

Abstract
Introduction
Obstructive sleep apnea (OSA) is associated with increased risk for cardiovascular and metabolic dysfunction in both adults and children. In adults with OSA, serum levels of macrophage migration inhibitory factor (MIF) are elevated. Therefore, we assessed plasma MIF levels and MIF allelic variant frequencies in children with and without OSA.
Methods
A total of 614 consecutive children ages 5–8 years were recruited. Children were divided into those with OSA and without OSA (NOSA) based on the apnea-hypopnea index (AHI). In addition to lipid profile, hsCRP, and fasting insulin and glucose levels, plasma MIF levels were assayed using ELISA, and 28 single-nucleotide polymorphisms (SNPs) covering the region were genotyped. Linkage disequilibrium and haplotype blocks were analyzed using Haploview version 4.2 software.
Results
Morning plasma MIF levels were increased in children with OSA. Of the 28 SNPs tested, the frequency of rs10433310 minor allele was significantly decreased in OSA. This SNP was also associated with reduced fasting insulin and hsCRP levels in OSA. The minor allele frequency of all other 27 SNPs was similar in OSA and NOSA groups.
Conclusions
Childhood OSA is associated with higher plasma MIF, hsCRP, and fasting insulin levels that promote cardiometabolic risk, and the MIF gene SNP rs10433310 may account for some the variance in such risk.
INTRODUCTION
Obstructive sleep apnea (OSA) is a highly prevalent and potentially serious health problem in children 1–3. Over the course of the last decade, it has become increasingly apparent that OSA in children is associated with an increased risk for metabolic and cardiovascular complications 4–7. Recently, we showed that a number of genes were differentially expressed in peripheral blood leukocytes of non-obese children with OSA, particularly those underlying inflammatory pathways, suggesting that OSA activates widespread pro-inflammatory networks that may play a role in the end-organ morbidity associated with this condition, 8,9. However, although the presence of OSA potentially increases the levels of inflammatory mediators that promote development of endothelial and metabolic dysfunction in children 10,11, not every child with OSA will present such abnormalities, suggesting that individual genetic susceptibility may underlie the variance of cardiometabolic and inflammatory phenotypes.
In adult OSA, a recent study showed that the plasma levels of macrophage migration inhibitory factor (MIF) are elevated and associated with OSA severity 12. MIF is an important pro-inflammatory cytokine involved in many acute and chronic inflammatory disorders including cardiovascular diseases 13. MIF has a chemokine-like function and promotes the directed migration and recruitment of leukocytes into infection and inflammatory sites 14. Furthermore, MIF is expressed by a broad variety of cells and tissues, including monocytes/macrophages and B-and-T cells 15. Increasing evidence suggests that MIF controls metabolic and inflammatory processes underlying the development of metabolic disorders such as glucose homeostasis during periods of stress, and macrophage infiltration into adipose tissue 16,17, i.e. major metabolic risk determinants for cardiovascular and metabolic disease 18,19. In addition, MIF plays a pivotal role in the pathogenesis of inflammatory disorders by promoting and amplifying involved inflammatory reactions such as monocyte/macrophage survival, MAPK signaling, or inflammatory cytokine release 13,15,20. Furthermore, MIF is a multifunctional cytokine, and has been implicated in a wide range of pathologies, such as inflammatory disorders and autoimmune, artherosclerosis, wound healing, and metabolic disorders 13,21,22. It has been shown that MIF can be induced by several mechanisms. For example, endotoxins and cytokines are the most powerful stimulators, but stress-induced activation of the hypothalamic– pituitary–adrenocortical (HPA)-axis also leads to substantial production of MIF 15,23. These multiple bidirectional activation mechanisms and functional pathways of MIF, particularly in the realm of the HPA-axis, make MIF an intriguing operator in the neuroendocrine-immune interface 24
The human MIF gene is located on chromosome 22q11.23 spanning approximately 1.24 kb and containing 3 axons and 2 intones. Several single nucleotide polymorphisms (SNPs) have been described in the MIF gene, and have been associated with circulating MIF levels and/or risk of cardiovascular diseases 25,26, and autoimmune diseases 27–29. Thus, changes in the expression and function of MIF protein as a result of changes in the sequence of the MIF gene alleles could induce differences in the clinical manifestations of pediatric OSA.
Based on aforementioned considerations, we hypothesized that OSA in children may be associated with increased plasma MIF levels, and that the presence of specific single nucleotide polymorphisms (SNPs) in the MIF gene in children with OSA may alter their cardiometabolic risk.
MATERAILS and METHODS
Subjects
Consecutive children who were referred for evaluation of habitual snoring and suspected sleep disordered breathing (SDB) to the University Of Louisville Pediatric Sleep Medicine Center were recruited. The study was approved by the University of Louisville Human Research Committee (protocol #474.99), and informed consent was obtained from the legal caregiver of each participant. As controls, we invited children from the community who had no history of any chronic or acute disorder and who did not snore. Of note, all children were otherwise healthy, and were representative of the demographic characteristics of the general population of the city of Louisville (http://ksdc.louisville.edu/sdc/census2000/cityprofiles/LouisvilleDP.pdf). Children were excluded if they were younger than 4 years or older than 10 years of age, had known diabetes or pre-diabetes (http://www.diabetes.org/pre-diabetes/pre-diabetes-symptoms.jsp), any defined genetic abnormality or underlying systemic disease including hypertension, or if they were within a month from any acute infectious process.
Overnight Polysomnography (NPSG)
All subjects underwent overnight polysomnography using standard techniques 30. Children were studied for up to 12 hours in a quiet, darkened room maintained at an ambient temperature of 24°C for only one night, as is customarily done in clinical settings. No medication was used to induce sleep. The following parameters were measured: chest and abdominal wall movement by inductance plethysmography, heart rate by electrocardiography, air flow was triply monitored with a side stream end-tidal capnograph that also provides breath-by-breath assessment of end-tidal carbon dioxide levels (BCI SC-300, Menomonee Falls, WI), a nasal pressure cannula, and an oronasal thermistor. Arterial pulse oxygen saturation (SpO2) was assessed by pulse oximetry [Nellcor N 100 (Nellcor Inc, Hayward, CA)], with simultaneous recording of the pulse waveform. The bilateral electro-oculogram, 8 channels of electroencephalogram (2 frontal, 2 occipital, 2 temporal, and 2 central leads), chin and anterior tibial electromyograms, and analog output from a body position sensor were also monitored. All measures were digitized using a commercially available system (Sandman, Nellcor Puritan Bennett, Kanata, ON, Canada, or Stellate Instruments, Montreal, QC, Canada). Tracheal sound was monitored with a microphone sensor, and a digital time-synchronized video recording was performed. The sleep technician followed patient behavior and confirmed sleep position by the infrared camera inside the room.
