

Abstract
Cell-penetrating peptides (CPPs) are a class of molecules that enable efficient internalization of a wide variety of cargo in diverse cell types, making them desirable for delivery of anticancer drugs to solid tumors. For CPPs to be useful, it is important to be able to turn their function on in response to an external trigger that can be spatially localized in vivo. Here we describe an approach to turning on CPP function by modulation of the local density of arginine (Arg) residues by temperature-triggered micelle assembly of diblock copolymer elastin-like polypeptides (ELPBCs). A greater than 8-fold increase in cellular uptake occurs when Arg residues are presented on the corona of ELPBC micelles, as compared to the same ELPBC at a temperature in which it is a soluble unimer. This approach is the first to demonstrate digital `off-on' control of CPP activity by an extrinsic thermal trigger in a clinically relevant temperature range by modulation of the interfacial density of Arg residues on the exterior of a nanoparticle.
Keywords: Cell-penetrating peptide, elastin-like polypeptide, micelle, self-assembly, cancer, drug delivery
Current approaches to cancer therapy rely heavily on chemotherapeutic agents whose cytotoxic actions are amplified in rapidly dividing cancer cells. Unfortunately, these agents also affect a subset of dividing healthy cells, resulting in toxic side effects that limit drug dose and ultimately hinder their therapeutic efficacy. Targeting systemically delivered drugs more precisely to the site of disease is a major goal in cancer chemotherapy so as to reduce off-target side effects and thereby increase their maximum tolerated dose and efficacy.
A number of active targeting strategies have been investigated that exploit unique features of tumors, such as overexpressed receptors,1,2 upregulated enzymes,3,4 depressed pH,5,6 and hypoxia,7,8 to enhance the systemic delivery of drug carriers to tumors. These approaches, however, are limited by several factors. First, overexpressed receptors or other cell surface proteins, although present at higher density in tumors, are also expressed by healthy cells, which leads to off-target accumulation and systemic toxicity. Second, many “tumor specific” features such as pH, hypoxia, and receptor expression exhibit great heterogeneity between different types of solid tumors, so that a strategy that seeks to exploit these features for targeted drug delivery is often applicable only to a narrow and specific subset of tumors.9 Third, even within a single tumor type, there is great heterogeneity in these features between patients.10–13
In contrast, cell-penetrating peptides (CPPs) are an attractive –and conceptually orthogonal–targeting approach for drug delivery because they do not rely on unique features of tumors to achieve efficient cellular uptake.14–17 These short peptides, such as TAT,18 penetratin,14 and oligoarginine19 achieve internalization by mechanisms that are believed to be initiated by the electrostatic interaction and hydrogen bonding between a CPP and the phospholipid and proteoglycan components of cell membranes.20,21 These putative interactions with ubiquitous components of the cell membrane enable CPPs to exhibit cellular uptake in a variety of cell lines.16,17 CPPs also enable efficient uptake of attached cargo, as CPPs conjugated to proteins,22 DNA,23 liposomes,24 and micelles25 lead to efficient internalization of these diverse cargo. CPPs thus provide an exciting opportunity to create functionalized drug carriers capable of achieving cellular uptake in a wide range of cancer types.
Unfortunately, their complete lack of cellular specificity is problematic, as systemically delivered CPPs can be taken up in a number of organs within the body.26,27 A `universal' tumor targeting system would thus combine the specificity of receptor-mediated targeting with the ubiquity and robustness of CPPs. This study focuses on solving this dilemma by the in situ triggered assembly of a CPP-presenting nanoparticle solely within a tumor. This approach builds upon the observation that oligoarginine CPPs display a strong cutoff effect in their cell penetration ability; below a threshold of six consecutive arginine (Arg) residues in the CPP, little internalization is observed, while above this threshold of Arg residues there is significant cell uptake.19 We hypothesized that it is not the absolute number of Arg residues, but instead the local Arg density that is responsible for this threshold effect in cell uptake. This hypothesis then suggests that triggered micelle assembly of a diblock copolymer, with less than six Arg residues on its hydrophilic terminus, should provide a system that exhibits digital `off-on' cellular uptake because of the large difference in the local Arg density between the unimer and the micelle.
