

Abstract
Diabetic retinopathy (DR) is the foremost cause of blindness in working-aged worldwide; it is characterized by vascular and neuronal degeneration. Features of DR include leukocyte adhesion, increased vascular permeability, neovascularization and neuronal cell death. Early diagnosis and intervention are important to prevent or at least ameliorate the development of DR. Recent reports indicate that pathophysiological mechanisms leading to diabetic retinopathy include oxidative stress and retinal cell death cascades. Circulating biomarkers of oxidative stress such as malondialdehyde (MDA), thiobarbituric acid reacting substances (TBARS), conjugated diene (CD), advanced oxidation protein products (AOPP), protein carbonyl, 8-hydroxydeoxyguanosin (8-OHdG), nitrotyrosine, and F(2) isoprostanes and pro-apoptosis molecules (caspase-3, Fas, and Bax) are associated with increased susceptibility to develop DR in diabetic subjects. Thus, identification of oxidative stress and cell death biomarkers in diabetic patients could be in favor of predicting, diagnosis, and prevention of DR, and to target for novel therapeutic interventions.
Keywords: Diabetic retinopathy, Oxidative stress, Apoptosis, Biomarkers
Diabetic retinopathy is a vision-threatening disease characterized by neurodegenerative features associated with extensive vascular changes. It remains unclear how these pathologies relate to each other and their net contribution to retinal damage. There are multiple biochemical pathways which contribute to the development of the neurovascular injury in DR. As a result, biomarkers which reflect different pathways are released locally and into the circulation. Early identification of these biomarkers could be in favor of predicting and efficient management of DR. Among these biomarkers are the ones related to inflammatory response, oxidative stress and retinal cell death. Diabetes increases oxidative stress, which plays a key regulatory role in the development of its complications [1, 2]. Hyperglycemia-induced reactive oxygen species (ROS) generation is considered a causal link between elevated glucose and the pathways of development of diabetic complications [3]. Oxidative stress may lead to cell death [4] via apoptotic means. Apoptosis of retinal neurons particularly ganglion cells [5–7] has been demonstrated in diabetic retinopathy as demonstrated by profound retinal abnormalities, evaluated by electroretinography, and potential visual changes evoked before the onset of the first vascular change is detectable in the diabetic retina [8, 9]. Retinal capillary cells also undergo accelerated apoptosis, which precedes the detection of any histopathological changes characteristic of diabetic retinopathy [10]. The current review discusses the markers of oxidative stress and retinal cell death associated with DR.
Oxidative stress and diabetic retinopathy
Chronic hyperglycemia plays a crucial role in the pathogenesis of diabetic retinopathy (DR). The mechanism of hyperglycemia-induced retinal damage is still to be elucidated. However, the oxidative stress which represents an imbalance between excess generation and/or impaired removal of reactive oxygen species (ROS) have been suggested to be the key events in the pathogenesis of diabetic retinopathy. The high content of polyunsaturated fatty acids, oxygen uptake and glucose oxidation make the retina more susceptible to oxidative stress than other tissues [11]. ROS are produced continuously in all cells to support normal cellular functions. However, excess production of ROS, or inefficient removal of ROS, could result in pathological conditions. In addition to ROS, reactive nitrogen species (RNS) are also a part of normal physiological function, and have great potential to contribute to oxidative stress [12]. In the presence of superoxide, nitric oxide spontaneously forms peroxynitrite. Peroxynitrite is much more reactive than superoxide and nitric oxide and can exert direct oxidative modifications through one- or two- electron oxidation processes [13, 14]. Thus, excessive abundance of ROS and RNS with concurrent dysfunction of antioxidant defense systems, which includes reducing enzymes such as superoxide dismutase (SOD), catalase and glutathione peroxidase (GSH) contributes to oxidative stress in diabetic retina. Chronic oxidative stress causes damage to DNA, lipids, proteins, and carbohydrates and disruption in cellular homeostasis resulting in many disease processes of clinical interest [15]. Accumulation of damaged molecules and ROS that are not easily removed contributes not only to the pathogenesis of DR but also to the resistance of retinopathy to reverse even after good glycemic control is reestablished- the metabolic memory phenomenon [16].
Sources of ROS in diabetic retina
Chronic exposure of retinal cells to hyperglycemia causes overproduction of ROS by activation of different enzymatic pathways which more likely interact to create the retinal damage seen in DR (Fig. 1). These sources include but not limited to NADPH oxidase, mitochondrial electron transport chain (ETC), formation of advanced glycation end products (AGEs), aldose reductase/polyol pathway, protein kinase C (PKC) activation and uncoupling of endothelial nitric oxide synthase (eNOS).
Fig. 1.
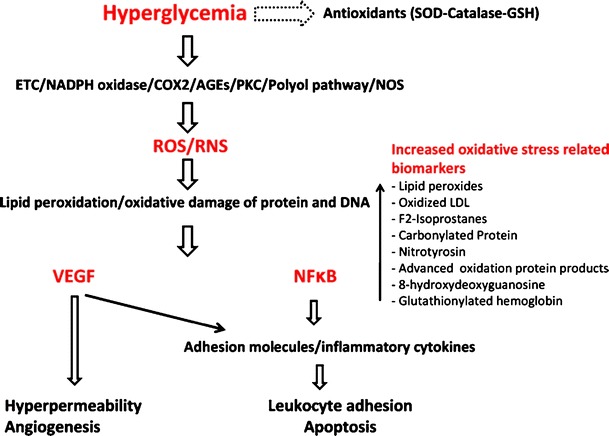
Hyperglycemia inhibits antioxidant enzymes (superoxide dismutase (SOD), catalase, glutathione (GSH)) and induces generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) from different sources such as mitochondrial electron transport chain (ETC), NADPH oxidase, cycloxygenase (COX2), advanced glycation end products (AGEs), protein kinase C (PKC), polyol pathway and nitric oxide synthase (NOS). Hyperglycemia-induced generated ROS and RNS, cause lipid peroxidation and oxidative damage of protein and DNA resulting in production of biomarkers such as lipid peroxides and nitrotyrosin. Lipid and protein modification by hyperglycemia-induced oxidative stress leads to upregulation of VEGF and activation of NFκB. VEGF and NFκB-dependent signaling pathway causes retinal vascular and neuronal damage associated with diabetic retinopathy such as hyperpermeability, leukostasis, apoptosis and angiogenesis
NADPH oxidase
Recent studies demonstrated that NADPH oxidase is a major source of ROS in diabetic patients and animals and retinal endothelial cells treated with high glucose [17–20]. ROS production by NADPH oxidase has been linked to the early signs of DR, including increases in ROS generation, vascular endothelial growth factor (VEGF) expression, leukostasis and vascular permeability[17, 21]. Additionally, increased NADPH oxidase activity and expression of its catalytic subunit NOX2 has been correlated to increased VEGF expression and retinal neovascularization in a mouse model for ischemic retinopathy [22]. Phagocyte NADPH oxidase consists of two membranous subunits, large catalytic subunit gp91phox (NOX2) and small p22phox as well as cytosolic regulatory subunits, p40 phox, p47phox and p67phox, and the low molecular weight G protein Rac-1 [23, 24]. Phosphorylation of p47phox is crucial for the assembly of the complex [25], while P67phox subunit mediates binding of the complex with activated Rac [26], both are critical for NADPH oxidase activation. Vascular endothelial cells express the same subunits as well as two NOX2 homologues, NOX1 and NOX4 [20]. Previous studies in animal and tissue culture models have shown that NOX2 is expressed at low levels in normal retinas and in retinal endothelial cells maintained under control conditions, but is substantially increased in retinal vessels of animals with diabetic or ischemic retinopathy and in retinal endothelial cells exposed to high glucose or hypoxia [17, 22]. During diabetes other cells in addition to endothelial cells become activated and become sources of NADPH oxidase derived ROS formation. For example, leukocytes are suggested to regulate retinal edema, ischemia and angiogenesis [27] and shown to be activated [28–30]and to become source of oxidative stress [31, 32].
