

Abstract
Diabetic nephropathy is a major complication of diabetes mellitus and the leading cause of end-stage renal disease. Both hyperglycemia and hypertension (systemic and/or intraglomerular) are established causal factors for diabetic nephropathy. Nonetheless, there is growing evidence that activated innate immunity and inflammation are also contributing factors to the pathogenesis of diabetic nephropathy. This notion is based on increasing evidence indicating that both cytokines-chemokines and pro-fibrotic growth factors are important players in the progression of diabetic nephropathy, effectively accelerating and exacerbating inflammatory and fibrotic processes leading to end-stage renal disease. In this review, we focus on several predominant cytokines-chemokines as potential predictive markers for diabetic nephropathy. These cytokines-chemokines may also be helpful as biomarkers to monitor the progression of the disease and the impact of interventional modalities aimed at halting eventual manifestation of end-stage renal disease in diabetic patients.
Keywords: Renal disease, Major complication of diabetes, Cytokines/chemokines, Advanced predictive diagnostics, Disease monitoring, Personalized patient treatment
Introduction
Nephropathy is a microvascular complication of both type 1 and type 2 diabetes mellitus [1]. It is the leading cause of end-stage renal disease (ESRD), and associated morbidity and mortality, worldwide [2–4]. The course of diabetic nephropathy remains unpredictable and the pathogenesis of progression is not completely understood. A number of risk factors have been suggested in the pathogenesis of diabetic nephropathy including low birth weight, endothelial dysfunction, smoking, renin-angiotensin system stimulation, obesity, hyperglycemia and hypertension (Fig. 1) [5–7]. The development of diabetic nephropathy is a gradual process which mainly starts with progression from normal albuminuria to microalbuminuria [urinary albumin excretion (UAE) 30–300 mg/24 hours] for 5–10 years [1, 8, 9]. Thus, microalbuminuria could be a predictive marker for development of overt diabetic nephropathy in the uncontrolled diabetic patient. Nonetheless, with progression of the disease, blood pressure increases associated with marked albuminuria (UAE > 300 mg/day) and a relentless decline in glomerular filtration rate (GFR) [8, 9].
Fig. 1.

A number of risk factors contribute to eventual manifestation of insulin resistance/hyperglycemia. In turn, hyperglycemia results in a myriad of metabolic and hemodynamic abnormalities that are intimately associated with microvascular complication of diabetes including nephropathy
Hyperglycemia is a well-recognized causal factor for development of diabetic microvascular complications including nephropathy [10–12]. Accordingly, it is suggested that hyperglycemia results in increased generation of superoxide from a number of sources including the mitochondria, the NADPH oxidase and the uncoupled nitric oxide synthase (NOS) [13–19]. Superoxide exerts a diverse array of effects that are considered detrimental to the cell. For example, superoxide could scavenge nitric oxide leading to the generation of peroxynitrite [20, 21]. This will have the dual effects of removing/attenuating the protective effects of nitric oxide on the vasculature and generation of peroxynitrite which could damage proteins and DNA [16, 21]. However, increased generation of superoxide and peroxynitrite also results in DNA damage and subsequent activation of the poly-ADP ribose polymerase [22, 23]. In turn, activation of poly-ADP ribose polymerase exerts a number of effects including poly-ADP ribosylation of other proteins including glycerladehyde-3-phosphate dehydrogenase. Subsequently, increased availability of glycolytic intermediates causes their diversion into other pathways including the polyol pathway, hexosamine pathway, protein kinase C (PKC) pathway and the advanced glycation end (AGE) products pathway which are ultimately linked to manifestation of diabetic microvascular and macrovascular complications [24–28]. For example, activation of PKC pathway is linked to activation of a myriad of downstream pathways. These include a) increased nuclear factor-κB (NF-κB) activation and subsequent pro-inflammatory gene expression [28–30], b) increased NADPH oxidase and subsequent reactive oxygen species (ROS) generation [28, 31–33], c) increased plasminogen activator inhibitor-1 (PAI-1)-induced reduction in fibrinolysis and consequent vascular occlusion [28, 34, 35], d) increased formation of vascular endothelial growth factor (VEGF) thereby resulting in vascular permeability changes and angiogenesis [28, 36, 37] and e) upregulation of transforming growth factor-β (TGF-β), collagen and fibronectin thereby promoting capillary occlusion and the increase in extracellular matrix deposition [28, 38–40].
Clearly, the pathogenic mechanisms of hyperglycemia-induced diabetic complications are complex and have been the focus of previous reviews [24, 28]. However, as alluded to earlier, one consequence of hyperglycemia-induced ROS generation is activation of pro-inflammatory cascades which, in turn, increase transcription of genes encoding cytokines-chemokines, growth factors and extracellular matrix proteins (Fig. 2) [41, 42]. Thus, this communication will focus on the emerging role of inflammatory cytokines-chemokines in relation to diabetic nephropathy. Given the devastating consequences of diabetic renal disease, early detection through identification of predictive biomarkers could potentially lead to prevention of disease progression, reduced morbidity and mortality and ultimately reduced healthcare costs.
Fig. 2.

Schematic diagram showing the proposed relationship between hyperglycemia, oxidative stress and inflammatory cytokines production in the pathogenesis and progression of diabetic nephropathy. ROS: reactive oxygen species; TNF-α: tumor necrosis factor-α; NFκB: nuclear transcription factor-κ; MCP-1: monocyte chemoattractant protein-1; CAMs: cellular adhesion molecules; NO: nitric oxide; TGF-β: transforming growth factor-β; CTGF: connective tissue growth factor; CRP: C-reactive protein
Inflammatory cytokines-chemokines and diabetic nephropathy
Cytokines are redundant secreted proteins that exert a myriad of effects including regulation and determination of the nature of immune responses, immune cell trafficking and cellular arrangements in immune organs [43]. Cytokines are produced de novo following an immune stimulus; in turn, they regulate proliferation of immune cells and their differentiation [43]. These include interleukin (IL)-1 (which activates T cells), IL-2 (which stimulates proliferation of antigen-activated T and B cells), IL-4, 5 and 6 (which stimulate proliferation and differentiation of B-cells) as well as IL-3 and IL-7 (which stimulate hematopoiesis), among others [44, 45].
Chemokines (chemotactic cytokines), also small secreted proteins (7–10 kDa), play crucial roles in many pathological processes including infection, allergic reaction, autoimmune disease and inflammation [46, 47]. A distinguishing feature of chemokines is the fact that they are the only members of the cytokine family that act on the superfamily of G-protein coupled serpentine receptors [46]. Once released, chemokines interact with specific cell surface receptors (e.g., CC, C, CXC and CX3C chemokine receptor subfamilies) of their target cells [48]. Consequently, there is recruitment and activation of leukocytes in preparation for mounting an immune response and initiation of wound healing [49]. Constitutive production of chemokines v is believed to regulate basal leukocytes trafficking. On the other hand, the induction of chemokines during inflammation (e.g., in response to early-response cytokines such as IL-1 and tumor necrosis factor) is crucial for recruitment of leukocytes to loci of immune reaction, a process regulated by mitogen-activated protein kinases [50, 51]. A noted feature of the process is the observation that the same stimulus can elicit production of multiple chemokines form the same cell thereby augmenting the inflammatory response. On the other hand, hemostatic chemokines are involved in adaptive immune responses such as lymphocyte trafficking, antigen sampling in secondary lymphoid tissues and immune surveillance [52].
Increasing evidence points to critical roles of pro-inflammatory cytokines in pathogenesis of diabetic nephropathy. For example, IL-1 is believed to increase vascular permeability and proliferation of mesangial cells and matrix deposition [4, 53]. On the other hand, IL-6 reportedly upregulates mesangial cell proliferation, increases fibronectin expression and affects extracellular matrix dynamics of mesangial cells and podocytes along with increased expression of adhesion molecules on endothelial cells and vascular smooth muscle cells [4, 54, 55]. Still other cytokines, such as tumor necrosis factor-α (TNF-α), impair balance among vasodilator and vasoconstriction mediators, upregulate production of ROS thereby contributing to alterations in glomerular capillary permeability barrier [4, 56]. Collectively, these effects are believed to contribute to the functional alterations associated with diabetic nephropathy such as albuminuria and dysregulation of sodium homeostasis.