All of the studies were initially scored by a certified technician and were then reviewed by a physician who was experienced in pediatric PSG and underwent training in an accredited fellowship program using defined criteria 30. Blood was drawn in the morning after the child completed the polysomnographic evaluation and after an overnight fast.
Demographics and definitions
All parents completed a detailed intake clinical questionnaire which included age, gender, ethnicity, and history of medication or of any other chronic disease. Height, weight and vital signs were recorded for each child, and body mass index (BMI) Z score was calculated using CDC 2000 growth standards (www.cdc.gov/growthcharts) and online software (www.cdc.gov/epiinfo). A BMI z-score>1.65 was considered as fulfilling obesity criteria. The diagnosis of OSA was defined by the presence of an obstructive apnea index ≥ 1/hour of total sleep time and an obstructive apnea-hypopnea index (AHI) ≥ 5/hour of total sleep time, respectively and a nadir oxyhemoglobin saturation <92% (23). Control children had AHI < 1/hour of total sleep time and no oxygen desaturations events during sleep.
Plasma Assays
Plasma MIF levels were measured using commercial Enzyme Linked Immunosorbant Assay (ELISA) kits (ALPCO Diagnostics, Salem, NH) following the manufacturer’s instructions. All assays were performed in duplicate and a calibration curve was included in each assay. The minimum detectable dose of MIF is typically < 6 pg/ml. The intra-assay and inter-assay coefficients of variability of plasma MIF were 10% and 12%, respectively. Plasma insulin levels were measured using a commercially available radioimmunoassay kit (Coat-A-Count Insulin; Diagnostic Products Inc). This method has a detection level of 1.2 μIU/mL and exhibits linear behavior up to 350 μIU/mL, with intra-assay and interassay coefficients of variability of 3.1% and 4.9%, respectively. Plasma glucose level was measured using a commercial kit based on the hexokinase-glucose-6-phosphate dehydrogenase method (Flex Reagent Cartridges; Dade Behring, Newark, DE). Insulin resistance was assessed by homoeostasis model assessment (HOMA) (insulin resistance index= [fasting glucose (mmol/L) × fasting insulin (U/mL)]/22.5) 31.
Plasma hsCRP levels were measured within 2–3 hours after collection using the Flex reagent Cartridge (Date Behring, Newark, DE). This method has a detection level of 0.05 mg/dL, and exhibits linear behavior up to 255 mg/dL, with intra-assay and inter-assay coefficients of variability of 9% and 18% respectively for hsCRP. Serum lipids including total cholesterol, high-density lipoprotein (HDL-C) cholesterol, calculated low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG) were also assessed using Flex Reagent Cartridges (Dade Behring).
DNA Extraction
Fasting peripheral blood samples were collected in vacutainer tubes containing EDTA (Becton Dickinson, Franklin Lakes, NJ, USA). All DNA samples were extracted using QIAmp DNA blood kit (Qiagen, Valencia, CA) according the manufacturer’s protocol. The concentration and quality of the DNA were determined using a ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). The purity of the DNA were determined by calculating the ratio of absorbance at 260/280 nm, and all DNA samples had a ratio of 1.8–1.9. The precise length of genomic DNA was determined by gel electrophoresis using 1% agarose gel. All the purified samples were stored at −80°C until further analyses.
Genotyping using Real-Time PCR
Genotyping was performed using the ABI PRISM 7500 Sequence Detection System for allelic discrimination following the manufacturer’s instructions (Applied Biosystems). Twenty-eight single nucleotide polymorphisms (SNPs) were examined in this study. All polymorphisms were genotyped using TaqMan technology (Applied Biosystems, Inc.). Two fluorogenic minor groove binder probes were used for eachlocus using the dyes 6-carboxyfluorescein (FAM; excitation, 494 nm) and VIC (excitation, 538 nm) which are easily differentiated in RT-PCR system. Real-time PCR reaction was performed using 12.5 μl of TaqMan 2x universal master mix (Applied Biosystems, CA), 1.25 μl of each primer, 10.25 μl of RNase and DNase-free water (Ambion, Austin, TX), and 1 μl of sample DNA, in a total volume of 25 μl per single well reaction. Two wells of a 96 well-plate (Applied Biosystems, CA) were used for each sample for each of the 4 SNPs. DNase-free water used as non-template control was included in each assay run. Assay conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Initially, the SNP assay was set up using SDS, version 2.1, software (AppliedBiosystems, CA) as an absolute quantification assay, but after assay completion the plate was read using the allelic discrimination settings. Post-assay analysis was performed using the SDS software. Hardy Weinberg equilibrium (HWE) for each SNP was verified using the equation p2 + q2 + 2pq = 1, where p and q represent the wild type and variant allele of a gene.
Data and Statistical Analysis
Data were expressed as mean±SD. Significant differences within groups were analyzed using ANOVA for continuous variables and chi-square tests for categorical variables. In addition, stepwise multivariate regression analyses were conducted while treating various metabolic or inflammatory variables as a dependent variable in relation to age, gender and BMI as covariates. Genotypic and allelic frequencies were compared using chi-square test. The differences in allelic and genotypic distributions between OSA and NOSA subjects were assessed using the χ2-test. Haplotype blocks of SNPs in strong Linkage Disequilibrium, delineated using the confidence interval method proposed by Gabriel et al 32 and implemented into Haploview. The Haploview version 4.2 software (hppt://www.borad.mit.edu/mpg/haploview) was then used to analyze the linkage disequilibrium structure, calculating D′ to define haplotype block 32 and to estimate haplotype frequencies. Haplotypes within these blocks were estimated using the estimation of maximization algorithum 33. Statistical analyses were performed using SPSS software (version 18.0; SPPS Inc., Chicago, IL.). All p-values reported are 2-tailed with statistical significance set at <0.05.
RESULTS
A total of 614 children were recruited and subdivided into 2 groups based on the presence or absence of OSA (OSA and NOSA), respectively. The demographic characteristics, their polysomnographic findings, and cardiometabolic markers of these subjects are shown in Table 1. Overall, children with OSA were more likely to have mildly reduced REM sleep and slow wave sleep and to display increased desaturations and alveolar hypoventilation, as well as increased arousals due to respiratory events (Table 1). The HOMA index was significantly higher (p-value <0.003) in children with OSA compared NOSA (Table 1). In addition, metabolic parameters such as HDL, LDL, as well as CRP levels were significantly higher in children with OSA (Table 1).
Table 1.
