

Abstract
Corneal epithelium relies on abundant glycogen stores as its primary energy source. MicroRNA-31 (miR-31), a corneal epithelial-preferred miRNA, negatively regulates factor inhibiting hypoxia-inducible factor-1 (FIH-1). Since HIF-1α is involved in anaerobic energy production, we investigated the role that miR-31 and FIH-1 play in regulating corneal epithelial glycogen. We used antagomirs (antago) to reduce the level of miR-31 in primary human corneal epithelial keratinocytes (HCEKs), and a miR-31-resistant FIH-1 to increase FIH-1 levels. Antago-31 raised FIH-1 levels and significantly reduced glycogen stores in HCEKs compared to irrelevant-antago treatment. Similarly, HCEKs retrovirally transduced with a miR-31-resistant FIH-1 had markedly reduced glycogen levels compared with empty vector controls. In addition, we observed no change in a HIF-1α reporter or known genes downstream of HIF-1α indicating that the action of FIH-1 and miR-31 on glycogen is HIF-1α-independent. An enzyme-dead FIH-1 mutation failed to restore glycogen stores, indicating that FIH-1 negatively regulates glycogen in a hydroxylase-independent manner. FIH-1 overexpression in HCEKs decreased AKT signaling, activated GSK-3β, and inactivated glycogen synthase. Treatment of FIH-1-transduced HCEKs with either a myristolated Akt or a GSK-3β inhibitor restored glycogen stores, confirming the direct involvement of Akt/GSK-3β signaling. Silencing FIH-1 in HCEKs reversed the observed changes in Akt-signaling. Glycogen regulation in a HIF-1α-independent manner is a novel function for FIH-1 and provides new insight into how the corneal epithelium regulates its energy requirements.—Peng, H., Hamanaka, R. B., Katsnelson, J., Hao, L., Yang, W., Chandel, N. S., Lavker, R. M. MicroRNA-31 targets FIH-1 to positively regulate corneal epithelial glycogen metabolism.
Keywords: Akt signaling, energy metabolism, keratinocytes
The outermost tissue of the cornea is a self-renewing, stratified squamous epithelium that functions to protect the underlying ocular structures (e.g., lens, iris, and retina) from the external environment, supports the tear film, and transmits light to the interior of the eye. The latter function is accomplished in concert with the lens to ensure that the correct refractive properties are established so that an accurate image is delivered to the retina (1). Since the cornea accounts for two-thirds of the refraction of light in the eye, maintenance of transparency is essential (2). A major factor in assuring corneal clarity necessary for vision is the absence of vessels (see ref. 3 and references therein). Therefore, when compared with other stratified squamous epithelia, the corneal epithelium is unique in that it is supported by an avascular stroma. A consequence of such avascularity is the well-established finding that corneal epithelial cells have large stores of glycogen, which serve as a primary energy source (4, 5).
Our understanding of corneal epithelial energy metabolism comes, in part, from investigations on corneal hypoxia and on the complications arising from diabetes. With respect to hypoxia, the corneal epithelium is exceptional in that it is routinely subjected to periods of hypoxia during normal eyelid closure when sleeping, as well as after extended contact lens wear. This can result in glycogen depletion, which leads to disruption of barrier function, inflammation, and/or infection (see refs. 6, 7 and references therein). Regarding diabetes, the corneal epithelium is one of the targets of diabetic complications (8); consequently, attention has been directed at understanding the effects that high glucose levels have on corneal epithelial physiology (9, 10). Common to hypoxia and diabetes is the hypoxia-inducible factor (HIF) family, which is the major effector of the hypoxic response (11) and may contribute to the pathogenesis of diabetes (12).
During normoxia, HIF-1α is polyubiquitinated and targeted for degradation by an E3 ubiquitin ligase complex that contains the von Hippel-Lindau tumor suppressor protein (pVHL), elongin B, elongin C, Cul2, and Rbx (13, 14). The binding of pVHL to HIF-1α is dependent on the hydroxylation of proline residues within the oxygen-dependent degradation (ODD) domain of HIF-1α (15, 16). In mammalian cells, HIF prolyl hydroxylation is carried out by a distinct family of prolyl hydroxylases (PHDs 1-3; refs. 17, 18). Oxygen tension also regulates the interaction of HIF-α with transcriptional coactivators p300 and CBP. Asparagine hydroxylation of HIF-1α by the enzyme factor-inhibiting HIF-1 (FIH-1) blocks the binding of p300 and CBP to HIF-1α, thus inhibiting HIF-mediated gene transcription (19, 20). As levels of hypoxia increase, the PHD- and FIH-1-induced hydroxylation becomes inactivated, and HIF-1α becomes stabilized (21, 22).
While oxygen levels are one means of regulating the activity of FIH-1, it has been reported that microRNA-31 (miR-31) could negatively regulate FIH-1, resulting in the transactivation of HIF in head and neck squamous cell carcinoma cell lines (23). Using human corneal epithelial keratinocytes (HCEKs), we demonstrate that FIH-1 is a direct target of miR-31. We also show that miR-31 loss of function in HCEKs results in an increase in FIH-1 along with a marked decrease in cellular glycogen. FIH-1's negative regulation of glycogen is independent of its hydroxylase activity, occurs in a HIF-1α-independent manner, and happens on a background of attenuated Akt signaling. Such attenuation of glycogen stores is a novel function for FIH-1 and provides new insights into how the corneal epithelium regulates its energy supply.