Importantly, chemokine-induced inflammatory cell recruitment into renal tissue is a critical feature of various forms of renal disease including diabetic nephropathy. For example, during early phase of diabetic nephropathy, monocyte chemoattractant protein-1 (MCP-1) induces macrophage recruitment and accumulation; this has been reported both for experimental models of diabetes and diabetic patients [57–59]. Chemokines also increase expression of intercellular and vascular cellular adhesion molecules (ICAM-1, VCAM-1) and E-selectin leading to recruitment of more monocytes and macrophages to site of inflammation [60]. In fact, chemokines-induced proinflammatory gene activation in diabetes leads to further production of cytokines such as TNF, interleukins, and interferon-γ thereby establishing a self-perpetuating cascade [61, 62]. The contributing factors that stimulate expression of these genes are components of the diabetic milieu including ROS, oxidized lipids, reduced nitric oxide (NO), increased angiotensin II, free fatty acids (FFA) and AGE products [4, 63]. Both endothelial cells and macrophages contribute to altered vascular reactivity and coagulation through increased expression of plasminogen activator-1 (PAI-1) and tissue factor as well as through platelet activation and increased generation of coagulation factors such as fibrinogen and factor VIII [64]. Importantly, a frequent accompanying feature of patients with type 2 diabetes is overweight/obesity and adipose tissue is known to contribute importantly to the inflammatory process in these individuals in both vascular and nonvascular tissues [64]. Pro-inflammatory and procoagulant mediators released by adipose cells in obese subjects exert both local and systemic effects on vascular metabolism and function [65]. The adipose tissue of obese subjects contains activated macrophages that together with adipocytes produce inflammatory mediators such as MCP-1, macrophage inhibitory factor (MIF), TNF-α, and IL-6 as well as vasoactive substances such as angiotensinogen and endothelin [65–67].
The following describes the most relevant inflammatory markers that are currently used or are under investigation as predictive/diagnostic markers for diabetic nephropathy.
C-reactive protein (CRP)
CRP is an acute phase protein produced by liver cells in response to various inflammatory stimuli; it is found in the blood [4]. CRP is a member of the pentraxin family of oligomeric proteins which is believed to play a fundamental role in natural host defense and innate immunity [4, 68]. As a member of the class of acute-phase reactants, plasma CRP level rises dramatically during acute inflammatory processes [69, 70]. This is primarily due to the rise in the serum concentration of macrophage-derived IL-6, the most important stimulator of CRP production. Thus, CRP is a very sensitive predictor of inflammation and it has been associated with various inflammatory diseases such as atherosclerosis, diabetes and myocardial infarction [4, 71]. Immunoregulatory functions of CRP include enhancement of leukocyte reactivity, complement fixation, modulation of platelet activation and clearance of cellular debris from sites of active inflammation. It is well known that serum CRP levels could serve as a sensitive circulating marker of inflammation as CRP is independently associated with an increased risk of cardiovascular diseases and diabetes mellitus [72, 73]. In addition, experimental and clinical studies suggest that CRP and IL-6 are sensitive physiological markers of subclinical systemic inflammation which are associated with insulin resistance and hyperglycemia [74]. Further, CRP level increases at early stages of diabetic nephropathy and is independently associated with biomarkers of glomerular and tubulointerestitial damage (e.g., urinary albumin excretion and urinary N-acetyl-β-glucosaminidase) [75–77]. Collectively, these observations suggest that assessment of serum CRP may serve as a useful predictive marker for detection and progression of diabetic nephropathy (Table 1).
Table 1.
Role of several inflammatory cytokines in the pathogenesis of diabetic nephropathy and their potential predictive roles
| Cytokine | Role in Diabetic Nephropathy | Possible prognostic Value |
|---|---|---|
| CRP | An acute phase reactant that plays a fundamental role in natural host defense and innate immunity during renal injury | Plasma CRP could be used as an indicator of early stages of diabetic nephropathy |
| TNF-α | Is involved in systemic inflammation via increasing the formation of lymphocyte chemoattractant factors and cell surface expression of intercellular adhesion molecule-1 which has been implicated in the development of diabetic nephropathy | Urinary TNF-α levels could be used as a marker for the development and progression of diabetic nephropathy |
| MCP-1 | Mediates macrophage infiltration and accumulation in diabetic nephropathy | Urinary MCP-1 levels is an important marker to detect the progression of later stages of diabetic nephropathy |
| ICAM-1 | Promotes leukocyte adhesion and is involved in renal infiltration of macrophages in diabetic nephropathy | Urinary ICAM-1 could be used as an early marker for monitoring the progression of diabetic nephropathy |
| TGF-β | TGF-β plays an important role in the development of glomerulosclerosis and interstitial fibrosis in diabetes via enhancing glomerular extracellular matrix formation. | Urinary TGF-β could be used as an early marker for the progression of diabetic nephropathy |
| CCN2 | CCN2 is the key factor in stimulating connective tissue cell proliferation, extracellular matrix production, and other profibrotic properties of TGF-β during diabetes | 1-Urinary CCN2 levels could be used as a marker of early stage of diabetic nephropathy |
| 2-Plasma CCN2 levels could be used as a marker for the late stage of diabetic nephropathy |
Tumor necrosis factor-alpha (TNF-α)
Monocytes and macrophages are key inflammatory cells and components of the innate immunity [78]. Infiltration of monocytes into renal tissue increases the release of inflammatory mediators, including TNF-α which is a pleiotropic cytokine, produced mainly in macrophages, involved in systemic inflammation [78]. It is well established that TNF-α plays a significant pathophysiological role in different experimental models of renal diseases such as lupus nephritis, crescentic glomerulonephritis, and the remnant kidney model of nephropathy [79–81]. In diabetic nephropathy, renal expression of TNF-α is increased compared to kidneys of non-diabetic animals [82]. Macrophages from obese db/db mice display enhanced expression of TNF-α. Glomerular and tubulointerestitial TNF-α gene expression is also increased in diabetic rats [77, 83]. Exposure of tubular epithelial cells to TNF-α increased the synthesis and secretion of lymphocyte chemoattractant factors as well as the cell surface expression of intercellular adhesion molecule-1 which has been implicated in the development of renal injury in diabetes [84]. It is important to note that TNF-α may not only be produced in the diabetic kidney by infiltrating macrophages but also intrinsically by renal cells such as endothelial, mesangial, glomerular and tubular epithelial cells [76, 77, 83]. Additionally, the cytotoxic effects of TNF-α can directly induce damage to glomerular, mesangial and epithelial cells [56, 77]. TNF-α also promotes the local generation of superoxide, which affects the barrier function of the glomerular capillary wall resulting in enhanced albumin permeability, independently of diabetic homodynamic effects and inflammatory cytokines activation [56]. TNF-α has stimulatory effects on sodium uptake by proximal tubule cells contributing to sodium retention and renal hypertrophy [85]. In experimental models of diabetic nephropathy, renal TNF-α gene expression was also increased in diabetic rats, with a significant correlation between renal TNF-α expression and urinary TNF-α level [83, 86]. The increase in renal TNF-α production in diabetes appears to be related to hyperglycemia and formation of AGE products [78]. diabetic patients had approximately 3-fold higher serum TNF-α than non-diabetic individuals; however, serum TNF-α concentration was increased only in diabetic patients with micro- or macro-albuminuria as well as in subjects with overt nephropathy and renal insufficiency suggesting a significant relationship between serum TNF-α and urinary albumin excretion [77]. Diabetic patients with micro- or macro-albuminuria had also elevated TNF-α expression and urinary concentrations of TNF-α [83, 86]. Urinary concentration of TNF-α may represent the local production of this cytokine within the kidney as experimental studies have demonstrated a direct and independent association between renal TNF-α expression and urinary TNF-α excretion with the severity of renal injury [87]. The elevation in urinary TNF-α concentration in diabetic patients with increased urinary albumin excretion was further exacerbated as diabetic nephropathy progressed [77]. Importantly, urinary TNF-α is significant and independently related to clinical markers of both glomerular and tubulointerestitial injury [77]. Thus, assessment of urinary TNF-α levels could be used as a predictive marker for the development and progression of renal injury in diabetic nephropathy (Table 1).