Demographic and metabolic data in children with OSA and matched controls (NOSA).
| NOSA | OSA | P-value | |
|---|---|---|---|
| Age (years) | 7.14 ± 0.1 | 7.03± 0.1 | 0.15 |
| BMI z score | 0.78 ± 1.2 | 1.11 ± 1.5 | 0.36 |
| AHI (h-1 TST) | 0.32 ± 0.0 | 8.13 ± 2.4 | 0.0001 |
| Sleep latency (min) | 17.6 ± 8.5 | 15.1 ± 8.2 | 0.28 |
| REM latency (min) | 117.7 ± 26.9 | 122.6 ± 39.8 | 0.41 |
| Slow wave sleep (%TST) | 24.1 ± 7.6 | 19.8 ± 9.1 | 0.035 |
| REM sleep (%TST) | 22.7 ± 5.6 | 18.4 ± 7.9 | 0.01 |
| Respiratory arousal index (h-1 TST) | 0.1 ± 0.0 | 6.7 ± 2.0 | 0.0001 |
| Triglycerides (mg/dL) | 69.1 ± 36.7 | 70.4 ± 40.3 | 0.41 |
| Total Cholesterol (mg/dL) | 155.7 ± 26.8 | 157.8 ± 23.9 | 0.28 |
| HDL (mg/dL) | 50.4 ± 12.7 | 47.2 ± 11.4 | 0.01 |
| LDL (mg/dL) | 91.5 ± 22.9 | 96 ± 22.7 | 0.05 |
| CRP (mg/dL) | 1.8 ± 3.4 | 2.7 ± 4.2 | 0.04 |
| Glucose (mg/dL) | 85.5 ± 11 | 86.5 ± 11 | 0.23 |
| Insulin (μU/mL) | 8.4 ± 8 | 9.8 ± 8.8 | 0.14 |
| HOMA-IR | 0.94 ± 1.3 | 1.45 ± 1.9 | 0.003 |
BMI, body masss index; AHI, apnea hypopnea index; h-1 TST, per hour of total sleep time; REM, rapid eye movement
Children with OSA exhibited significantly higher MIF plasma levels compared to NOSA (Figure 1). Furthermore, MIF levels exhibited weak correlations with the obstructive AHI (r: 0.287; p<0.01), stronger associations with hsCRP (r:0.436; p<0.001) and with HOMA values (0.465; p<0.001), but absence of any significant correlations with HDL or LDL.
Figure 1. Fasting morning concentrations of MIF plasma levels in NOSA and OSA subjects.
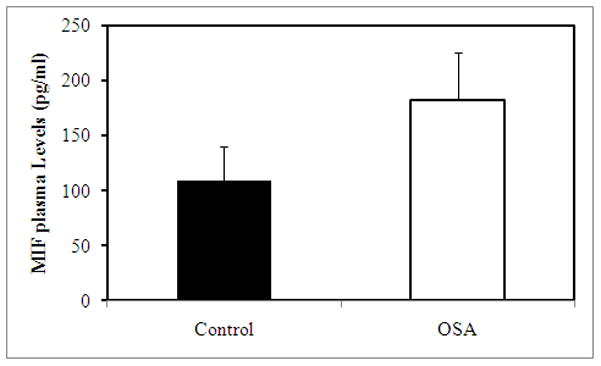
OSA vs. NOSA:p<0.02
The 28 SNPs examined were used to assess linkage disequilibrium (LD) using haploview software for both OSA and NOSA subjects. This software visualizes patterns of LD, performs association studies between SNPs, and estimates haplotype frequencies. In NOSA subjects, the software generated two haplotype blocks, which are outlined in black triangular regions (Figure 2, Panel A). In OSA subjects, LD of the 28 SNPs showed the presence of only one block (Figure 2, Panel B). The haplotype of these blocks and their frequencies in NOSA and OSA are shown in Figures 3 Panels A and B, respectively. Thus, the data in Figures 2 and 3 show that the patterns of LD and haplotype frequencies differ between NOSA and OSA, suggesting that some of these SNPs could participate in variance in metabolic dysfunction among children with OSA. The general information on the 28 SNPs, such as allele positions and functions is shown in Table 2. Among the identified SNPs for the MIF gene, rs10433310, showed significant differences in its frequency between OSA and NOSA (Table 3). The frequencies of AA, AG and GG genotypes were 13 % (42), 47 % (151) and 40 % (130) in NOSA children, and 7 % (9), 41 % (54) and 52 % (68) in OSA (Table 3). Indeed, children with OSA had significantly lower variant allele frequency of rs10433310 than NOSA (p=0.03), and this pattern became even more apparent when alleles A and G were compared between the two groups (p<0.005; Table 3). The allelic data were further analyzed in conjunction with the metabolic profiles among both NOSA and OSA subjects for the significant SNP (i.e., rs10433310), as shown in Table 4. We found that the GG genotype had a significant inverse association with LDL (p=0.02), CRP (p=0.02), insulin (p=0.006) and HOMA-IR (p=0.01) (Table 4). In particular, insulin levels in OSA subjects with the variant allele of rs10433310 were significantly lower than in OSA subjects without the variant allele (p<0.03). However, no associations emerged between rs10433310 and sleep measures. No other SNPs showed any identifiable pattern for cardiometabolic risk markers. Similarly, the presence of rs10433310 was associated with reduced hsCRP levels (p<0.02). All significant associations identified for rs10433310 remained significant even after inclusion of BMI as a co-variate in the stepwise linear regression model.
Figure 2. Pairwise linkage disequilibrium (LD) structure and 28 SNPS of the MIF gene.