MATERIALS AND METHODS
Human and mouse corneas
Normal human corneal and limbal epithelia were obtained from the Midwest Eye Banks (Ann Arbor, MI, USA). Wild-type female Balb/c mice were obtained from Charles River. Eyes from ob/ob mice and diet-induced obesity (DIO) mice were kindly provided by Dr. Amy S. Paller (Department of Dermatology, Northwestern University, Chicago, IL, USA).
Laser capture microdissection
Eyes from 13-wk-old female Balb/c mice were embedded in optimal cutting temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA)and stored at −80°C until sectioning. Limbal and corneal epithelium from 5-μm frozen sections were isolated and captured using a PALM laser capture system (Carl Zeiss Instruments, Bernreid, Germany), as described previously (24).
Cell culture
Primary HCEKs and limbal epithelial keratinocytes (HLEKs) were isolated from cadaver corneas provided by Midwest Eye Banks and cultured in CnT-20 medium with supplements (CellnTech, Bern, Switzerland) on collagen IV-coated plates (BD Biosciences, San Jose, CA, USA), as described previously (25). All experiments were performed using keratinocytes with one passage.
Target prediction
We utilized Pictar (New York University, New York, NY, USA) and TargetScan (Massachusetts Institute of Technology, Cambridge, MA, USA) to survey the potential targets of miR-31.
Reagents, constructs, and oligonucleotides
The following chemicals were used in this study: the 3′ untranslated region (UTR) of the human FIH-1 mRNA 5′ glycogen synthase kinase 3 (GSK-3) inhibitor X (0.5 μM; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and dimethyloxalyglycine (DMOG; 0.5 mM; Sigma-Aldrich, St. Louis, MO, USA). The following primers were used for amplifying the 3′ UTR of the human FIH-1 mRNA: 5′-ATTCAACTAGTTCCTGCCAGGTGACTGCTATCC and 3′-CTATTAAGCTTGGGGGCTCACACTGTACTG.
The 3′ UTR of the human FIH-1 mRNA was cloned in between the SpeI and HindIII sites of pMIR-Report. A cDNA encoding miR-31-resistant FIH-1 (FIH-1-cds) was ligated between BamHI and XhoI sites of the retroviral expression plasmid LZRS. The enzyme-dead FIH-1 mutant [D201A; dominant/negative FIH-1 (dnFIH-1)] construct was produced from the LZRS-FIH-1-cds construct using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The primers used were forward, 5′-GCTCACTATGCTGAGCAGCAGAAC-3′, and reverse, 5′-CTGCTGCTCAGCATAGTGAGCAGG-3′. The following constructs were used in this study: pGF1-HIF1 transcription reporter and pGF1-mCMV control (SBI, Mountain View, CA, USA); FIH-1 shRNA expression lentiviral vector pGIPZ-shRNA-FIH-1 (Open Biosystems, Huntsville, AL, USA). A constitutively active form of Akt (pBABE-Myr-Akt) was provided by Navdeep Chandel (Northwestern University; ref. 26). Antagomirs were synthesized by Dharmacon (Lafayette, CO, USA). Sequences were as follows: antagomir to miR-31 (antago-31), 5′-mAsmGsmCsmUmAmUmGmCmCmAmGmCmAmUmCmUmUmGsmCsmCsmU-Chol 3′, and irrelevant antagomir (ir-antago), 5′-mGsmGsmCsmAmUmUmCmAmCmCmGmCmGmUmGmCmCsmUsmUsmA-Chol-3′, where mN represents 2′-O-methyl-modified oligonucleotide, s represents a phosphorothioate linkage, and Chol represents linked cholesterol.
Transfections and antagomirs
For retroviral infections, keratinocytes were transduced with retroviral supernatants for 6 h. Viruses were produced in Phoenix amphotropic packaging cells (Genprice Inc., San Jose, CA, USA) as described previously (27). For lentiviral infections, keratinocytes were transduced with lentiviral supernatants for 6 h, according to the manufacturer's instructions, and switched to fresh culture medium overnight. For antagomir treatments, keratinocytes were grown in normal culture medium to confluence and then switched to culture medium containing antagomirs at a final concentration of 1000 nM for 48 h.
Western blot analysis
Western blots were performed as described previously (25). The following antibodies were used: GSK-3β (Santa Cruz Biotechnology), FIH-1, p-Akt (S473), p-Akt (T308), Akt, p-GSK-3β, p-glycogen synthase (p-GS; Cell Signaling Technologies, Beverly, MA, USA), GAPDH (Abcam, Cambridge, MA, USA), and α-tubulin (Invitrogen, Carlsbad, CA, USA).
Immunohistochemistry and periodic acid-Schiff (PAS) staining
Paraffin sections were processed for immunohistochemistry, as described previously (27). For FIH-1 staining, sections were incubated for 1 h with FIH-1 rabbit polyclonal antibody (1:300; Santa Cruz Biotechnology). Sections were counterstained with hematoxylin to visualize morphology. PAS staining was performed as described previously (28). Images were taken using a Zeiss Axioplan 2 microscope system (Carl Zeiss). Quantification was carried out using a Zeiss Axiovision program. To determine significance, Student's t test was applied to the data.