Mitochondrial Electron Transport Chain (ETC)
Mitochondria are the principal endogenous source of superoxide. Hyperglycemia disrupts the normal ETC electron flow leading to electron leak and superoxide generation which induces mutations in mitochondrial DNA resulting in defective subunits of the electron transport complexes causing increased superoxide production at physiological concentrations of glucose [2, 33]. Moreover, mitochondrial ROS suppresses the antioxidant defense mechanisms through inhibition of SOD and GSH leading to enhanced sensitivity of retinal cells to oxidative stress since they cannot scavenge ROS effectively [34–36]. The critical role of mitochondrial superoxide in DR has been demonstrated by recent studies using transgenic mice that overexpress mitochondrial SOD. Overexpression of mitochondrial SOD protected the diabetic retina from oxidative damage and capillary degeneration [36]. This indicates that hyperglycemia-induced oxidative stress can modulate mitochondria function, thus contributing to the development of DR.
Endothelial Nitric Oxide Synthase (eNOS)
eNOS uncoupling is an additional source of superoxide generation in diabetic retina. This occurs, when there is limited amount of its substrate L-arginine or its co-factor tetrahydrobiopterin. Uncoupled eNOS use molecular oxygen as a substrate producing superoxide instead of nitric oxide (NO) [37–39]. Several studies demonstrated the role of eNOS uncoupling in vascular dysfunctions associated with diabetes which reversed by supplemental L-arginine treatment [40, 41]. The role of eNOS in DR has been also demonstrated in human as certain eNOS polymorphism are associated with DR [42].
Polyol pathway
The polyol pathway involves the conversion of glucose into sorbitol by aldose reductase. Sorbitol is then oxidized to form fructose by sorbitol dehydrogenase. Increased polyol pathway in diabetes causes depletion of NADPH. Since NADPH is required for regenerating the intracellular antioxidant, GSH, increased polyol pathway in diabetes enhances oxidative stress [43]. The role of this pathway in the development of DR has been reported in animal model of experimental diabetes and high glucose-treated endothelial cells [44, 45].
Advanced glycation end products
The AGEs are produced from strong glycating dicarbonyl compounds such as methylglyoxal and glyoxal [46]. Chronic hyperglycemia favors glycation reactions and nonenzymatic glycation leading to cross-linking of proteins and in turn alterations in their function, activity, and physical properties. Diabetes, increases production of AGE and its receptor, RAGE, in the retinal microvasculature which becomes irreversible In the late stages of DR [47]. Interaction of AGEs with RAGE activates PKC-δ and subsequently NADPH oxidase to enhance oxidative stress in diabetic retina [48]. ROS generation by AGEs leads to the activation of nuclear transcriptional factor, NF-kB, [49] which has been linked to retinal capillary cell apoptosis [50] vascular inflammation [51, 52] and neovascularization [53, 54].
Protein kinase C activation
Activation of PKC is also considered as a major pathway implicated in the pathogenesis of diabetic retinopathy [55, 56]. High glucose levels increase the release of ROS and the synthesis of diacylglycerol (DAG) increasing the activity of PKC [57]. Activated PKC has been implicated in changes characteristic of diabetic retinopathy such as increased vascular permeability, neovascularization, endothelial proliferation and apoptosis, and regulating the action of VEGF [58–60]. There are cumulative evidences supporting the role of PKC in ROS-mediated diabetic complications. For example, diabetes-induced oxidative stress has been shown to be prevented by PKCβ specific inhibitor (LY53331), and in mice lacking PKCβ isoform [61–63].
Oxidative stress and vascular changes in diabetic retinopathy
Diabetic retinopathy is associated with alterations in the structure and function of retinal microvasculature. For example, basement membrane thickening, pericyte and endothelial cell loss, acellular capillary formation [64, 65], microaneurysm, altered blood flow, leukocyte adhesion, hyperpermeability and neovascularization. Many of these changes have been linked to the oxidative stress-induced biochemical changes. ROS mediate these changes by both direct and indirect mechanisms which include increases in the transcriptional activation of VEGF expression [66], inflammatory mediators [67, 68], AGEs formation on collagen which results in basement membrane thickening [69] and modulation of vasoactive effector molecules such as endothelin-1 and NOS which contribute to changes in blood flow and vascular permeability [70, 71]. VEGF, a proangiogenic factor, is known to play a pivotal role in the development of vascular changes associated with diabetic retinopathy [72, 73]. Hyperglycemia-induced oxidative stress plays a role in mediating both the expression and pathological effect of VEGF [17, 74, 75]. For example, our subsequent studies demonstrated the implication of NADPH oxidase-derived ROS in the development of vascular dysfunctions in ischemic retinopathy. We reported abrogation in ROS formation, retinal expression of VEGF and new vessel formation in mouse model of ischemic retinopathy when treated by NADPH oxidase inhibitor [22]. Further studies on experimental diabetes demonstrated decreased ROS formation, VEGF expression leukostasis, and vascular permeability by NADPH oxidase inhibitor or deletion of its catalytic subunit, NOX2, [21]. Other studies have reported the beneficial effect of antioxidants such as N-acetylcysteine in preventing pericyte loss and endothelial/perivascular cell changes occur early in the pathogenesis of DR [76]. Moreover, PEDF, an essential antiangiogenic factor that produced by different retinal cells has been reported to prevent neuronal derangements, vascular hyperpermeability and leukostasis and VEGF expression in diabetic retinopathy via inhibition of ROS formation [77]. Taken together, these findings confirm the crucial role of ROS in the development of vascular dysfunction in DR.
Markers of oxidative stress
Oxidative stress induces biochemical changes in the serum and target organs of diabetic patients including severe lipid peroxidation, protein oxidation, and oxidative DNA damage and changes in their antioxidant status (Fig. 1). Thus, identification of the by-products induced by oxidative stress in serum, vitreous body, aqueous humor, fibrovascular tissue on the retinal surfaces and urine of diabetic patients could be in favor of predicting, diagnosis and prevention of microvascular complications of diabetes such as retinopathy and nephropathy. Oxidative stress parameters include malondialdehyde (MDA), thiobarbituric acid reacting substances (TBARS), conjugated diene (CD), advanced oxidation protein products (AOPP), protein carbonyl, 8-hydroxydeoxyguanosin (8-OHdG), nitrotyrosine, and F(2) isoprostanes. Several studies demonstrated a marked increase in these products in serum/retinas of diabetic patients or experimental animals and significantly higher in patients or animals with DR than in diabetic patients/animals without DR. Additionally, amelioration of the antioxidant status plays a crucial role in development of DR.