To create a targeted system with tumor-specific assembly of a functional CPP, we chose heat as the trigger and thermally responsive elastin-like polypeptides (ELPs) as the actuator to modulate the local density of Arg residues by an external thermal stimulus. Heat is an interesting trigger for targeted drug delivery because focused mild hyperthermia –wherein a specific region of the body is heated to a maximum in vivo temperature of approximately 42 °C– can be carried out in a clinical setting using microwaves,28 radio-frequency,29 or ultrasound.30 Mild hyperthermia is of particular interest in oncology because compared to surrounding normal tissue, tumors have poor heat exchange properties because of their tortuous vasculature and low blood flow rates,31,32 so that mild hyperthermia using an external applicator can be confined to solid tumors.33
We chose ELPs as the actuator to build a digital `off-on' switch of CPP activity because ELPs are peptide polymers34 that exhibit lower critical solution temperature (LCST) phase transition behavior, such that below their LCST, or inverse transition temperature (Tt), they exist as soluble unimers, while above their Tt they coacervate into insoluble micron-size aggregates.35 Diblock copolymer ELPs (ELPBCs), composed of hydrophobic and hydrophilic ELP segments, are capable of temperature-triggered micelle assembly.36–38 Upon thermally triggered selective desolvation of the hydrophobic segment, the amphiphilic ELPBCs self-assemble into spherical micelles with a core consisting of the hydrophobic segment and a corona composed of the hydrophilic segment.
To exploit temperature-triggered micelle assembly of ELPBCs to modulate the local Arg density, ELPBCs were recombinantly designed to incorporate five Arg residues –below the threshold necessary for efficient cell uptake– at the terminus of the hydrophilic block (Arg5-ELPBC) (Figure 1A). Below their critical micelle temperature (CMT), we show that Arg5-ELPBCs exist as soluble unimers, whose interfacial Arg density is below the threshold necessary for cell uptake, preventing efficient cellular internalization in this `off state'. Above the CMT Arg5-ELPBCs self-assemble into spherical micelles that display an increased interfacial density of Arg residues on the hydrated surface of the micelle corona. We hypothesize that this dramatic increase in the local interfacial density of Arg residues exceeds the threshold necessary for uptake leading to efficient cellular internalization of the micelles in this `on state' (Figure 1B). We specifically chose a CMT in the range of 39–42 °C, which is typical of temperatures achieved with mild clinical hyperthermia.39,40 The established clinical use of externally applied mild hyperthermia as adjuvant treatment for chemotherapy or external beam irradiation makes this approach practical to translate to the clinic.40
Figure 1.
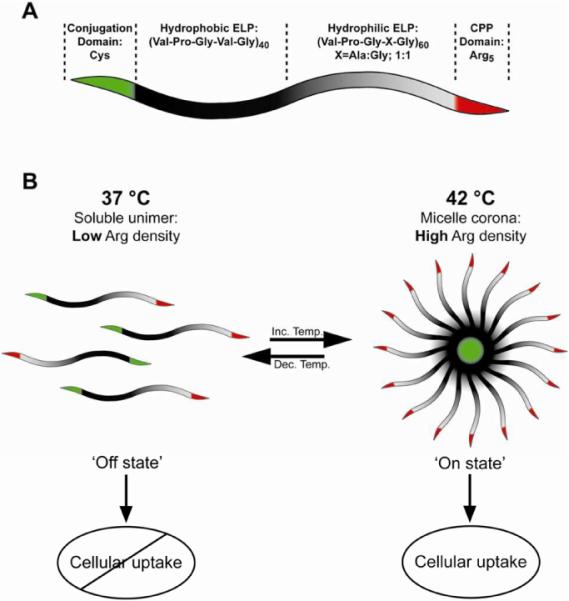
(A) Controlled cellular uptake by modulation of local Arg density is achieved by temperature-triggered micelle assembly of a genetically encoded Arg5-ELPBC. (B) `Off state': At 37 °C, below the CMT, Arg5-ELPBCs exist as soluble unimers, whose limited number of Arg residues is below the threshold necessary for efficient cellular uptake. `On state': Above the CMT at 42 °C (a temperature that can be achieved with mild clinical hyperthermia) Arg5-ELPBCs assemble into spherical micelles. The high density of Arg decorating the micelle corona exceeds the threshold of Arg residues necessary for internalization, leading to cellular uptake of the micelles.
To achieve mild hyperthermia-triggered micelle assembly, the sequence, molecular weight (MW), and ratio of the hydrophobic and hydrophilic segments of the ELPBC have to be precisely tuned to ensure that ELPBCs are soluble unimers at 37 °C and that temperature-triggered micelle assembly occurs between 39 and 42 °C. The design of an ELPBC with a specified CMT in such a narrow temperature range is not trivial for two reasons. First, although the Tt of a single segment ELP can be predicted with precision as a function of length and concentration for a given composition,41 the CMT of an ELPBC is difficult to predict a priori from the Tt of its constituent blocks due to the interactions between these blocks.38 The CMT of the ELPBC is also influenced by the addition of functional peptide sequences at the ELPBC's N- and C-terminus, due to the peptides' effect on the Tt of their adjacent block.