Northern blot analysis and real-time quantitative PCR (qPCR) analysis
Total RNA from cells was harvested using TRIzol (Invitrogen). Northern blots were performed as described previously (29) using a probe against miR-31 (Exiqon, Woburn, MA, USA). For real-time qPCR, total RNAs were cleaned up by the RNeasy kit (Qiagen, Valencia, CA, USA). cDNA was prepared using Superscript III reverse transcription kit (Invitrogen). Real-time qPCR was performed on an Applied Biosystems 7000 Real Time PCR System (Applied Biosystems, Foster City, CA, USA) using the quantitative SYBR green PCR kit (Qiagen). Primer sequences used in this study were as follows: carbonic anhydrase 9 (CA9), forward 5′-TGGAAGAAATCGCTGAGGAAGGCT-3′, reverse 5′-AGCACTCAGCATCACTGTCTGGTT-3′; vascular endothelial growth factor (VEGF), forward 5′-ACACATTGTTGGAAGAAGCAGCCC-3′, reverse 5′-AGGAAGGTCAACCACTCACACACA-3′; GAPDH, forward 5′-TCGACAGTCAGCCGCATCTTCTTT-3′, reverse 5′-ACCAAATCCGTTGACTCCGACCTT-3′. For microRNA real-time qPCR, total RNAs were isolated by miRNeasy kit (Qiagen), according to the manufacturer's instructions. Taqman microRNA Assays (Applied Biosystems) was performed according to the manufacturer's instructions.
Reporter assay
HCEKs were maintained to confluence in normal culture medium and transduced by either pGF1-HIF1 transcription reporter or pGF1-mCMV control. After keratinocytes were either treated with antagomirs (48 h) or transduced by FIH-1-cds (3 d), luciferase assay was performed as described previously (30). Luciferase activity was normalized to total protein levels between the samples.
RESULTS
miR-31 expression is correlated with FIH-1 levels in vitro and in vivo
Expression profiling indicated that with respect to the ocular anterior segmental epithelium, miR-31 is a corneal epithelial preferred miRNA (29). Consistent with this earlier observation, expression of miR-31 in primary cultures of HCEKs was greater than in primary cultures of HLEKs (Fig. 1A). FIH-1 is a predicted target of miR-31 (Pictar and TargetScan). A reciprocal relationship was noted between FIH-1 protein and miR-31, with low levels of FIH-1 in HCEKs and higher levels in HLEKs (Fig. 1B). We extend this work to the in vivo situation, using laser capture microdissection to isolate relatively pure populations of “resting” adult mouse limbal and corneal epithelial cells from frozen sections of the anterior ocular segmental epithelia. miR-31 was detected in the isolated limbal epithelial cells; however, expression was significantly greater (3-fold) in the corneal epithelial cells (Fig. 1C). Not surprisingly, FIH-1 was barely detected immunocytochemically in the human (Fig. 1D) and mouse corneal epithelia (Fig. 1F), whereas FIH-1 was more readily detected in limbal epithelial cells (Fig. 1D, E). Despite the differential expression of FIH-1 protein between the limbal and corneal epithelia, FIH-1 mRNA levels were similar for both epithelia (Supplemental Fig. S1A). This suggests a regulatory role for miR-31 on FIH-1 protein translation.
Figure 1.

miR-31 and FIH-1 are reciprocally expressed in corneal and limbal epithelia. A) Northern blots of miR-31 and U6 expression in HCEKs and HLEKs. B) Western blots of FIH-1, keratin 3, and GAPDH in HCEKs and HLEKs. C) RNA samples prepared from laser-capture microdissected populations of resting adult limbal and corneal epithelia were subjected to real-time PCR. miR-31 levels were significantly higher in corneal epithelium. D–F) FIH-1 immunostaining of human (D) and mouse (E, F) limbal (E; Lim) and corneal (F; Cor) epithelia. PC, peripheral cornea.
To formally confirm that FIH-1 was a direct target of miR-31, we used luciferase assays to examine the effects of endogenous miR-31 on FIH-1 in HeLa cells. Endogenous miR-31 reduced luciferase activity of the FIH-1wt construct (∼30%) but did not affect the activity of the FIH-1mut (Supplemental Fig. S1B, C). HeLa cells have low endogenous levels of most miRNAs (31); however, these cells have high levels of miR-31. Thus to further validate the mutation data, we transfected the FIH-1wt construct into HeLa cells treated with antago-31 to knock down miR-31. This had no effect on luciferase activity (Supplemental Fig. S1D), indicating that restoration of luciferase activity following transfection of the FIH-1mut (Supplemental Fig. S1C) was due to disruption of the miR-31 binding site. To confirm and extend the HeLa cell findings, we investigated the relationship between miR-31 and FIH-1 in primary cultures of HCEKs. Loss of miR-31 function studies with antago-31 resulted in a marked reduction in endogenous levels of miR-31 (Supplemental Fig. S1E) along with an increase in FIH-1 levels when compared with irrelevant antagomir-treated HCEKs (Supplemental Fig. S1F). Collectively, these findings establish that FIH-1 is a target of miR-31 in cultured corneal epithelial keratinocytes.
