

Abstract
Endothelial dysfunction is a predisposing factor for vascular disease in diabetes, which contributes significantly to the mortality of diabetic patients. The currently utilized assessment methods of endothelial function/dysfunction in humans are associated with various limitations. Circulating endothelial-derived/associated markers have been proposed as potential alternatives for evaluation of the endothelium in condition of vascular disorders. These indicators include von Willebrand factor, soluble thrombomodulin, soluble E-selectin, asymmetric dimethylarginine, tissue plasminogen activator, endothelial microparticles, circulating endothelial cells and circulating endothelial progenitor cells. While tentative evidence is available for most of these biomarkers to serve as reliable sources of information, their usefulness for routine clinical applications has not yet been established. Thus, circulating endothelial markers are currently the subject of intense research interest and it is anticipated that as more information becomes available their improved quantification will provide a suitable diagnostic and prognostic tool for vascular events in diabetes and related diseases.
Keywords: Vascular disease; Insulin resistance; Improved technology limitations; Circulating markers; Endothelial-associated diagnostic, prognostic tools
Introduction
Diabetes mellitus is a common chronic metabolic disease with a growing prevalence rate worldwide. The World Health Organization (WHO) estimates that, in 2000, at least 171 million people worldwide had diabetes and this figure is expected to double by 2030 [1]. About 3.2 million deaths annually are attributable to complications of diabetes. Depending upon regional prevalence of the disease and the treatments available, healthcare costs for diabetes are estimated to range from 2.5%–15% of annual healthcare budgets [1]. The costs of lost production due to diabetes are reported to reach 5 times the healthcare costs. The United States (US) is considered among the top 10 countries in the world regarding the number of people suffering from diabetes. According to the American Diabetes Association, presently, there are nearly 24 million Americans with diabetes and this figure appears to be significantly higher than what was reported a decade ago [2]. Unfortunately, of these individuals, about one-quarter are unaware that they have the disease.
Several forms of diabetes mellitus are known to exist but two are recognized as the major ones. Type 1 diabetes is characterized by an absolute insulin deficiency attributable to pancreatic insufficiency caused by autoimmune destruction of β cells [3]. It usually involves children and accounts for 5% to 10% of all diagnosed cases of diabetes in the US. On the other hand, type 2 diabetes is characterized by insulin resistance associated with a reduced glucose uptake during insulin exposure, thus representing a deficiency in the effectiveness of insulin despite high plasma level of the hormone. It eventually can lead to an absolute insulin deficiency due to the progressive dysfunction and failure of the β cells [4]. Genetic predisposition is considered to be a key factor for developing type 2 diabetes, but it may progress with additional risk factors such as obesity [5, 6] and sedentary lifestyle [7, 8]. Type 2 diabetes usually manifests itself later in life following slowly developing metabolic processes that include a phase of glucose intolerance/prediabetes. Type 2 diabetes accounts for approximately 90% to 95% of all diagnosed cases of diabetes. Increasing incidences of type 2 diabetes is a major contributory factor to the overall rise in the number of diabetic patients worldwide.
Both type 1 and type 2 diabetes being associated with hyperglycemia and related disturbances in body’s metabolism can lead to serious secondary complications, such as cardiovascular disorders, nephropathy, retinopathy and neuropathy [9]. Heart disease and stroke are the major cardiovascular disorders manifested in both types of diabetes. Diabetes-induced reduced blood flow and neuropathy in the foot leads to increased risk of foot ulcers and eventual amputation. Diabetic retinopathy occurring as a consequence of damage to the small blood vessels in the retina is a major cause of blindness. The effect of diabetes on the kidney (i.e., nephropathy) results in kidney failure and death in 10–20% of people with diabetes [10].
While diabetes is known to be associated with a number of complications, vascular disease is a prominent feature of both type 1 and type 2 diabetes. It is reported that as high as 80% of the diabetes mortality is a consequence of this disorder [11]. Although the reason(s) for diabetic vasculopathy (angiopathy) is incompletely understood, it is now established that the vascular endothelium plays a central role [12]. As described in some details below, the endothelium is critical for proper functioning of blood vessels and, hence, target organs. Accordingly, endothelial dysfunction or injury predisposes diabetic patients to both microangiopathy and macroangiopathy leading to target organ damage [12]. Microangiopathy in diabetes is a hallmark feature of retinopathy, neuropathy and nephropathy whereas macroangiopathy is more closely linked to accelerated atherosclerosis. Therefore, maintaining the function of the endothelium is of prime importance for healthy vasculature and proper tissue/organ functions. Determining the status of the endothelium using appropriate techniques can be instrumental for this assessment.
Although strict glycemic control can lower the incidence of diabetic complications, this is often difficult to achieve and is not also sufficient to avert the vascular problem [13]. There is also the question of reliability/sensitivity of the existing diagnostic tests for glucose [14]. Evidence is also available indicating that endothelial dysfunction precedes the development of overt hyperglycemia in patients with type 2 diabetes [15]. On the other hand, impairment of the endothelium in type 1 diabetes has been demonstrated to occur much later after a period of exposure to hyperglycemia [16]. In view of these observations, endothelial dysfunction, rather than blood glucose levels, appears attractive for consideration as a critical early target for detecting vascular disease in diabetes. However, assessing the status of the endothelium in human subjects for this application using the currently utilized functional methods appears problematic for routine purposes (see below for details). Alternatively, there are emerging assay methods based on the measurements of circulating endothelial markers which have the potential to be developed as viable alternatives for routine human use. Nonetheless, many of these assays presently are used mainly for research purposes.
This review article presents a synopsis of currently available circulating endothelial biomarkers with the potential to be utilized as convenient and reliable predictors of endothelial dysfunction in diabetic patients. To put the review in perspective, it is relevant to briefly review, as background information, the function of the endothelium under normal and diabetic conditions, and the methods that have been used commonly for evaluating endothelial function/dysfunction in humans.
Background on endothelial function/dysfunction and its assessment in humans
General remarks on endothelial function
The endothelium is a mono-layer of cells that line the lumen of all the blood vessels in the body. It provides an interface between circulating blood and the underlying vascular smooth muscle. Besides serving as a physical barrier between the blood and tissues, the endothelium is involved in multiple other functions, including regulation/maintenance of vascular tone, coagulation, fibrinolysis, cell growth and platelet and leukocyte adherence [17, 18]. The type and location of the blood vessel can determine the interplay between the endothelium and the vessel. For instance, endothelial cells in conduit arteries limit activation of clotting and pro-inflammatory factors, while they contribute to the regulation of blood flow in resistance vessels and are involved in the transport and distribution of nutrients and hormones in precapillary arterioles [17, 18].
The important role of the endothelium in the regulation of vascular tone, as a vasodilator, was demonstrated almost three decades ago by the 1998 Nobel Prize-winner Robert Furchgott [19]. Its vasodilatory function was shown to be mediated by what was known as endothelium-derived relaxing factor (EDRF), which was subsequently identified as nitric oxide (NO). Nitric oxide is now considered a key vasodilator substance generated in the vascular endothelium [20]. To make this possible, endothelial cells constitutively express the enzyme NO synthase, more specifically NO synthase-type III (eNOS). For initiation of NO formation, the first step is activation of the endothelial cells thereby leading to an increase in intracellular Ca2+concentration. The eNOS enzyme, with Ca2+/calmodulin binding, then generates NO using L-arginine as a substrate together with reduced nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide/mononucleotide (FAD/FMN), and tetrahydrobiopterin (BH4) as cofactors [20]. In addition, more recently it was shown that NO itself can activate eNOS in endothelial cells, suggesting its involvement in a positive feedback process to further increase its own release [21]. The NO produced in the endothelial cells rapidly diffuses into underlying vascular smooth muscle cells and binds to a heme group of soluble guanylate cyclase (sGC), resulting in activation of the enzyme and formation of cyclic GMP (cGMP) [22]. Cyclic GMP in turn activates a cGMP-dependent protein kinase, leading to decreased cytosolic Ca2+ in vascular smooth muscle cells and inhibition of the contractile machinery followed by vasodilatation [23] (Fig. 1).