Lipid peroxidation
Lipid peroxidation of cellular structures, an oxidative stress-induced activity, is thought to play an important role in complications of diabetes mellitus, especially in diabetic retinopathy. Measurement of TBARS, MDA or CD provides a convenient assessment of lipid hydroperoxide (LHP) concentration in serum. CD is the initial formation of lipid peroxide while MDA is a decomposition product of peroxidized polyunsaturated fatty acids. The elevated levels of Poly Unsaturated Fatty Acids (PUFAs) and increased rate of glucose oxidation in diabetes make the retina susceptible for oxidative stress and lipid peroxidation. Increased lipid peroxides has been demonstrated in serum of diabetic patients with vascular complications [78–85]. Lipid peroxidation increases also in retinas of diabetic animals and mouse model of ischemic retinopathy [17, 22]. Compared with healthy subjects, diabetic patients demonstrate significantly higher levels of lipid peroxidation products [86–90]. Additionally, patients with features of DR are found to have much more lipid peroxidation products compared with those without DR [85, 86]. Studies by Armstrong et al., demonstrated a statistically significant correlation between the increasing serum lipid peroxidation levels and prevalence of retinopathy in diabetic patient [91] and reduction in retinal cell number in streptozotocin (STZ)-induced diabetic rat model [92]. Similar results have observed by Augustin et al. [93, 94] in patients with PDR where they demonstrated a significant increase in lipid peroxidation in the vitreous and epiretinal membranes.
In addition to the previous markers of lipid peroxidation several studies have demonstrated the role of oxidative stress-induced modification of low density lipoprotein in the pathogenesis of DR [95–97]. Fredrikson et al. [98] reported that patients affected by retinopathy had significantly higher levels of autoantibodies against apolipoprotein B peptides than patients without retinopathy. Thus, autoantibodies against oxidized LDL antigens might also reflect disease severity and the risk of developing vascular complication of diabetes including DR.
A major step regarding the measurement of lipid peroxidation products came with the discovery of F2-isoprostanes [99, 100]. The F2-isoprostanes are a family of PG (prostaglandin)-like compounds produced by non-enzymatic peroxidation of arachidonic acid. Of these, 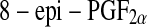 (also known as
(also known as 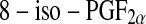 or 15-F2t-isoprostane) has received most attention because it has been shown to possess certain adverse biological activities [101–104]. Circulating
or 15-F2t-isoprostane) has received most attention because it has been shown to possess certain adverse biological activities [101–104]. Circulating 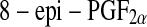 is mainly present bound to phospholipids in situ and is released by the action of phospholipase A2 [105] and has proved a reliable marker of oxidative stress in human body. However, the quantification of urinary
is mainly present bound to phospholipids in situ and is released by the action of phospholipase A2 [105] and has proved a reliable marker of oxidative stress in human body. However, the quantification of urinary 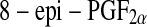 has been proposed as being superior to the measurement of circulating
has been proposed as being superior to the measurement of circulating 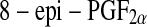 levels, as it is believed to represent a more accurate systemic index of oxidative stress [106]. In their study on human retinas obtained from healthy donors Nourooz-Zadeh and Pereira provided the first direct evidence for free-radical-catalyzed peroxidation of AA in retinal tissue by detecting esterified F(2)-IPs in human retina. Thus they proposed analysis of F(2)-IPs for studying the implications of oxidative injury in various diseases of retina, including diabetic retinopathy and age-related macular degeneration [107].
levels, as it is believed to represent a more accurate systemic index of oxidative stress [106]. In their study on human retinas obtained from healthy donors Nourooz-Zadeh and Pereira provided the first direct evidence for free-radical-catalyzed peroxidation of AA in retinal tissue by detecting esterified F(2)-IPs in human retina. Thus they proposed analysis of F(2)-IPs for studying the implications of oxidative injury in various diseases of retina, including diabetic retinopathy and age-related macular degeneration [107].
Biomarkers of DNA and protein oxidative damage
8-hydroxydeoxyguanosine (8-OHdG)
ROS can cause DNA damage and base modifications. 8-OHdG, an oxidized nucleoside of DNA, results from oxidation of guanine residues and is frequently used as DNA lesion marker. Higher levels of 8-OHdG were observed in the mononuclear cells, urine, pancreatic islet and mitochondrial DNA from diabetic patients [108–112] and in kidney tissues of streptozotocin-induced diabetic rats [113]. Moreover, the content of 8-OHdG in the urine, serum and mononuclear cells of patients with type 2 diabetes with either retinopathy or nephropathy were much higher than those in patients without complications. [86, 114, 115]. Therefore, the contents of 8-OHdG in serum could act as a sensitive biomarker for the diabetic retinopathy.
Carbonylated proteins
Proteins can also be modified and become cytotoxic by ROS and RNS, thus contributing to the development of DR. There is association between the elevated biomarkers of protein oxidation with aging and different diseases such as Alzheimer’s, diabetes mellitus and cancer [86, 116, 117]. Protein carbonylation and nitration have attracted a great deal of attention due to its irreversible and unrepairable nature which may serve as an important biomarker in DR.
Carbonylated proteins are marked for proteolysis by the proteasome but can escape degradation and form high-molecular-weight aggregates that accumulate with age. Such carbonylated aggregates can become cytotoxic and have been associated with a large number of diseases and age-related disorders, including Parkinson’s disease, Alzheimer’s disease, cancer, cataractogenesis, diabetes, and sepsis [118, 119], for review see [120]. The implication of protein carbonylation in reflecting the severity of diabetes and its complication has been investigated by many researchers [86, 121, 122]. There is a positive correlation between protein carbonylation and the clinical severity of DR. For example, compared to healthy individuals diabetic patients have significantly increased levels of carbonylated proteins. Furthermore, patients with DR show higher level of protein carbonylation more than in diabetic patients without DR [86, 123].
Protein nitration
Nitrotyrosin is additional marker for oxidative damage of protein that disrupts its normal functions. Several studies have demonstrated that diabetes increases protein nitration in retinas of experimental animals [17, 124, 125]. Nitrotyrosine is an oxidative protein modification which formed from the reaction of free or protein-bound tyrosine with reactive nitrogen species such as free radical nitrogen dioxide [126] and peroxynitrite [127] and has been used as a marker of oxidative stress in retina of experimental animals [17, 124]. Although tyrosine nitration has been associated with development of diabetic and ischemic retinopathies little is known about the targets of tyrosine nitration in retina and the role nitration may play in the pathophysiology of diabetic retinopathy. Recently, Zhan et al. identified some proteins that are nitrated in diabetic rat retina and in rat retinal Müller cells grown in high glucose which could be linked to retinal neovascularization[125]. Additionally, subsequent studies by El-remessy et al., have shown that peroxynitrite-induced tyrosine nitration correlates with accelerated retinal endothelial cell death, breakdown of the brain-retinal barrier, and accelerated neuronal cell death in models of experimental diabetes and neurotoxicity and human diabetes [124, 128–130]. Recent reports demonstrated that accumulation of nitrosylated protein in peripheral blood monocyte of diabetic patients is an emerging marker that reflects oxidative stress-induced protein damage. [131, 132]. Julius et al., 2009 reported that diabetic subjects accumulate significant amount of nitrosylated protein in peripheral blood monocyte and this is positively correlated with the severity of diabetic microangiopathy such as nephropathy and retinopathy [132]. Thus, targeting tyrosine nitration represents potentially effective therapeutic strategy in attenuating retinal vascular neurodegenerative diseases.