Down-regulation of miR-31 depresses glycogen deposition in HCEKs via FIH-1
Anaerobic energy production is one of the functions of HIF-1α (32), and the corneal epithelium is dependent on glycogen for its energy supply (5). Therefore, we reasoned that since down-regulation of miR-31 expression in HCEKs increases FIH-1 protein (Supplemental Fig. S1E, F), this should repress HIF-1α function and result in a change in HCEK glycogen stores. Such a FIH-glycogen relationship has not been investigated in keratinocytes. Toward this end, we treated HCEKs with antago-31 for 48 h and assessed glycogen levels using the PAS/amylase reaction. Ir-antago-treated HCEKs contained abundant amounts of PAS-positive amylase-sensitive material (glycogen; Fig. 2A). In contrast, antago-31 treatment resulted in a significant decrease in intracellular glycogen (Fig. 2A). Quantification of the staining reaction using computer-assisted image analysis (Fig. 2B) indicated that antago-31 treatment reduced glycogen by >48%, which was statistically significant (P<0.01).
Figure 2.

Down-regulation of mir-31 or ectopic FIH-1 reduces glycogen. A) HCEKs treated with an ir-antago or antago-31 for 48 h, stained with PAS with and without amylase to digest glycogen. B) Glycogen density was quantified using computer-assisted image analysis. C) HCEKs transduced with LZRS or LZRS-FIH-1 for 3 d, stained to detect glycogen. D) Glycogen density was quantified as in B. Error bars = sd derived from 4 experiments in triplicate.
To validate that the miR-31-induced decrease in glycogen was working through FIH-1, HCEKs were transduced with a retrovirus containing FIH-1 cDNA, but lacking the endogenous 3′ UTR sequences (FIH-1-cds) and thus making it miR-31 resistant. An increase in FIH-1 protein was observed in the HCEKs transduced with the FIH-1-cds when compared with HCEKs transduced with the empty LZRS vector, indicative that the construct was working (Supplemental Fig. S1G). HCEKs transduced with FIH-1-cds contained undetectable amounts of glycogen, whereas HCEKs transduced with the empty vector had the usual amount of PAS-positive/amylase-sensitive stainable material (Fig. 2C, D), confirming a miR-31/FIH-1/ glycogen relationship. These findings indicate that FIH-1 is a negative regulator of glycogen stores in HCEKs.
FIH-1's action on glycogen is independent of HIF-1α activity
To ascertain whether the observed FIH-1-mediated decrease in glycogen involved HIF-1α, we investigated changes in CA9 and VEGF-known genes downstream of HIF-1α (33), following treatment of HCEKs with FIH-1-cds. Such treatment did not result in significant decreases in the levels of either gene under normoxic conditions (Fig. 3A, B). To further confirm that FIH-1's action on glycogen was independent of HIF-1α, we transduced FIH-1-cds into HCEKs containing a HIF-1 reporter. After 3 d, no change was detected in HIF-1 transcriptional activity under normoxic conditions (Fig. 3C). As a positive control, we treated FIH-1-transduced HCEKs with the cell-permeable hydroxylase inhibitor DMOG (19) for 2 d and observed a marked increase in CA9 and VEGF, as well as HIF-1 transcriptional activity (Fig. 3D–F). Glycogen regulation in a HIF-1α-independent manner is a novel function for FIH-1.
Figure 3.

miR-31/FIH-1 regulates glycogen in a HIF-1α- and hydroxylase-independent manner. A, B) qRT-PCR assessment of CA9 (A) and VEGF (B) levels in HCEKs transduced with FIH-1 or an empty vector for 3 d. C) No changes were noted in the relative luciferase activity of a HIF-1 reporter in FIH-1-transduced HCEKs. D, E) qRT-PCR assessment of CA9 (D) and VEGF (E) mRNA levels in HCEKs treated with DMOG or a vehicle control for 2 d, showing marked increase in CA9 and VEGF. F) DMOG, a hydroxylase inhibitor, markedly increased luciferase activity of HIF-1 reporter-transduced HCEKs. G, H) qRT-PCR assessment of CA9 (G) and VEGF (H) mRNA levels in HCEKs treated with antago-31 or an irrelevant antagomir for 48 h. I) No changes were noted in the relative luciferase activity of a HIF-1 reporter in HCEKs treated with antago-31 or an ir-antago. A–I) J) HCEKs transduced with LZRS, LZRS-FIH-1-cds, or LZRS dnFIH-1 for 3 d were stained with PAS with and without amylase to digest glycogen. K) Glycogen density was quantified using computer-assisted image analysis. Both FIH-1-cds and dnFIH-1 significantly decreased glycogen, as determined by Student's t test. Error bars = sd derived from 3 experiments.
We also determined whether miR-31's positive effect on glycogen was independent of HIF-1α. No significant decrease was noted in CA9, VEGF, or HIF-1 transcriptional activity in HCEKs treated with antago-31 when compared with controls (Fig. 3G–I), indicative that miR-31 is independent of HIF-1α under normoxic conditions.