Intercellular adhesion molecules-1 (ICAM-1)
ICAM-1 is a cell-surface protein with five immunoglobulin-like domains [3]. Its production is induced by inflammatory cytokines such as TNF-α, interleukin-1, and interferon-γ [88]. Activation of PKC and shear stress in diabetes also induce ICAM-1 [89, 90]. ICAM-1 expression on the vascular endothelium and its binding to β2 leukocyte integrins promotes leukocyte adhesion to the endothelium [3]. Clinically, ICAM-1 expression is upregulated in renal diseases such as human glomerulonephritis and in experimental animal models of renal diseases such as glomerulonephritis, renal ablation and ischemia/reperfusion injury [91–94]. Renal ICAM-1 expression is increased during diabetic nephropathy [95, 96]. For example, studies demonstrated that macrophage infiltration and expression of adhesion molecules including ICAM-1 and selectins increase in the kidneys of patients with diabetic nephropathy, streptozotocin (STZ)-induced diabetic rats, and in type 2 diabetes [96–98]. Blocking ICAM-1 signaling, with anti–ICAM-1 antibody, prevented leukocyte influx into the glomeruli and reduced renal injury in diabetes [99]. Glomerular infiltration of macrophages and renal and glomerular hypertrophy increased in ICAM-1+/+ diabetic mice and these changes were accompanied with overproduction of TGF-β, accumulation of glomerular type IV collagen, and albuminuria [3]. ICAM-1−/− diabetic mice showed a decrease in glomerular macrophage infiltration and hypertrophy and reduced glomerular TGF-β and type IV collagen when compared with ICAM-1+/+ diabetic mice [3]. Infiltration of CD4+ cells was decreased in the glomeruli of diabetic ICAM-1-deficient db/db mice compared with ICAM-1-intact db/db mice suggesting that ICAM-1 is critically involved in renal infiltration of macrophages in diabetic nephropathy [100]. Clinically, it has been shown that patients with both type 1 and type 2 diabetes mellitus and diabetic nephropathy have elevated concentrations of plasma ICAM-1 compared with subjects without renal injury indicating that assessment of plasma ICAM-1 levels can be used as a predictor of the development of renal damage [101, 102]. Recent studies also showed that renal ICAM-1 levels increased in diabetic patients and in experimental models of diabetic nephropathy [103, 104]. These findings suggest that early assessment of plasma and urinary ICAM-1 levels could be a useful marker for diagnosis of diabetic nephropathy and preventing disease progression (Table 1). Importantly, modulation of ICAM-1 activity may be a new venue for treating diabetic nephropathy especially with the current trials developing ICAM-1antagonists [105].
Monocyte chemoattractant protein-1 (MCP-1)
Chemokine (C-C motif) ligand 2 (CCL2) is a small cytokine belonging to the CC chemokine family that is also known as MCP-1 [106]. MCP-1 mediates macrophage migration and recruits monocytes, memory T cells, and dendritic cells to sites of tissue injury and infection [106, 107]. It is a monomeric polypeptide, with a molecular weight of approximately 13 kDa [108]. As with many other CC chemokines, CCL2 is located on chromosome 17 in humans [108]. The cell surface receptors that bind CCL2 are chemokine receptor 2 and 4 (CCR2 and CCR4) [109].
The role of MCP-1 in the pathogenesis of diabetic renal complications is clearly established. For example, high concentrations of glucose and AGE products stimulate MCP-1 production from cultured mesangial cells, podocytes, and renal tubular epithelial cells [110–112] in vitro. Increased glomerular macrophages infiltration was detected before deposition of extracellular matrix component [113] and MCP-1 was upregulated and has been shown to mediate macrophage infiltration and accumulation during the development of early diabetic nephropathy [7, 114]. In addition, increased amounts of MCP-1 were detected in renal biopsies and urine of patients with diabetic nephropathy [58, 115, 116]. MCP-1 knockout mice have recently been shown to be protected from streptozotocin (STZ) induced diabetic nephropathy and glomerular and tubulointerestitial injury were abrogated in these mice [114]. Blockade of the MCP-1/CCR2 also reduced glomerulosclerosis in STZ-induced diabetic mice [117]. These data suggest that MCP-1 plays a crucial role in the progression of diabetic nephropathy and inhibition of MCP-1 signaling could be an important therapeutic goal in the treatment of diabetic nephropathy.
Urinary MCP-1 was elevated in patients with diabetic nephropathy, but with different patterns and implications for progression of renal disease [7]. Elevated urinary MCP-1 levels in diabetic patients with macroalbuminuria were prognostic for deterioration in kidney function [7]. It was found that urinary MCP-1 levels were better correlated with the rate of deterioration of glomerular filtration rate (GFR) than urinary protein/creatinine ratio suggesting that urinary MCP-1 levels could provide a better prognostic tool in the latter stage of diabetic nephropathy where patients have symptoms of macroalbuminuria and worsening of GFR [7] (Table 1). As indicated earlier, hypertension and poor glycemic control are established factors in the progression of diabetic nephropathy. However, neither HbA1c nor blood pressure assessment showed a significant correlation with urinary MCP-1 levels in patients with diabetic nephropathy [7]. Therefore, measurement of urinary MCP-1 levels could provide more accurate prognostic information for the progression of diabetic nephropathy than blood pressure and glycemic control assessment.
Transforming growth factor-β (TGF-β)
Glomerular hypertrophy is a characteristic feature of diabetic nephropathy [118]. Recent evidence suggests that pro-fibrotic growth factors are involved in the pathogenesis of diabetic glomerular hypertrophy; these include increased mesangial matrix that is mainly composed of type IV collagen (Fig. 3) [118, 119]. This may relate to the fact that diabetes mellitus upregulates TGF-β expression and signaling, leading to increased collagen formation [120]. TGF-β, a prosclerotic cytokine, is a member of the transforming growth factor superfamily that controls proliferation and cellular differentiation; three isozymes have been identified and designated as TGF-β1, TGF-β2 and TGF-β3. Several experimental and clinical studies demonstrate a major role for TGF-β in development of glomerulosclerosis and interstitial fibrosis in diabetes [121] as TGF-β plays a central role in the enhancement of glomerular extracellular matrix production in diabetic nephropathy [120–122]. This is evident from studies indicating that TGF-β expression and activity increases in glomerular mesangial cells and proximal tubule cells during diabetes [123–125]. TGF-β mRNA also increased in mesangial cells, podocytes and tubular epithelial cells in diabetic nephropathy and was further increased with the disease progression especially in the glomeruli [121]. TGF-β is also involved in interstitial fibrosis, another important histopathological change that correlates with diabetic renal dysfunction [42, 126]. Thus, as would be expected, inhibition of TGF-β prevented glomerular enlargement and reduced fibrosis in experimental models of diabetic nephropathy [127, 128]. The observation that urinary TGF-β excretion is elevated in diabetic patients with micro- and macroalbuminuria, it suggests a role for TGF-β in the development of diabetic nephropathy [129, 130]. Thus assessment of urinary TGF-β levels could serve as a predictive marker for early detection and monitoring of the progression of diabetic nephropathy (Table 1).
Fig. 3.
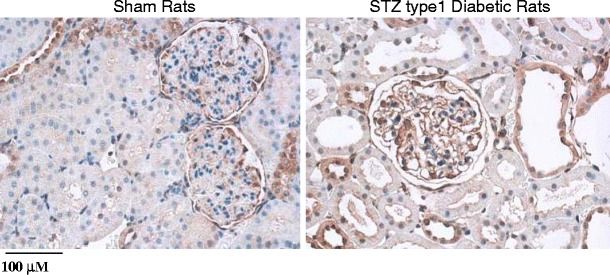
shows immuno-histochemical assessment of collagen IV deposition in the kidney section from streptozotocin-induced type 1 diabetic rats and sham control rat. Collagen IV deposition (dark brown) increased in the kidney section from diabetic vs. sham control rat. Images are shown at 200×
Connective tissue growth factor (CTGF)
CCN2, formerly named connective tissue growth factor (CTGF), is now known to be a major downstream effector of TGF-β signaling [131]. CCN2 is a 36- to 38-kDa protein that was first identified in conditioned media of endothelial cells as a polypeptide containing chemotactic activity towards fibroblasts [132]. CCN2 is a crucial factor in extracellular matrix production and other profibrotic activity mediated by TGF-β [132, 133]. It also plays an important role in angiogenesis, cell adhesion, migration, proliferation and differentiation [134]. Recently, CCN2 has been shown to play an important role in the pathogenesis of diabetic nephropathy [132]. High concentrations of glucose and AGE products stimulated the production of both TGF-β and CCN2 in mesangial cell cultures [135, 136]. CCN2 is involved in diabetes-induced pathophysiological changes such as extracellular matrix synthesis, cell migration, and epithelial-to-mesenchymal transition [137, 138]. Furthermore, upregulation of CCN2 has also been demonstrated in human and experimental models of diabetic nephropathy [139–141]. Inhibition of CCN2 signaling preserved the structure and function of the kidney in diabetic mice [142].