Our strategy for the synthesis of a suitable ELPBC involved an iterative design cycle that required systematic adjustment in each design cycle of ELPBC parameters including ELP sequence and block length. The design process is described in detail in the Supporting Information. After three iterative design cycles, which involved the synthesis and screening of approximately seven ELPBCs, we identified one block copolymer that exhibited the desired CMT between 39 °C and 42 °C. This optimized ELPBC was composed of a hydrophobic (Val-Pro-Gly-Val-Gly)40 segment and a hydrophilic (Val-Pro-Gly-X-Gly)60 segment, in which the guest residue X is Ala or Gly at a ratio of 1:1, and is referred to as Arg5-ELPBC in the remainder of this paper.
The thermal behavior of Arg5-ELPBC was characterized by temperature-programmed turbidimetry and dynamic light scattering (DLS) (Figure 2). The first transition temperature of the ELPBC was observed at 38 °C as a modest increase in optical density, which is caused by its transition from unimer to micelle, and is hence defined as the CMT. The second transition temperature of the ELPBC, indicated by a large increase in optical density, is consistent with a transition from micelles to micron-scale aggregates at approximately 56 °C. The hydrodynamic radius (Rh), measured by DLS as a function of temperature, agreed with the temperature-programmed turbidimetry, indicating a transition of ELPBC from unimer (Rh <10 nm) to micelle (Rh ~20 nm) at 39 °C.
Figure 2.
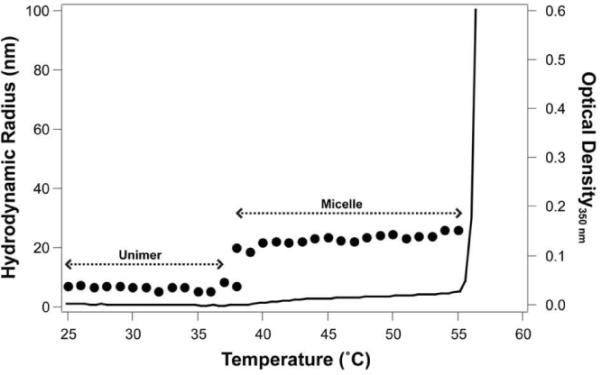
Arg5-ELPBC was characterized by temperature-programmed turbidimetry and DLS. The optical density at 350 nm (black line) indicated that Arg5-ELPBCs existed as soluble unimers with a Rh of <10 nm as determined by DLS (dots) up to 39 °C. At 39 °C Arg5-ELPBCs self-assembled into micelles with a Rh of approximately 20 nm that persisted up to 56 °C. Above 56 °C, Arg5-ELPBC formed micron-size aggregates.
In addition to the Arg5-ELPBC, two control ELPs were synthesized. A control ELPBC was designed with the same ELPBC sequence as the Arg5-ELPBC, but without terminal Arg residues. This ELPBC control served to deconvolute the role of micelle assembly from that of local Arg density on cellular uptake. Additionally, a single segment ELP was synthesized with the same composition as the hydrophilic domain of the ELPBC and was functionalized with five Arg residues at its C-terminus. This Arg5-ELP control was designed to identify the role of Arg on cellular uptake, independent of micelle formation, as it was incapable of self-assembly and had a sufficiently high Tt, such that it existed as a soluble unimer at both 37 °C and 42 °C. All constructs contained an N-terminal cysteine residue on the opposite end of the C-terminal Arg5 peptide, for conjugation of a maleimide derivative of Alexa Fluor 488. The MW and Rh of the three ELPs following fluorophore conjugation are listed in Table 1.
Table 1.
