

Abstract
Objective:
To characterize the molecular basis of a novel fast-channel congenital myasthenic syndrome.
Methods:
We used the candidate gene approach to identify the pathogenic mutation in the acetylcholine receptor (AChR) ɛ subunit, genetically engineered the mutant AChR into HEK cells, and evaluated the level of expression and kinetic properties of the mutant receptor.
Results:
An 8-year-old boy born to consanguineous parents had severe myasthenic symptoms since birth. He is wheelchair bound and pyridostigmine therapy enables him to take only a few steps. Three similarly affected siblings died in infancy. He carries a homozygous p.W55R mutation at the α/ɛ subunit interface of the AChR agonist binding site. The mutant protein expresses well in HEK cells. Patch-clamp analysis of the mutant receptor expressed in HEK cells reveals 30-fold reduced apparent agonist affinity, 75-fold reduced apparent gating efficiency, and strikingly attenuated channel opening probability (Popen) over a range agonist concentrations.
Conclusion:
Introduction of a cationic Arg into the anionic environment of α/ɛ AChR binding site hinders stabilization of cationic ACh by aromatic residues and accounts for the markedly perturbed kinetic properties of the receptor. The very low Popen explains the poor response to pyridostigmine and the high fatality of the disease.
Congenital myasthenic syndromes (CMS) are heterogeneous disorders caused by defects in endplate (EP)-associated proteins.1 No fewer than 13 CMS disease proteins have been identified to date and mutations occurring in subunits of AChR are the commonest cause of CMS. AChR is a ligand-gated ion channel with a pentameric structure consisting of 4 homologous subunits with a stoichiometry of α2βδγ in fetal and α2βδɛ in adult receptor. Each subunit is composed of 2 extracellular domains at its N- and C-terminal ends, an intracellular domain, and 4 transmembrane domains connected by intracellular and extracellular linkers.2 Two agonist binding pockets of AChR are present at the α/δ and α/ɛ subunit interfaces (figure 1A). The principal face of each binding site is formed by the α subunit, which contributes peptide loops A–C, while the complimentary face is formed by either the δ or the ɛ subunit, which contribute loops D–G3 (figure 1B). In each binding pocket, the cationic agonist ACh is stabilized by interaction with 5 conserved aromatic residues. We report and evaluate the consequences of a spontaneous mutation of an aromatic residue at the α/ɛ agonist binding site of AChR (figure 1C).
Figure 1. Agonist binding sites of acetylcholine receptor (AChR).

(A) Structural model of extracellular domains of human AChR viewed from the synaptic space indicating positions of Trp residues at the α/δ and α/ɛ binding sites. (B) Side view of the α and ɛ subunits showing position of loops E, D, G, and F in the ɛ subunit, and loops A, B, and C in the α subunit. (C) Stereo view of the binding site showing positions of aromatic residues shrouding the binding pocket. In each panel the mutated ɛTrp55 at the α/ɛ binding site is highlighted in red. (Based on the crystal structure of the ACh binding protein [PDB 1I9B] and lysine scanning mutagenesis delineating the structure of the human AChR binding domain.13)
METHODS
Protocol approval and consents.
The investigations described in this study were approved by the Institutional Review Board of the Mayo Clinic. The patient's father gave informed consent for the genetic studies.
Molecular genetic studies.
Molecular genetic studies were performed by previously described methods.4 We directly sequenced CHRNA1, CHRNB1, CHRND, and CHRNE using genomic DNA extracted from blood. The p.Trp55Arg mutation detected in CHRNE was traced with allele-specific PCR in family members and was not present in 400 normal alleles of 200 unrelated controls. Expression of the mutant AChR in HEK cells is described in appendix e-1 on the Neurology® Web site at www.neurology.org.
Patch-clamp recordings from AChRs expressed in HEK cells at low and high concentrations of ACh were obtained in the cell-attached configuration at a membrane potential of −80 mV at 22°C and with bath and pipette solutions containing (mM) KCl 142, NaCl 5.4, CaCl2 1.8, MgCl2 1.7, HEPES 10, pH 7.4.4 The recordings and their analyses were performed by previously described methods (see reference 4 and appendix e-2).
RESULTS
Clinical data.
An 8-year-old boy born to asymptomatic second-degree cousins had severe myasthenic symptoms since birth. He is wheelchair bound; pyridostigmine therapy enables him to take a few steps. He has a 19% decremental EMG response in the trapezius muscle, and single fiber EMG reveals a mean jitter of 102 μs (normal <50 μs). Three similarly affected siblings died in infancy, 1 similarly affected brother is alive, and 2 other siblings are unaffected.