Because CCN2 is a secreted protein that can be detected in biological fluids, recent studies suggest that urinary and plasma CCN2 could serve as a predictive marker for diabetic nephropathy. This hypothesis was supported by the findings that both urinary CCN2 excretion and plasma CCN2 levels are elevated in patients with diabetic nephropathy [143, 144]. Although healthy individuals excrete low levels of urinary CCN2, patients with diabetic nephropathy and experimental animal models of diabetic nephropathy exhibit significant CCN2 excretion [143, 145, 146]. Interestingly, urinary CCN2 levels were highest at the early stage of diabetic nephropathy when patients were microalbuminuric and prospective follow up showed that raised urinary CCN2 predicted worsening of microalbuminuria [7]. In the streptozotocin-induced type 1 diabetes, urinary CCN2 excretion was elevated as early as 2 weeks following the development of diabetes, peaked during the early progression of diabetic nephropathy, and then decreased as animals became proteinuric [145]. The early increase in the urinary CCN2 in diabetic nephropathy supports the idea that renal fibrosis starts early in the pathogenesis of diabetic nephropathy, and that CCN2 is an important triggering factor in this process [7].
Plasma CCN2 was also evaluated as a possible marker for diabetic nephropathy and was found to be higher in patients with diabetic nephropathy than in patients with normoalbuminuria [132]. Plasma CCN2 level correlated with rate of decline in GFR and was an independent predictor of both ESRD and mortality in patients with type 1 diabetic nephropathy [132]. These data suggest that plasma CCN2 is associated with decline in renal function in type 1 diabetic patients with severe proteinuria than in those with mild proteinuria and assessment of plasma CCN2 has unique potential as a prognostic biomarker of renal function decline, especially in diabetic patients with severe proteinuria. Thus, assessment of plasma CCN2 levels could be used to improve prediction of ESRD and mortality in patients with type 1 diabetic nephropathy. The unique predictive value of plasma CCN2 in the progression of diabetic nephropathy, particularly in patients with severe proteinuria, suggests that CCN2 could also be used as a biomarker not only in diabetic patients who are likely to develop clinical nephropathy, but also in those who will exhibit rapid disease progression despite receiving appropriate treatment (Table 1).
Inflammatory cytokines; lessons from a diabetic animal model
The obese Zucker rat (OZR) has an autosomal recessive mutation of the fa gene encoding the leptin receptor [147, 148]. As a result, it manifests marked obesity which becomes increasingly prominent, starting at about 6 weeks of age, compared to their lean counterparts [148]. As shown in Fig. 4, both fasting plasma glucose and insulin levels are (significantly) higher in (6-month-old) OZR than lean Zucker rats (LZR) suggestive of marked insulin resistance of OZR; this is reflected from the calculated homeostatic model assessment (HOMA) index of insulin resistance, based on fasting plasma glucose and insulin levels, which is (significantly) higher for the OZR than LZR [148].
Fig. 4.
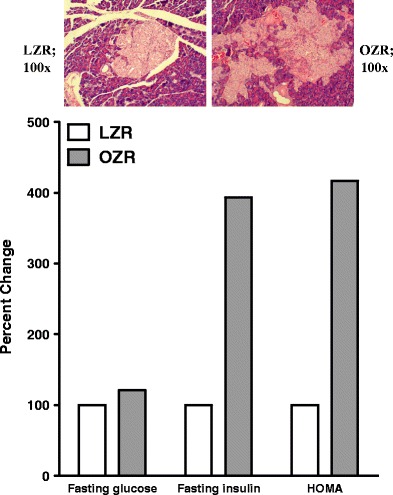
Bar graphs show fasting plasma glucose and insulin levels as well as the homeostatic model assessment (HOMA) insulin resistance index of (6-month-old) OZR, expressed as percent of values of LZR. Also shown are hematoxylin-eosin stained sections of pancreas from LZR and OZR rats (insets, 100×)
The marked compensatory hyperinsulinemia of OZR is associated with prominent structural changes of the endocrine pancreas. Microscopic examination of the pancreas from OZR reveals markedly enlarged hyperplastic islets of Langerhans exhibiting benign cellular proliferation which results in markedly irregular and jagged peripheral outline compared to those of the LZR which show sharply demarcated, small islets of Langerhans (Fig. 4). We have shown that pancreatic islets of OZR display prominent intercellular deposits of collagen fibers [148]. Collectively, these histological findings are consistent with the interpretation of islet hyperplasia with interstitial fibrosis of pancreatic islets of OZR.
A noted physical feature of the OZR is marked accumulation of fat within the abdominal cavity and consequent effective encasement of the kidney with fat deposits. Interestingly, local release of autocrine and paracrine factors is believed to contribute importantly to the pathogenesis of cardiac abnormalities in animals with dietary fat-induced obesity [149]. Nonetheless, the relevance of this mechanism for encapsulated organs, such as the kidney, remains to be established. More importantly, however, is the observation that OZR kidney displays numerous lipid droplets compared to LZR kidney (Fig. 5, panels a and b for LZR and OZR kidneys, respectively). Ectopic lipid accumulation is suggested to contribute importantly to organ dysfunction in obesity/type diabetes (i.e., lipotoxicity) as adipocytes are sources of a myriad factors including those promoting inflammatory changes [150]. Indeed, renal tissue of OZR shows foci of interstitial CD68 positive cells; whereas, examination of renal tissue from LZR does not show CD68 positive cells (Fig. 5, panels c and d for LZR and OZR kidneys, respectively). Consistent with the notion of upregulation of inflammatory processes associated with type 2 diabetes/ obesity, the OZR group showed (significant) elevation in urinary excretion of MCP-1 compared to LZR (Fig. 6a) [148]. Interestingly, however, plasma CRP was similar between OZR and LZR [0.62 ± 0.04 (n = 8) vs. 0.58 ± 0.07 (n = 5) ng/ml, respectively). As described earlier, plasma CRP is an index of early changes in inflammatory status providing a likely explanation for lack of a significant differential between LZR and OZR at 6 months of age. Taken together, these observations reinforce the notion that while plasma CRP may serve as a useful predictive index at early stages of obesity/type 2 diabetes, urinary excretion of MCP-1 may be helpful in monitoring the progression of renal disease and the efficacy of interventional modalities. It is noteworthy that the kidney of OZR displays increased levels of ICAM-1 compared to that of LZR (Fig. 6b). This finding is consistent with our recent demonstration that other indices of inflammation and oxidative stress (i.e., tissue cyclooxygenase-2 and nitrotyrosine) are increased in kidneys of OZR than LZR [148]. As a functional correlate, Fig. 6a shows that OZR display (significant) elevation in urinary albumin excretion [148]. Interestingly, however, blood pressure was similar between OZR and LZR thereby suggestive of lack of a differential contribution to genesis of proteinuria (although greater sensitivity of OZR kidney to the same level of blood pressure cannot be ruled out) [148]. Nonetheless, it is important to note that multiple mechanisms contribute to genesis of proteinuria including oxidative stress and inflammatory chemokines and cytokines some of which were significantly elevated in OZR than LZR as described above [148].
Fig. 5.

Oil-Red-O stained kidney tissue from (6-month-old) OZR show more numerous lipid droplets compared to age-matched LZR (panels a and b; 400×). Also shown are immunostaining for CD 68 positive cells (arrows) in kidney tissue from experimental groups (panels c and d; 200×)
Fig. 6.