Advanced oxidation protein products (AOPP)
Serum level of AOPP is also a novel protein oxidation marker which has been described by different investigators [133–136]. They are elevated in patients with renal insufficiency and diabetes mellitus [137–139]. Additionally, the level AOPP in diabetic patients with features of DR was reported to be significantly higher than in diabetic patients without DR [86, 136].
In addition to the previous markers of oxidative damage of protein and DNA in diabetic patient, mass spectrometric quantification of amino acid oxidation products and glutathionylated hemoglobin has been proposed to serve as markers for the oxidative stress in diabetic patients, in particular those with complications such as DR [140, 141].
Antioxidant status
The antioxidant status of diabetic patient plays a crucial role in producing oxidative stress and development of vascular complications. ROS generation is inhibited by an intracellular enzymatic system composed mainly of glutathione peroxidase (GPx), SOD and catalase (CAT). All serum antioxidants are usually measured together as total antioxidant status (TAS). The reports about the status of antioxidants and antioxidant enzymes in diabetic patients are very contradictory, both increases and decreases of antioxidant activity have been reported [142–145]. Generally, TAS has been shown to be significantly lower in patients with proliferative retinopathy than in diabetics not developing retinopathy [146]. Antioxidants SOD and vitamin C decrease with the progression of the disease, however GPx tends to increase in the later part of the disease [147]. Kesavulu et al., in their study they found that among the erythrocyte antioxidant enzymes, CAT activity was significantly increased in all the diabetics compared to controls, however, there was no difference in the CAT activity between the diabetic patients with or without microvascular complications (MVC) such as diabetic retinopathy or nephropathy. In contrast, GPx activity was significantly decreased in the diabetic patients compared to controls, and further decreased in the diabetics with MVC compared to those without the complications. The SOD activity in diabetic patients was not much different from that of non diabetic controls, but a significant decrease in SOD activity was observed in diabetic patients with MVC compared to those without these complications [85]. The total thiol level as a marker of antioxidant status in diabetics has also shown to be significantly decreased in patient with DR than those without DR [136]. An inverse relation between the level of GSH and HbA1c was also reported in diabetic patients indicating reduction in the antioxidant status in poorly controlled diabetics [141]. Additionally, nonenzymic antioxidants such as vitamin C, vitamin E, and β-carotene that exist biologically for the regulation of redox homeostasis are also depressed by diabetes [148]. Therefore, the level and activity of specific antioxidant enzymes compared to other enzymes that do not match to development of DR could be predictive for the susceptibility to develop DR in diabetic patients.
Oxidative stress related gene polymorphism
There are several studies showing that vascular complications of diabetes develop only in genetically susceptible patients [149–151] and hyperglycemia by itself is not sufficient for the development of diabetic vascular disease. There is tendency to have familial clustering of diabetic nephropathy and coronary vascular disease (CVD). Additionally, there is a significant ethnic difference in the incidence of CVD in subjects with diabetes that could not be explained by differences in conventional cardiovascular risk factors (UK Prospective Diabetes Study Group 1998). Various candidate genes that affect the oxidative stress such as manganese mitochondrial superoxide dismutase (Mn-SOD), endothelial nitric oxide synthase (eNOS), angiotensin-converting enzyme, glutathione peroxidase, superoxide dismutase and haptoglobin demonstrate functional polymorphisms which correlated with the increased susceptibility to vascular diseases and diabetes [152–156].
Manganese superoxide dismutase (MnSOD) polymorphism
Hovnik et al, 2009 investigated polymorphic markers in genes encoding enzymes regulating production of reactive oxygen species in association with diabetic retinopathy (DR) or diabetic nephropathy (DN) in one hundred and twenty four patients with type 1 diabetes and matched for sex, age and duration of diabetes. A positive association of MnSOD genotype Val/Val with diabetic retinopathy but not with diabetic nephropathy was demonstrated [157]. Moreover, the association of diabetic retinopathy with single nucleotide polymorphisms (SNPs) on MnSOD was investigated and a statistically significant association of MnSOD Ala16Val polymorphism with diabetic retinopathy was found compared with the diabetic control. Additionally, the Ala(-9)Val [156] and VV genotype of the V16A [155] polymorphism of the Mn-SOD gene are also reported to be significantly higher in patients with DR retinopathy in Chinese and Caucasians type 2 diabetic patients respectively.
Haptoglobin polymorphism
Haptoglobin (Hp) is a hemoglobin-binding serum protein. It is found at levels of 30–300 mg/dL in normal human serum [158]. However, serum level of Hp shows 3- to 8-fold increase during the acute phase reaction and in response to injury [159]. Free hemoglobin (Hb) is considered as an extremely potent pro-oxidant agent which catalyzes various oxidative and peroxidative reactions [160]. Haptoglobin (Hp) plays an essential role in capturing free Hb, thus preventing oxidative tissue damage mediated by free Hb. For example, Hp has been demonstrated to inhibit Hb-induced linolenic acid and low-density lipoprotein (LDL) oxidation [161]. Additionally, Hp knockout mice have shown to be more prone to oxidative tissue damage such as renal damage and endothelial dysfunction [162]. Recent reports demonstrated that a polymorphism in the haptoglobin (Hp) gene is an independent risk factor for diabetic vascular disease [153, 154]. In man, there are two alleles for Hp, denoted 1 and 2, giving rise to three major phenotypes. Individuals homozygous for the 1 allele express the Hp1-1 phenotype at the protein level. Individuals homozygous for the 2 allele express the Hp2-2 phenotype, whereas heterozygotes express the Hp2-1 phenotype. Nakhoul et al., 2000 have observed a marked decrease in the prevalence of diabetic retinopathy in patients with Hp1-1 phenotype compared with patients with the Hp2-1 and Hp2-2 phenotypes [163]. Hp1-1and Hp1-2 types have been found to have significantly higher levels of serum vitamin C [164], lower serum iron, and lower circulating oxidized LDL (oxLDL)/LDL ratios compared with those with the Hp2-2 [165, 166]. Thus, antioxidant therapy may be more beneficial in diabetic patients with the Hp2-2 phenotype as compared with those with the Hp1-1 phenotype. Furthermore, a highly significant increase in retinal capillary basement membrane thickness was observed in mice with streptozotocin-induced diabetes with the Hp 2 genotype. These data provide important support for association studies done in humans showing an increased prevalence of diabetic retinopathy in individuals with the Hp 2 genotype [167].
eNOS gene polymorphism
Polymorphisms of eNOS gene have been reported to be associated with multiple health conditions including DR, hypertension, nephropathy, and cardiovascular diseases in several ethnic groups. Several studies demonstrated the association between certain eNOS polymorphism with DR in human [42, 168, 169]. For example, the eNOS 4b/b [42] or 4a/b [168] polymorphism demonstrate increased eNOS expression and activity and associated with sever DR. On the other hand, homozygous deletion of eNOS 4a/a causes reduction in eNOS expression and activity and susceptibility to DR [170].
Inflammation related markers
In addition to oxidative stress related biomarkers, generation of ROS in diabetic patients is known to initiate a cascade of signaling pathways responsible for the inflammatory component of DR. This includes activation of NFκB and up-regulation of genes responsive to inflammatory processes such as adhesion molecules [21, 52, 171, 172]. Golubnitschaja et al., 2005 have studied the alteration in inflammatory related genes in mononuclear blood cells (MBCs) isolated from diabetic patients with and without active retinopathy. They found significant increase in the expression of recoverin in diabetic patients compared to the control group. “Real-Time”-PCR showed that recoverin levels were positively correlated with the stage of DR and with the activity of serum matrix metalloprotinases (MMPs) [173]. Thus, identification of inflammation related biomarkers in isolated MBCs and serum of diabetic subjects may be helpful for the prediction of active proliferative retinopathy in diabetics.