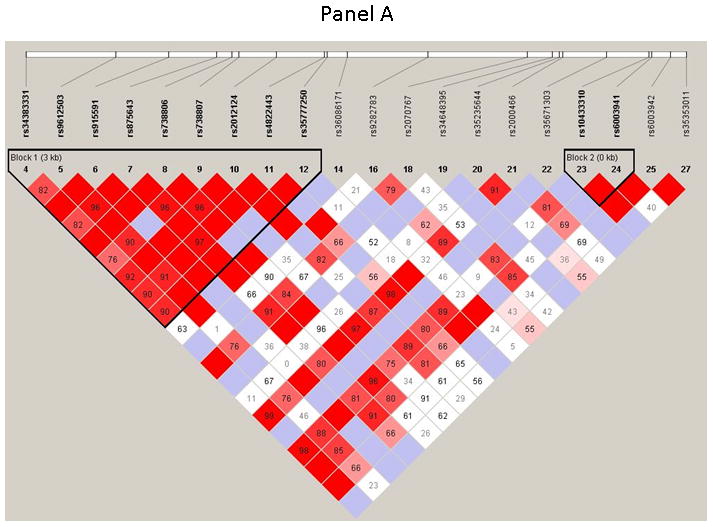
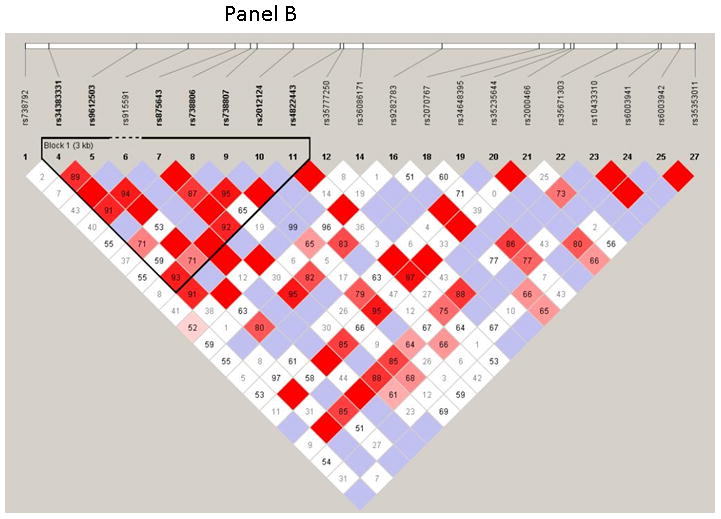
Panel (A) represents control children (NOSA), and Panel (B) represents children with OSA. LD map of MIF gene region chr22:22556987–22576986 was depicted using 28 SNPs (MAF >0.05) in HapMap database. The plot was generated using Haploview 4.2 with D′ Color Scheme (D′=0, D′<1 and D′=1 were shown by white, shades of pink and red (respectively) and pairwise r2 values shown in diamonds. The value within each diamond represents the pair-wise LD (correlation, measured as D′) between the two SNPs defined by the top left and the top right of the diamond. Solid lines represent SNPs that were used in the haplotype analysis and are part of the haplotype from SNP block whereas dashed lines represent SNPs that were used in the analysis, but were not part of the haplotype.
Figure 3. Haplotype frequencies in NOSA and OSA children.

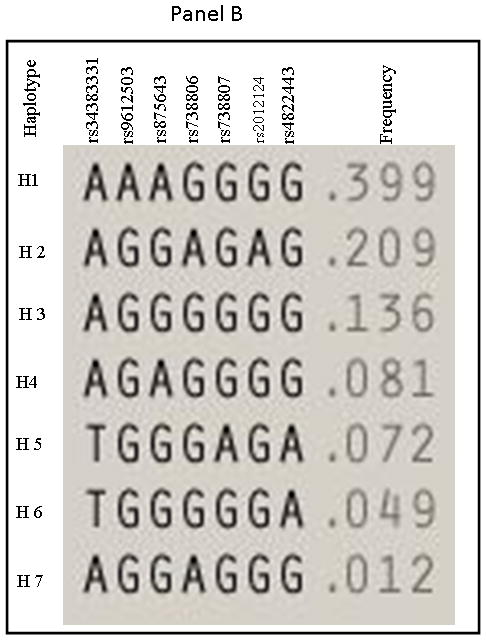
Panel A represents haplotype for NOSA and Panel B represents haplotype for OSA children.
TABLE 2.
List of single nucleotide polymorphisms (SNPs) for the MIF gene examined in this study.
| Numbers | Name | Alleles | Position | MAF % | Function |
|---|---|---|---|---|---|
|
| |||||
| 1 | rs738792 | C/T | 38 | 17 | missense |
| 2 | rs28363646 | A/G | 44 | 0 | missense |
| 3 | rs17004038 | A/C | 152 | 0 | missense |
| 4 | rs34383331 | C/T | 306 | 14 | UTR-5 |
| 5 | rs9612503 | A/T | 1311 | 42 | NA* |
| 6 | rs915591 | C/T | 1898 | 5 | NA |
| 7 | rs875643 | C/T | 2434 | 49 | NA |
| 8 | rs738806 | A/G | 2608 | 23 | NA |
| 9 | rs738807 | C/T | 2681 | 10 | NA |
| 10 | rs2012124 | C/T | 3100 | 21 | near UTR-5 |
| 11 | rs4822443 | A/G | 3634 | 16 | near UTR-5 |
| 12 | rs35777250 | C/T | 3666 | 6 | near UTR-5 |
| 13 | rs35178050 | C/G | 3856 | 0 | near UTR-5 |
| 14 | rs36086171 | A/G | 3891 | 6 | near UTR-5 |
| 15 | rs35006085 | A/G | 4514 | 0 | UTR-3 |
| 16 | rs9282783 | C/G | 4795 | 3 | UTR-3 |
| 17 | rs35833535 | C/T | 5652 | 0 | UTR-5 |
| 18 | rs2070767 | C/T | 5899 | 21 | UTR-5 |
| 19 | rs34648395 | C/G | 6182 | 1 | near UTR-3 |
| 20 | rs35235644 | C/G | 6258 | 2 | near UTR-3 |
| 21 | rs2000466 | G/T | 6298 | 17 | near UTR-3 |
| 22 | rs35671303 | A/T | 6787 | 0.8 | NA |
| 23 | rs10433310 | A/G | 7259 | 28 | NA |
| 24 | rs6003941 | G/T | 7293 | 20 | NA |
| 25 | rs6003942 | C/T | 7501 | 9 | NA |
| 26 | rs35112599 | C/T | 7659 | 0 | NA |
| 27 | rs35353011 | G/T | 7675 | 1 | NA |
| 28 | rs17004044 | C/T | NA | 27 | NA |
Notice: NA means data not available.
TABLE 3.
Distributions of genotype and allelic frequencies in NOSA and OSA subjects for SNP rs10433310 of the MIF gene.
| Alleles | NOSA n=323 | OSA n=131 | p-value | ||
|---|---|---|---|---|---|
| n | % | N | % | 0.03 | |
| AA | 42 | 13 | 9 | 7 | |
| AG | 151 | 47 | 54 | 41 | |
| GG | 130 | 40 | 68 | 52 | |
| Allele A | 235 | 36 | 72 | 26 | 0.005 |
| Allele G | 411 | 64 | 198 | 74 | |
TABLE 4.