To investigate whether the decreases observed in glycogen were the result of FIH-1's hydroxylase ability, we transfected HCEKs with an enzyme-dead FIH-1 mutant (D201A; dnFIH-1) for 3 d and compared glycogen levels with wild-type FIH-1. Interestingly, the dnFIH-1 treatment resulted in a similar decrease in glycogen compared with the wild-type FIH-1 (Fig. 3J, K). This suggests that the negative effect of FIH-1 on corneal glycogen stores occurs in a hydroxylase-independent manner, which is a novel function for this protein.
FIH-1 attenuates Akt signaling
It is well established that GSK-3, a multifunctional serine threonine kinase, is a key enzyme involved in glycogen synthesis (34). We observed that FIH-1 overexpression in HCEKs decreased levels of p-Akt (Fig. 4A). Attenuation of Akt signaling by FIH-1 also resulted in activation of GSK-3β, as indicated by a decrease in p-GSK-3β (34) and inactivation of GS, as indicated by an increase in p-GS levels (Fig. 4A). Moreover, silencing FIH-1 led to increases in p-Akt and p-GSK-3β levels (Fig. 4B). Treatment of HCEKs with antago-31 increased FIH-1 levels and negatively affected the Akt signaling pathway, leading to inactivation of GS in a manner similar to the ectopic expression of FIH-1 in these cells (Fig. 4C). To confirm that the Akt/GSK-3β signaling pathway was directly involved in FIH-1-mediated changes in glycogen stores, FIH-1-transduced HCEKs were treated with either a myristolated Akt (Fig. 5A) or a GSK-3β inhibitor (Fig. 5C) and the amounts of amylase-resistant, PAS staining material (glycogen) were determined. FIH-1 transduced HCEKs had significantly reduced amounts of glycogen (Fig. 5), which were restored following treatment with either the myristolated Akt or the GSK-3β inhibitor (Fig. 5). The amount of glycogen present in control-transduced HCEKs was significantly less than in HCEKs exposed to the GSK-3β inhibitor (Fig. 5C, D), reflecting the dampening effects of the endogenous levels of GSK-3β on glycogen synthesis in the control HCEKs. Taken together, these findings indicate that FIH-1 attenuates Akt/GSK-3β signaling, which contributes to the decrease in HCEK glycogen stores.
Figure 4.
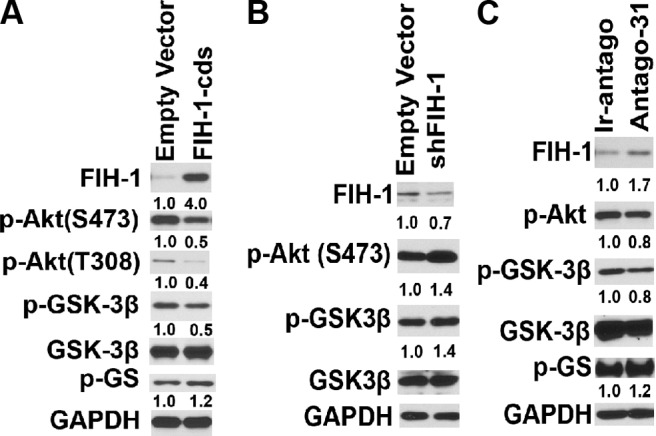
FIH-1 inactivates Akt signaling. Immunoblotting of FIH-1, p-Akt (S473), p-GSK-3β, GSK-3β, and GS in FIH-1-transduced (A), shFIH-1- transduced (B), and antago-31-treated HCEKs (C). Numbers below panels represent the normalized expression signals of proteins.
Figure 5.

Akt/GSK-3β signaling is directly affected by FIH-1. A) HCEKs transduced with LZRS or LZRS-FIH-1-cds were treated with a myristolated Akt for 3 d and stained with PAS with and without amylase to digest glycogen. B) Glycogen density was quantified using computer-assisted image analysis. FIH-1-cds significantly decreased glycogen stores, whereas myristolated Akt or myristolated Akt in combination with FIH-1-cds rescued glycogen. C) HCEKs transduced with LZRS or LZRS-FIH-1-cds were treated with a GSK-3β inhibitor for 1 d and stained with PAS with and without amylase to digest glycogen. D) Glycogen density was quantified using computer-assisted image analysis. GSK-3β inhibitor significantly increased glycogen in HCEKs transduced with either LZRS or FIH-1-cds.
FIH-1 diminishes glycogen stores in vivo
Many of the changes seen in HCEKs with excess FIH-1 (e.g., decreased p-Akt) are also associated with diabetic corneas (9, 10). Thus, we investigated the status of FIH-1 in the corneal epithelium from DIO mice and the ob/ob mouse, well-recognized models for diabetes (35–37). FIH-1 expression was increased in the corneal epithelium from both of these mouse models (Fig. 6A–D). Consistent with this increase in FIH-1 protein, we observed a decrease in miR-31 expression in diabetic mouse corneas (Fig. 6E). These changes were correlated with a decrease in glycogen stores within the corneal epithelium from both mouse models (Fig. 6F–H). Our finding of decreased corneal epithelial glycogen parallels the coordinated increase in FIH-1 and decrease in miR-31 expression in these tissues and mirrors what we observed for HCEKs in vitro.
Figure 6.