Neuronal death and diabetic retinopathy
The preceding section of this chapter describes the complex vascular changes that accompany diabetic retinopathy. For many years, the disease was considered primarily vascular in nature. Beginning in the late 1990s, however, information about the neuronal components of diabetic retinopathy emerged. Several excellent reviews published in the last decade have summarized the evidence that neurons, particularly retinal ganglion cells, are vulnerable and die in diabetic retinopathy [5–7, 174, 175]. Loss of color [176, 177] and contrast [178, 179] sensitivity are early signs of neural retinal dysfunction in humans that occur within 2 years of diabetes onset. Bresnick proposed that diabetic retinopathy is a primary neurosensory disorder that precedes vasculopathy by many years [8, 180]. It is noteworthy that as early as the 1960’s Wolter [181] and Bloodworth [182] described loss of retinal neurons in diabetic patients. However, owing to the more easily recognizable vascular features as viewed via ophthalmoscopy, these observations were largely overlooked. More sophisticated electrophysiological studies using macular or focal ERG (FERG), which can detect electrical responses of ganglion cells, showed ganglion cell dysfunction early in diabetes [183]. Though FERG is not used widely clinically, important information has been forthcoming from several studies. Greco et al. [184] used FERG in prepubescent diabetic children and found that ganglion cell function was significantly impaired, while other cells such as photoreceptors appeared unaffected. Others have reported similar findings, including ganglion cell dysfunction in IDDM patients without vasculopathy [185, 186]. Other investigations using ERG, dark adaptation, contrast sensitivity, and color vision tests have provided convincing functional evidence that neural retina function is compromised oftentimes before the onset of vascular lesions in humans [187–192].
Markers for apoptosis in human tissues
The relevance of this functional evidence of neuronal degeneration in diabetic retinopathy has been borne out by molecular and biochemical analyses of the retinas of human patients. Barber et al. used post-mortem tissues from human patients with diabetic retinopathy and reported for the first time that ganglion cells die in this disease and do so via an apoptotic mechanism [193]. Subsequent studies by Abu El-Asrar used eyes from human diabetic patients and confirmed the expression of several pro-apoptosis molecules (caspase-3, Fas, and Bax) emphasizing the vulnerability of these cells [194]. They also found strong immunoreactivity for ERK1/2 in the inner nuclear layer of diabetic retinas as well as the nerve fiber and ganglion cell layers. This group extended their findings by examining additional anti- and pro-apoptotic markers in humans and showed that ganglion cells in diabetic and non-diabetic retinas express the anti-apoptotic molecules Cox-2, Akt, and Mcl-1. Retinal ganglion cells express the proapoptotic molecule Bad in response to diabetes-induced neuronal injury; diabetic retinas also show upregulation of the mitochondrial proteins cytochrome c, and AIF (Fig. 2) [195]. Human tissue from diabetic patients has also been shown to express connective tissue growth factor (CTGF), CD105 and gelatinase B, and ganglion cells and microglia were the sites of immunoreactivity [196]. Other investigators have provided additional support that the death of neurons in human diabetic retinas is mediated via mitochondria- and caspase-dependent cell-death pathways [197].
Fig. 2.

Table depicting biomarkers of neuronal death that have been reported in human patients with diabetic retinopathy and in rodent models of this disease
These data collected from human tissues provide strong evidence that neurons die in diabetic retinopathy and that they do so via an apoptotic mechanism, however no comprehensive reports are available indicating specific markers that predict neuronal cell death in human subjects. By definition, biomarkers should reflect a change in expression or state of a protein that correlates with the risk or progression of disease, or with the susceptibility of the disease to a given treatment. The field of biomarker research is just emerging and the published reports on biomarkers for neuronal cell death, especially in humans with diabetic retinopathy, are limited.
Model systems to investigate biomarkers of neuronal death in diabetic retinopathy
To understand genes/proteins whose expression is changed in diabetic retinopathy, investigators have relied extensively on the use of model systems including in vivo animal models and in vitro systems. Much of the work has focused on ganglion cells since they are vulnerable in human diabetic retinopathy. Schmidt and colleagues postulate that ganglion cells are particularly susceptible to neurodegenerative disease because of their high metabolic rate [198]. They note that ganglion cells have very long axons extending from the cell soma, through the lamina cribrosa (LC) and beyond. Moreover, unlike many neurons they are unmyelinated from the retina to the LC and only thereafter become myelinated. This reversed myelination pattern leads to an impedance mismatch that consumes considerable energy.
Studies in diabetic rats
Animal models of diabetic retinopathy include the well-characterized streptozotocin-induced diabetic rat. In rats, diabetes accelerates retinal neuronal cell death and inhibits neurite regeneration via an apoptotic mechanism that involves upregulation of Bax, a pro-apoptotic gene [199]. In the rat model, a number of inflammatory markers have been shown to be upregulated concomitant with ganglion cell death. These markers include interleukin (IL)-18 [200], IL-1β [124, 200], vascular endothelial growth factor (VEGF) [124], tumor necrosis factor-α (TNF-α) [124, 200], activated p38 MAP kinase [124]. Related to these observations are the data showing a significant ∼30% increase in glucosylceramide levels in diabetic rats compared to controls [201]. These investigators were the first to demonstrate that augmented glycosphingolipid metabolism may contribute to the neuronal pathology of diabetic retinopathy and may serve as an important and relevant biomarker in human patients. Nakazawa et al have shown that anti-inflammatory agents (statins) can prevent ganglion cell death in ischemic/reperfusion rat models [202]. Zhang and co-workers observed an increase in the receptor for erythropoietin, a glycoprotein hormone that controls erythropoiesis in diabetic rats [203]. Another interesting molecule whose expression is decreased in the diabetic rat retina is SERPINA3K, an extracellular serine proteinase inhibitor (Fig. 2) [204]. This observation is noteworthy because SERPINA3K prevents intracellular calcium overload and calcium is well known to trigger neuronal death. Several groups have demonstrated that insulin mediates a pro-survival pathway in retinal neurons. It has been demonstrated that insulin receptor signaling diminishes with increased diabetes-induced apoptosis [205]. Very recently Fort and colleagues have applied complementary proteomic and genomic techniques and discovered that the proteins of the crystallin superfamily are increased dramatically in early diabetic retinopathy [206]. Their data suggest that the changes observed in crystallins are greater than other changes described so far in diabetic retinopathy. The work also emphasizes the power of the ‘omic’ approaches (genomic, proteomic, etc) to uncovering biomarkers for diabetic retinopathy.
As data are accumulated about molecules whose expression changes in diabetic retinopathy it is tempting to target them as a means to protect against deleterious consequences of diabetes. However, caution must be exercised regarding this strategy. A case in point is VEGF. VEGF plays key role in vasculogenesis and has been an anti-angiogenic target in treating a number of blinding diseases [207]. Recently VEGF-A has been recognized as an important neuroprotectant in the central nervous system [208]. Nishijima and co-workers used a model of ischemia-reperfusion injury and found that VEGF-A exposure resulted in a dose-dependent reduction in retinal neuron apoptosis [209]. Indeed, chronic inhibition of VEGF-A function in normal adult animals led to a significant loss of retinal ganglion cells yet had no observable effect on several vascular parameters. Their findings have important implications for both neural pathologies and ocular vascular diseases, particularly diabetic retinopathy.