Metabolic data in relation to SNP rs10433310 alleles of the MIF gene among OSA and NOSA children.
| rs10433310 A/G | NOSA | OSA | p-value of NOSA vs OSA | |||||
|---|---|---|---|---|---|---|---|---|
| A+ | A− | p-value | A+ | A− | p- value | A+ | A− | |
| Triglycerides | 68.83 ± 35.94 | 69.36 ± 36.59 | 0.45 | 71.58 ± 43.17 | 70.92 ± 41.03 | 0.46 | 0.34 | 0.28 |
| Total Cholesterol | 156.52 ± 27 | 151.74 ± 23 | 0.04 | 158.86 ± 25 | 157.92 ± 24 | 0.42 | 0.15 | 0.18 |
| HDL | 51.16 ± 14 | 50.23 ± 13 | 0.28 | 48.12 ± 13 | 47.04 ± 11 | 0.3 | 0.02 | 0.09 |
| LDL | 89.28 ± 20 | 91.01 ± 21 | 0.24 | 90.1 ± 22 | 96.84 ± 23 | 0.05 | 0.16 | 0.02 |
| CRP | 2.10 ± 4.4 | 1.87 ± 3.51 | 0.31 | 2.91 ± 4.13 | 2.93 ± 4.50 | 0.49 | 0.16 | 0.02 |
| Glucose | 86.40 ± 9.5 | 85.28 ± 11 | 0.19 | 85.43 ± 13.18 | 87.03 ± 11.23 | 0.23 | 0.29 | 0.12 |
| Insulin | 8.40 ± 5.57 | 8.88 ± 8.33 | 0.42 | 6.69 ± 3.25 | 11.47 ± 8.91 | 0.01 | 0.12 | 0.006 |
| HOMA-IR | 0.69 ±1.41 | 0.78 ± 1.45 | 0.50 | 0.83 ± 2.08 | 1.69 ± 2.02 | 0.03 | 0.15 | 0.01 |
DISCUSION
In this study, we found that plasma MIF levels are significantly higher in subjects with OSA when compared with NOSA. Furthermore, the frequency of the rs10433310 SNP in the MIF gene was reduced in OSA compared to NOSA, and seemingly contributes to lower serum levels of known cardiometabolic risk markers such as hsCRP and HOMA, while the frequencies of all other SNPs tested for the MIF gene were comparable in OSA and NOSA.
In the context of the present work, some methodological issues need to be mentioned. We a priori excluded any child with known diabetes, hypertension or another chronic disease condition associated with either OSA or obesity, and this approach could therefore have artificially reduced the potential impact of such conditions on plasma MIF levels, as well as the magnitude of the association of any given MIF allelic variant with cardiometabolic markers. Therefore, it will be important to explore such potential relationships in larger and non-restricted clinical referral pediatric cohorts being assessed for OSA. We also narrowed the age range of the current cohort to match the period in life that is associated with significant changes in food consumption patterns 34. Such approach aimed to minimize as much as possible any confounding factors that might be operational across a wide age range, and particularly reduce the impact of adolescence. Thirdly, we identified and recruited closely matching control children and thus reduce modifying factors that could be involved in the process of subject selection. Finally, while we did not examine the 24-hour MIF plasma level variability, all of the samples were collected at the same time, immediately after awakening in the morning after the sleep study, and therefore, circadian influences should have been prevented by such approach 12. Although the precise mechanisms underlying the occurrence of cardiovascular and metabolic morbidities in the context of childhood OSA remain to be fully elucidated, it has become apparent that several pathways are operational 35 and that the presence of OSAS induces activation of several inflammatory cascades, which are central to the initiation and progression of disease morbidity. After our initial study showed the presence of significant associations between log CRP levels and polysomnographic markers of OSA severity 36, similar findings were reported by several investigators, except one 37–41. Furthermore, in a study aiming to examine the metabolic implications of OSAS in children, we found that both elevations in CRP levels and serum lipid alterations occurred in both non-obese untreated OSAS and in obese untreated OSAS children 10. Our current findings in this additional large cohort of matched children with and without OSA further confirm all previous reports.
MIF has been proposed as a bridge between inflammatory processes and other biological pathways related to the metabolic syndrome and cardiovascular disease 7,13. Moreover, MIF has the potential to be involved in the pathogenesis of a range of immune-mediated inflammatory diseases affecting multiple organ systems 42. Several studies have demonstrated that MIF participates in both experimental and human atherosclerosis 20,43,44. Its expression was up-regulated in endothelial cells, vascular smooth muscle cells, and macrophages, and up-regulation of vascular MIF was associated with macrophage adhesion, accumulation, and foam cell transformation during atherogenesis 43,45. Furthermore, blockade of MIF using a neutralizing MIF antibody will reduce vascular inflammation, cell proliferation, and neointimal thickening 46. In an animal model, deficiency of MIF attenuates atherogenesis in low density lipoprotein receptor-deficient mice 47. It has been suggested that MIF may directly affect endothelial-monocyte adhesion since MIF triggers monocyte arrest under flow conditions on aortic endothelial cells exposed to oxidized LDL 48. MIF is an essential, upstream component of the inflammatory cascade, and MIF will up-regulate TNF-α secretion by macrophages 49 and other cytokines such as IL-1 and IL6. Recent epidemiological data provide support for a role for MIF in the development of insulin resistance in humans. Herder et al. (2006) reported a strong positive association between systemic concentrations of MIF and impaired glucose tolerance and type 2 diabetes. Another study showed that the MIF genotype rs1007888CC was associated with increased circulating MIF levels and increased type 2 diabetes 50.
Several animal models and clinical studies have reported that MIF plays an important role in the pathophysiology of inflammatory and autoimmune diseases 15,51. One of the physiologic functions of MIF is to counter-regulate glucocorticoid suppression of immune cell responses 52, which is very important for the regulation of the systemic inflammatory response. In a recent study by Edwards and colleagues, MIF was strongly associated with the hypothalamic pituitary adrenal (HPA) axis and postulated to play a key role in regulation of the inflammatory response 12. It also appears that MIF may be playing a dual role in optimizing inflammatory activity (i.e. increase IL-8 and TNF-α levels), while indirectly inhibiting maximal anti-inflammatory glucocorticoid activity 53,54
In this study, MIF plasma levels were increased in OSA when compared to NOSA, and such effect persisted when their BMI z score was incorporated into the analyses as a co-variate. A significant association between the severity of OSA and MIF plasma levels emerged in children, and therefore, our findings agree with the only other published study in adults in which the presence of OSA was not only a risk factor for elevated plasma MIF levels but the latter also correlated with degree of severity in OSA12. Indeed, Edwards et al (2011) reported that plasma MIF levels were significantly associated with AHI and arousal index, independent of age and obesity. Plasma MIF levels were also associated with increased insulin resistance, independent of age and adiposity 55, a finding that we have also recently corroborated in children at risk for OSA56.