FIH-1 and miR-31 are associated with diabetic corneas and a proposed model for FIH-1 and miR-31 in corneal epithelial homeostasis. A–D) Corneas from normal mice (A, C), DIO mice (B), and ob/ob mice (D) stained for FIH-1. E) miR-31 levels in normal and DIO mouse corneal epithelia assessed by qRT-PCR. F) Age-matched wild-type and ob/ob mouse corneal epithelia treated with or without amylase to digest glycogen. G, H) Glycogen density in ob/ob (G) and DIO (H) corneal epithelia was quantified using computer-assisted image analysis. I) FIH-1 is highly expressed in the limbal region, where it impairs glycogen stores and could contribute to lower limbal glycogen levels. miR-31 levels are high in corneal epithelium, where it negatively regulates FIH-1, thus preserving glycogen stores and protects the cornea from hypoxic stress.
DISCUSSION
The ability of FIH-1 to interact with and mediate the repression of HIF-1α has been appreciated for more than a decade, and its greater affinity for oxygen has led to the well-accepted idea that this hydroxylase is an oxygen sensor (20, 38, 39). In addition to HIF-1α, FIH-1 has been shown to hydroxylate ankyrin repeat domain (ARD)-containing proteins, such as Notch (21, 22). Recently, FIH-1 has been shown to regulate respiration, energy balance, and lipid metabolism in vivo (40). Despite the pleiotropic nature of FIH-1, its roles in stratified squamous epithelia in general, and specifically in corneal/limbal epithelia, have not been considered. We now show that FIH-1 is a negative regulator of corneal epithelial glycogen stores, regulates glycogen in a HIF-1α-independent manner, decreases glycogen in a hydroxylase-independent fashion, and attenuates Akt signaling, which results in a decrease in glycogen synthesis. Collectively, these findings delineate a novel function for FIH-1 and provide new insight into how the limbal and corneal epithelia regulate their energy needs.
The normal corneal epithelium has large amounts of readily available glycogen, which is used as a primary energy source (5). The metabolic support provided by glycogen is critical for proper corneal epithelial proliferation and migration, factors central to homeostasis and tissue regeneration (41). For example, corneal epithelial cells use glycogen while migrating or “sliding” during regeneration (5); however, within 1 d following reepithelialization, glycogen stores are at similar levels to the unwounded corneal epithelium (5). Perturbation of rabbit corneal epithelium by 6- and 18-h exposure to a hard contact lens resulted in a ∼4-fold decrease in glycogen levels (5). Thus, the corneal epithelial cell can rapidly alter glycogen synthesis and breakdown. We propose that such metabolism is regulated, in part, via miR-31's action on FIH-1.
It is also well established that glycogen content is lower in the limbal epithelium compared with the corneal epithelium (42). Such differential expression of glycogen can be explained, in part, by the reciprocal relationship between miR-31 and FIH-1, where miR-31 levels are low in the limbal epithelium and high in the corneal epithelium; conversely, FIH-1 protein is high in the limbal epithelium and low in the corneal epithelium. The fact that FIH-1 mRNA expression is similar in limbal and corneal epithelia (Supplemental Fig. S1A) suggests that miR-31 plays an important role in maintaining corneal epithelial glycogen levels via a suppression in FIH-1 translation. Presently, it is not clear whether miR-31 is the only miRNA that regulates FIH-1. However, since FIH-1 is low in the corneal epithelium, we searched the predicted binding sites of corneal-preferred miRNAs in the FIH-1 3′UTR. Interestingly, miR-184, which is a highly corneal epithelial-preferred miRNA (29), was also predicted to target FIH-1 (TargetScan). Luciferase assays showed that miR-184 does not target FIH-1 (data not shown).
The in vivo reciprocal relationship between miR-31 and FIH-1 is maintained in vitro, where HCEKs and HLEKs are cultured under the same conditions. This suggests that the high-miR-31/low-FIH-1 phenotype of corneal epithelial cells is an innate mechanism of these cells to maintain glycogen stores. Likewise, we propose that the high limbal FIH-1 is responsible for the lower glycogen content of this epithelium (Fig. 6I). It is interesting that neonatal cardiomyocytes treated with DMOG to inhibit the PHDs (as well as FIH-1; ref. 19) had significantly higher glycogen stores compared with vehicle treatment (43). These authors postulated that the PHD oxygen-sensing pathway (and by implication, FIH-1) protects cells against hypoxic stress. Lowering miR-31 levels in HCEKs with anatgo-31, which increases FIH-1 and depletes corneal epithelial glycogen stores, is the reciprocal situation to hydroxylase inhibition by DMOG. Thus, one function of miR-31 in corneal epithelium appears to be the negative regulation of FIH-1, which results in the accumulation of corneal epithelial glycogen. Increased glucose in the form of glycogen may be a mechanism by which the corneal epithelium is able to withstand periods of hypoxia during eyelid closure or extended contact lens wear. Thus, miR-31 may function as a novel means of protecting the corneal epithelium from hypoxic stress (Fig. 6I).
Supplementary Material
Acknowledgments
The authors thank Nihal Kaplan for critical reading of the manuscript and helpful discussions. The Northwestern University Skin Disease Research Center Pathology Core assisted in morphological analyses, and the DNA/RNA Delivery Core assisted with retroviral and lentiviral constructs with support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, grant AR057216.