Studies in diabetic mice
Mice constitute a powerful model system for understanding disease mechanisms, particularly the influence of genetic mutations on the progression of pathology. The field of diabetic retinopathy is no exception and a number of mouse models have proven informative with respect to gene/protein changes that occur in this disease. One of the most useful models is the Ins2Akita/+ mouse model, which has proven relevant to and useful for studies of diabetic retinopathy [210–214]. The Ins2Akita/+ mouse has a point mutation of the Insulin2 gene leading to hypoinsulinemia and hyperglycemia in heterozygous mice by ∼4 weeks [215]. Homozygosity for the mutation is embryonically lethal. Over a period of several weeks (∼10–25 weeks), heterozygous mice demonstrate marked retinal alterations including ∼20–25% reduction inner plexiform layer thickness, a ∼16% reduction of inner nuclear layer thickness and a ∼25% reduction in the number of cell bodies in the RGC layer [211, 212, 214]. The cell death appears to occur via apoptosis as evidenced by data from TUNEL assays [214] and detection of active caspase 3 [211]. Oshitara and colleagues have utilized the Ins2Akita/+ mouse to examine the role of ER stress in diabetic retinopathy [174]. ER stress refers to the phenomenon whereby misfolded proteins aggregate in the ER lumen activating the unfolded protein response (UPR). The aim of the UPR is to alleviate ER stress, restore ER homeostasis and prevent cell death. Activation of the response is mediated by GPR78 (BiP, binding protein), which dissociates from three resident proteins: inositol requiring ER-to-nucleus signal kinase (IRE)1, activating transcription factor (ATF) 6 and soluble-stranded RNA-activated kinase (PKR)-like ER kinase (PERK) [216]. Studies of the Ins2Akita/+ mouse demonstrated that ER stress is involved in the death of both retinal neurons and vascular cells in diabetic eyes as manifested by increased expression of BiP as well as PERK, ATF6, IRE1 (Fig. 2) [174]. These data are particularly interesting to our group because of our work showing that (+)-pentazocine, a ligand for sigma receptor 1 (σR1), affords profound neuroprotection in the Ins2Akita/+ mouse [214]. σR1 is a molecular chaperone protein that resides at the ER-mitochondrial membrane and interacts with BiP. We are currently examining the expression of ER-stress related genes and our preliminary data suggest that expression of PERK, ATF6 and IRE1 as well as downstream effectors CHOP, BCL2 are increased in the diabetic retina, but expression returns to normal upon sustained treatment with (+)-pentazocine (manuscript in preparation). ER stress genes/proteins may prove to be important additional biomarkers of neuronal death in diabetic retinopathy and reducing or blocking ER stress may be a potential therapy for preventing the onset and the progression of this disease.
Other mouse models, in which diabetes occurs endogenously, have demonstrated neuronal degeneration as well. The retinas of hyperglycemic, hyperlipidemic (HGHL, apolipoprotein E(−/−) db/db) have been examined for advanced glycation endproducts (AGE) and receptors for these endproducts (RAGE) [217, 218]. The mice demonstrate early inner retinal neuronal dysfunction, manifested by prolonged latencies of the oscillatory potentials and b-wave as well as acellular capillaries and pericyte ghosts. The investigators found AGEs in the vitreous and associated with the inner limiting membrane formed by the Müller cell endfeet. They noted that attenuation of the RAGE axis with soluble RAGE ameliorated neuronal dysfunction and reduced the development of capillary lesions in these mice. The KKAY mouse is another model that is useful for studies of Type 2 diabetes. As with the Ins2Akita/+ and the db/db mice, the KKAY mouse develops retinal neuropathy and microangiopathy [219], and hence should prove useful for analyses of biomarkers especially in determining whether there are differences in gene expression associated with this model of Type 2 diabetes versus those that more closely mimic Type 1. Other groups have induced diabetes in mice using streptozotocin and examined effects on neuronal function [220]. When the mice are not maintained on insulin, which is known for its neuroprotective properties [205, 221, 222], death of ganglion cells has been documented [220]. Using this model Kurihara and colleagues examined the relationship of the rennin-angiotensin system (RAS) with synaptophysin, a synaptic vesicle protein [222]. They examined the streptozotocin-induced mouse model have examined synergistic effects of diabetes neuronal activity in streptozotocin-induced diabetic mice by ERG. Using a variety of molecular and biochemical approaches they found that diabetes was associated with a significant increase in retinal production of angiotensin II and AT1R together with ERK activation. They observed ERG changes and a reduction in the level of synaptophysin protein (but not mRNA levels). Based on these observations, they treated the diabetic mice with the angiotensin II type 1 receptor (AT1R) blocker telimsartan or valsartan and found that they could reverse the ERG changes and the decreased synaptophysin. Other studies have used and other genetic defects. For example, mice with a defect in the cystathionine-β-synthase gene develop hyperhomocysteinemia [223]. Increased levels of homocysteine are implicated in several ocular diseases including diabetic retinopathy [224–226] and glaucoma [227, 228]. In the cbs+/− mutant mice, retinal ganglion cells die by apoptosis [229] and diabetes accelerates the retinal neuronal loss in these mice [230].
Studies in cell lines and purified ganglion cells
While in vivo models are critical for advancing our understanding of biomarkers in neuronal death associated with diabetic retinopathy, there is a place also for studies using a single cell type. Indeed initial screenings to detect biomarkers and postulate therapies are well-served by the availability of appropriate in vitro model systems. Fortunately, the field of diabetic retinopathy research has several models to exploit for these purposes.
The rat R28 retinal precursor cells, immortalized with the 12S, nontumorigenic portion of the adenovirus E1A gene, provide researchers with an inexhaustible supply of cells for studying structural and functional differentiation in the retina. The R28 cells were developed in the laboratory of Dr. Gail Seigel and have been used extensively to analyze mechanisms of diabetes-induced neuronal cell death [231]. Notable among the studies are discoveries of several biomarkers for retinal neuronal cell death that may have implications for diabetic retinopathy including sphingolipids [201], p44/p42 mitogen-activated protein kinase [232], glucosamine-induced PERK activation [233], hexosamines [233], advanced glycation end products (AGE) [234], IL-1β [235], and glutamate receptor/calcium homeostasis [236] to name a few.
Useful also for studies of retinal neuronal cell death is the RGC-5 cell line. In 2001, Krishnamoorthy and colleagues published the development of the RGC-5 cell line [193]. It was reportedly derived by transforming postnatal day 1 rat retinal cells with ψ2 E1A virus [237]. Recent re-characterization of the RGC-5 cell line suggests that it is actually of mouse origin [238]. When originally described, the RGC-5 cells expressed Thy-1.2, Brn-3C, neuritin, NMDA-R1 and GABA receptors, which are neuronal markers characteristic of intact ganglion cells. RGC-5 cells do not express glial fibrillary acidic protein (GFAP) a marker of glial cells. In early passage, the cells were sensitive to glutamate (5 mM) and that sensitivity could be increased if the cells were treated with succinyl concanavalin A (S Con A) [237]. Interestingly, recent studies by Van Bergen and co-workers showed that RGC-5 cells no longer express the ganglion cell marker protein Thy 1.2, whether differentiated using S Con A or not [238]. Our laboratory obtained this cell line shortly after it was first available in 2001 and our stock continues to express Thy1.2, however the sensitivity to homocysteine, glutamate and oxidative stress is diminished [239]. There are over 100 papers published since 2001 using the RGC-5 cells. Many studies explore alterations of genes/proteins associated with diabetic retinopathy. Indeed, in the last 12 months, there have been over 30 publications using these cells for such analyses. Many of these reports used the RGC-5 cell to determine gene/protein alterations or therapeutic intervention strategies under conditions intended to mimic diabetes. For example, Osborne et al. exposed the cells to retinal ischemia and oxidative stress to determine effects of latanoprost acid [240]. Other studies have examined the PI3-kinase/Akt pathway [241], 5-S-GAD as a neuroprotectant [242], bevacizumab toxicity [243], the ERK1/2 pathways [244], and tafluprost [245].