In addition, we found that of the 28 MIF SNPs examined, which were selected to cover the whole genomic sequence of MIF, only the rs10433310 polymorphism emerged as being significantly less prevalent among OSA children. In a previous study assessing genomic variability in the MIF gene in relation to obesity, a significant association was identified for allelic variants in the promoter region of the gene and the risk for obesity 57. In a pediatric population, those individuals carrying the G-173C, rs755622 polymorphisms did not exhibit any functional alterations in relation to their response to therapy in the context of nephritic syndrome 58. In another study examining the potential associations between MIF and gestational diabetes and metabolic syndrome, the polymorphism rs1007888 was associated with increased risk for these complications of pregnancy 59. Similarly, The C allele of SNP rs1007888 (3.8 kb 3′ of the translation termination codon) was associated with increased circulating MIF levels and with an increased risk of type 2 diabetes in women 50. Although multiple studies have investigated putative associations between selected SNPs in the MIF gene and several disease conditions, such as malaria, childhood asthma and bronchopulmonary dysplasia, non-alcoholic liver disease, coronary complications in Kawasaki disease, inflammatory bowel disease and peptic ulcer disease 60–67, we are unaware of any report on the exploration of MIF gene polymorphisms and sleep apnea. Promoter polymorphisms of the MIF gene are associated with increased production of MIF and have been found to confer increased risk of susceptibility to chronic inflammatory diseases. The present study identifies not only the elevation of MIF levels among children with OSA, and as such corroborates previous findings in adults with OSA 12, but further indicates that some of the variance in metabolic and cardiovascular risk associated with pediatric OSA may be accounted for a specific allelic variant in the MIF gene, namely the rs10433310 polymorphism. To better understand the potential impact of genetic variation in the MIF gene variant (rs10433310) that was significantly associated with metabolic profiles, we confirmed that the variant allele of rs10433310 was associated with reduced insulin resistance, as indicated by the lower HOMA-IR. Thus, allelic variants that reduce plasma MIF expression or activity should be metabolically protective, while those associated with higher plasma MIF levels may increase susceptibility. Taken together, these initial findings in a pediatric population suggest that MIF genomic variance may be an important determinant for cardiometabolic risk and those reductions in risk seems to be present among the rs10433310 allelic variant carriers.
Study Limitations
Studying MIF plasma levels and SNP variance in children with OSA should help to improve our understanding of the pathogenesis of OSA disease and its morbidities, and provide potential novel therapeutic targets. The main aims of this study were to test MIF SNP frequencies, MIF plasma levels and the metabolic profiles in children with OSA as compared to NOSA children. In a future study, we will be attempting to conduct a post-hoc analysis based on the phenotypes and determine the frequency of SNP re10433310 in children with similar OSA but manifesting distinct metabolic profiles. Confirmatory evidence of the putative implications defined in the current study will then enable assessment of the functional role of this SNP in the transcriptional regulation of the MIF gene.
In summary, we have shown that young school-age children with OSA exhibit higher circulating levels of the pro-inflammatory cytokine MIF, and have also confirmed the previously reported higher hsCRP levels and lipid alterations in the context of OSA. All these in turn, may contribute to the risk for systemic inflammation and metabolic dysfunction. Furthermore, a specific allelic variant in MIF gene appears to attenuate the pro-inflammatory or diabetogenic potential of OSA during childhood.
Acknowledgments
Funding Sources: DG is supported by National Institutes of Health grants HL-065270 and HL-086662.
References
- 1.Capdevila OS, Dayyat E, Kheirandish-Gozal L, Gozal D. Prevalence of epileptiform activity in healthy children during sleep. Sleep Med. 2008;9(3):303–309. doi: 10.1016/j.sleep.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 2.Gozal D. Obstructive sleep apnea in children: implications for the developing central nervous system. Semin Pediatr Neurol. 2008;15(2):100–106. doi: 10.1016/j.spen.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dayyat E, Kheirandish-Gozal L, Gozal D. Childhood Obstructive Sleep Apnea: One or Two Distinct Disease Entities? Sleep Med Clin. 2007;2(3):433–444. doi: 10.1016/j.jsmc.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kheirandish-Gozal L, Bhattacharjee R, Kim J, Clair HB, Gozal D. Endothelial progenitor cells and vascular dysfunction in children with obstructive sleep apnea. Am J Respir Crit Care Med. 2010;182(1):92–97. doi: 10.1164/rccm.200912-1845OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharjee R, Kheirandish-Gozal L, Pillar G, Gozal D. Cardiovascular complications of obstructive sleep apnea syndrome: evidence from children. Prog Cardiovasc Dis. 2009;51(5):416–433. doi: 10.1016/j.pcad.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Kheirandish-Gozal L, Bhattacharjee R, Gozal D. Autonomic alterations and endothelial dysfunction in pediatric obstructive sleep apnea. Sleep Med. 2010;11(7):714–720. doi: 10.1016/j.sleep.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharjee R, Kim J, Kheirandish-Gozal L, Gozal D. Obesity and obstructive sleep apnea syndrome in children: a tale of inflammatory cascades. Pediatr Pulmonol. 2011;46(4):313–323. doi: 10.1002/ppul.21370. [DOI] [PubMed] [Google Scholar]