This research is supported by U.S. National Institutes of Health grants EY06769, EY017536, and EY019463 (R.M.L.), 1F32HL099007 (R.B.H.), and 5P30AR057216 (N.S.C.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- antago-31
- antagomir to miR-31
- CA9
- carbonic anhydrase 9
- DIO
- diet-induced obesity
- DMOG
- dimethyloxalyglycine
- dnFIH-1
- dominant/negative FIH-1
- FIH-1
- factor inhibiting hypoxia-inducible factor-1
- GS
- glycogen synthase
- GSK-3
- glycogen synthase kinase 3
- HCEK
- human corneal epithelial keratinocyte
- HIF
- hypoxia-inducible factor
- HLEK
- human limbal epithelial keratinocyte
- ir-antago
- irrelevant antagomir
- miR-31
- microRNA-31
- PAS
- periodic acid-Schiff
- PHD
- prolyl hydroxylase
- UTR
- untranslated region
- VEGF
- vascular endothelial growth factor
REFERENCES
- 1. Piatigorsky J. (2008) Lens and cornea: the “refracton hypothesis”. Semin. Cell Dev. Biol. 19, 69–70 [DOI] [PubMed] [Google Scholar]
- 2. Sun L., Sun T.-T., Lavker R. M. (1999) Identification of a cytosolic NADP+-dependent isocitrate dehydrogenase that is preferentially expressed in bovine corneal epithelium: a corneal epithelial crystallin. J. Biol. Chem. 274, 17334–17341 [DOI] [PubMed] [Google Scholar]
- 3. Cursiefen C., Chen L., Saint-Geniez M., Hamrah P., Jin Y., Rashid S., Pytowski B., Persaud K., Wu Y., Streilein J. W., Dana R. (2006) Nonvascular VEGF receptor 3 expression by corneal epithelium maintains avascularity and vision. Proc. Natl. Acad. Sci. U. S. A. 103, 11405–11410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thoft R. A., Friend J. (1975) Biochmical aspects of contact lens wear. Am. J. Ophthalmol. 80, 139–145 [DOI] [PubMed] [Google Scholar]
- 5. Thoft R. A., Friend J. (1977) Biochemical transformation of regenerating ocular surface epithelium. Invest. Ophthalmol. Vis. Sci. 16, 14–20 [PubMed] [Google Scholar]
- 6. Kimura K., Teranishi S., Kawamoto K., Nishida T. (2010) Protection of human corneal epithelial cells from hypoxia-induced disruption of barrier function by hepatocyte growth factor. Exp. Eye Res. 90, 337–343 [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto N., Jester J. V., Petroll W. M., Cavanagh H. D. (2006) Prolonged hypoxia induces lipid raft formation and increases Pseudomonas internalization in vivo after contact lens wear and lid closure. Eye Contact Lens 32, 114–120 [DOI] [PubMed] [Google Scholar]
- 8. Kaji Y. (2005) Prevention of diabetic keratopathy. Br. J. Ophthalmol. 89, 254–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu K., Yu F. S. (2011) Impaired epithelial wound healing and EGFR signaling pathways in the corneas of diabetic rats. Invest. Ophthalmol. Vis. Sci. 52, 3301–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu K. P., Li Y., Ljubimov A. V., Yu F. S. (2009) High glucose suppresses epidermal growth factor receptor/phosphatidylinositol 3-kinase/Akt signaling pathway and attenuates corneal epithelial wound healing. Diabetes 58, 1077–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semenza G. L. (2001) HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107, 1–3 [DOI] [PubMed] [Google Scholar]
- 12. Gunton J. E., Kulkarni R. N., Yim S., Okada T., Hawthorne W. J., Tseng Y. H., Roberson R. S., Ricordi C., O'Connell P. J., Gonzalez F. J., Kahn C. R. (2005) Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 122, 337–349 [DOI] [PubMed] [Google Scholar]
- 13. Ohh M., Park C. W., Ivan M., Hoffman M. A., Kim T. Y., Huang L. E., Pavletich N., Chau V., Kaelin W. G. (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2, 423–427 [DOI] [PubMed] [Google Scholar]
- 14. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 15. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 16. Masson N., Willam C., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 20, 5197–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- 18. Bruick R. K., McKnight S. L. (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 [DOI] [PubMed] [Google Scholar]
- 19. Lando D., Peet D. J., Gorman J. J., Whelan D. A., Whitelaw M. L., Bruick R. K. (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mahon P. C., Hirota K., Semenza G. L. (2001) FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cockman M. E., Webb J. D., Ratcliffe P. J. (2009) FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N. Y. Acad. Sci. 1177, 9–18 [DOI] [PubMed] [Google Scholar]
- 22. Coleman M. L., McDonough M. A., Hewitson K. S., Coles C., Mecinovic J., Edelmann M., Cook K. M., Cockman M. E., Lancaster D. E., Kessler B. M., Oldham N. J., Ratcliffe P. J., Schofield C. J. (2007) Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J. Biol. Chem. 282, 24027–24038 [DOI] [PubMed] [Google Scholar]
