarization. These results are in agreement to that obtained with GA in human hepatoma cells [12].
Since cluvenone was found to depolarize mitochondria, it was hypothesized that it induced apoptosis through the mitochondrial pathway. Hence, the classic indicators of the mitochondrial pathway of apoptosis including the release of Cyt c and activation of caspase 9, were examined. Cyt c was measured by an ELISA in both the cytosolic and mitochondrial fractions of cell lysates obtained from CEM cells treated with 1 μM cluvenone or 0.2% DMSO for 7 h. The results demonstrated that the levels of both cytosolic and mitochondrial Cyt c were increased at least 4-fold in CEM cells treated with cluvenone (Figure 4). This observation indicated that cluvenone increased the expression of Cyt c of which, a portion, was released into the cytoplasm as part of the process of apoptosis. This finding is supported by Sanchez-Alcazar et al. [13] who reported that teniposide and various other chemotherapeutic agents induced a dose-dependent increase in the expression of Cyt c and other mitochondrial respiratory chain proteins, and that Cyt c was released into the cytoplasm only after maximal levels had been reached in the mitochondria. An immuno-fluorescence based assay was performed to further confirm that Cyt c was released from the mitochondria. In these experiments, HeLa cells treated with 1 μM cluvenone for 4h were stained for the mitochondrial marker Tom20 and Cyt c. While Tom20 staining revealed extensively damaged, rounded, and swollen mitochondria, Cyt c appeared as diffuse cytosolic staining (data not shown).
Figure 4.
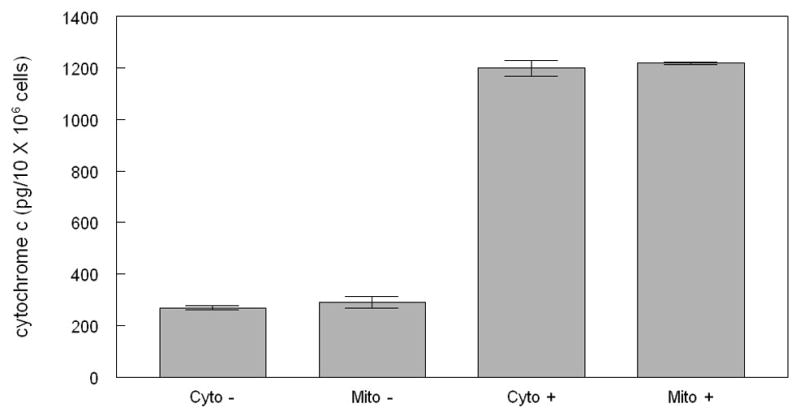
Cluvenone increases total Cyt c levels and induces cytosolic release in CEM cells. Cells were treated with 1 μM cluvenone or 0.1% DMSO for 7 h, followed by subcellular fractionation using a digitonin-based extraction method. Cytochrome c in cell extracts was measured by an ELISA assay with absorbance measured at 405 nm with correction at 580 nm. All conditions were performed in duplicate. Levels of Cyt c in samples was determined by comparison to standards and all values were reported per 10 × 106 cells. Cyto −, cytosolic extract from untreated cells; Mito −, mitochondrial extract from untreated cells; Cyto +, cytosolic extract from cells treated with cluvenone; Mito +, mitochondrial extract from cells treated with cluvenone.
The activity of caspase 9, one of the hallmarks of the mitochondrial pathway of apoptosis, was measured in CEM cells cultured in the presence and absence of cluvenone. After treatment of cells for 5 h with 1 μM cluvenone, caspase-9 activity was measured in a colorimetric assay using the caspase 9-specific substrate, LEHD-pNA. The results presented in Figure 5 demonstrated that the activity of caspase-9 was increased 2.6-fold upon treatment of cells with cluvenone for 5 h. These results confirmed those of an earlier experiment in which cluvenone induced the activity of caspase-9 3.4-fold (data not shown), slightly greater the induction (2.3- fold induction) by an equal concentration of the serine/threonine kinase inhibitor and inducer of apoptosis, UCN01.
Figure 5.