Characterization of Alexa 488-labeled ELP constructs.
| Construct | ELP | C-terminal functionality | MW (kDa) | Rh (nm) 37 °C1 | Rh (nm) 42 °C1 |
|---|---|---|---|---|---|
| Arg5-ELPBC | (Val-Pro-Gly-Val-Gly)40-(Val-Pro-Gly-X-Gly)60 [X=Ala:Gly] | Arg5 | 40.6 | 5.5 ± 0.1 | 21.0 ± 1.3 |
| ELPBC | (Val-Pro-Gly-Val-Gly)40-(Val-Pro-Gly-X-Gly)60 [X=Ala:Gly] | none | 39.8 | 5.8 ± 0.2 | 20.5 ± 0.6 |
| Arg5-ELP | (Val-Pro-Gly-X-Gly)100 [X=Ala:Gly] | Arg5 | 39.2 | 5.4 ± 0.2 | 5.2 ± 0.1 |
ELP at 10 μM in PBS. Data represents the average of 3 replicates ± SEM.
The cellular uptake of all three ELPs was examined in vitro with live cell confocal fluorescence microscopy. Human cervical cancer HeLa cells were incubated with Alexa 488- labeled ELP at 10 μM for 1 hour at 37 °C or 42 °C. There was no significant uptake by cells incubated with Arg5-ELPBC, ELPBC, or Arg5-ELP at 37 °C (Figure 3A–C), a temperature at which all constructs were soluble unimers. In agreement with previous studies,19 the limited number of Arg residues on the Arg5-ELPBC unimer is below the threshold necessary for cellular internalization, thereby leading to minimal cell uptake in this `off state'. At 42 °C the self-assembled Arg5-ELPBC micelle demonstrated significantly increased cellular uptake, as seen by punctate green fluorescence within the cell boundaries at this `on state' (Figure 3D). This internalized Arg5-ELPBC appeared to localize to the perinuclear space (Figure 3D – box). In contrast, the ELPBC and Arg5-ELP controls (Figure 3E–F) showed no increase in cellular uptake at 42 °C, as compared to 37 °C. These results support our hypothesis that it is not micelle assembly alone, or the presentation of Arg residues under hyperthermic conditions, that is responsible for the enhanced cellular uptake evident with the Arg5-ELPBC micelles. Cellular uptake thus requires the high –local– interfacial Arg density on the corona of the self-assembled Arg5-ELPBC micelle.
Figure 3.

Digital switching of cellular uptake by modulation of Arg density was investigated by live cell confocal microscopy. No significant cellular uptake was evident after incubation with Arg5-ELPBC (A), ELPBC (B), or Arg5-ELP (C) at 37 °C, a temperature at which all constructs existed as soluble unimers. At 42 °C temperature-triggered micelle assembly of Arg5-ELPBC increased the local density of Arg on the micelle corona, resulting in enhanced cellular uptake, evident as punctate green intracellular fluorescence (D). This internalized Arg5-ELPBC localized to the perinuclear space (D – box). No increase in cellular uptake was observed with the ELPBC (E) or Arg5-ELP (F) controls at 42 °C. Green: ELP; Red: cell membrane; Blue: cell nuclei; scale bars 25 μm.
Live-cell flow cytometry was performed at different times post-incubation, to quantify the effect of enhanced Arg density on cellular uptake. Over the course of one hour at 42 °C, cells exhibited increasing cellular fluorescence when incubated with Arg5-ELPBCs, which display a high interfacial Arg density on the corona of their self-assembled micelles (Figure 4A). A minimal increase in cellular fluorescence was observed at 37 °C when cells were incubated with Arg5-ELPBC, which exists as a soluble unimer at 37 °C. Cellular fluorescence was low and approximately invariant over the course of one hour at 37 °C and 42 °C for ELPBC (Figure 4B) and Arg5-ELP (Figure 4C) controls. This data clearly shows that there was a dramatic, approximately 8-fold, difference in cellular uptake between 37 °C and 42 °C for Arg5-ELPBC as compared to either ELPBC or Arg5-ELP controls at all time points.
Figure 4.
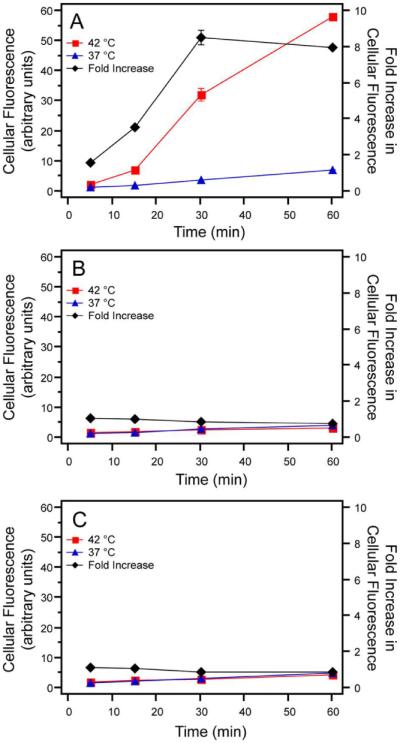
Cellular uptake was quantified by measurement of fluorescence with live-cell flow cytometry. Fluorescence of cells incubated with Arg5-ELPBC at 42 °C increased significantly over the course of one hour, while those incubated at 37 °C showed little enhancement in fluorescence (A). ELPBC (B) and Arg5-ELP (C) controls showed minimal change in uptake over the course of an hour and showed little difference in uptake between 37 °C and 42 °C at each time point. The fold increase in cellular fluorescence (black line), calculated as the fluorescence at 42 °C divided by the fluorescence at 37 °C, reached approximately 8-fold for Arg5-ELPBC within one hour. Data represents the average of two replicates ± SEM.