Fig. 1.
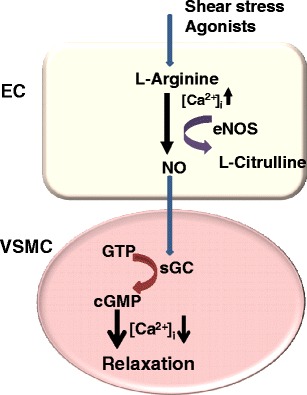
Endothelial cell as a regulator of vascular smooth muscle tone. Endothelial cells synthesize NO which diffuses into adjacent smooth muscle cells to activate sGS, generating cGMP. cGMP, through a reduction in intracellular calcium (Ca2+i), causes relaxation of smooth muscle cells. Abbreviations: NO: nitric oxide; sGS: soluble guanylate cyclase; GTP: guanosine triphosphate; cGPM: cyclic GMP; eNOS: NO synthase-type III; EC: endothelial cell; VSMC: vascular smooth muscle cell
Different agonists activating receptor-operated mechanisms in endothelial cells are known to cause the release of NO. Examples include acetylcholine (Ach), bradykinin, serotonin and substance P [23, 24]. Nitric oxide is also released by receptor-independent mechanisms, such as physical stimuli (e.g., shear stress, ischemia), most likely independent of the involvement of Ca2+/calmodulin [25]. The release of NO by endothelial cells also takes place under baseline conditions and this plays a role in the regulation of blood pressure. Consequently, many studies have demonstrated that eNOS inhibitors or mechanisms that impair NO generation elevate blood pressure. Besides causing vasodilation, NO also mediates many other functions, which appear to be protective. For example, NO by inhibiting expression of pro-inflammatory cytokines, chemokines and leukocyte adhesion molecules, limits vascular recruitment of leukocytes and platelets, thus limiting associated disorders such as atherosclerosis and thrombosis [26]. The molecule also inhibits vascular smooth muscle cell proliferation, which is an early sign of vasculopathy such as atherosclerosis [26].
It is predictable that endothelial dysfunction can take place at any level in the vasculature and, as noted above, this problem is considered a major risk factor for the development of vascular diseases and target organ damage. Such a dysfunction occurs with different pathophysiological conditions including diabetes and related disorders such as insulin resistance/glucose intolerance/prediabetes [27]. In the healthy vasculature, there is a balance between vasodilatation and vasoconstriction. Endothelial NO is a key factor for vascular tone regulation by inducing vasodilatation, although prostacyclin and endothelium-derived hyperpolarizing factor (EDHF) also play similar roles (albeit to a lesser extent). On the other hand, there are opposing endothelium-generated factors that promote vasoconstriction in blood vessels. These factors are produced in abnormally high amounts under certain pathological states that affect blood vessels, one being diabetes. The main factor in this regard is endothelin-1, a potent vasoconstrictor [28]. Besides causing vasoconstriction on its own, endothelin-1 also augments the vascular action of other vasoactive contractile substances, such as norepinephrine, angiotensin II, thromboxane A2 and prostaglandin H2. Accordingly, the chain of events that leads from endothelial dysfunction to overt vasculopathy can be related to an imbalance in the production of these mediators. This abnormal state may affect the critical role of the endothelium in overall homeostasis, predisposing it to pro-angiopathic factors [24, 28].
A number of studies identify impairment of NO production and/or loss of its biological activity as the immediate culprit lesion for the dysfunction of the endothelium [29]. With the availability of suboptimal concentrations of substrate and/or cofactors for NO synthesis, eNOS becomes uncoupled thereby impairing the synthesis of the molecule. This situation leads to the generation of reactive oxygen species (ROS), such as superoxide anion and hydrogen peroxide, a condition collectively defined as oxidative stress [30]. The excessive ROS produced during oxidative stress cannot be completely scavenged by the available mechanisms which function efficiently under normal conditions. With excess ROS, the scavenger system becomes ineffective, enabling NO to rapidly react with superoxide to form peroxynitrite, which further exacerbates the oxidative stress [31, 32].
Effect of diabetes on endothelial function
A large body of clinical and experimental evidence indicates that both type 1 and type 2 diabetes, depending upon the severity and duration of the disease, cause dysfunction of the endothelium, with or without overt vasculopathy [12, 24, 27]. As an example, in Fig. 2 is shown impairment of endothelium-dependent relaxation of aortas from streptozotocin-treated diabetic rats (with type 1-like diabetes) of 14 weeks duration. As stated earlier, there is also compelling evidence for the hypothesis that endothelial dysfunction can precede type 2 diabetes, as evidenced, for example, by the finding of impaired vascular endothelial responsiveness in subjects with normal glucose tolerance and with a family history of diabetes (32). In this regard, endothelial dysfunction may be a critical early target for preventing overt vascular disease that may subsequently develop as diabetes ensues. It is also mentioned that in many cases of type 1 diabetes, the development of endothelial dysfunction has been observed to occur after a certain period of exposure to diabetic milieu [15, 16, 22]. Whichever way it may occur, endothelial dysfunction, once established, can induce further alterations in blood vessels that continue to worsen the disorder. As pointed out above, endothelial dysfunction linked to diabetes is associated with reduced production of NO. This effect of the disease may be coupled to the development of oxidative stress [15, 22, 29, 30]. In addition to the documentation of biochemical changes, dysfunction of the endothelium in diabetes has also been shown to be associated with morphological/structural changes (injury) of the endothelium, particularly at advanced stages of the insult. Although it has not been determined systematically, the literature generally indicates that, depending upon the type and/or degree of insult, endothelial dysfunction can be more of local or systemic [22, 33, 34]. The nature of the dysfunction also determines whether or not it is reversible.
Fig. 2.
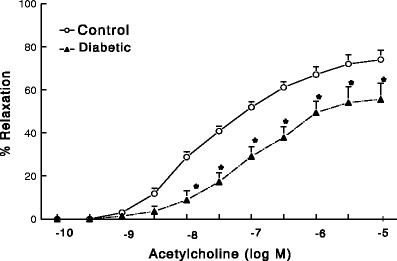
Effect of type 1-like diabetes of 14 weeks duration on acetylcholine-mediated, endothelium-dependent relaxant responses of aortas from male streptozotocin-treated rats (n = 6 rats/group). *Significantly different from corresponding control responses (P < 0.05)
Basic mechanisms underlying endothelial dysfunction in diabetes
The basic mechanisms underlying endothelial dysfunction in diabetes and related disorders are very complex and incompletely understood. However, the mechanisms described below have been demonstrated to be involved in this dysfunction. While only a brief account of these mechanisms is given here, readers are referred to other available sources for additional information [15, 22, 30].
Mechanisms related to hyperglycemia
Hyperglycemia is a characteristic feature of both type 1 and type 2 diabetes. It plays a pivotal role in diabetes-associated microvascular complications. Hyperglycemia and its immediate biochemical sequelae have been shown to directly affect endothelial function. The example in Fig. 3 demonstrates that incubation of rat aortas with high concentration of glucose (45 mM) for 3 h causes inhibition of endothelium-dependent relaxation.
Fig. 3.
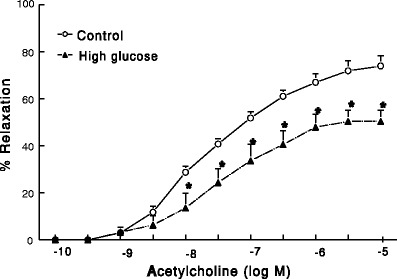
Effect of high-glucose (45 mM for 3 h) on acetylcholine-mediated, endothelium-dependent relaxant responses of aortas from male rats (n = 6 rats/group). *Significantly different from corresponding control responses (P < 0.05)
Glucose transport into endothelial cells occurs by facilitated diffusion that is not dependent on insulin. This transport is not also autoregulated in endothelial cells, in which case an increase in blood glucose concentration leads to increased intracellular accumulation of glucose and its metabolites [15, 30, 35–37].