Mutation analysis.
Sequencing of the each AChR subunit gene revealed a homozygous replacement of Trp by Arg in CHRNE at codon 55: p.W55R (c.163T>C) (reference sequence NM_000080.3). The mutated Trp is conserved across the AChR ɛ, γ, and δ subunits of all species and is located at the α/ɛ agonist binding site interface (figure 1). No DNA was available from other family members but homozygosity of the proband is strong evidence for the asymptomatic parents being carriers and for transmission by recessive inheritance.
Expression studies.
125I-α-bungarotoxin binding studies revealed that the mutant receptor expressed at 86% of wild-type. Hence the deleterious effects of the mutation likely arise from altered AChR activation kinetics.
To examine kinetic consequences of the ɛW55R mutation, we recorded single channel currents from HEK cells expressing mutant and wild-type AChR at a low concentration of ACh (50 nM). The resulting channel events appear as isolated openings or as several openings in quick succession, called bursts (figure 2 and table 1). The burst duration histograms reveal 2 exponential components for ɛW55R-AChR and 3 for wild-type AChR. For wild-type AChR, the longest component of bursts has a mean duration of 3.31 msec, but for ɛW55R-AChR the corresponding value is only 0.37 msec, predicting fast decay of the synaptic current.
Figure 2. Single-channel currents elicited from human embryonic kidney fibroblast cells transfected with wild-type acetylcholine receptor (AChR) and ɛW55R-AChR.

Left: Representative channel openings elicited by 50 nM ACh. Right: Logarithmically binned burst duration histograms fitted to the sum of exponentials. Arrows indicate mean duration of burst components.
Table 1.
Open intervals and burst durations of wild-type and mutant AChRs in HEK cellsa
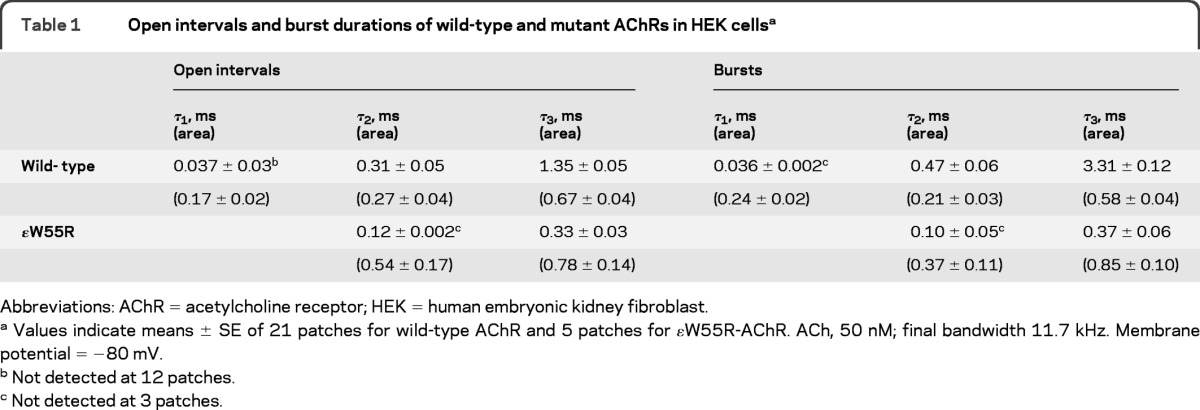
Abbreviations: AChR = acetylcholine receptor; HEK = human embryonic kidney fibroblast.
Values indicate means ± SE of 21 patches for wild-type AChR and 5 patches for ɛW55R-AChR. ACh, 50 nM; final bandwidth 11.7 kHz. Membrane potential = −80 mV.
Not detected at 12 patches.
Not detected at 3 patches.
To identify kinetic steps in AChR activation altered by ɛW55R, we recorded channel openings over a range of ACh concentrations and constructed histograms of open and closed dwell times (figure 3). Wild-type AChR generates well defined clusters of openings with 10 μM ACh but ɛW55R-AChR does so only with 100 μM and higher concentrations of ACh. The mutant clusters have longer closed times with fewer and briefer openings than wild-type. For both wild-type and ɛW55R-AChRs, the longest closed-time component shifts to the left with increasing ACh concentration, but the mutant histograms shift less than the wild-type.
Figure 3. Activation kinetics of wild-type and ɛW55R-acetylcholine receptor (AChR) and channel open probabilities.