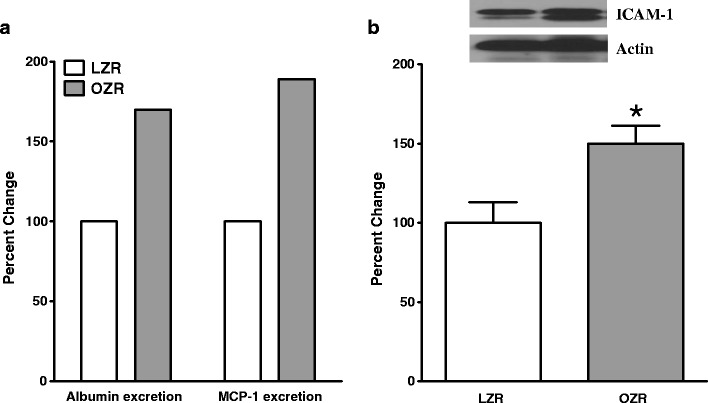
Bar graphs show urinary excretions of albumin and monocyte chemoattractant protein-1 (MCP-1) expressed as the percent of the LZR group (panel A). Panel B shows that renal tissue from (6-month-old) OZR displayed higher level of intercellular adhesion molecule-1 (ICAM-1) compared to age-matched LZR. Also shown are representative blots for ICAM-1 and β-actin (i.e., loading control). Data are means ± SEM of 7–9 animals/group. *p < 0.05 compared to LZR
Conclusion
Over the past decade, a close association between inflammation and diabetes has become increasingly clear. It is also well-established that diabetic nephropathy is a devastating complication of diabetes that often leads to ESRD. The relationship between inflammatory cytokines-chemokines levels and the development and progression of diabetic nephropathy is very complex and requires further elucidation. However, it is increasingly becoming clear that many inflammatory cytokines-chemokines play significant roles in this scenario, including adipokines, chemoattractant cytokines, adhesion molecules, and pro-fibrotic cytokines. Because current treatment of diabetic nephropathy is still suboptimal, early assessment of levels of some inflammatory cytokines could potentially help with early diagnosis and halting of disease progression to ESRD. Also, a better understanding of the role of inflammatory cytokines-chemokines in the progression of diabetic nephropathy should facilitate the development of novel treatment(s) and improvement of current therapeutic strategies. Nonetheless, given the complex web of mechanisms that contribute to the genesis of diabetic nephropathy, a pressing challenge is to identify predictive markers for early detection of the disease. However, it is unlikely that this important task can be relegated to a single marker. Rather, in order to increase the power of predictive strategies, multiple markers will be required to increase the likelihood of early detection of the disease process. Another important consideration is the need for to identify minimally- or non-invasive sources for obtaining these biomarkers such as those from the blood (or its components) and/or urine. Clearly, temporal changes in the levels of these biomarkers from such sources should reflect changes occurring in target organs such as the kidney. To that end, the OZR (and other relevant animals models) can serve as valuable investigational tools to compare and contrast the levels of molecular biomarkers in the organ of interest to those of blood (or its components) and/or urine.
Acknowledgments
This review was supported, in part, by a grant from the National Institutes of Health (MSM).
References
- 1.Batlle D. Clinical and cellular markers of diabetic nephropathy. Kidney Int. 2003;63:2319–2330. doi: 10.1046/j.1523-1755.2003.00053.x. [DOI] [PubMed] [Google Scholar]
- 2.Ohga S, Shikata K, Yozai K, et al. Thiazolidinedione ameliorates renal injury in experimental diabetic rats through anti-inflammatory effects mediated by inhibition of NF-kappaB activation. Am J Physiol Renal Physiol. 2007;292:F1141–1150. doi: 10.1152/ajprenal.00288.2005. [DOI] [PubMed] [Google Scholar]
- 3.Okada S, Shikata K, Matsuda M, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–2593. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 4.Rivero A, Mora C, Muros M, Garcia J, Herrera H, Navarro-Gonzalez JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci (Lond) 2009;116:479–492. doi: 10.1042/CS20080394. [DOI] [PubMed] [Google Scholar]
- 5.Rossing P. Prediction, progression and prevention of diabetic nephropathy. The Minkowski Lecture 2005. Diabetologia. 2006;49:11–19. doi: 10.1007/s00125-005-0077-3. [DOI] [PubMed] [Google Scholar]
- 6.Rossing P. Diabetic nephropathy: worldwide epidemic and effects of current treatment on natural history. Curr Diab Rep. 2006;6:479–483. doi: 10.1007/s11892-006-0083-y. [DOI] [PubMed] [Google Scholar]
- 7.Tam FW, Riser BL, Meeran K, Rambow J, Pusey CD, Frankel AH. Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine. 2009;47:37–42. doi: 10.1016/j.cyto.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Hovind P, Rossing P, Tarnow L, Smidt UM, Parving HH. Progression of diabetic nephropathy. Kidney Int. 2001;59:702–709. doi: 10.1046/j.1523-1755.2001.059002702.x. [DOI] [PubMed] [Google Scholar]
- 9.Parving HH. Diabetic nephropathy: prevention and treatment. Kidney Int. 2001;60:2041–2055. doi: 10.1046/j.1523-1755.2001.00020.x. [DOI] [PubMed] [Google Scholar]
- 10.Brown WV. Microvascular complications of diabetes mellitus: renal protection accompanies cardiovascular protection. Am J Cardiol. 2008;102:10L–13L. doi: 10.1016/j.amjcard.2008.09.068. [DOI] [PubMed] [Google Scholar]
- 11.Steinke JM. The natural progression of kidney injury in young type 1 diabetic patients. Curr Diab Rep. 2009;9:473–479. doi: 10.1007/s11892-009-0077-7. [DOI] [PubMed] [Google Scholar]
- 12.Lewko B, Stepinski J. Hyperglycemia and mechanical stress: targeting the renal podocyte. J Cell Physiol. 2009;221:288–295. doi: 10.1002/jcp.21856. [DOI] [PubMed] [Google Scholar]
- 13.Munusamy S, MacMillan-Crow LA. Mitochondrial superoxide plays a crucial role in the development of mitochondrial dysfunction during high glucose exposure in rat renal proximal tubular cells. Free Radic Biol Med. 2009;46:1149–1157. doi: 10.1016/j.freeradbiomed.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Serpillon S, Floyd BC, Gupte RS, et al. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol. 2009;297:H153–162. doi: 10.1152/ajpheart.01142.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidig P, McMaster D, Bayraktutan U. High glucose mediates pro-oxidant and antioxidant enzyme activities in coronary endothelial cells. Diabetes Obes Metab. 2004;6:432–441. doi: 10.1111/j.1462-8902.2004.00364.x. [DOI] [PubMed] [Google Scholar]
- 16.Zheng L, Kern TS. Role of nitric oxide, superoxide, peroxynitrite and PARP in diabetic retinopathy. Front Biosci. 2009;14:3974–3987. doi: 10.2741/3505. [DOI] [PubMed] [Google Scholar]
- 17.Thum T, Fraccarollo D, Schultheiss M, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–674. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- 18.San Martin A, Foncea R, Laurindo FR, Ebensperger R, Griendling KK, Leighton F. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products. Free Radic Biol Med. 2007;42:1671–1679. doi: 10.1016/j.freeradbiomed.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 19.San Martin A, Du P, Dikalova A, et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am J Physiol Heart Circ Physiol. 2007;292:H2073–2082. doi: 10.1152/ajpheart.00943.2006. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Li Y, Qi J, Wang H, Liu K. Peroxynitrite plays a key role in glomerular lesions in diabetic rats. J Nephrol. 2009;22:800–808. [PubMed] [Google Scholar]
- 21.Liang JH, Li YN, Qi JS, Jia XX. Peroxynitrite-induced protein nitration is responsible for renal mitochondrial damage in diabetic rat. J Endocrinol Invest. 2009. [DOI] [PubMed]
- 22.Narasimhan P, Fujimura M, Noshita N, Chan PH. Role of superoxide in poly(ADP-ribose) polymerase upregulation after transient cerebral ischemia. Brain Res Mol Brain Res. 2003;113:28–36. doi: 10.1016/S0169-328X(03)00062-7. [DOI] [PubMed] [Google Scholar]
- 23.Ahmad R, Rasheed Z, Ahsan H. Biochemical and cellular toxicology of peroxynitrite: implications in cell death and autoimmune phenomenon. Immunopharmacol Immunotoxicol. 2009;31:388–396. doi: 10.1080/08923970802709197. [DOI] [PubMed] [Google Scholar]
- 24.Szabo C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br J Pharmacol. 2009;156:713–727. doi: 10.1111/j.1476-5381.2008.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammes HP. Pathophysiological mechanisms of diabetic angiopathy. J Diabetes Its Complicat. 2003;17:16–19. doi: 10.1016/S1056-8727(02)00275-1. [DOI] [PubMed] [Google Scholar]