The fold-increase in uptake in the `on state' versus `off state' was calculated as the ratio of mean fluorescence intensity at 42 °C and 37 °C. This fold-increase in fluorescence serves as an in vitro metric to predict targeted cellular uptake by cells within a heated tumor compared to the cellular uptake in off-target tissues that are at physiological temperature. This metric, as well as the overall cellular uptake, is important to characterize the ability of Arg5-ELPBC to achieve sufficient uptake at the heated disease site while minimizing uptake at off-target tissues. Cells incubated with Arg5-ELPBC demonstrated a greater than 8-fold maximum increase in fluorescence at 42 °C, compared to 37 °C, after approximately 30 minutes of incubation (Figure 4A – black line). Furthermore, the maximal fold-increase in uptake between 42 °C and 37 °C is achieved within the standard one hour duration of mild clinical hyperthermia.39
To examine the utility of thermally triggered uptake of Arg5-ELPBCs across cell types, we quantified cellular internalization in two different tumor cell lines –HeLa (human cervical cancer) and MCF7 (human breast cancer)– as well as primary human umbilical vein endothelial cells (HUVECs), because endothelial cells in the tumor vasculature are the first cells that Arg5-ELPBCs will encounter when they are self-assembled into micelles as they circulate through a heated tumor. Arg5-ELPBC showed a significantly greater fold-increase in cellular fluorescence between 42 °C and 37 °C, as compared to ELPBC or Arg5-ELP controls, in all three cell lines, though the effect in HUVECs was more modest (Figure 5). This suggests that uptake in the tumor vasculature is likely to only be modestly enhanced, with the major effect of increased uptake likely to be observed in tumor cells.
Figure 5.
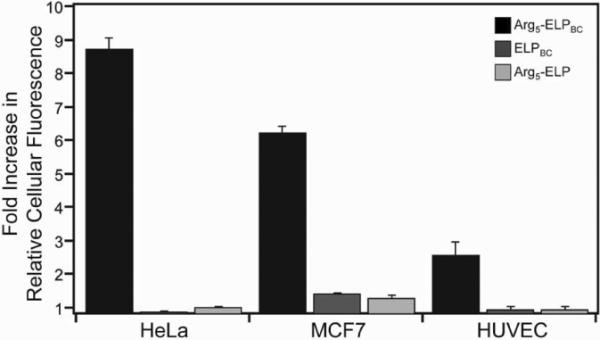
Cellular uptake was quantified in three cell lines by live-cell flow cytometry. The fold-increase in fluorescence at 42 °C, as compared to 37 °C, was measured for HeLa cervical cancer cells, MCF7 breast cancer cells, and primary HUVECs. All cell lines demonstrated the effect of Arg density modulation by temperature-triggered micelle assembly in controlling cellular uptake, as the fold-increase in cellular fluorescence of Arg5-ELPBC was significantly greater than ELPBC or Arg5-ELP controls within each cell line. Data represents the average of 3 experiments ± SEM.
We next investigated the mechanism of cellular uptake. Although the exact mechanism of CPP-mediated cellular uptake is yet to be precisely and unequivocally elucidated, and is likely to depend upon the appended cargo, nanoparticles functionalized with Arg-rich CPPs have largely been shown to preferentially utilize endocytosis pathways other than clathrin-mediated endocytosis.42,43 This is in contrast to carriers that utilize receptor-ligand interactions for cell uptake, which typically lead to clathrin-mediated endocytosis. To investigate the mechanism of uptake of ELP constructs, HeLa cells were co-incubated at 42 °C with ELP and dansylcadaverine, genistein, or amiloride, to inhibit clathrin-dependent endocytosis, caveolae-dependent endocytosis, or macropinocytosis, respectively (Figure 6A). After one hour, cells were analyzed by flow cytometry.