(A, B) Left column shows individual clusters of single-channel currents recorded at indicated ACh concentrations from human embryonic kidney fibroblast (HEK) cells. Right columns show histograms of closed and open durations at each ACh concentration with superimposed probability density functions (smooth curves) generated from a global fit of the scheme to dwell times obtained for the entire range of ACh concentrations. Fitted rate constants are shown in table 2. (C) Channel open probability (Popen) as function of ACh concentration. Symbols and vertical lines indicate means and standard deviations. Smooth curves are Popen predicted by the fitted rate constants in table 2.
To determine the consequences of the mutations on rate constants underlying receptor activation, we analyzed the global set of open and closed dwell times according to Scheme 1 shown below:
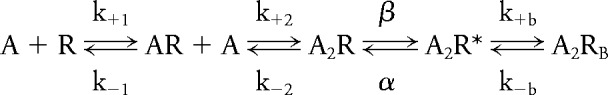 |
Here, 2 agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2, and dissociate with rate constants k−1 and k−2. The receptor with 2 agonists bound opens with rate constant β and closes with rate constant α. Asterisk indicates the open state and RB indicates the blocked state. At high concentrations, ACh blocks the open channel with rate constant k+b, and the channel unblocks with rate constant k-b. The fitted rate constants allow calculation of the equilibrium dissociation constants so that K1 = k−1/k+1, K2 = k−2/k+2, KB = k−b/k+b, and the channel gating equilibrium constant θ = β/α. For wild-type human AChR, the α/δ binding site has a higher agonist affinity than the α/ɛ binding site,5 so that the first binding step occurs at the α/δ site. For the mutant receptor, we constrained k+1 and k−1 to the wild-type values with the assumption that the 2 binding sites are independent and affinity of ACh for the α/δ binding site is unchanged by the mutation. This scheme together with fitted rate constants describes well the closed and open duration histograms of wild-type and mutant receptors.
The fitted rate constants in table 2 indicate that the ɛW55R increases the dissociation constant of the diliganded receptor 30-fold by decreasing the forward binding rate constant 10-fold and increasing the dissociation rate constant 3-fold, and reduces gating efficiency 75-fold mainly by decreasing the channel opening rate constant 46-fold. Because Scheme 1 does not include the recently detected primed state between closed and open states,6,7 the fitted rate constants are apparent rate constants. Nevertheless, the open probability (Popen) within defined clusters is not affected by this omission, and is computed from the sum of open times divided by the sum of open and closed times. A plot of Popen over a range of ACh concentrations reveals a marked decrease and shift to the right of Popen values for the mutant receptor (figure 3C). The plotted points are well described by Popen curves computed from the fitted rate constants and support the validity of the estimated rate constants shown in table 2.
Table 2.
Kinetic parameters of wild-type and mutant AChRs expressed in HEK cells
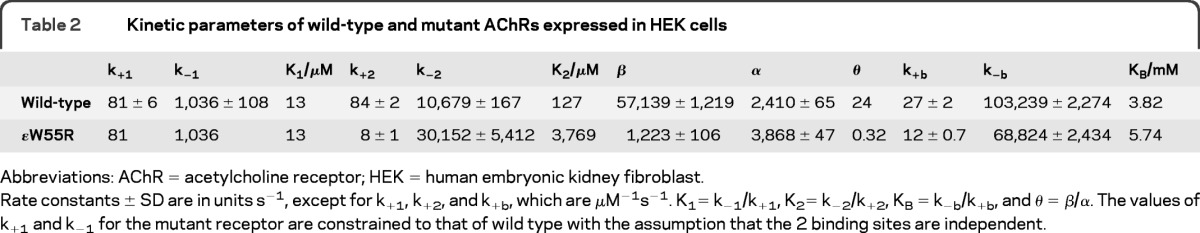
Abbreviations: AChR = acetylcholine receptor; HEK = human embryonic kidney fibroblast.
Rate constants ± SD are in units s−1, except for k+1, k+2, and k+b, which are μM−1s−1. K1= k−1/k+1, K2= k−2/k+2, KB = k−b/k+b, and θ = β/α. The values of k+1 and k−1 for the mutant receptor are constrained to that of wild type with the assumption that the 2 binding sites are independent.
DISCUSSION
The correct diagnosis of a CMS is important because it dictates appropriate therapy. In most CMS clinical, EMG, and molecular genetic studies point to the correct diagnosis but diagnosis of a fast-channel CMS requires in vitro microelectrode or single-channel patch-clamp recordings. Moreover, only single-channel patch-clamp recordings can elucidate the mechanistic consequences of the identified mutation.