- 26.Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care. 2003;26:1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 27.Forbes JM, Fukami K, Cooper ME. Diabetic nephropathy: where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes. 2007;115:69–84. doi: 10.1055/s-2007-949721. [DOI] [PubMed] [Google Scholar]
- 28.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 29.Kumar A, Hawkins KS, Hannan MA, Ganz MB. Activation of PKC-beta(I) in glomerular mesangial cells is associated with specific NF-kappaB subunit translocation. Am J Physiol Renal Physiol. 2001;281:F613–619. doi: 10.1152/ajprenal.2001.281.4.F613. [DOI] [PubMed] [Google Scholar]
- 30.Park CW, Kim JH, Lee JH, et al. High glucose-induced intercellular adhesion molecule-1 (ICAM-1) expression through an osmotic effect in rat mesangial cells is PKC-NF-kappa B-dependent. Diabetologia. 2000;43:1544–1553. doi: 10.1007/s001250051567. [DOI] [PubMed] [Google Scholar]
- 31.Wei XF, Zhou QG, Hou FF, Liu BY, Liang M. Advanced oxidation protein products induce mesangial cell perturbation through PKC-dependent activation of NADPH oxidase. Am J Physiol Renal Physiol. 2009;296:F427–437. doi: 10.1152/ajprenal.90536.2008. [DOI] [PubMed] [Google Scholar]
- 32.Thallas-Bonke V, Thorpe SR, Coughlan MT, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–469. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 33.Tojo A, Asaba K, Onozato ML. Suppressing renal NADPH oxidase to treat diabetic nephropathy. Expert Opin Ther Targets. 2007;11:1011–1018. doi: 10.1517/14728222.11.8.1011. [DOI] [PubMed] [Google Scholar]
- 34.Baccora MH, Cortes P, Hassett C, Taube DW, Yee J. Effects of long-term elevated glucose on collagen formation by mesangial cells. Kidney Int. 2007;72:1216–1225. doi: 10.1038/sj.ki.5002517. [DOI] [PubMed] [Google Scholar]
- 35.Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005;10(Suppl):S7–10. doi: 10.1111/j.1440-1797.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- 36.Xia L, Wang H, Munk S, et al. Reactive oxygen species, PKC-beta1, and PKC-zeta mediate high-glucose-induced vascular endothelial growth factor expression in mesangial cells. Am J Physiol Endocrinol Metab. 2007;293:E1280–1288. doi: 10.1152/ajpendo.00223.2007. [DOI] [PubMed] [Google Scholar]
- 37.Frank RN. Treating diabetic retinopathy by inhibiting growth factor pathways. Curr Opin Investig Drugs. 2009;10:327–335. [PubMed] [Google Scholar]
- 38.Wu D, Peng F, Zhang B, et al. PKC-beta1 mediates glucose-induced Akt activation and TGF-beta1 upregulation in mesangial cells. J Am Soc Nephrol. 2009;20:554–566. doi: 10.1681/ASN.2008040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meier M, Menne J, Park JK, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol. 2007;18:1190–1198. doi: 10.1681/ASN.2005070694. [DOI] [PubMed] [Google Scholar]
- 40.Meier M, Park JK, Overheu D, et al. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes. 2007;56:346–354. doi: 10.2337/db06-0891. [DOI] [PubMed] [Google Scholar]
- 41.Wolf G. New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur J Clin Invest. 2004;34:785–796. doi: 10.1111/j.1365-2362.2004.01429.x. [DOI] [PubMed] [Google Scholar]
- 42.Kanwar YS, Wada J, Sun L, et al. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood) 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 43.Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. 2009;94:3171–3182. doi: 10.1210/jc.2008-2534. [DOI] [PubMed] [Google Scholar]
- 44.Sarafidis PA, Ruilope LM. Insulin resistance, hyperinsulinemia, and renal injury: mechanisms and implications. Am J Nephrol. 2006;26:232–244. doi: 10.1159/000093632. [DOI] [PubMed] [Google Scholar]
- 45.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 46.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2005;288:L3–15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity. 2009;31:711–721. doi: 10.1016/j.immuni.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2:123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- 49.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 50.Keane MP, Strieter RM. Chemokine signaling in inflammation. Crit Care Med. 2000;28:N13–26. doi: 10.1097/00003246-200004001-00003. [DOI] [PubMed] [Google Scholar]
- 51.Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 52.Ward SG, Marelli-Berg FM. Mechanisms of chemokine and antigen-dependent T-lymphocyte navigation. Biochem J. 2009;418:13–27. doi: 10.1042/BJ20081969. [DOI] [PubMed] [Google Scholar]
- 53.Sedor JR, Konieczkowski M, Huang S, et al. Cytokines, mesangial cell activation and glomerular injury. Kidney Int Suppl. 1993;39:S65–70. [PubMed] [Google Scholar]
- 54.Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am J Physiol. 1989;257:L399–410. doi: 10.1152/ajplung.1989.257.6.L399. [DOI] [PubMed] [Google Scholar]
- 55.Dalla Vestra M, Mussap M, Gallina P, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16(Suppl 1):S78–82. doi: 10.1681/ASN.2004110961. [DOI] [PubMed] [Google Scholar]
- 56.McCarthy ET, Sharma R, Sharma M, et al. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J Am Soc Nephrol. 1998;9:433–438. doi: 10.1681/ASN.V93433. [DOI] [PubMed] [Google Scholar]
- 57.Chiarelli F, Cipollone F, Mohn A, et al. Circulating monocyte chemoattractant protein-1 and early development of nephropathy in type 1 diabetes. Diabetes Care. 2002;25:1829–1834. doi: 10.2337/diacare.25.10.1829. [DOI] [PubMed] [Google Scholar]
- 58.Morii T, Fujita H, Narita T, et al. Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J Diabetes ItsComplicat. 2003;17:11–15. doi: 10.1016/S1056-8727(02)00176-9. [DOI] [PubMed] [Google Scholar]
- 59.Takebayashi K, Matsumoto S, Aso Y, Inukai T. Association between circulating monocyte chemoattractant protein-1 and urinary albumin excretion in nonobese Type 2 diabetic patients. J Diabetes Its Complicat. 2006;20:98–104. doi: 10.1016/j.jdiacomp.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci. 2009;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hohmeier HE, Tran VV, Chen G, Gasa R, Newgard CB. Inflammatory mechanisms in diabetes: lessons from the beta-cell. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S12–16. doi: 10.1038/sj.ijo.0802493. [DOI] [PubMed] [Google Scholar]
- 62.Kim KA, Lee MS. Recent progress in research on beta-cell apoptosis by cytokines. Front Biosci. 2009;14:657–664. doi: 10.2741/3271. [DOI] [PubMed] [Google Scholar]
- 63.Rudijanto A. The expression and down stream effect of lectin like-oxidized low density lipoprotein 1 (LOX-1) in hyperglycemic state. Acta Med Indones. 2007;39:36–43. [PubMed] [Google Scholar]
- 64.Fonseca VA. Rationale for the use of insulin sensitizers to prevent cardiovascular events in type 2 diabetes mellitus. Am J Med. 2007;120:S18–25. doi: 10.1016/j.amjmed.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 65.Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17:319–326. doi: 10.1016/j.numecd.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 66.Karalis KP, Giannogonas P, Kodela E, Koutmani Y, Zoumakis M, Teli T. Mechanisms of obesity and related pathology: linking immune responses to metabolic stress. FEBS J. 2009;276:5747–5754. doi: 10.1111/j.1742-4658.2009.07304.x. [DOI] [PubMed] [Google Scholar]
- 67.Vernochet C, Peres SB, Farmer SR. Mechanisms of obesity and related pathologies: transcriptional control of adipose tissue development. FEBS J. 2009;276:5729–5737. doi: 10.1111/j.1742-4658.2009.07302.x. [DOI] [PubMed] [Google Scholar]
- 68.Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118:2243–2251. doi: 10.1161/CIRCULATIONAHA.108.814251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Correale M, Brunetti ND, Gennaro L, Biase M. Acute phase proteins in atherosclerosis (acute coronary syndrome) Cardiovasc Hematol Agents Med Chem. 2008;6:272–277. doi: 10.2174/187152508785909537. [DOI] [PubMed] [Google Scholar]