Figure 6.
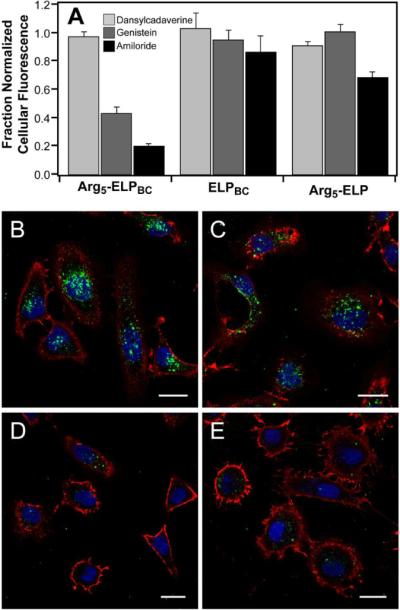
The mechanism of cellular internalization was investigated by flow cytometry and confocal microscopy of cells co-incubated with ELP and endocytosis inhibitors at 42 °C. Cellular uptake, as measured by cellular fluorescence, significantly decreased for Arg5-ELPBC micelles co-incubated with genistein or amiloride, but not dansylcadaverine (A). This effect was more pronounced for Arg5-ELPBC as compared to ELPBC or Arg5-ELP controls. This suggests that caveolae-mediated endocytosis and macropinocytosis play important roles in the internalization of Arg5-ELPBCs in their micelle state. Confocal microscopy of cells incubated with Arg5-ELPBC micelles alone (B), or in combination with dansylcadaverine (C), genistein (D), or amiloride (E) support the conclusions drawn from flow cytometry. Data represents average of 3 experiments ± SEM. Green – ELP; Red – cell membrane; blue – cell nuclei; scale bars 25 μm. Signal from the ELP is shown as a maximum intensity projection, demonstrating cellular uptake throughout the volume of the cell.
Cellular fluorescence in the presence of endocytosis inhibitors was normalized to the fluorescence of cells treated with the same ELP in the absence of that inhibitor. Dansylcadaverine had little effect on the cellular internalization of Arg5-ELPBC, ELPBC, or Arg5-ELP, suggesting that clathrin-mediated endocytosis is not the primary endocytosis pathway for these polypeptides. In contrast, genistein and amiloride decreased cellular internalization of Arg5-ELPBC by 57% and 80%, respectively, as compared to uptake in the absence of inhibitor. ELPBC and Arg5-ELP controls showed no significant change in cellular uptake in the presence of these inhibitors, with the exception of a decrease in uptake by 31% of Arg5-ELP co-incubated with amiloride. These results agree with previous studies that carriers functionalized with oligoarginine CPPs exploit macropinocytosis as a predominant mechanism of cellular uptake.44 Additionally, the enhanced effect of amiloride in decreasing cellular uptake of Arg5-ELPBC, when compared with the Arg5-ELP control, demonstrated the increase in macropinocytosis as the internalization mechanism with increasing density of Arg, similar to results observed for other Arg-decorated drug delivery vehicles.44 However, Arg5-ELPBC appears to exploit more than one pathway of internalization, as a decrease in cellular uptake in the presence of genistein implicates caveolae-mediated endocytosis as a secondary mechanism for cellular uptake of Arg5-ELPBC.
These results were qualitatively confirmed by confocal microscopy of HeLa cells incubated with Arg5-ELPBC alone or in combination with dansylcadaverine, genistein, or amiloride at 42 °C (Figure 6B–E). Internalization of Arg5-ELPBC was observed in cells receiving no inhibitor treatment and uptake was not affected by the presence of dansylcadaverine. This was in contrast to cells incubated with genistein or amiloride, in which uptake was largely eliminated. These results confirmed that both caveolae-mediated endocytosis and macropinocytosis are important pathways for the uptake of Arg5-ELPBC micelles in their `on state'. These results are interesting as these mechanisms offer advantages for uptake of drug delivery vehicles, as they are less degradative routes of cellular entry compared to clathrin-mediated endocytosis. Studies have shown that caveolae-mediated endocytosis avoids intracellular lysosomal trafficking,45 while macropinocytosis involves inherently leaky vesicles that permit some release of cargo, as well as intracellular routing that, in part, avoids fusion with lysosome compartments.46 Together, these pathways provide routes of cellular entry that are likely to be less harmful to labile therapeutic cargo such as biologic drugs.