Various possibilities have been forwarded to explain how hyperglycemia causes endothelial dysfunction in diabetes. Normally, glucose undergoes cellular metabolism through the glycolytic pathway. Under hyperglycemia four major pathways are proposed to affect the function of the endothelium. Accordingly, hyperglycemia-induced increased generation of reactive oxygen species (e.g., superoxide) causes exacerbated oxidative DNA damage and subsequent activation of the poly (ADP-ribose) polymerase (PARP). This, in turn, results in PARP-induced addition of ADP-ribose polymers onto the glyceraldehydes-3 phosphate dehydrogenase (GAPDH) thereby reducing its activity [15, 22, 30, 38] (Fig. 4). Consequently, greater substrate availability, upstream of GAPDH step in the glycolytic pathway, leads to activation of the four pathways that are briefly described below.
The sorbitol/polyol pathway: In this pathway, excess glucose is metabolized to sorbitol and fructose by aldose reductase and sorbitol dehydrogenase. This pathway may impair endothelial function through two primary mechanisms: increased osmotic stress and oxidative stress, although other related mechanisms are also suggested to be involved [15, 22, 30].
The hexosamine pathway: In this pathway, fructose-6-phosphate is converted to fructosamine 6-phosphate by the enzyme glutamine:fructose 6-phosphate amidotrasferase and subsequently into N-acetlyglucosamine. This pathway has been shown to be involved in the augmentation of transcription of plasminogen activator inhibitor-1 (PAI-1) and transforming growth factor β1 (TGF-β1) and modification of protein kinase C (PKC) and eNOS [15, 22, 30].
The protein kinase C (PKC) pathway: The mechanism responsible for the activation of PKC by hyperglycemia is related to de novo synthesis of the PKC activator diacylglycerol (DAG) from a stepwise acylation of glycerol 3-phosphate and phosphatidic acid. The effect of PKC activation include inhibition of eNOS activity, increased endothelin-1 synthesis, basement membrane thickening (via TGF-β-mediated increased synthesis of type IV collagen and fibronectin), impaired fibrinolysis (via tissue plasminogen activator (PAI-1)) and increased oxidative stress (via NADPH regulation) [15, 22, 30].
The glycosylation pathway: This pathway involves a non-enzymatic reaction of glucose and glucose-derived dicabonyl compounds with the basic amino acids lysine and arginine in proteins to form advanced glycation end (AGE) products both extra- and intracellularly. The formation of AGE products leads to a number of adverse effects that can affect endothelial cells and other tissues. These include structural modifications of proteins, increase in oxidative stress, activation of transcription factors, increased expression of cytokines and growth factors, among others [15, 22, 30].
Mechanisms related to action of insulin
Fig. 4.
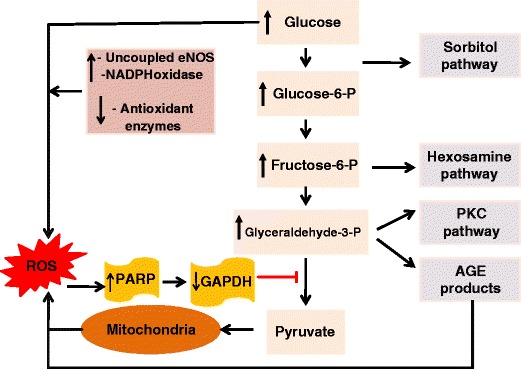
Proposed mechanisms by which hyperglycemia and subsequent overproduction of reactive oxygen species such as superoxide (from mitochondria and other sources) cause oxidative DNA damage and activation of PARP. In turn, increased PARP activity causes addition of ADP-ribose polymer onto the GAPDH resulting in its inhibition. Inhibition of GAPDH will result in build up of glycolytic intermediates and their subsequent diversion into and augmentation of the following four pathways: the sorbitol/polyol, hexosamine, PKC and glycosylation pathways. Abbreviations: NADPH: reduced nicotinamide adenine dinucleotide phosphate; eNOS: nitric oxide synthase-type III; PKC: protein kinase C; ROS: reactive oxygen species; PARP: poly(ADP-ribose) polymerase; GAPDH: glyceraldehydes-3-phosphate dehydrogenase; AGEs: advanced glycation end (products)
Insulin is a vasoactive hormone which can increase blood flow through processes involving binding to insulin receptors on endothelial cell membrane. By this process, insulin increases the production of NO and induces endothelium-dependent vasorelaxation, although the molecular pathway has not yet been fully elucidated. However, it has been suggested to involve the activation of the intracellular enzymes phosphatidylinositol-3-kinase (PI3K) and Akt, which ultimately lead to phosphorylation and activation of eNOS. Besides the generation of NO, insulin, via another pathway (mitogen-activated protein kinase (MAPK) pathway), causes the release of endothelin-1 by endothelial cells, and growth and migration of vascular smooth muscle cells [15, 22, 27, 39, 40] (Fig. 5).
Fig. 5.
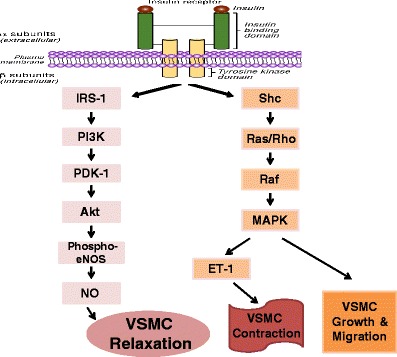
Insulin signaling pathways in endothelial cells. The PI3K/Akt branch regulates NO production in endothelial cells and vasodilation in vascular smooth muscle cells. The MAPK branch controls secretion of endothelin-1 in the endothelium, and also regulates growth and mitogenesis. Under normal conditions, there is an optimum balance between these pathways. Abbreviations: IRS-1: insulin receptor substrate protein-1; PI3K: phosphatidylinositol-3-kinase; PDK-1: phospho-3’-phosphoinositide-dependent kinase-1; NO: nitric oxide; eNOS: NO synthase-type III; MAPK: mitogen-activated protein kinase; Et-1: endothelin-1; VSMC: vascular smooth muscle cell
The vasodilator action of insulin is impaired under conditions of insulin-resistance, more notably prediabetes, type 2 diabetes, obesity and hypertension. It is also decreased by mediators closely associated with insulin-resistance, namely tumor necrosis factor-α (TNF-α), non-esterified fatty acids (NEFAs) and an impaired NO-dependent action [15, 22]. Similarly, when there is an absolute lack or impairment of insulin production as in type 1 diabetes, effects mediated by insulin per se in endothelial cells (and elsewhere) will be lacking, potentially shifting the balance in favor of vasoconstriction. In support of impaired vascular relaxation in insulin resistance, endothelin-1 released from the endothelium by increased plasma insulin level has been suggested to have a contributory role [22, 39].
Another important aspect of insulin resistance/type 2 diabetes is the consideration that it is linked to elevated plasma free fatty acids which can generate ROS via mitochondrial oxidation [18, 23, 26]. The ROS produced subsequently undergo similar metabolic pathways as shown for hyperglycemia (Fig. 4). However, unlike the role of hyperglycemia, this mechanism is proposed to be involved more in diabetic macrovascular dysfunction [30].
Mechanisms related to overproduction of growth factors and cytokines
Diabetes has been established to be associated with overproduction of growth factors and cytokines which have an influence on endothelial cell function. Hyperglycemia is known to contribute to this effect by inducing increased synthesis of these factors in different types of cells [15, 27, 41]. Among others, the mechanisms for this effect involve hyperglycemia-induced PKC activation, AGE production and cytokine activation of endothelia cells. Hyperglycemia being a common factor, such processes can take place in both type 1 and type 2 diabetes.
Overproduction of growth factors has been implicated as a link between diabetes and proliferation of endothelial cells and vascular smooth muscle cells, possibly promoting neovascularization [15, 27, 41]. Among the growth factors that play important roles in diabetes complications, TGF-β1 and vascular endothelial growth factor (VEGF) have greater influence on endothelial cells. The receptors for both hormones are expressed in endothelial cells [15, 27]. Both growth factors regulate multiple cellular functions in different tissues. For TGF-β1, these include cell growth, cell death/apoptosis, cellular differentiation and extracellular matrix protein synthesis. Similarly, VEGF stimulates cell differentiation, survival, migration, and proliferation, tubulogenesis and vascular permeability [15, 27]. However, although the levels of these growth factors have been shown to increase in animal models of diabetes, the temporal sequence is not well defined, implying that the roles of growth factors in diabetes require further investigation.