- 70.Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 71.Blake GJ, Ridker PM. Tumour necrosis factor-alpha, inflammatory biomarkers, and atherogenesis. Eur Heart J. 2002;23:345–347. doi: 10.1053/euhj.2001.2905. [DOI] [PubMed] [Google Scholar]
- 72.Buckley DI, Fu R, Freeman M, Rogers K, Helfand M. C-reactive protein as a risk factor for coronary heart disease: a systematic review and meta-analyses for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:483–495. doi: 10.7326/0003-4819-151-7-200910060-00009. [DOI] [PubMed] [Google Scholar]
- 73.Greenfield JR, Campbell LV. Relationship between inflammation, insulin resistance and type 2 diabetes: ‘cause or effect’? Curr Diabetes Rev. 2006;2:195–211. doi: 10.2174/157339906776818532. [DOI] [PubMed] [Google Scholar]
- 74.Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 75.Saraheimo M, Teppo AM, Forsblom C, Fagerudd J, Groop PH. Diabetic nephropathy is associated with low-grade inflammation in Type 1 diabetic patients. Diabetologia. 2003;46:1402–1407. doi: 10.1007/s00125-003-1194-5. [DOI] [PubMed] [Google Scholar]
- 76.Navarro JF, Mora C, Maca M, Garca J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am J Kidney Dis. 2003;42:53–61. doi: 10.1016/S0272-6386(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 77.Navarro JF, Mora C, Muros M, Garcia J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant. 2006;21:3428–3434. doi: 10.1093/ndt/gfl469. [DOI] [PubMed] [Google Scholar]
- 78.Sugimoto H, Shikata K, Wada J, Horiuchi S, Makino H. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia. 1999;42:878–886. doi: 10.1007/s001250051241. [DOI] [PubMed] [Google Scholar]
- 79.Javaid B, Quigg RJ. Treatment of glomerulonephritis: will we ever have options other than steroids and cytotoxics? Kidney Int. 2005;67:1692–1703. doi: 10.1111/j.1523-1755.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- 80.Aringer M, Smolen JS. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res Ther. 2008;10:202. doi: 10.1186/ar2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feldmann M, Pusey CD. Is there a role for TNF-alpha in anti-neutrophil cytoplasmic antibody-associated vasculitis? Lessons from other chronic inflammatory diseases. J Am Soc Nephrol. 2006;17:1243–1252. doi: 10.1681/ASN.2005121359. [DOI] [PubMed] [Google Scholar]
- 82.Fornoni A, Ijaz A, Tejada T, Lenz O. Role of inflammation in diabetic nephropathy. Curr Diabetes Rev. 2008;4:10–17. doi: 10.2174/157339908783502361. [DOI] [PubMed] [Google Scholar]
- 83.Navarro JF, Milena FJ, Mora C, et al. Tumor necrosis factor-alpha gene expression in diabetic nephropathy: relationship with urinary albumin excretion and effect of angiotensin-converting enzyme inhibition. Kidney Int. 2005;68(Suppl):S98–102. doi: 10.1111/j.1523-1755.2005.09918.x. [DOI] [PubMed] [Google Scholar]
- 84.Ishikura H, Takahashi C, Kanagawa K, Hirata H, Imai K, Yoshiki T. Cytokine regulation of ICAM-1 expression on human renal tubular epithelial cells in vitro. Transplantation. 1991;51:1272–1275. doi: 10.1097/00007890-199106000-00024. [DOI] [PubMed] [Google Scholar]
- 85.DiPetrillo K, Coutermarsh B, Gesek FA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol. 2003;284:F113–121. doi: 10.1152/ajprenal.00026.2002. [DOI] [PubMed] [Google Scholar]
- 86.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 2003;64:1208–1213. doi: 10.1046/j.1523-1755.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 87.Aso Y, Inukai T, Tayama K, Takemura Y. Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol. 2000;37:87–92. doi: 10.1007/s005920070025. [DOI] [PubMed] [Google Scholar]
- 88.Wertheimer SJ, Myers CL, Wallace RW, Parks TP. Intercellular adhesion molecule-1 gene expression in human endothelial cells. Differential regulation by tumor necrosis factor-alpha and phorbol myristate acetate. J Biol Chem. 1992;267:12030–12035. [PubMed] [Google Scholar]
- 89.Lane TA, Lamkin GE, Wancewicz E. Modulation of endothelial cell expression of intercellular adhesion molecule 1 by protein kinase C activation. Biochem Biophys Res Commun. 1989;161:945–952. doi: 10.1016/0006-291X(89)91334-X. [DOI] [PubMed] [Google Scholar]
- 90.Ballermann BJ, Dardik A, Eng E, Liu A. Shear stress and the endothelium. Kidney Int Suppl. 1998;67:S100–108. doi: 10.1046/j.1523-1755.1998.06720.x. [DOI] [PubMed] [Google Scholar]
- 91.Kawasaki K, Yaoita E, Yamamoto T, Tamatani T, Miyasaka M, Kihara I. Antibodies against intercellular adhesion molecule-1 and lymphocyte function-associated antigen-1 prevent glomerular injury in rat experimental crescentic glomerulonephritis. J Immunol. 1993;150:1074–1083. [PubMed] [Google Scholar]
- 92.Wada J, Shikata K, Makino H, et al. The critical role of intercellular adhesion molecule-1 in Masugi nephritis in rats. Nephron. 1996;73:264–272. doi: 10.1159/000189050. [DOI] [PubMed] [Google Scholar]
- 93.Miyatake N, Shikata K, Sugimoto H, et al. Intercellular adhesion molecule 1 mediates mononuclear cell infiltration into rat glomeruli after renal ablation. Nephron. 1998;79:91–98. doi: 10.1159/000044997. [DOI] [PubMed] [Google Scholar]
- 94.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsui H, Suzuki M, Tsukuda R, Iida K, Miyasaka M, Ikeda H. Expression of ICAM-1 on glomeruli is associated with progression of diabetic nephropathy in a genetically obese diabetic rat, Wistar fatty. Diabetes Res Clin Pract. 1996;32:1–9. doi: 10.1016/0168-8227(96)01209-0. [DOI] [PubMed] [Google Scholar]
- 96.Coimbra TM, Janssen U, Grone HJ, et al. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000;57:167–182. doi: 10.1046/j.1523-1755.2000.00836.x. [DOI] [PubMed] [Google Scholar]
- 97.Lavaud S, Michel O, Sassy-Prigent C, et al. Early influx of glomerular macrophages precedes glomerulosclerosis in the obese Zucker rat model. J Am Soc Nephrol. 1996;7:2604–2615. doi: 10.1681/ASN.V7122604. [DOI] [PubMed] [Google Scholar]
- 98.Hirata K, Shikata K, Matsuda M, et al. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia. 1998;41:185–192. doi: 10.1007/s001250050888. [DOI] [PubMed] [Google Scholar]
- 99.Sugimoto H, Shikata K, Hirata K, et al. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes. 1997;46:2075–2081. doi: 10.2337/diabetes.46.12.2075. [DOI] [PubMed] [Google Scholar]
- 100.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–1722. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 101.Clausen P, Jacobsen P, Rossing K, Jensen JS, Parving HH, Feldt-Rasmussen B. Plasma concentrations of VCAM-1 and ICAM-1 are elevated in patients with Type 1 diabetes mellitus with microalbuminuria and overt nephropathy. Diabet Med. 2000;17:644–649. doi: 10.1046/j.1464-5491.2000.00347.x. [DOI] [PubMed] [Google Scholar]
- 102.Guler S, Cakir B, Demirbas B, et al. Plasma soluble intercellular adhesion molecule 1 levels are increased in type 2 diabetic patients with nephropathy. Horm Res. 2002;58:67–70. doi: 10.1159/000064664. [DOI] [PubMed] [Google Scholar]
- 103.Xiang G, Schinzel R, Simm A, Sebekova K, Heidland A. Advanced glycation end products impair protein turnover in LLC-PK1: amelioration by trypsin. Kidney Int Suppl. 2001;78:S53–57. doi: 10.1046/j.1523-1755.2001.07840.x. [DOI] [PubMed] [Google Scholar]
- 104.Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. 2006;17:368–377. doi: 10.1681/ASN.2005080859. [DOI] [PubMed] [Google Scholar]
- 105.Anderson ME, Siahaan TJ. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: designing peptide and small molecule inhibitors. Peptides. 2003;24:487–501. doi: 10.1016/S0196-9781(03)00083-4. [DOI] [PubMed] [Google Scholar]
- 106.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci USA. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol. 1996;60:365–371. doi: 10.1002/jlb.60.3.365. [DOI] [PubMed] [Google Scholar]
- 108.Mehrabian M, Sparkes RS, Mohandas T, Fogelman AM, Lusis AJ. Localization of monocyte chemotactic protein-1 gene (SCYA2) to human chromosome 17q11.2-q21.1. Genomics. 1991;9:200–203. doi: 10.1016/0888-7543(91)90239-B. [DOI] [PubMed] [Google Scholar]