For CPPs to be useful for systemically delivered and tumor targeted drug delivery, their function must be precisely controlled outside of the disease site. Several approaches to this challenge have involved the unmasking of CPPs by removal of stealth polymers that coat the CPP,24,25,47,48 dissociation of CPPs from anionic inhibitors,49,50 or triggered presentation of CPPs beyond a stealth coating using molecular actuator tethers.51 All of these approaches depend on triggers intrinsic to solid tumors, such as pH52 or enzymes,53 to achieve CPP activation. These methods for controlling CPP function provide useful alternatives to traditional active targeting, but may be limited by several factors including their reliance on intrinsic tumor triggers, which can be heterogeneous across tumor type, and the irreversibility of CPP activation, which can result in off-target accumulation if the activated CPP-functionalized carrier leaks out of the tumor.
The approach presented here has the potential to surmount these limitations by exploiting an extrinsic thermal trigger to control CPP activation by the interfacial –local– modulation of Arg density on the exterior of a self-assembled nanoparticle. This approach circumvents the heterogeneity of tumor phenotypes by avoiding the use of intrinsic tumor triggers (such as pH or protease levels) while providing a digital `off-on' switch for CPP activation by focused mild hyperthermia in a narrow, clinically relevant, temperature window. The digital switching of Arg density also biases uptake towards endocytic pathways that avoid lysosomal accumulation and thereby prevent premature degradation of labile cargo. Together, these properties provide a new nanoscale approach to harness the power of CPPs for targeted drug delivery.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by NIH grant R01EB007205 (A.C.) and by the NSF through the Research Triangle MRSEC (NSF DMR-11-21107).
Footnotes
SUPPORTING INFORMATION. Experimental methods and detailed description of ELPBC design are provided. This material is available and free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nature nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- (2).Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Molecular membrane biology. 2010;27:286–298. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vartak DG, Gemeinhart RA. J Drug Target. 2007;15:1–20. doi: 10.1080/10611860600968967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).MacEwan SR, Callahan DJ, Chilkoti A. Nanomedicine. 2010;5:793–806. doi: 10.2217/nnm.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lee ES, Gao Z, Bae YH. J Control Release. 2008;132:164–170. doi: 10.1016/j.jconrel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Manchun S, Dass CR, Sriamornsak P. Life sciences. 2012;90:381–387. doi: 10.1016/j.lfs.2012.01.008. [DOI] [PubMed] [Google Scholar]
- (7).Kizaka-Kondoh S, Inoue M, Harada H, Hiraoka M. Cancer Sci. 2003;94:1021–1028. doi: 10.1111/j.1349-7006.2003.tb01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wilson WR, Hay MP. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- (9).Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Anal Biochem. 2005;338:284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- (10).Muss HB, Thor AD, Berry DA, Kute T, Liu ET, Koerner F, Cirrincione CT, Budman DR, Wood WC, Barcos M, et al. N Engl J Med. 1994;330:1260–1266. doi: 10.1056/NEJM199405053301802. [DOI] [PubMed] [Google Scholar]
- (11).Fink-Retter A, Gschwantler-Kaulich D, Hudelist G, Mueller R, Kubista E, Czerwenka K, Singer CF. Oncol Rep. 2007;18:299–304. [PubMed] [Google Scholar]
- (12).Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Cancer Sci. 2008;99:929–935. doi: 10.1111/j.1349-7006.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dexter DL, Leith JT. J Clin Oncol. 1986;4:244–257. doi: 10.1200/JCO.1986.4.2.244. [DOI] [PubMed] [Google Scholar]
- (14).Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- (15).Kerkis A, Hayashi MA, Yamane T, Kerkis I. IUBMB life. 2006;58:7–13. doi: 10.1080/15216540500494508. [DOI] [PubMed] [Google Scholar]
- (16).Mueller J, Kretzschmar I, Volkmer R, Boisguerin P. Bioconjug Chem. 2008;19:2363–2374. doi: 10.1021/bc800194e. [DOI] [PubMed] [Google Scholar]