Although certain cytokines appear to play significant roles in endothelial functions, many cytokines produce effects to which endothelial cells respond variably. In short, in the vasculature, inflammatory cytokines enhance vascular permeability, alter vasoregulatory responses, increase leukocyte adhesion to endothelium, facilitate procoagulant activity, and regulate gene expression in endothelial cells, among other effects [15, 27]. Of the many known cytokines, TNF-α is clearly implicated as a link between insulin resistance, diabetes, and endothelial dysfunction. In view of the association between obesity and insulin resistance, increased expression of TNF-α (and others) in human obesity supports the hypothesis that its elevation induces insulin resistance. TNF-α also induces the synthesis of other cytokines, such as interleukin (IL)-1, which in turn regulates the expression of c-reactive protein (CRP), an important downstream marker of inflammation [15, 27]. These mediators, alone or in concert with each others, contribute to artherothrombosis by altering endothelial function.
More recently a strong association has been found between obesity and insulin resistance/type 2 diabetes. Accordingly, endothelial dysfunction occurring in obese subjects or animal models of obesity can be related to insulin resistance/type 2 diabetes, although obesity can also be an independent risk factor for this dysfunction and associated disorders. This relationship is believed to be, at least in part, the result of overproduction of chemical mediators from adipose tissue in the obese. Evidence has been accumulating that visceral adipose tissue is specially a highly active endocrine organ that releases a variety of substances, including certain hormones, cytokines and enzymes that are capable of attenuating insulin sensitivity [32, 40, 42]. Of these products, cytokines (for example, TNF-α, IL-6, PAI-1) and adipokines (for example, adiponectin, lepin) have been proposed to play a role in inflammation, insulin resistance and vascular disease [27, 32, 40]. Studies have shown that while some of these mediators are protective, others are associated with various adverse effects, as noted earlier. On balance, however, visceral obesity gives rise to insulin resistance and endothelial dysfunction primarily through a cascade of secreted proinflammatory substances, such as cytokines and CRP. Furthermore, insulin resistance by causing hyperglycemia, may perpetuate inflammation via augmented oxidative stress [30, 32, 40].
Assessment of endothelial function/dysfunction in humans
Since the initial report by Furchgott and Zawadzki [19] revealing the role of the endothelium in vasorelaxation, the function of the endothelium has been a focus in the study of vascular disease. With the initial understanding that derangement in NO formation contributes to the development of artherosclerosis, various techniques for assessing endothelial function/dysfunction in humans in relation to the release of NO in coronary circulation as well as in the systemic circulation have been developed. Most of the endothelial functional tests performed are meant to evaluate abnormalities in the regulation of the lumen of blood vessels [22, 43–46]. However, this approach may not necessarily be reflective of the function of the endothelium at a given time point because (a) the lumen of blood vessels can also be influenced by factors other than the endothelium, such as smooth muscle alterations independent of the endothelium and (b) endothelial cells may affect one or more functions in the vasculature, either simultaneously or in a temporal sequence, and as such cannot always be considered a single and discrete functional entity [22, 43, 45]. Thus, with the assessment of the above parameter (reflection of lumen size), it is not always possible to reliably define the status of the endothelium. This requires the need for developing techniques that can be more precise, selective and sensitive for measuring endothelial function.
While the current methods for assessing endothelial function in humans are used alone in most cases, sometimes they are complemented by measurements of plasma concentrations of biomarkers (e.g., nitrite assays), or an index of the structure of the vessel such as assessment of carotid intima media thickness [43–54]. The following is a brief description of the methods that are commonly utilized to measure endothelial function/dysfunction in humans as an outcome of endothelial cell activity.
Use of plethysmography
This method is based on strain-gauge venous impedance plethysmography for studying endothelium-dependent as well as independent vasodilatation in the peripheral circulation [22, 43, 45, 46]. It is especially designed for mapping dose-response relationships of stimulators and inhibitors of endothelial cell activity. This technique often uses direct infusion (by arterial cannulation) of acetylcholine, or other muscarinic receptor agonists (as a selective activator of endothelial cells) in the brachial artery and determines the vasodilator responses in the forearm resistance vessels. Sodium nitroprusside is usually administered as a control agent in order to assess endothelium-independent vasodilatation. The determination of changes in blood flow (reflective of lumen size), contributed by the whole limb portion indicates a measure of endothelial function. However, the invasive nature of the technique limits its repeatability and prohibits its use in larger studies. In addition, the results are also difficult to standardize because resistance vessel tone is variable, in addition to the general problems mentioned above in connection to the measurement of lumen size [22, 43–46].
Use of ultrasound and doppler
In coronary circulation, non-invasive methods for assessing endothelial function, including doppler echocardiography, positron emission tomography and phase-contrast magnetic resonance imaging, were described before the development of most of the currently used methods [43, 44, 46, 47]. However, in view of technical difficulties and high-costs associated with these techniques, their uses have remained very limited. Subsequently, quantitative coronary angiography with an intra-coronary ultrasound and doppler transducer has been developed and used more often to assess endothelial function in the coronary circulation [22, 43–47]. By a graded administration of acetylcholine and sodium nitroprusside, endothelium-dependent and independent vasodilatation, respectively, can be quantified using this approach. Nonetheless, in addition to being invasive, this technique is also technically complicated for routine application. This leads to the emergence of a simpler and a noninvasive method based on flow-mediated vasodilatation (FMD) in brachial artery, which is more suitable for repeated studies and for evaluating large groups of patients [22, 43, 45, 47, 48]. This method uses ultrasound for detection of changes in artery diameter (lumen) and blood flow. Flow-mediated vasodilatation thus measured in brachial artery has been found to correlate well with endothelial function in the coronary circulation [49]. Because of the above advantages, this method has been widely used, and in fact, it is considered to represent the gold standard for clinical research on conduit artery endothelial biology [50]. Flow-mediated vasodilatation is based on the theory of shear stress, whereby a short period (for example, 5 min) of arterial occlusion increases flow in the artery. This procedure is believed to induce the endothelium to release NO with subsequent vasodilatation. Nitroglycerin (or another nitrate) is administered sublingually/orally to evaluate endothelium-independent vasorelaxant responses. In shear stress, no specific receptor signaling pathway has been reported to be activated or inhibited. Therefore, the precise mechanisms for the detection of shear stress and the subsequent signal transduction to modulate vasomotor tone are not fully understood [51].
Brachial artery FMD has been studied widely in clinical research because, among others, it enables serial assessment of young subjects, and also permits testing of lifestyle and pharmacological interventions on the endothelium at an early stage of a disease (i.e., when the disease process is most likely to be reversed) [22, 43, 46]. However, the equipments used in this technique are still expensive and the procedures involved are also technically demanding, although the protocol can be reliably standardized to yield reproducible results that correlate with coronary vascular endothelial function [43, 46].
Circulating endothelial markers relevant to diabetes
In view of the significance of assessing the status of the endothelium in vasculopathy and the limitations of the above techniques, more recent efforts have been directed towards exploring other approaches of evaluation based on measurement of circulating endothelial markers. The advantages of measuring such circulating indicators may include (a) simplicity of quantification with current immunological techniques for most of them, (b) the release of most of the biomarkers specifically from the vascular endothelium and the possibility of providing information on the status of the endothelium directly and/or (c) their involvement (directly or indirectly) in the function or regulation of endothelial cells. However, it is noteworthy that the usefulness of this approach has not yet been fully verified for clinical applications and some of the procedures involved may also require delicate conditions of sampling and/or analysis [43, 44, 46]. Nonetheless, there appears to be a general consensus that circulating endothelial biomarkers have the potential to be used for routine clinical testing of endothelial dysfunction in patients with possible vascular disorders. Thus, we will describe some biomarkers which are believed to be relevant for assessing endothelial function/dysfunction in diabetes (and other similar conditions) (Fig. 6). Except endothelial progenitor cells (EPCs), these biomarkers have the common property of being released specifically from the vascular endothelium. Endothelial progenitor cells are also closely associated with the endothelium in a different way, as detailed below. It should be noted that there are several other circulating biomarkers which are not specific to the endothelium and/or their assays are less sensitive to detect subtle changes in the endothelium. Examples of such biomarkers include P-selectin, adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), endothelin-1 and NO/nitrite [43–47, 52] (Fig. 6).