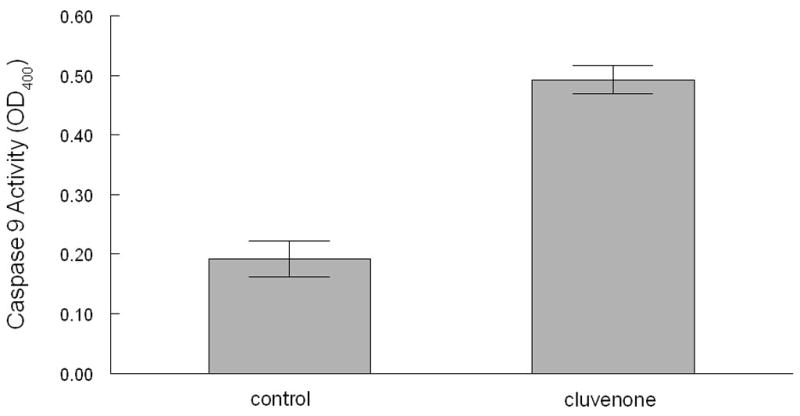
Activation of caspase 9 by cluvenone in CEM cells. CEM cells were treated with 0.1% DMSO or 1 μM cluvenone for 5 h and cell protein was extracted. Caspase 9 activity was measured using a colorimetric assay in which cell lysates were incubated with caspase−9 substrate for 2 h at 37°C and absorbance was measured at 405 nm. All conditions were performed in triplicate and error bars represent SD.
To further confirm that cluvenone induces apoptosis through the mitochondrial pathway, a caspase-9 inhibitor was used to block the induction of apoptosis by cluvenone. In these experiments, PC3 cells were pre-treated with increasing concentrations of the caspase-9 inhibitor, Z-LEHD-FMK, followed by treatment with 1.5μM cluvenone for 7 h prior to measuring apoptosis by a photometric enzyme-immunoassay which measures cytoplasmic histone-associated-DNA fragments. The results presented in Figure 6 demonstrated that the caspase-9 inhibitor blocked cluvenone-induced apoptosis in a dose-dependent manner. Apoptosis was completely blocked with 10 μM of the caspase-9 inhibitor and was equal to background levels of control cells (0.2% DMSO ± Z-LEHD-FMK). These results suggest that the mitochondrial pathway is the predominant mechanism of apoptosis elicited by cluvenone in PC3 cells and, presumably, in Hela cells as well since cluvenone clearly localized in the mitochondria (Figures 1, 2). Whether the mitochondrial pathway is the main mechanism of apoptosis induced by cluvenone in other types of tumor cells remains to be determined.
Figure 6.
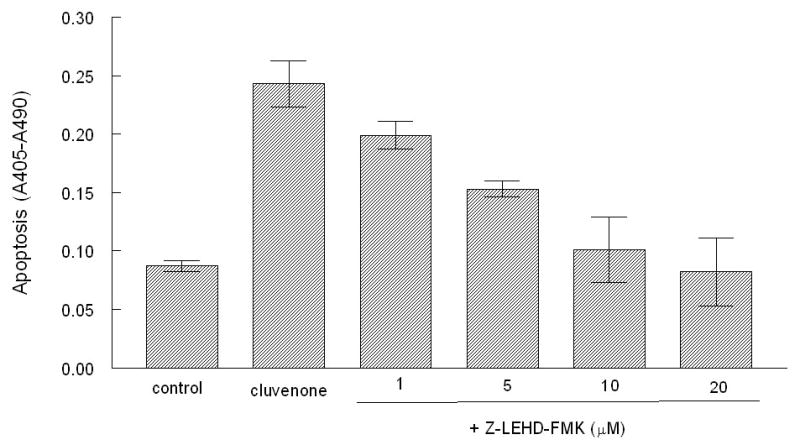
The caspase-9 specific inhibitor blocks apoptosis induced by cluvenone. PC3 cells were pre-treated with increasing concentrations of the caspase 9 inhibitor, Z-LEHD-FMK for 30 min prior to treatment with 1.5 μM cluvenone for 7 h. Control cells received 0.2% DMSO, cluvenone alone, or Z-LEHD-FMK alone. Apoptosis was measured by the Cell Death Detection ELISAplus kit. All conditions were performed in triplicate and error bars represent SD.
Discussion
Our current results demonstrate that cluvenone localizes in the mitochondria by displacing GA-Bodipy and disrupts the function of mitochondria as evidenced by loss of mitochondrial structural integrity and membrane potential. In agreement with these results, cluvenone induced the release of Cyt c into the cytoplasm and activated caspase-9. Furthermore, blocking the activity of caspase-9 with the caspase-9 specific inhibitor, Z-LEHD-FMK, inhibited the induction of apoptosis by cluvenone. Collectively, these results confirm that cluvenone induces apoptosis through the mitochondrial pathway. More importantly, however, our results indicate for the first time that the direct target(s) of cluvenone resides within the mitochondria and is shared with GA.