The RGC-5 cell lines proliferate quickly and are not neuronal in appearance. However, when incubated with the protein kinase inhibitor staurosporine, a more neuronal phenotype is achieved. RGC-5 cells treated with 316 nM staurosporine express neurites, become postmitotic and nonapoptotic, and alter their kinase phosphorylation patterns [246, 247]. In addition, they are positive for a number of neuronal markers including Thy 1.2 [246]. Staurosporine-induced differentiated RGC-5 cells are sensitive to oxidative stress induced by hydrogen peroxide, ischemia, glucose deprivation and plasminogen activators [248, 249]. Recent studies in our lab examined the susceptibility of these differentiated RGC-5 cells to homocysteine. We observed that when differentiated, the cells increase expression of active caspase-3, which is not observed in non-staurosporine-treated cells [239]. The staurosporine-treated cells were not susceptible to homocysteine until applied in exceedingly high dosages. Thus, RGC-5 cells are a useful neuronal model, but data must be interpreted cautiously acknowledging the limitations of the in vitro system.
A more labor-intensive method to study proteins/genes in ganglion cells that are affected by diabetic retinopathy is to purify the cells from the intact animal. Our laboratory has adapted methodology from the Barres laboratory [250] to immunopan ganglion cells from neonatal mice [251]. The cells are viable for 22 days when maintained in a supplemented neurobasal media [251]. They extend neurite processes characteristic of ganglion cells. Using these cells we have been able to demonstrate exquisite sensitivity to oxidative stress, a factor implicated in diabetic retinopathy, that leads to upregulation of the xCT protein component of the glutamate-cystine exchanger system x−c [251]. Owing to the role of this exchanger in generation of the antioxidant glutathione, it likely represents a mechanism to enhance ganglion cell survival under stress. The primary ganglion cell cultures are sensitive to physiologically-relevant levels of glutamate and homocysteine; they die by an apoptotic mechanism when exposed to these excitotoxins, a phenomenon that is inhibited by treatment with the σR1 ligand (+)-pentazocine [252]. We have preliminary evidence that when primary ganglion cells are treated with the oxidative stressor xanthine:xanthine oxidase, numerous pro-apoptotic genes are upregulated including Bad, Bak, Fas-L and Trail [Ha et al., manuscript in preparation]. These data are noteworthy since xanthine oxidase is involved in free radical production in type 1 diabetic human patients [253]. We have used the primary ganglion cells to explore expression of a number of genes/proteins and found them to reflect the expression in intact mouse retina faithfully [254–257]. Studies are underway to explore ER stress genes whose expression is altered as a consequence of factors associated with diabetic retinopathy.
In Summary, diabetic retinopathy in human patients is characterized by neuronal and vascular alterations that can ultimately lead to devastating vision loss. Oxidative stress is crucial in mediating vascular dysfunctions associated with DR via modification of retinal lipids, proteins and DNA. This effect is associated with switching on various signaling pathways that include but not limited to upregulation of angiogenic factors, activation of NF-kB and production of inflammatory mediators. Therefore, early detection of the biochemical effect of oxidative stress such as by-products of lipid peroxidation, DNA and protein oxidative damage in diabetic patients could serve as biomarkers which may be effective in early prediction and treatment of DR. Interestingly, variations in oxidative stress related genes among diabetic patients plays also a role as an additional risk factor that makes some patients more vulnerable to develop DR than others. Thus, gene polymorphism could be also used as predictive marker for increased susceptibility to develop DR in diabetic patient. There is strong evidence from analysis of human tissue that retinal ganglion cells are particularly susceptible in diabetic retinopathy. Investigators are using a variety of model systems, both in vivo (rat and mouse) and in vitro (ganglion cell lines and primary cultures) to determine key genes/proteins whose expression is changed under diabetic conditions with the aim of uncovering the most promising targets for successful intervention. Using relevant in vitro models and confirming the data in relevant in vivo models should pave the way for important biomarkers to be identified and targeted in treating this disease.
Acknowledgements
This work is supported by AHA00104 (MA) and NIH R01 EY01456 (SS). We also thank the Dr. Nasser Al-Rashid Research Chair and Ms. Stacy Deppeler from Opthalmology at King Saud University for the editing help during preparation and submission of this manuscript.
References
- 1.Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. Vii. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–42. doi: 10.2337/diabetes.50.8.1938. [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 3.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–34. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 4.Giardino I, Fard AK, Hatchell DL, et al. Aminoguanidine inhibits reactive oxygen species formation, lipid peroxidation, and oxidant-induced apoptosis. Diabetes. 1998;47:1114–20. doi: 10.2337/diabetes.47.7.1114. [DOI] [PubMed] [Google Scholar]
- 5.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:283–90. doi: 10.1016/S0278-5846(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 6.Antonetti DA, Barber AJ, Bronson SK, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–11. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 7.Smith S. The impact of diabetes on neuronal, glial and vascular cells of the retina: implications for the pathogenesis of diabetic retinopathy. In: Barnstable CJ, editor. Retinal degenerations: biology, diagnostics, and therapeutics. 2007. Totowa: Humana Press; 2007. [Google Scholar]
- 8.Bresnick GH, Korth K, Groo A, et al. Electroretinographic oscillatory potentials predict progression of diabetic retinopathy. Preliminary report. Arch Ophthalmol. 1984;102:1307–11. doi: 10.1001/archopht.1984.01040031057023. [DOI] [PubMed] [Google Scholar]
- 9.Lopes de Faria JM, Katsumi O, Cagliero E, et al. Neurovisual abnormalities preceding the retinopathy in patients with long-term type 1 diabetes mellitus. Graefes Arch Clin Exp Ophthalmol. 2001;239:643–8. doi: 10.1007/s004170100268. [DOI] [PubMed] [Google Scholar]
- 10.Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–90. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson RE, Rapp LM, Wiegand RD. Lipid peroxidation and retinal degeneration. Curr Eye Res. 1984;3:223–7. doi: 10.3109/02713688408997203. [DOI] [PubMed] [Google Scholar]
- 12.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 13.Saran M, Michel C, Bors W. Reaction of no with o2-. Implications for the action of endothelium-derived relaxing factor (edrf) Free Radic Res Commun. 1990;10:221–6. doi: 10.3109/10715769009149890. [DOI] [PubMed] [Google Scholar]
- 14.Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195–9. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 15.Mohr S, Xi X, Tang J, et al. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes. 2002;51:1172–9. doi: 10.2337/diabetes.51.4.1172. [DOI] [PubMed] [Google Scholar]