- 23. Liu C. J., Tsai M. M., Hung P. S., Kao S. Y., Liu T. Y., Wu K. J., Chiou S. H., Lin S. C., Chang K. W. (2010) miR-31 ablates expression of the HIF regulatory factor FIH to activate the HIF pathway in head and neck carcinoma. Cancer Res. 70, 1635–1644 [DOI] [PubMed] [Google Scholar]
- 24. Zhou M., Li X. M., Lavker R. M. (2006) Transcriptional profiling of enriched populations of stem cells versus transient amplifying cells. A comparison of limbal and corneal epithelial basal cells. J. Biol. Chem. 281, 19600–19609 [DOI] [PubMed] [Google Scholar]
- 25. Yu J., Peng H., Ruan Q., Fatima A., Getsios S., Lavker R. M. (2010) MicroRNA-205 promotes keratinocyte migration via the lipid phosphatase SHIP2. FASEB J. 24, 3950–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., Chandel N. S. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U. S. A. 107, 8788–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Getsios S., Simpson C. L., Kojima S., Harmon R., Sheu L. J., Dusek R. L., Cornwell M., Green K. J. (2009) Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J. Cell Biol. 185, 1243–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei Z. G., Sun T. T., Lavker R. M. (1996) Rabbit conjunctival and corneal epithelial cells belong to two separate lineages. Invest. Ophthalmol. Vis. Sci. 37, 523–533 [PubMed] [Google Scholar]
- 29. Ryan D. G., Oliveira-Fernandes M., Lavker R. M. (2006) MicroRNAs of the mammalian eye display distinct and overlapping tissue specificity. Mol. Vis. 12, 1175–1184 [PubMed] [Google Scholar]
- 30. Yu J., Ryan D. G., Getsios S., Oliveira-Fernandes M., Fatima A., Lavker R. M. (2008) MicroRNA-184 antagonizes microRNA-205 to maintain SHIP2 levels in epithelia. Proc. Natl. Acad. Sci. U. S. A. 105, 19300–19305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barad O., Meiri E., Avniel A., Aharonov R., Barzilai A., Bentwich I., Einav U., Gilad S., Hurban P., Karov Y., Lobenhofer E. K., Sharon E., Shiboleth Y. M., Shtutman M., Bentwich Z., Einat P. (2004) MicroRNA expression detected by oligonucleotide microarrays: system establishment and expression profiling in human tissues. Genome Res. 14, 2486–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weidemann A., Johnson R. S. (2008) Biology of HIF-1α. Cell Death Differ. 15, 621–627 [DOI] [PubMed] [Google Scholar]
- 33. Bartrons R., Caro J. (2007) Hypoxia, glucose metabolism and the Warburg's effect. J. Bioenerg. Biomembr. 39, 223–229 [DOI] [PubMed] [Google Scholar]
- 34. Rayasam G. V., Tulasi V. K., Sodhi R., Davis J. A., Ray A. (2009) Glycogen synthase kinase 3: more than a namesake. Br. J. Pharmacol. 156, 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rebuffe-Scrive M., Surwit R., Feinglos M., Kuhn C., Rodin J. (1993) Regional fat distribution and metabolism in a new mouse model (C57BL/6J) of non-insulin-dependent diabetes mellitus. Metabolism 42, 1405–1409 [DOI] [PubMed] [Google Scholar]
- 36. Surwit R. S., Kuhn C. M., Cochrane C., McCubbin J. A., Feinglos M. N. (1988) Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37, 1163–1167 [DOI] [PubMed] [Google Scholar]
- 37. Buchanan J., Mazumder P. K., Hu P., Chakrabarti G., Roberts M. W., Yun U. J., Cooksey R. C., Litwin S. E., Abel E. D. (2005) Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349 [DOI] [PubMed] [Google Scholar]
- 38. Koivunen P., Hirsila M., Gunzler V., Kivirikko K. I., Myllyharju J. (2004) Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904 [DOI] [PubMed] [Google Scholar]
- 39. Stolze I. P., Tian Y. M., Appelhoff R. J., Turley H., Wykoff C. C., Gleadle J. M., Ratcliffe P. J. (2004) Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (HIF) in regulating HIF transcriptional target genes. J. Biol. Chem. 279, 42719–42725 [DOI] [PubMed] [Google Scholar]
- 40. Zhang N., Fu Z., Linke S., Chicher J., Gorman J. J., Visk D., Haddad G. G., Poellinger L., Peet D. J., Powell F., Johnson R. S. (2010) The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 11, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu L., Reinach P. S., Kao W. W. (2001) Corneal epithelial wound healing. Exp. Biol. Med. (Maywood) 226, 653–664 [DOI] [PubMed] [Google Scholar]
- 42. Kinoshita S., Kiorpes T. C., Friend J., Thoft R. A. (1982) Limbal epithelium in ocular surface wound healing. Invest. Ophthalmol. Vis. Sci. 23, 73–80 [PubMed] [Google Scholar]
- 43. Sridharan V., Guichard J., Bailey R. M., Kasiganesan H., Beeson C., Wright G. L. (2007) The prolyl hydroxylase oxygen-sensing pathway is cytoprotective and allows maintenance of mitochondrial membrane potential during metabolic inhibition. Am. J. Physiol. Cell Physiol. 292, C719–C728 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.