Fig. 6.
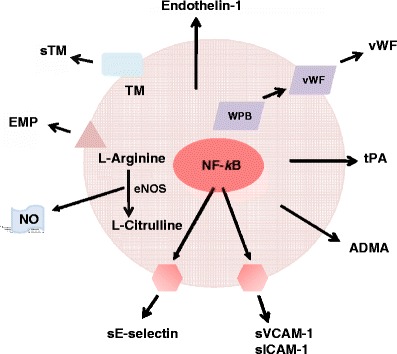
Major endothelial-derived circulating biomarkers. While vWF, sTM, ADMA, EMP, sE-selectin, tPA and endotheline-1 are released into the circulation specifically by endothelial cells, the other biomarkers shown are also released by other types of cells, making assay of the latter less specific to precisely reflect the status of the endothelium. In addition, the assay method available for endothelin-1 is not sensitive enough to reveal relatively subtle changes in the endothelium. Abbreviations: NO: nitric oxide; eNOS: NO synthase-type III; NF-kB: nuclear factor kB; EMP: endothelial microparticle; TM: thrombomodulin; sTM: soluble TM; vWF: von Willebrand factor; WPB: Weibel-Palade body; tPA: tissue plasminogen activator; ADMA: asymmetric dimethylarginine; sVCAM: soluble vascular cell adhesion molecule-1; sICAM: soluble intracellular adhesion molecule
Von Willebrand factor
Von Willebrand factor (vWF) is a glycoprotein released from the endothelium [43, 52]. It is a large multimeric structure consisting of over 80 subunits of 250 kDa each. Its presence in the circulatory system is important for platelet aggregation and adhesion and for factor VII survival [43, 52]. Only the large multimers are believed to be functional. As such, vWF plays a vital role in mediating platelet adhesion to damaged arterial walls, thereby causing localized blood clots or thrombus formation.
Von Willebrand factor is produced and stored in endothelial Weibel-Palade bodies and secreted into blood upon endothelial cell injury (Fig. 6). It is considered a gold standard in the measurement of endothelial damage. Its plasma level has been shown to increase in vascular disease such as that associated with diabetes or insulin resistance [43, 52]. Plasma vWF can be determined by enzyme-linked immunosorbent assay (ELISA) kit which makes use of a monoclonal antibody.
Soluble thrombomodulin
Thrombomodulin (TM) (CD141/BDCA-3) is a membrane protein expressed on the surface of endothelial cells [43, 52]. It has a molecular mass of 74 KDa, consisting of a single chain with five distinct domains. It serves as a cofactor for protein C activation in anticoagulant pathway involving thrombin.
Thrombomodulin, like vWF, can be released from injured endothelial cells, and its soluble form (soluble thrombomodulin, sTM) has been demonstrated to be a specific biomarker of endothelial cell injury (Fig. 6). Plasma sTM concentration can be measured reliably by ELISA kits [43, 52].
Soluble E-selectin
E-selectin is a member of the selectin family adhesion molecules, which is specifically expressed on the surface of cytokine-activated endothelial cells [43, 44, 52]. In this regard, it was observed that maximal expression of E-selectin on the surface of human umbilical vein endothelial cells (HUVECs) occurred 6 h after stimulation by TNF-α or IL-1 and the level remained sustained for at least 24 h [43, 44]. E-selectin plays an important part in inflammation by enabling the recruitment of leukocytes to the site of cell injury.
A circulating or soluble form of E-selectin (sE-selectin) is released from endothelial cells by enzymatic cleavage or as a result of shedding of damaged or activated cells [43, 44, 52] (Fig. 6). It has been shown that the plasma concentration of sE-selectin is correlated to its expression on the surface of HUVECs [43, 44]. Hence, plasma level of sE-selectin has been proposed to serve as a marker of endothelial cell damage and/or activation. The role of E-selectin in vascular disease has been extensively investigated in recent years and it appears that there is the possibility of developing an E-selectin assay system for routine clinical application [43, 44]. Soluble E-selectin in the plasma can be detected by a sensitive ELISA kit [43, 44].
Asymmetric dimethylarginine
Asymmetric dimethylarginine (ADMA) is a molecule considered to be an endogenous inhibitor of eNOS, thereby reducing the level of endothelial NO [43, 44, 46, 53]. It is a metabolic product of protein modifications and is closely related to L-arginine. Following its formation, ADMA migrates into the extracellular space and then into the blood (Fig. 6). Evidence is also available that the molecule may cause eNOS uncoupling, thereby switching eNOS from a NO- to superoxide-generating enzyme [43–46, 53].
Elevated plasma ADMA levels have been associated with many cardiovascular risk factors including diabetes and insulin resistance [43–46, 53]. A direct correlation has also been observed between the concentration of ADMA and the severity of cardiovascular diseases. For instance, the level of ADMA was shown to be doubled when endothelial function was further impaired in diabetic patients after a high-fat meal [43–46, 53]. As a result, in recent years, considerable attention has been placed on ADMA as a diagnostic indicator and as a mediator of endothelial dysfunction. Asymmetric dimethylarginine (ADMA) can be measured by HPLC [43–46, 53].
Tissue plasminogen activator
Tissue plasminogen activator (tPA) is a protein (serine protease) released by vascular endothelial cells [43, 44]. It is involved in breakdown of blood clots by catalyzing the conversion of plasminogen to plasmin. It also plays a role in cell migration and tissue remodeling. Being specifically released by endothelial cells, circulating tPA has been suggested to serve as a suitable biomarker of endothelial cells (Fig. 6). It has been shown to be elevated in hypercholestrolaemia and subclinical conditions of stroke, myocardial infarction and atheosclerososis [43, 44]. Studies also indicate that the production of tPA is higher in atheromatous arteries than in normal blood vessels [43, 44]. From these observations, it has been suggested that increased tPA levels may reflect excessive endothelial cell activation and/or damage. However, as yet, no reports have been published regarding the role tPA in diabetic vascular disease.
Circulating tPA levels have been demonstrated to increase rapidly with local adrenergic or cholinergic receptor stimulation [54]. This suggests that assessment of tPA release after adrenergic or cholinergic receptor stimulation may be a valuable tool for evaluating endothelial function.
Tissue plasminogen activator (tPA) can be measured by using ELISA or by measuring its activity, both assays providing closely similar results [43].
Endothelial microparticles
Endothelial microparticles (EMPs) have recently been identified to be circulating phospholipids-rich submicron vesicles that are released from membranes of endothelial cells as a consequence of activating or injurious external stimuli, although some studies suggest that similar particles are also released by platelets and T lymphocytes under certain conditions [43, 46, 52, 55, 56] (Fig. 6). These microparticles consist of plasma membrane structures containing receptors and other cell surface molecules which enable the identification of their endothelial origin. This allows them to be distinguished from microparticles from other cell types, if present. Although circulating EMPs can be found in the blood of normal subjects, increased number of the microparticles have been measured in individuals with diseases affecting blood vessels including diabetes [43, 46, 52, 55, 56]. As a result, EMPs have gained greater considerations to be utilized as an index of changes in endothelial function/structure for such disease states. Also, endothelial changes that may be correlated with levels of circulating EMPs can serve to indicate the severity of the disease per se.
With regard to diabetes, in a number of studies, increased release of EMPs has been shown along with reductions in endothelium-dependent vasorelaxation and NO production [55, 56]. Corroborating with this observation, a correlation has also been seen between EMPs and reduced FMD of arteries in diabetic patients with renal failure. In another group of patients with diabetes, augmented circulating EMPs was measured together with an increase in ICAM-1 [55, 56]. From these studies, it appears that the more severe the diabetes state is, the greater the amount of EMP in the circulation, both of which correlate with the degree of dysfunction of the endothelium.