- 8.Khalyfa A, Capdevila OS, Buazza MO, Serpero LD, Kheirandish-Gozal L, Gozal D. Genome-wide gene expression profiling in children with non-obese obstructive sleep apnea. Sleep Med. 2009;10(1):75–86. doi: 10.1016/j.sleep.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Khalyfa A, Gharib SA, Kim J, Capdevila OS, Kheirandish-Gozal L, Bhattacharjee R, Hegazi M, Gozal D. Peripheral blood leukocyte gene expression patterns and metabolic parameters in habitually snoring and non-snoring children with normal polysomnographic findings. Sleep. 2011;34(2):153–160. doi: 10.1093/sleep/34.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gozal D, Capdevila OS, Kheirandish-Gozal L. Metabolic alterations and systemic inflammation in obstructive sleep apnea among nonobese and obese prepubertal children. Am J Respir Crit Care Med. 2008;177(10):1142–1149. doi: 10.1164/rccm.200711-1670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Lee S, Bhattacharjee R, Khalyfa A, Kheirandish-Gozal L, Gozal D. Leukocyte Telomere Length and Plasma Catestatin and Myeloid Related Protein 8/14 Concentrations in Children with Obstructive Sleep Apnea. Chest. 2010 doi: 10.1378/chest.09-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards KM, Tomfohr LM, Mills PJ, Bosch JA, Ancoli-Israel S, Loredo JS, Dimsdale J. Macrophage migratory inhibitory factor (MIF) may be a key factor in inflammation in obstructive sleep apnea. Sleep. 2011;34(2):161–163. doi: 10.1093/sleep/34.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117(12):1594–1602. doi: 10.1161/CIRCULATIONAHA.107.729125. [DOI] [PubMed] [Google Scholar]
- 14.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 15.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleemann R, Bucala R. Macrophage migration inhibitory factor: critical role in obesity, insulin resistance, and associated comorbidities. Mediators Inflamm. 2010;2010:610479. doi: 10.1155/2010/610479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toso C, Emamaullee JA, Merani S, Shapiro AM. The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia. 2008;51(11):1937–1946. doi: 10.1007/s00125-008-1063-3. [DOI] [PubMed] [Google Scholar]
- 18.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, Bucala R. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 2000;106(10):1291–1300. doi: 10.1172/JCI9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5(5):399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 21.Bifulco C, McDaniel K, Leng L, Bucala R. Tumor growth-promoting properties of macrophage migration inhibitory factor. Curr Pharm Des. 2008;14(36):3790–3801. doi: 10.2174/138161208786898608. [DOI] [PubMed] [Google Scholar]
- 22.Nishihira J, Mitsuyama K. Overview of the role of macrophage migration inhibitory factor (MIF) in inflammatory bowel disease. Curr Pharm Des. 2009;15(18):2104–2109. doi: 10.2174/138161209788489113. [DOI] [PubMed] [Google Scholar]
- 23.Tierney T, Patel R, Stead CA, Leng L, Bucala R, Buckingham JC. Macrophage migration inhibitory factor is released from pituitary folliculo-stellate-like cells by endotoxin and dexamethasone and attenuates the steroid-induced inhibition of interleukin 6 release. Endocrinology. 2005;146(1):35–43. doi: 10.1210/en.2004-0946. [DOI] [PubMed] [Google Scholar]
- 24.Baugh JA, Donnelly SC. Macrophage migration inhibitory factor: a neuroendocrine modulator of chronic inflammation. J Endocrinol. 2003;179(1):15–23. doi: 10.1677/joe.0.1790015. [DOI] [PubMed] [Google Scholar]
- 25.Flex A, Gaetani E, Angelini F, Sabusco A, Chilla C, Straface G, Biscetti F, Pola P, Castellot JJ, Jr, Pola R. Pro-inflammatory genetic profiles in subjects with peripheral arterial occlusive disease and critical limb ischemia. J Intern Med. 2007;262(1):124–130. doi: 10.1111/j.1365-2796.2007.01791.x. [DOI] [PubMed] [Google Scholar]
- 26.Flex A, Gaetani E, Papaleo P, Straface G, Proia AS, Pecorini G, Tondi P, Pola P, Pola R. Proinflammatory genetic profiles in subjects with history of ischemic stroke. Stroke. 2004;35(10):2270–2275. doi: 10.1161/01.STR.0000140740.19421.fe. [DOI] [PubMed] [Google Scholar]
- 27.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3(3):170–176. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 28.Barton A, Lamb R, Symmons D, Silman A, Thomson W, Worthington J, Donn R. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun. 2003;4(7):487–491. doi: 10.1038/sj.gene.6364014. [DOI] [PubMed] [Google Scholar]
- 29.Donn R, Alourfi Z, Zeggini E, Lamb R, Jury F, Lunt M, Meazza C, De Benedetti F, Thomson W, Ray D. A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum. 2004;50(5):1604–1610. doi: 10.1002/art.20178. [DOI] [PubMed] [Google Scholar]
- 30.Montgomery-Downs HE, O’Brien LM, Gulliver TE, Gozal D. Polysomnographic characteristics in normal preschool and early school-aged children. Pediatrics. 2006;117(3):741–753. doi: 10.1542/peds.2005-1067. [DOI] [PubMed] [Google Scholar]
- 31.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 32.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 33.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70(2):425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, Minokoshi Y, Kahn BB, Parker RA, Hotamisligil GS. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1(2):107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Gozal D, Kheirandish-Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. Am J Respir Crit Care Med. 2008;177(4):369–375. doi: 10.1164/rccm.200608-1190PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tauman R, Ivanenko A, O’Brien LM, Gozal D. Plasma C-reactive protein levels among children with sleep-disordered breathing. Pediatrics. 2004;113(6):e564–569. doi: 10.1542/peds.113.6.e564. [DOI] [PubMed] [Google Scholar]
- 37.Larkin EK, Rosen CL, Kirchner HL, Storfer-Isser A, Emancipator JL, Johnson NL, Zambito AM, Tracy RP, Jenny NS, Redline S. Variation of C-reactive protein levels in adolescents: association with sleep-disordered breathing and sleep duration. Circulation. 2005;111(15):1978–1984. doi: 10.1161/01.CIR.0000161819.76138.5E. [DOI] [PubMed] [Google Scholar]
- 38.Kaditis AG, Alexopoulos EI, Kalampouka E, Kostadima E, Germenis A, Zintzaras E, Gourgoulianis K. Morning levels of C-reactive protein in children with obstructive sleep-disordered breathing. Am J Respir Crit Care Med. 2005;171(3):282–286. doi: 10.1164/rccm.200407-928OC. [DOI] [PubMed] [Google Scholar]
- 39.Kheirandish L, Gozal D. Neurocognitive dysfunction in children with sleep disorders. Dev Sci. 2006;9(4):388–399. doi: 10.1111/j.1467-7687.2006.00504.x. [DOI] [PubMed] [Google Scholar]
- 40.Villa MP, Ianniello F, Tocci G, Evangelisti M, Miano S, Ferrucci A, Ciavarella GM, Volpe M. Early cardiac abnormalities and increased C-reactive protein levels in a cohort of children with sleep disordered breathing. Sleep Breath. 2011 doi: 10.1007/s11325-010-0462-0. [DOI] [PubMed] [Google Scholar]
- 41.Goldbart AD, Levitas A, Greenberg-Dotan S, Ben Shimol S, Broides A, Puterman M, Tal A. B-type natriuretic peptide and cardiovascular function in young children with obstructive sleep apnea. Chest. 2010;138(3):528–535. doi: 10.1378/chest.10-0150. [DOI] [PubMed] [Google Scholar]
- 42.Hoi AY, Iskander MN, Morand EF. Macrophage migration inhibitory factor: a therapeutic target across inflammatory diseases. Inflamm Allergy Drug Targets. 2007;6(3):183–190. doi: 10.2174/187152807781696455. [DOI] [PubMed] [Google Scholar]