In recent years several unrelated natural products have been found to elicit their cytotoxic effects by targeting mitochondria, thereby acting as a mitocans. For instance, GA and α-mangostin from the Garcinia genus of plants are known to activate the mitochondrial pathway of apoptosis by disrupting mitochondrial function [12, 14]. Furthermore, unrelated natural products or their analogs including betulinic acid [15], fenretinide [16], α-tocopheryl succinate (α-TOS) [17], and resveratrol [18] have also been found to destabilize mitochondria and induce apoptosis. Mitocans as such can target mitochondria at various sites to disrupt their function. These targets include the Bcl-2 family of proteins, the mitochondrial inner membrane, voltage-dependent anion channels, electron transport chain proteins, hexokinases I and II, and several others [4].
One of the best studied mitocans whose targets have been clearly identified includes the vitamin E analog, α-TOS. α-TOS targets complex II of the respiratory chain to displace ubiquinone binding thus interrupting electron flow and resulting in the generation of ROS, which in turn, destabilizes the mitochondria [17, 19]. In addition, α-TOS has also been found to bind the Bcl-2 family of proteins thus acting as a BH3 mimetic [20]. Interestingly, GA was also recently reported to bind to the Bcl-2 family of mitochondrial proteins [21] and neutralize their effects. Because cluvenone not only localizes in the mitochondria and appears to share targets with GA, but like α-TOS and GA, also induces ROS formation and destabilizes mitochondria to activate the mitochondrial pathway of apoptosis, the complexes of the electron transport chain and the Bcl-2 family of proteins are likely targets of cluvenone. The fact that cluvenone induced ROS formation requires more than 2 h [10] in contrast to the agent elesclomol [22] which induces ROS within 30 min, presumably by directly stimulating ROS-generating enzymes in the cytoplasm, is consistent with the induction of ROS formation by cluvenone as a consequence of the disruption of the mitochondria, specifically, in the electron transport chain. The role of cluvenone as a BH3 mimetic has yet to be demonstrated and may be secondary to its role as an inducer of ROS but would enhance cluvenone’s apoptogenic potential. We are currently investigating the potential specific targets of cluvenone in the mitochondria.
Our current findings, in combination with those of our previous report, highlight the potential of cluvenone as an effective chemotherapeutic agent and provide a potential basis for cluvenone’s cancer cell selectivity and ability to circumvent chemo-resistance. The induction of ROS has been shown to initiate apoptosis preferentially in cancer cells since, unlike normal cells, cancer cells are under high oxidative stress often having limited anti-oxidant capacity and thus, have a lower tolerance to oxidative stress [6]. Furthermore, ROS production can activate the proapoptotic proteins Bax and Bak by allowing them to dimerize and mobilize to the mitochondrial outer membrane [23]. Lastly, the potential activity of cluvenone as a BH3 mimetic would allow the release of Bax and Bak from the Bcl-2 family of proteins known to be over expressed in many cancer cells providing a source of chemo-resistance. In conclusion, our results indicate that cluvenone, an agent with potent and selective cytotoxicity against multi-drug resistant cancer cells, has direct targets in mitochondria underscoring the therapeutic value of mitochondrial targets in the treatment of refractory cancers, thus, setting precedence for drug discovery efforts against these targets.
Acknowledgments
We gratefully acknowledge Drs. Robert Naviaux, Gordon Alton, and James Zapf for their critical review of the manuscript and Lisa Barnhill for her technical assistance.
This work was supported by an NIH grant (CA 133002) awarded to Emmanuel Theodorakis and, in part, by fund from Academia Sinica (A. L. Yu).