More recent investigations have revealed that EMPs impair endothelium-dependent relaxation and NO production in the rat aorta [55, 56]. This effect may be related to an increase in superoxide anion production, which might reduce the bioavailability of NO. Therefore, microparticles of endothelial origin may reflect cellular injury in this way too, and, as such, may serve as circulating markers for endothelial/vascular dysfunction that are easily measured in the blood [55, 56].
Endothelial microparticles can be assayed by analyzing the expression of CD31+/CD41-, CD144 and CD62e+ on circulating cell surface by flow cytometry [55, 56]. Among these surface markers, CD62e+ is more specific to endothelial cells; the expression of this marker is enhanced by cytokine. Phenotype analysis of EMPs is based on size characterization and fluorescence measurement, which ultimately enables determination of EMP levels [55, 56]. Another less widely utilized method is functional assay performed using the pro-coagulant property of EMPs [55, 56]. However, due to lack of standardization, the measurement of EMPs by both methods remains a technical challenge.
Circulating endothelial cells
Circulating endothelial cells (CEC) are mature cells expressing the membrane glycoproteins CD146, CD31 and CD144 [46, 52, 56, 57]. They are rarely found in the blood of healthy subjects but are elevated in disease conditions associated with cardiovascular and inflammatory disorders. They present a heterogeneous size ranging from 10 to 50 micron. These cells are negative for leukocyte markers and, unlike EPCs, do not express immature markers such as CD133 and do not rise with high proliferative potential [56, 57]. Circulating endothelial cells (CEC) may arise from damaged endothelium, although the mechanisms of endothelial cell detachment are multiple and not exclusive. Denudation of the vessels may be triggered by mechanical injury, defective adhesion, protease- or cytokine-mediated detachment or activation of apoptosis, among other possible reasons. A major problem associated with CEC quantification is their low number in blood (i.e., approximately one per one million nucleated cells). To circumvent this limitation, CECs first have to be enriched (using CD146 coated magnetic microbeads) and then phenotyped using monoclonal antibodies to cell surface glycoproteins [52, 56, 57]. This process is known as immuno-magnetic separation (IMS) assay, which has now become a consensus protocol for CEC quantification. Alternative method for CEC counting includes flow cytometry; however, in most cases, this method provides inconsistent results relative to the IMS assay.
The use of CECs as a biomarker of vascular endothelial dysfunction and the associated risk factors such as diabetes and hypertension has recently been reviewed [56, 57]. In fact, an inverse correlation was found between CEC levels and FMD, indicating a positive relationship between endothelial dysfunction and increased CEC number. A positive correlation was also shown between CECs and vWF but not between CECs and sE-selectin, suggesting that CECs may reflect endothelial damage rather than endothelial activation [56, 57]. Furthermore, CECs levels have been observed to positively relate to the degree of coronary syndrome and peripheral arterial disease. Besides their role in diagnosis, CECs have also been proposed as a novel prognostic indicator since raised CEC levels could independently predict both death and adverse cardiovascular events before they could occur [52, 56, 57].
Circulating endothelial progenitor cells
Recently, endothelial progenitor cells (EPCs) released from bone marrow into the circulation have been found to be critical for maintenance and repair of the vascular endothelium [58] (Fig. 7). In this process, EPCs undergo differentiation into mature ECs following recruitment. It is therefore plausible that endothelial dysfunction that occurs in diabetes may be linked to alterations of EPCs. This observation has been explored to find out if EPCs can be used as a viable biomarker for risk assessment or prediction of vascular disorders and target organ deficiencies in diabetes (and other disease) affecting the vasculature [44, 52, 53, 56] (Fig. 7).
Fig. 7.
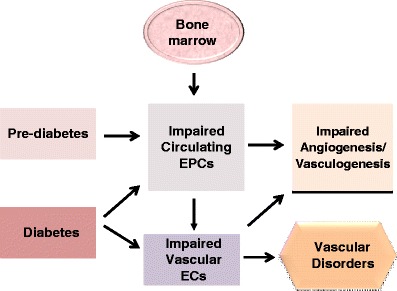
Proposed role of circulating EPCs in endothelial function/dysfunction and disorder of the vasculature in diabetes. EPCs released from bone marrow are involved in the repair of endothelial cells under normal conditions and their levels in the circulation have been shown to decrease significantly when there is impairment or damage to the endothelium. Abbreviations: EPCs: endothelial progenitor cells; ECs: endothelial cells
Endothelial progenitor cells (EPCs) are a group of heterogeneous bone marrow-originated cells that can be characterized by expression of surface markers, including CD34+, DC133+ and vascular endothelial growth receptor-2, by acetylated low density lipoprotein (LDL) uptake and by lecitin binding [59–61]. With cultivation of mononuclear cells (MNCs) from human peripheral blood, these cells attach and develop an endothelium-like phenotype in blood vessels. After being transported, the cells can incorporate into sites of active neovascularization.
Cardiovascular risk factors, including diabetes, that cause endothelial dysfunction, have been shown to be associated with decreased number and functional ability of circulating EPCs [59–61]. Such an inverse association between circulating EPCs and endothelial dysfunction has been observed in human patients as well as animal models of both type 1 and type 2 diabetes (58–60). In EPC colonies from culture of MNCs, diabetes and hyperglycemia-induced functional impairments have been reported to be mediated via mechanisms involving chemokines (e.g., growth factors, stromal-derived factor (SDF-1), granulocyte-monocyte stimulating factor (GM-SCF)), eNOS/NO, and matrix metalloproteinase-9 (MMP-9) [59–63]. From these and other related findings, low circulating EPCs have been proposed to represent a useful indicator for future cardiovascular events. Other studies have also related impairment of EPC functionality to cardiovascular disorders [59–61]. It is noteworthy that most studies on EPCs reported to date in relation to diabetes have been conducted under conditions favoring or associated with overt vascular endothelial dysfunction. This implies that the assessment of the effects of diabetes/prediabetes on circulating EPCs prior to the development of detectable endothelial dysfunction remains to be performed. Data from such studies can help to determine whether or not predictions of future cardiovascular outcomes are possible based on the status of EPCs.
A number of chemokines and related factors appear to regulate the release and function of EPCs [59–61]. Vascular endothelial growth factor (VEGF), which regulates vasculogenesis and angiogenesis, is involved in the mobilization of EPCs from bone marrow. Nitric oxide, in addition to modulating various aspects of endothelial function has been shown to have a significant role in the activities of EPCs [60]. Impaired MMP-9 activity has been demonstrated to contribute to reduced EPC mobilization in such cardiovascular conditions as post-myocardial infarction [61], but this has not been determined for blood vessels. Furthermore, SDF-1 has been reported to be involved in the pathophysiology of EPCs and promotion of angiogenesis by enhancing EPC release and its incorporation into new vessels [64, 65]. Granulocyte-monocyte stimulating factor (GM-SCF) is another substance with significant ability to stimulate the mobilization of EPCs from bone marrow, thereby leading to homage to regions of vascular damage for regeneration [59–63]. Thus, the consideration of the above factors is important when studying the role of EPCs in relation to endothelial function/dysfunction in diabetes and other diseases having an effect on the vasculature/endothelium.
Circulating EPCs can be quantified in whole blood using the technique of flow cytometry. For this, MNCs, (identified by CD34/KDR and CD133/KDR double positive cells) are isolated using density-gradient centrifugation followed by flow cytometry assay [66]. These cells can also be studied directly for their proliferating property. The functional abilities of EPCs can be determined in some detail ex-vivo using expanded cell culture by the application of different assay methods. For culturing, the MNCs are usually separated in histopaque-1083 density gradient medium, and then expanded using appropriate media and conditions [66]. The number of cell colonies obtained from a sufficient number of wells is determined manually under a microscope by a trained observer. Commonly performed assays to evaluate the functionalities of EPCs include chemotaxis, cell proliferation, eNOS activity/expression, senescence-associated β-galactosidase activity, apoptosis/cell survival rate and matrigel tube formation [62, 63, 67–69]. The measurement of these parameters enables to determine if there is a correlation between the number and functions of EPCs and the status of the endothelium in health and disease. If a correlation is established using accessible methodologies, the opportunity exists for EPCs to be used routinely as an index of endothelial dysfunction associated with vascular diseases.