- 43.Burger-Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105(13):1561–1566. doi: 10.1161/01.cir.0000012942.49244.82. [DOI] [PubMed] [Google Scholar]
- 44.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24(11):1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 45.Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R, Lau CP, Lan HY. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res. 2000;87(12):1202–1208. doi: 10.1161/01.res.87.12.1202. [DOI] [PubMed] [Google Scholar]
- 46.Chen Z, Sakuma M, Zago AC, Zhang X, Shi C, Leng L, Mizue Y, Bucala R, Simon D. Evidence for a role of macrophage migration inhibitory factor in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24(4):709–714. doi: 10.1161/01.ATV.0000119356.35748.9e. [DOI] [PubMed] [Google Scholar]
- 47.Pan JH, Sukhova GK, Yang JT, Wang B, Xie T, Fu H, Zhang Y, Satoskar AR, David JR, Metz CN, Bucala R, Fang K, Simon DI, Chapman HA, Libby P, Shi GP. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109(25):3149–3153. doi: 10.1161/01.CIR.0000134704.84454.D2. [DOI] [PubMed] [Google Scholar]
- 48.Schober A, Bernhagen J, Thiele M, Zeiffer U, Knarren S, Roller M, Bucala R, Weber C. Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation. 2004;109(3):380–385. doi: 10.1161/01.CIR.0000109201.72441.09. [DOI] [PubMed] [Google Scholar]
- 49.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33(47):14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 50.Herder C, Klopp N, Baumert J, Muller M, Khuseyinova N, Meisinger C, Martin S, Illig T, Koenig W, Thorand B. Effect of macrophage migration inhibitory factor (MIF) gene variants and MIF serum concentrations on the risk of type 2 diabetes: results from the MONICA/KORA Augsburg Case-Cohort Study, 1984–2002. Diabetologia. 2008;51(2):276–284. doi: 10.1007/s00125-007-0800-3. [DOI] [PubMed] [Google Scholar]
- 51.Greven D, Leng L, Bucala R. Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets. 2010;14(3):253–264. doi: 10.1517/14728220903551304. [DOI] [PubMed] [Google Scholar]
- 52.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377(6544):68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 53.Metz CN, Bucala R. Role of macrophage migration inhibitory factor in the regulation of the immune response. Adv Immunol. 1997;66:197–223. doi: 10.1016/s0065-2776(08)60598-2. [DOI] [PubMed] [Google Scholar]
- 54.Morand EF, Leech M. Glucocorticoid regulation of inflammation: the plot thickens. Inflamm Res. 1999;48(11):557–560. doi: 10.1007/s000110050503. [DOI] [PubMed] [Google Scholar]
- 55.Lam DC, Xu A, Lam KS, Lam B, Lam JC, Lui MM, Ip MS. Serum adipocyte-fatty acid binding protein level is elevated in severe OSA and correlates with insulin resistance. Eur Respir J. 2009;33(2):346–351. doi: 10.1183/09031936.50075408. [DOI] [PubMed] [Google Scholar]
- 56.Khalyfa A, Bhushan B, Hegazi M, Kim J, Kheirandish-Gozal L, Bhattacharjee R, Capdevila OS, Gozal D. Fatty-acid binding protein 4 gene variants and childhood obesity: potential implications for insulin sensitivity and CRP levels. Lipids Health Dis. 2010;9:18. doi: 10.1186/1476-511X-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakaue S, Ishimaru S, Hizawa N, Ohtsuka Y, Tsujino I, Honda T, Suzuki J, Kawakami Y, Nishihira J, Nishimura M. Promoter polymorphism in the macrophage migration inhibitory factor gene is associated with obesity. Int J Obes (Lond) 2006;30(2):238–242. doi: 10.1038/sj.ijo.0803148. [DOI] [PubMed] [Google Scholar]
- 58.Choi HJ, Cho HY, Ro H, Lee SH, Han KH, Lee H, Kang HG, Ha IS, Choi Y, Cheong HI. Polymorphisms of the MDR1 and MIF genes in children with nephrotic syndrome. Pediatr Nephrol. 2011 doi: 10.1007/s00467-011-1903-0. [DOI] [PubMed] [Google Scholar]
- 59.Aslani S, Hossein-Nezhad A, Maghbooli Z, Mirzaei K, Karimi F. Genetic Variation in Macrophage Migration Inhibitory Factor Associated with Gestational Diabetes Mellitus and Metabolic Syndrome. Horm Metab Res. 2011 doi: 10.1055/s-0031-1275706. [DOI] [PubMed] [Google Scholar]
- 60.Simonini G, Corinaldesi E, Massai C, Falcini F, Fanti F, De Martino M, Cimaz R. Macrophage migration inhibitory factor -173 polymorphism and risk of coronary alterations in children with Kawasaki disease. Clin Exp Rheumatol. 2009;27(6):1026–1030. [PubMed] [Google Scholar]
- 61.Awandare GA, Ouma C, Keller CC, Were T, Otieno R, Ouma Y, Davenport GC, Hittner JB, Ong’echa JM, Ferrell R, Perkins DJ. A macrophage migration inhibitory factor promoter polymorphism is associated with high-density parasitemia in children with malaria. Genes Immun. 2006;7(7):568–575. doi: 10.1038/sj.gene.6364332. [DOI] [PubMed] [Google Scholar]
- 62.Prencipe G, Auriti C, Inglese R, Devito R, Ronchetti MP, Seganti G, Rava L, Orzalesi M, De Benedetti F. A polymorphism in the macrophage migration inhibitory factor promoter is associated with bronchopulmonary dysplasia. Pediatr Res. 2011;69(2):142–147. doi: 10.1203/PDR.0b013e3182042496. [DOI] [PubMed] [Google Scholar]
- 63.Shiroeda H, Tahara T, Shibata T, Nakamura M, Yamada H, Nomura T, Hayashi R, Saito T, Fukuyama T, Otsuka T, Yano H, Ozaki K, Tsuchishima M, Tsutsumi M, Arisawa T. Functional promoter polymorphisms of macrophage migration inhibitory factor in peptic ulcer diseases. Int J Mol Med. 2010;26(5):707–711. doi: 10.3892/ijmm_00000517. [DOI] [PubMed] [Google Scholar]
- 64.Martin RJ, Savage DA, Carson DJ, McKnight AJ, Maxwell AP, Patterson CC. Polymorphisms of the macrophage migration inhibitory factor gene in a UK population with Type 1 diabetes mellitus. Diabet Med. 2010;27(2):143–149. doi: 10.1111/j.1464-5491.2009.02916.x. [DOI] [PubMed] [Google Scholar]
- 65.Palomino-Morales R, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Torres O, Miranda-Filloy JA, Llorca J, Martin J, Gonzalez-Gay MA. Lack of association between macrophage migration inhibitory factor-173 gene polymorphism with disease susceptibility and cardiovascular risk in rheumatoid arthritis patients from northwestern Spain. Clin Exp Rheumatol. 2010;28(1):68–72. [PubMed] [Google Scholar]
- 66.Akyildiz M, Gunsar F, Nart D, Sahin O, Yilmaz F, Akay S, Ersoz G, Karasu Z, Ilter T, Batur Y, Berdeli A, Akarca U. Macrophage migration inhibitory factor expression and MIF gene -173 G/C polymorphism in nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2010;22(2):192–198. doi: 10.1097/MEG.0b013e328331a596. [DOI] [PubMed] [Google Scholar]
- 67.Wu J, Fu S, Ren X, Jin Y, Huang X, Zhang X, Bai J. Association of MIF promoter polymorphisms with childhood asthma in a northeastern Chinese population. Tissue Antigens. 2009;73(4):302–306. doi: 10.1111/j.1399-0039.2008.01206.x. [DOI] [PubMed] [Google Scholar]