We detected a homozygous missense mutation of an aromatic Trp residue in the ɛ subunit at the α/ɛ binding site of AChR in a highly fatal form of CMS. To our knowledge, no spontaneous mutation of an aromatic residue in the binding pocket of AChR has been reported to date. ɛW55R-AChR expresses well in HEK cells, has very low apparent ACh affinity for the α/ɛ site, and exhibits markedly reduced apparent gating efficiency. The findings underline the importance of the aromatic residues that shroud the AChR agonist binding sites.8–10 A contribution of Trp55 in the γ, δ, and ɛ subunits to agonist binding has been previously emphasized.10–12 Single-channel recordings from mouse ɛW55R-AChR expressed in HEK cells yielded a very low Popen, although kinetic analysis was not performed.12
Introduction of a cationic Arg residue into the aromatic-rich anionic α/ɛ binding site of AChR hinders stabilization of cationic ACh and markedly reduces apparent agonist affinity. In addition, the mutation hinders isomerization of the receptor from the closed to the open state, slows the apparent opening rate, speeds the apparent closing rate, and reduces open channel probability. The altered channel kinetics predict a short duration and low amplitude of the endplate potential that falls short of the threshold for activating postsynaptic voltage-gated Na channels. Finally, the very low opening probability of the mutant receptor over a range of ACh concentrations explains the limited clinical response to pyridostigmine.
Supplementary Material
GLOSSARY
- AChR
acetylcholine receptor
- CMS
congenital myasthenic syndrome
- EP
endplate
- HEK
human embryonic kidney fibroblast
- Popen
channel open probability
Footnotes
Editorial, page 404
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
X.-M. Shen contributed to study concept, data acquisition, analysis, and interpretation. J. Brengman contributed to data acquisition. S. Edvardson contributed to data acquisition. S. Sine contributed to interpretation of data. A. Engel contributed to study concept, data acquisition, analysis, and interpretation.
DISCLOSURE
X.-M. Shen, J. Brengman, and S. Edvardson report no disclosures. S. Sine is supported by a research grant from NIH. A. Engel serves as an Associate Editor of Neurology® and is supported by research grants from NIH and the Muscular Dystrophy Association. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Engel AG. Congenital myasthenic syndromes. In: Aminoff MJ, ed. Handbook of Clinical Neurology Series: Disorders of Neuromuscular Transmission. New York: Elsevier; 2008: 285– 332 [DOI] [PubMed] [Google Scholar]
- 2.Galzi JL, Revah F, Bessis A, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: from electric organ to brain. Annu Rev Pharmacol Toxicol 1991; 31: 37– 72 [DOI] [PubMed] [Google Scholar]
- 3.Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol 2002; 53: 431– 446 [DOI] [PubMed] [Google Scholar]
- 4.Shen X-M, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR δ subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest 2008; 118: 1867– 1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sine SM, Shen X-M, Wang H-L, et al. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol 2002; 120: 483– 496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 2009; 459: 451– 454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature 2008; 454: 722– 727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brejc K, van Dij WV, Schuurmans M, van der Oost J, Smit AB, Sixma TK. Crystal structure of ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001; 411: 269– 276 [DOI] [PubMed] [Google Scholar]
- 9.Chiara DC, Middleton RE, Cohen JB. Identification of tryptophan 55 as the primary site of 3[H]nicotine photoincorporation in the gamma subunit of Torpedo nicotinic acetylcholine receptor. FEBS Lett 1998; 423: 223– 226 [DOI] [PubMed] [Google Scholar]
- 10.Akk G. Contribution of the non-alpha subunit residues (loopD) to agonist binding and channel gating in the muscle nicotinic acetylcholine receptor. J Physiol 2002; 544: 695– 705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Y, Cohen JB. Contribution of Torpedo nicotinic acetylcholine receptor gamma Trp-55 and delta Trp-55 to agonist and competitive antagonist function. J Biol Chem 2001; 276: 2417– 2426 [DOI] [PubMed] [Google Scholar]
- 12.Bafna PA, Jha A, Auerbach A. Aromatic residues ɛTrp-55 and δTrp-57 and the activation of acetylcholine receptor channels. J Biol Chem 2009; 284: 8582– 8588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sine SM, Wang H-L, Bren N. Lysine scanning mutagenesis delineates structure of nicotinic receptor binding domain. J Biol Chem 2002; 277: 29210– 29223 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.