Conclusion
Diabetes is associated with vascular complications, which contribute to the death of as high as 80% of diabetic patients. Endothelial dysfunction plays a critical and an initiating role in vascular disorders in both type 1 and type 2 diabetes and insulin resistance/prediabetes. Assessing the status of the endothelium can serve as a valuable diagnostic and prognostic tool for vascular disease in diabetes. Endothelial function/dysfunction has been assessed in humans using techniques utilizing the measurement of the functional consequences of endothelial activity (ie., relaxation/lumen size changes of blood vessels). The commonly utilized techniques in this regard apply plethysmography, ultrasound and/or doppler to measure blood flow or lumen size in coronary arteries and peripheral blood vessels (e. g., brachial arteries). While these techniques are used primarily for research purposes, some of them appear to be clinically relevant only for a limited scope of evaluation. Certain limitations associated to the methods, particularly as related to technical difficulties and costs, have precluded their routine clinical application.
Recently, a number of endothelial-derived circulating markers have been found to more directly reflect the status of the endothelium. These include vWF, sTM, sE-selectin, ADMA, tPA and EMPs. While the relative significance of each substance (or their combinations) for endothelial evaluation is undefined presently, in most cases, these biomarkers provide good indications of endothelial damage/activation, as it occurs in disease states, such as diabetes, which adversely affect the vasculature. Nonetheless, the measurement of these circulating indicators for diagnosis and/or prognosis of endothelial dysfunction and the associated vascular disorders has not yet been established for routine clinical use. Other indicators of endothelial function/dysfunction are CECs and circulating EPCs, which have the potential to offer additional information on the status of the endothelium. However, only very few studies have investigated the clinical application of these cellular biomarkers, indicating limitation of information on their usefulness for diagnostic and/or prognostic purposes. Taken together, currently, the issue of circulating endothelial markers is an area of intense research interest with the potential to lead to the development of clinically relevant assessment techniques. It is hoped that as more data become available, the improved measurement of circulating endothelial indicators will make it possible to obtain valuable diagnostic and prognostic information on vascular events in diabetes and related conditions for routine clinical testing.
Acknowledgements
The data reported in Figs. 2 and 3 are from studies that were supported by the Combined Intramural Grant Program of the Medical College of Georgia Research Institutes and the Dental Research Center of the School of Dentistry of Medical College of Georgia (WA).
References
- 1.World Health Organization, www.who.int/mediacentre/factsheets/fs312/en/print.html
- 2.American Diabetes Association, www.diabetes.org
- 3.Korc M. Update on diabetes mellitus. Disease Markers. 2004;20:161–165. doi: 10.1155/2004/945073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar V, Abbas A, Fausto N. The endocrine pancreas. In: Robbins and Cotran editors. Pathological Basis of Disease. 7th ed. Elsevier Saunders; 2003.
- 5.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–62. [PMC free article] [PubMed] [Google Scholar]
- 6.Hartz AJ, Rupley DC, Kalkhoff RD, et al. Relationship of obesity to diabetes: influence of obesity level and body fat distribution. Prev Med. 1983;12:351–357. doi: 10.1016/0091-7435(83)90244-X. [DOI] [PubMed] [Google Scholar]
- 7.Ragland HSP, Leung RW DR, et al. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus. N Engl J Med. 1991;325:147–152. doi: 10.1056/NEJM199107183250302. [DOI] [PubMed] [Google Scholar]
- 8.Kriska AM, Bennett PH. An epidemiological perspective of the relationship between physical activity and NIDDM: from activity assessment to intervention. Diabetes Metab Rev. 1992;8:355–72. doi: 10.1002/dmr.5610080404. [DOI] [PubMed] [Google Scholar]
- 9.Martin JH, Mangiafico S, Kelly DJ. Role of statins in diabetes complications. Curr Diabetes Rev. 2009;5:165–170. doi: 10.2174/157339909788920901. [DOI] [PubMed] [Google Scholar]
- 10.Ulrich B. Diabetes and kidney disease. Nephrol Nurs J. 2009;36:461. [PubMed] [Google Scholar]
- 11.Cooper ME, Bonnet F, Oldfield M, et al. Mechanisms of diabetic vasculopathy: an overview. Am. J. Hypertension. 2001;14:475–486. doi: 10.1016/S0895-7061(00)01323-6. [DOI] [PubMed] [Google Scholar]
- 12.Potenza MA, Gagliardi S, Nacci C, et al. Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr Med Chem. 2009;16:94–112. doi: 10.2174/092986709787002853. [DOI] [PubMed] [Google Scholar]
- 13.UK prospective Diabetes Study (UKPDS) Group Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. The Lancet. 1998;352:837–853. doi: 10.1016/S0140-6736(98)07019-6. [DOI] [PubMed] [Google Scholar]
- 14.Koehn J, Tirhani D, krapfenbauer K. Potential predictive biomarkers for the dysfunction of β-cell in type 2 diabetes. In Predictive diagnostic and Personalized treatment, Nova Scientific publishers, Inc.; 2009. p. 183–212
- 15.Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond) 2005;109:143–159. doi: 10.1042/CS20050025. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald SM, Brands MW. Nitric oxide may be required to prevent hypertension at the onset of diabetes. Am J Physiol Endocrinol Metab. 2000;279:E762–E768. doi: 10.1152/ajpendo.2000.279.4.E762. [DOI] [PubMed] [Google Scholar]
- 17.Szmitko PE, Wang CH, Weisel RD, et al. New markers of inflammation and endothelial cell activation: part I. Circulation. 2003;108:1917–1923. doi: 10.1161/01.CIR.0000089190.95415.9F. [DOI] [PubMed] [Google Scholar]
- 18.Szmitko PE, Wang CH, Weisel RD, et al. Biomarkers of vascular disease linking inflammation to endothelial activation: part II. Circulation. 2003;108:2041–2048. doi: 10.1161/01.CIR.0000089093.75585.98. [DOI] [PubMed] [Google Scholar]
- 19.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 20.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 21.Zhang J, Xie Z, Dong Y, et al. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 2008;283:27452–27461. doi: 10.1074/jbc.M802578200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Nathanson D, Nyström T. Hypoglycemic pharmacological treatment of type 2 diabetes: targeting the endothelium. Mol Cell Endocrinol. 2009;297(1-2):112–126. doi: 10.1016/j.mce.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 23.Mombouli JV, Vanhoutte PM. Endothelial dysfunction: from physiology to therapy. J Mol Cell Cardiol. 1999;31(1):61–74. doi: 10.1006/jmcc.1998.0844. [DOI] [PubMed] [Google Scholar]
- 24.Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation. 2002;105(5):546–549. doi: 10.1161/hc0502.104540. [DOI] [PubMed] [Google Scholar]
- 25.Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res. 1999;43:532–541. doi: 10.1016/S0008-6363(99)00094-2. [DOI] [PubMed] [Google Scholar]
- 26.Furchgott RF. The 1996 Albert Lasker medical research awards. The discovery of endothelium-derived relaxing factor and its importance in the identification of nitric oxide. JAMA. 1996;276:1186–1188. doi: 10.1001/jama.276.14.1186. [DOI] [PubMed] [Google Scholar]
- 27.Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev. 2001;22:36–52. doi: 10.1210/er.22.1.36. [DOI] [PubMed] [Google Scholar]
- 28.Tesfamariam B, Jakubowski JA, Cohen RA. Contraction of diabetic rabbit aorta caused by endothelium-derived PGH2-TxA2. Am J Physiol. 1989;257:H1327–H1333. doi: 10.1152/ajpheart.1989.257.5.H1327. [DOI] [PubMed] [Google Scholar]