- 109.Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006;25:611–619. doi: 10.1007/s10555-006-9027-x. [DOI] [PubMed] [Google Scholar]
- 110.Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol. 2002;13:894–902. doi: 10.1681/ASN.V134894. [DOI] [PubMed] [Google Scholar]
- 111.Yamagishi S, Inagaki Y, Okamoto T, et al. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J Biol Chem. 2002;277:20309–20315. doi: 10.1074/jbc.M202634200. [DOI] [PubMed] [Google Scholar]
- 112.Gu L, Hagiwara S, Fan Q, et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol Dial Transplant. 2006;21:299–313. doi: 10.1093/ndt/gfi210. [DOI] [PubMed] [Google Scholar]
- 113.Young BA, Johnson RJ, Alpers CE, et al. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 1995;47:935–944. doi: 10.1038/ki.1995.139. [DOI] [PubMed] [Google Scholar]
- 114.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 115.Mezzano S, Aros C, Droguett A, et al. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505–2512. doi: 10.1093/ndt/gfh207. [DOI] [PubMed] [Google Scholar]
- 116.Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000;58:684–690. doi: 10.1046/j.1523-1755.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- 117.Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2008;294:F697–701. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 118.Chen S, Jim B, Ziyadeh FN. Diabetic nephropathy and transforming growth factor-beta: transforming our view of glomerulosclerosis and fibrosis build-up. Semin Nephrol. 2003;23:532–543. doi: 10.1053/S0270-9295(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 119.Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res. 2009;41:585–593. doi: 10.1055/s-0029-1220752. [DOI] [PubMed] [Google Scholar]
- 120.Yokoyama H, Deckert T. Central role of TGF-beta in the pathogenesis of diabetic nephropathy and macrovascular complications: a hypothesis. Diabet Med. 1996;13:313–320. doi: 10.1002/(SICI)1096-9136(199604)13:4<313::AID-DIA56>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 121.Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]
- 122.Nicholas SB. Advances in pathogenetic mechanisms of diabetic nephropathy. Cell Mol Biol (Noisy-le-grand) 2003;49:1319–1325. [PubMed] [Google Scholar]
- 123.Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. doi: 10.2337/diabetes.44.10.1139. [DOI] [PubMed] [Google Scholar]
- 124.Leehey DJ, Singh AK, Alavi N, Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl. 2000;77:S93–98. doi: 10.1046/j.1523-1755.2000.07715.x. [DOI] [PubMed] [Google Scholar]
- 125.Phillips AO. Diabetic nephropathy: the modulating influence of glucose on transforming factor beta production. Histol Histopathol. 1998;13:565–574. doi: 10.14670/HH-13.565. [DOI] [PubMed] [Google Scholar]
- 126.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–1637. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- 127.Benigni A, Zoja C, Campana M, et al. Beneficial effect of TGFbeta antagonism in treating diabetic nephropathy depends on when treatment is started. Nephron Exp Nephrol. 2006;104:e158–168. doi: 10.1159/000094967. [DOI] [PubMed] [Google Scholar]
- 128.Gagliardini E, Benigni A. Role of anti-TGF-beta antibodies in the treatment of renal injury. Cytokine Growth Factor Rev. 2006;17:89–96. doi: 10.1016/j.cytogfr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 129.Rivarola EW, Moyses-Neto M, Dantas M, Da-Silva CG, Volpini R, Coimbra TM. Transforming growth factor beta activity in urine of patients with type 2 diabetes and diabetic nephropathy. Braz J Med Biol Res. 1999;32:1525–1528. doi: 10.1590/S0100-879X1999001200011. [DOI] [PubMed] [Google Scholar]
- 130.Sharma K, McGowan TA. TGF-beta in diabetic kidney disease: role of novel signaling pathways. Cytokine Growth Factor Rev. 2000;11:115–123. doi: 10.1016/S1359-6101(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 131.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 132.Nguyen TQ, Tarnow L, Jorsal A, et al. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care. 2008;31:1177–1182. doi: 10.2337/dc07-2469. [DOI] [PubMed] [Google Scholar]
- 133.Locatelli F, Pozzoni P, Vecchio L. Renal replacement therapy in patients with diabetes and end-stage renal disease. J Am Soc Nephrol. 2004;15(Suppl 1):S25–29. doi: 10.1097/01.ASN.0000093239.32602.04. [DOI] [PubMed] [Google Scholar]
- 134.Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 135.Riser BL, Denichilo M, Cortes P, et al. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol. 2000;11:25–38. doi: 10.1681/ASN.V11125. [DOI] [PubMed] [Google Scholar]
- 136.Blom IE, Dijk AJ, Weger RA, Tilanus MG, Goldschmeding R. Identification of human ccn2 (connective tissue growth factor) promoter polymorphisms. Mol Pathol. 2001;54:192–196. doi: 10.1136/mp.54.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Blom IE, Dijk AJ, Wieten L, et al. In vitro evidence for differential involvement of CTGF, TGFbeta, and PDGF-BB in mesangial response to injury. Nephrol Dial Transplant. 2001;16:1139–1148. doi: 10.1093/ndt/16.6.1139. [DOI] [PubMed] [Google Scholar]
- 138.Burns WC, Twigg SM, Forbes JM, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- 139.Adler SG, Kang SW, Feld S, et al. Glomerular mRNAs in human type 1 diabetes: biochemical evidence for microalbuminuria as a manifestation of diabetic nephropathy. Kidney Int. 2001;60:2330–2336. doi: 10.1046/j.1523-1755.2001.00073.x. [DOI] [PubMed] [Google Scholar]
- 140.Ito Y, Aten J, Bende RJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998;53:853–861. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- 141.Roestenberg P, Nieuwenhoven FA, Joles JA, et al. Temporal expression profile and distribution pattern indicate a role of connective tissue growth factor (CTGF/CCN-2) in diabetic nephropathy in mice. Am J Physiol Renal Physiol. 2006;290:F1344–1354. doi: 10.1152/ajprenal.00174.2005. [DOI] [PubMed] [Google Scholar]
- 142.Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–3368. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]
- 143.Gilbert RE, Akdeniz A, Weitz S, et al. Urinary connective tissue growth factor excretion in patients with type 1 diabetes and nephropathy. Diabetes Care. 2003;26:2632–2636. doi: 10.2337/diacare.26.9.2632. [DOI] [PubMed] [Google Scholar]
- 144.Roestenberg P, Nieuwenhoven FA, Wieten L, et al. Connective tissue growth factor is increased in plasma of type 1 diabetic patients with nephropathy. Diabetes Care. 2004;27:1164–1170. doi: 10.2337/diacare.27.5.1164. [DOI] [PubMed] [Google Scholar]
- 145.Riser BL, Cortes P, DeNichilo M, et al. Urinary CCN2 (CTGF) as a possible predictor of diabetic nephropathy: preliminary report. Kidney Int. 2003;64:451–458. doi: 10.1046/j.1523-1755.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 146.Nguyen TQ, Tarnow L, Andersen S, et al. Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care. 2006;29:83–88. doi: 10.2337/diacare.29.01.06.dc05-1670. [DOI] [PubMed] [Google Scholar]
- 147.Chander PN, Gealekman O, Brodsky SV, et al. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol. 2004;15:2391–2403. doi: 10.1097/01.ASN.0000135971.88164.2C. [DOI] [PubMed] [Google Scholar]
- 148.Mozaffari MS, Abdelsayed R, Liu JY, Wimborne H, El-Remessy A, El-Marakby A. Effects of chromium picolinate on glycemic control and kidney of the obese Zucker rat. Nutr Metab (Lond) 2009;6:51. doi: 10.1186/1743-7075-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Swifka J, Weiss J, Addicks K, Eckel J, Rosen P. Epicardial fat from guinea pig: a model to study the paracrine network of interactions between epicardial fat and myocardium? Cardiovasc Drugs Ther. 2008;22:107–114. doi: 10.1007/s10557-008-6085-z. [DOI] [PubMed] [Google Scholar]
- 150.Szendroedi J, Roden M. Ectopic lipids and organ function. Curr Opin Lipidol. 2009;20:50–56. doi: 10.1097/MOL.0b013e328321b3a8. [DOI] [PubMed] [Google Scholar]