Abbreviations
- GA
gambogic acid
- MMP
mitochondrial membrane potential
- Cyt
cytochrome
- ROS
reactive oxygen species
Footnotes
Conflict of Interest: None
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong JS. Mitochondria: a target for cancer therapy. Br J Pharmacol. 2006;147:239–248. doi: 10.1038/sj.bjp.0706556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Moreno-Sánchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol Aspects Med. 2010;31:29–59. doi: 10.1016/j.mam.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Ralph SJ, Neuzil J. Mitochondria as targets for cancer therapy. Mol Nutr Food Res. 2009;53:9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- 5.Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Saavedra E, Moreno-Sánchez R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation – Why mitochondria are targets for cancer therapy. Mol Aspects Med. 2010;31:145–170. doi: 10.1016/j.mam.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Borrello S, De Leo ME, Galeotti T. Defective gene expression of MnSOD in cancer cells. Mol Aspects Med. 1993;14:253–8. doi: 10.1016/0098-2997(93)90012-3. [DOI] [PubMed] [Google Scholar]
- 7.Deorukhkar A, Krishnan S, Sethi G, Aggarwal BB. Back to basics: how natural products can provide the basis for new therapeutics. Expert Opin Investig Drugs. 2007;16:1753–73. doi: 10.1517/13543784.16.11.1753. [DOI] [PubMed] [Google Scholar]
- 8.Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. Chemistry and biology of the caged Garcinia xanthones. Chemistry. 2010;16:9944–62. doi: 10.1002/chem.201000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chantarasriwong O, Cho WC, Batova A, Chavasiri W, Moore C, Rheingold AL, et al. Evaluation of the pharmacophoric motif of the caged Garcinia xanthones. Org Biomol Chem. 2009;7:4886–94. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batova A, Altomare D, Chantarasriwong O, Ohlsen KL, Creek KE, Lin YC, et al. The synthetic caged garcinia xanthone cluvenone induces cell stress and apoptosis and has immune modulatory activity. Mol Cancer Ther. 2010;9:2869–78. doi: 10.1158/1535-7163.MCT-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batova A, Lam T, Wascholowski V, Yu AL, Giannis A, Theodorakis EA. Synthesis and evaluation of caged Garcinia xanthones. Org Biomol Chem. 2007;5:494–500. doi: 10.1039/b612903j. [DOI] [PubMed] [Google Scholar]
- 12.Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu W, et al. Reactive oxygen species accumulation contributes to gambogic acid-induced apoptosis in human hepatoma SMMC-7721 cells. Toxicology. 2009;260:60–7. doi: 10.1016/j.tox.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Sánchez-Alcázar JA, Khodjakov A, Schneider E. Anticancer drugs induce increased mitochondrial cytochrome c expression that precedes cell death. Cancer Res. 2001;61:1038–1044. [PubMed] [Google Scholar]
- 14.Matsumoto K, Akao Y, Yi H, Ohguchi K, Ito T, Tanaka T, et al. Preferential target is mitochondria in alpha-mangostin-induced apoptosis in human leukemia HL60 cells. Bioorg Med Chem. 2004;12:5799–806. doi: 10.1016/j.bmc.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 15.Fulda S, Kroemer G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov Today. 2009;14:885–90. doi: 10.1016/j.drudis.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Hail N, Jr, Kim HJ, Lotan R. Mechanisms of fenretinide-induced apoptosis. Apoptosis. 2006;11:1677–1694. doi: 10.1007/s10495-006-9289-3. [DOI] [PubMed] [Google Scholar]
- 17.Neuzil J, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, et al. Mitocans as anti-cancer agents targeting mitochondria: lessons from studies with vitamin E analogues, inhibitors of complex II. J Bioenerg Biomembr. 2007;39:65–72. doi: 10.1007/s10863-006-9060-z. [DOI] [PubMed] [Google Scholar]
- 18.Dörrie J, Gerauer H, Wachter Y, Zunino SJ. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 2001;61:4731–9. [PubMed] [Google Scholar]
- 19.James AM, Cochemé HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 20.Shiau CW, Huang JW, Wang DS, Weng JR, Yang CC, Lin CH, et al. alpha-Tocopheryl succinate induces apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 function. J Biol Chem. 2006;281:11819–25. doi: 10.1074/jbc.M511015200. [DOI] [PubMed] [Google Scholar]
- 21.Zhai D, Jin C, Shiau CW, Kitada S, Satterthwait AC, Reed JC. Gambogic acid is an antagonist of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2008;7:1639–46. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7:2319–27. doi: 10.1158/1535-7163.MCT-08-0298. [DOI] [PubMed] [Google Scholar]
- 23.D'Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C, et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 2005;19:1504–6. doi: 10.1096/fj.04-3329fje. [DOI] [PubMed] [Google Scholar]