- 29.Harrison DG. Endothelial function and oxidant stress. Clin Cardiol. 1997;20(11 Suppl 2):11–17. [PubMed] [Google Scholar]
- 30.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 31.Tesfamariam B, Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321–H326. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- 32.Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res. 2003;11:1278–1289. doi: 10.1038/oby.2003.174. [DOI] [PubMed] [Google Scholar]
- 33.Vriese AS, Verbeuren TJ, Voorde J, et al. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–74. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrews KL, Pannirselvam M, Anderson TJ, et al. The vascular endothelium in diabetes: a practical target for drug treatment? Expert Opin Ther Targets. 2005;9:101–17. doi: 10.1517/14728222.9.1.101. [DOI] [PubMed] [Google Scholar]
- 35.Baumgartner-Parzer SM, Wagner L, Pettermann M, et al. High-glucose-triggered apoptosis in cultured endothelial cells. Diabetes. 1995;44:1323–1327. doi: 10.2337/diabetes.44.11.1323. [DOI] [PubMed] [Google Scholar]
- 36.McGinn S, Saad S, Poronnik P, et al. High-glucose-mediated effects on endothelial cell proliferation occur via p38 MAP kinase. Am J Physiol. 2003;285:E708–E717. doi: 10.1152/ajpendo.00572.2002. [DOI] [PubMed] [Google Scholar]
- 37.Graier WF, Grubenthal I, Dittrich P, et al. Intracellular mechanism of high D-glucose-induced modulation of vascular cell proliferation. Eur J Pharmacol. 1995;294:221–229. doi: 10.1016/0014-2999(95)00534-X. [DOI] [PubMed] [Google Scholar]
- 38.Mozaffari MS, Abdelsayed R, Schaffer SW. Diabetic complications: pathogenic mechanisms and prognostic indicators. In: Golubnitschaja O, editor. Predictive diagnostics and personalized treatment: dream or reality? New York: Nova Science Publishers; 2009. [Google Scholar]
- 39.Kim JA, Montagnani M, Koh KK, et al. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–7904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 40.Bakker W, Eringa EC, Sipkema P, et al. Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 2009;335:165–189. doi: 10.1007/s00441-008-0685-6. [DOI] [PubMed] [Google Scholar]
- 41.Kofler S, Nickel T, Weis M. The role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clin Sci (Lond) 2005;108:205–213. doi: 10.1042/CS20040174. [DOI] [PubMed] [Google Scholar]
- 42.Arner P. The adipocyte in insulin resistance: key molecules and the impact of the thiazolidinediones. Trends Endocrinol Metab. 2003;14:137–145. doi: 10.1016/S1043-2760(03)00024-9. [DOI] [PubMed] [Google Scholar]
- 43.Raitakari OT, Celermajer DS. Testing for endothelial dysfunction. Ann Med. 2000;32:293–304. doi: 10.3109/07853890008995931. [DOI] [PubMed] [Google Scholar]
- 44.Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 45.Goligorsky MS. Clinical assessment of endothelial dysfunction: combine and rule. Curr Opin Nephrol Hypertens. 2006;15:617–624. doi: 10.1097/01.mnh.0000247497.62505.72. [DOI] [PubMed] [Google Scholar]
- 46.Münzel T, Sinning C, Post F, et al. Pathophysiology diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40:180–196. doi: 10.1080/07853890701854702. [DOI] [PubMed] [Google Scholar]
- 47.Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. 2007;3:853–876. [PMC free article] [PubMed] [Google Scholar]
- 48.Celermajer DS, Sorensen KE, Gooch VM, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–1115. doi: 10.1016/0140-6736(92)93147-F. [DOI] [PubMed] [Google Scholar]
- 49.Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. doi: 10.1016/0735-1097(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 50.Bots ML, Westerink J, Rabelink TJ, et al. Assessment of flow-mediated vasodilatation (FMD) of the brachial artery: effects of technical aspects of the FMD measurement on the FMD response. Eur Heart J. 2005;26:363–368. doi: 10.1093/eurheartj/ehi017. [DOI] [PubMed] [Google Scholar]
- 51.Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial artery reactivity task force; International Brachial artery reactivity task force. J Am Coll Cardiol. 2002;39:257–265. doi: 10.1016/S0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 52.Constans J, Conri C. Circulating markers of endothelial function in cardiovascular disease. Clin Chim Acta. 2006;368:33–47. doi: 10.1016/j.cca.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 53.Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag. 2005;1:55–71. doi: 10.2147/vhrm.1.1.55.58939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salomaa V, Stinson V, Kark JD, et al. Association of fibrinolytic parameters with early atherosclerosis. The ARIC study. Atherosclerosis risk in communities study. Circulation. 1995;91(2):284–290. doi: 10.1161/01.cir.91.2.284. [DOI] [PubMed] [Google Scholar]
- 55.Benameur T, Andriantsitohaina R, Martínez MC. Therapeutic potential of plasma membrane-derived microparticles. Pharmacol Rep. 2009;61:49–57. doi: 10.1016/s1734-1140(09)70006-4. [DOI] [PubMed] [Google Scholar]
- 56.Sabatier F, Camoin-Jau L, Anfosso F, et al. Circulating endothelial cells, microparticles and progenitors: key players towards the definition of vascular competence. J Cell Mol Med. 2009;13:454–471. doi: 10.1111/j.1582-4934.2008.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erdbruegger U, Haubitz M, Woywodt A. Circulating endothelial cells: a novel marker of endothelial damage. Clin Chim Acta. 2006;373:17–26. doi: 10.1016/j.cca.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 58.Werner N, Kosiol S, Schiegl T, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 59.Kalka C, Masuda H, Takahashi T, et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ Res. 2000;86:1198–1202. doi: 10.1161/01.res.86.12.1198. [DOI] [PubMed] [Google Scholar]
- 60.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 61.Maclauchlan S, Skokos EA, Agah A, et al. Enhanced angiogenesis and reduced contraction in thrombospondin-2-null wounds is associated with increased levels of matrix metalloproteinases-2 and -9, and soluble VEGF. J Histochem Cytochem. 2009;57:301–313. doi: 10.1369/jhc.2008.952689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee LC, Chen CS, Choong PF et al. Time-dependent dynamic mobilization of circulating progenitor cells during percutaneous coronary intervention in diabetics. Int. J. cardiology Jan 19. 2009; [Epub ahead of print]. [DOI] [PubMed]
- 63.Vöö S, Dunaeva M, Eggermann J, et al. Diabetes mellitus impairs CD133+ progenitor cell function after myocardial infarction. J Intern Med. 2009;265:238–249. doi: 10.1111/j.1365-2796.2008.02011.x. [DOI] [PubMed] [Google Scholar]
- 64.Hu CH, Li ZM, ZM DU, et al. Human umbilical cord-derived EPCs promote growth cytokines-mediated neorevascularization in rat myocardial infarction. Chin Med J (Engl) 2009;122:548–555. [PubMed] [Google Scholar]
- 65.Seandel M, Butler J, Lyden D, et al. Catalytic role for proangiogenic marrow-derived cells in tumor neovascularization. Cancer Cell. 2008;13:181–183. doi: 10.1016/j.ccr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao EH, Fukuda N, Matsumoto T, et al. Effects of the antioxidative beta-blocker celiprolol on endothelial progenitor cells in hypertensive rats. Am J Hypertens. 2008;21:1062–1068. doi: 10.1038/ajh.2008.233. [DOI] [PubMed] [Google Scholar]
- 67.Wu X, Wang K, Cui L, et al. Effect of granulocyte-colony stimulating factor on the repair of balloon-injured arteries. Pathology. 2008;40:513–519. doi: 10.1080/00313020802197947. [DOI] [PubMed] [Google Scholar]
- 68.Rousseau A, Ayoubi F, Deveaux C et al. Impact of age and gender interaction on circulating endothelial progenitor cells in healthy subjects. Fertil Steril Dec 24. 2008; [Epub ahead of print]. [DOI] [PubMed]
- 69.Xiao Q, Xu Q. Caspase-8: a double-edged sword for EPC functioning. Arterioscler Thromb Vasc Biol. 2009;29:444–446. doi: 10.1161/ATVBAHA.108.183087. [DOI] [PubMed] [Google Scholar]