- (17).Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, Gross LA, Tsien RY. Integr Biol (Camb) 2009;1:382–393. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Frankel AD, Pabo CO. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- (19).Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- (21).Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K, Negishi M, Nomizu M, Sugiura Y, Futaki S. Biochemistry. 2007;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- (22).Wadia JS, Dowdy SF. Adv Drug Deliv Rev. 2005;57:579–596. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- (23).Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR, Juliano RL. Pharm Res. 2002;19:744–754. doi: 10.1023/a:1016136328329. [DOI] [PubMed] [Google Scholar]
- (24).Kale AA, Torchilin VP. J Drug Target. 2007;15:538–545. doi: 10.1080/10611860701498203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sethuraman VA, Lee MC, Bae YH. Pharm Res. 2008;25:657–666. doi: 10.1007/s11095-007-9480-4. [DOI] [PubMed] [Google Scholar]
- (26).Aguilera TA, Olson ES, Timmers MM, Jiang T, Tsien RY. Integr Biol (Camb) 2009;1:371–381. doi: 10.1039/b904878b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Miyaji Y, Walter S, Chen L, Kurihara A, Ishizuka T, Saito M, Kawai K, Okazaki O. Drug metabolism and disposition: the biological fate of chemicals. 2011;39:1946–1953. doi: 10.1124/dmd.111.040725. [DOI] [PubMed] [Google Scholar]
- (28).Jones E, Thrall D, Dewhirst MW, Vujaskovic Z. Int J Hyperthermia. 2006;22:247–253. doi: 10.1080/02656730600765072. [DOI] [PubMed] [Google Scholar]
- (29).James JR, Gao Y, Soon VC, Topper SM, Babsky A, Bansal N. Int J Hyperthermia. 2010;26:79–90. doi: 10.3109/02656730903373509. [DOI] [PubMed] [Google Scholar]
- (30).O'Neill BE, Li KC. Int J Hyperthermia. 2008;24:506–520. doi: 10.1080/02656730802093661. [DOI] [PubMed] [Google Scholar]
- (31).Alpard SK, Vertrees RA, Tao W, Deyo DJ, Brunston RL, Jr., Zwischenberger JB. Perfusion. 1996;11:425–435. doi: 10.1177/026765919601100602. [DOI] [PubMed] [Google Scholar]
- (32).Thrall DE. Radiation and environmental biophysics. 1980;17:229–232. doi: 10.1007/BF01323649. [DOI] [PubMed] [Google Scholar]
- (33).Ranjan A, Jacobs GC, Woods DL, Negussie AH, Partanen A, Yarmolenko PS, Gacchina CE, Sharma KV, Frenkel V, Wood BJ, Dreher MR. J Control Release. 2012;158:487–494. doi: 10.1016/j.jconrel.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tatham AS, Shewry PR. Trends Biochem Sci. 2000;25:567–571. doi: 10.1016/s0968-0004(00)01670-4. [DOI] [PubMed] [Google Scholar]
- (35).Urry DW. J. Phys. Chem. B. 1997;101:11007–11028. [Google Scholar]
- (36).Lee TAT, Cooper A, Apkarian RP, Conticello VP. Adv. Mater. 2000;12:1105–1110. [Google Scholar]
- (37).Wright ER, Conticello VP. Adv Drug Deliv Rev. 2002;54:1057–1073. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- (38).Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. J Am Chem Soc. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Roemer RB. Annu Rev Biomed Eng. 1999;1:347–376. doi: 10.1146/annurev.bioeng.1.1.347. [DOI] [PubMed] [Google Scholar]
- (40).Falk MH, Issels RD. Int J Hyperthermia. 2001;17:1–18. doi: 10.1080/02656730150201552. [DOI] [PubMed] [Google Scholar]
- (41).Meyer DE, Chilkoti A. Biomacromolecules. 2004;5:846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- (42).Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- (43).Jones SW, Christison R, Bundell K, Voyce CJ, Brockbank SM, Newham P, Lindsay MA. British journal of pharmacology. 2005;145:1093–1102. doi: 10.1038/sj.bjp.0706279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Khalil IA, Kogure K, Futaki S, Harashima H. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- (45).Shin JS, Abraham SN. Science. 2001;293:1447–1448. doi: 10.1126/science.1061079. [DOI] [PubMed] [Google Scholar]
- (46).Khalil IA, Kogure K, Akita H, Harashima H. Pharmacological reviews. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- (47).Mok H, Bae KH, Ahn CH, Park TG. Langmuir. 2009;25:1645–1650. doi: 10.1021/la803542v. [DOI] [PubMed] [Google Scholar]
- (48).Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. Bioconjug Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BL. Bioconjug Chem. 2006;17:787–796. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- (50).Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Proc Natl Acad Sci U S A. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. J Control Release. 2008;129:228–236. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cardone RA, Casavola V, Reshkin SJ. Nat Rev Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- (53).Roy R, Yang J, Moses MA. J Clin Oncol. 2009;27:5287–5297. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.