- 16.Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–23. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 17.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Role of nadph oxidase and stat3 in statin-mediated protection against diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:3231–8. doi: 10.1167/iovs.08-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoguchi T, Sonta T, Tsubouchi H, et al. Protein kinase c-dependent increase in reactive oxygen species (ros) production in vascular tissues of diabetes: role of vascular nad(p)h oxidase. J Am Soc Nephrol. 2003;14:S227–32. doi: 10.1097/01.ASN.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- 19.Sonta T, Inoguchi T, Tsubouchi H, et al. Evidence for contribution of vascular nad(p)h oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–23. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Griendling KK, Sorescu D, Ushio-Fukai M. Nad(p)h oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 21.Al-Shabrawey M, Rojas M, Sanders T, et al. Role of nadph oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008;49:3239–44. doi: 10.1167/iovs.08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of nad(p)h oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams ER, Dratz EA, Gizachew D, et al. Interaction of human neutrophil flavocytochrome b with cytosolic proteins: Transferred-noesy nmr studies of a gp91phox c-terminal peptide bound to p47phox. Biochem J. 1997;325(Pt 1):249–57. doi: 10.1042/bj3250249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenger RH, Marti HH, Schuerer-Maly CC, et al. Hypoxic induction of gene expression in chronic granulomatous disease-derived b-cell lines: Oxygen sensing is independent of the cytochrome b558-containing nicotinamide adenine dinucleotide phosphate oxidase. Blood. 1996;87:756–61. [PubMed] [Google Scholar]
- 25.Heyworth PG, Curnutte JT, Nauseef WM, et al. Neutrophil nicotinamide adenine dinucleotide phosphate oxidase assembly. Translocation of p47-phox and p67-phox requires interaction between p47-phox and cytochrome b558. J Clin Invest. 1991;87:352–6. doi: 10.1172/JCI114993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koga H, Terasawa H, Nunoi H, et al. Tetratricopeptide repeat (tpr) motifs of p67(phox) participate in interaction with the small gtpase rac and activation of the phagocyte nadph oxidase. J Biol Chem. 1999;274:25051–60. doi: 10.1074/jbc.274.35.25051. [DOI] [PubMed] [Google Scholar]
- 27.Ishida S, Yamashiro K, Usui T, et al. significance of leukocytes in the regulation of retinal edema, ischemia, and angiogenesis. Nippon Ganka Gakkai Zasshi. 2004;108:193–201. [PubMed] [Google Scholar]
- 28.Miyamoto K, Hiroshiba N, Tsujikawa A, et al. In vivo demonstration of increased leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 1998;39:2190–4. [PubMed] [Google Scholar]
- 29.Kinukawa Y, Shimura M, Tamai M. Quantifying leukocyte dynamics and plugging in retinal microcirculation of streptozotosin-induced diabetic rats. Curr Eye Res. 1999;18:49–55. doi: 10.1076/ceyr.18.1.49.5389. [DOI] [PubMed] [Google Scholar]
- 30.Nonaka A, Kiryu J, Tsujikawa A, et al. Pkc-beta inhibitor (ly333531) attenuates leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 2000;41:2702–6. [PubMed] [Google Scholar]
- 31.Wong RK, Pettit AI, Davies JE, et al. Augmentation of the neutrophil respiratory burst through the action of advanced glycation end products: a potential contributor to vascular oxidant stress. Diabetes. 2002;51:2846–53. doi: 10.2337/diabetes.51.9.2846. [DOI] [PubMed] [Google Scholar]
- 32.Mohanty P, Hamouda W, Garg R, et al. Glucose challenge stimulates reactive oxygen species (ros) generation by leucocytes. J Clin Endocrinol Metab. 2000;85:2970–3. doi: 10.1210/jc.85.8.2970. [DOI] [PubMed] [Google Scholar]
- 33.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 34.Kanwar M, Chan PS, Kern TS, et al. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–11. doi: 10.1167/iovs.06-1280. [DOI] [PubMed] [Google Scholar]
- 35.Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006;47:1594–9. doi: 10.1167/iovs.05-1276. [DOI] [PubMed] [Google Scholar]
- 36.Kowluru RA, Kowluru V, Xiong Y, et al. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006;41:1191–6. doi: 10.1016/j.freeradbiomed.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Heinzel B, John M, Klatt P, et al. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J. 1992;281(Pt 3):627–30. doi: 10.1042/bj2810627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer B, Heinzel B, Klatt P, et al. Nitric oxide synthase-catalyzed activation of oxygen and reduction of cytochromes: Reaction mechanisms and possible physiological implications. J Cardiovasc Pharmacol. 1992;20(Suppl 12):S54–6. doi: 10.1097/00005344-199204002-00016. [DOI] [PubMed] [Google Scholar]
- 39.Presta A, Liu J, Sessa WC, et al. Substrate binding and calmodulin binding to endothelial nitric oxide synthase coregulate its enzymatic activity. Nitric Oxide. 1997;1:74–87. doi: 10.1006/niox.1996.0110. [DOI] [PubMed] [Google Scholar]
- 40.Tanner FC, Noll G, Boulanger CM, et al. Oxidized low density lipoproteins inhibit relaxations of porcine coronary arteries. Role of scavenger receptor and endothelium-derived nitric oxide. Circulation. 1991;83:2012–20. doi: 10.1161/01.cir.83.6.2012. [DOI] [PubMed] [Google Scholar]
- 41.Jay MT, Chirico S, Siow RC, et al. Modulation of vascular tone by low density lipoproteins: effects on l-arginine transport and nitric oxide synthesis. Exp Physiol. 1997;82:349–60. doi: 10.1113/expphysiol.1997.sp004030. [DOI] [PubMed] [Google Scholar]
- 42.Taverna MJ, Elgrably F, Selmi H, et al. The t-786c and c774t endothelial nitric oxide synthase gene polymorphisms independently affect the onset pattern of severe diabetic retinopathy. Nitric Oxide. 2005;13:88–92. doi: 10.1016/j.niox.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Miwa K, Nakamura J, Hamada Y, et al. The role of polyol pathway in glucose-induced apoptosis of cultured retinal pericytes. Diabetes Res Clin Pract. 2003;60:1–9. doi: 10.1016/S0168-8227(02)00248-6. [DOI] [PubMed] [Google Scholar]
- 44.Sun W, Oates PJ, Coutcher JB, et al. A selective aldose reductase inhibitor of a new structural class prevents or reverses early retinal abnormalities in experimental diabetic retinopathy. Diabetes. 2006;55:2757–62. doi: 10.2337/db06-0138. [DOI] [PubMed] [Google Scholar]
- 45.El-Remessy AB, Abou-Mohamed G, Caldwell RW, et al. High glucose-induced tyrosine nitration in endothelial cells: role of enos uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003;44:3135–43. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 46.Glomb MA, Monnier VM. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the maillard reaction. J Biol Chem. 1995;270:10017–26. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 47.Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol. 2003;75:95–108. doi: 10.1016/S0014-4800(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Renier G. Activation of nicotinamide adenine dinucleotide phosphate (reduced form) oxidase by advanced glycation end products links oxidative stress to altered retinal vascular endothelial growth factor expression. Metabolism. 2006;55:1516–23. doi: 10.1016/j.metabol.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 49.Mohamed AK, Bierhaus A, Schiekofer S, et al. The role of oxidative stress and nf-kappab activation in late diabetic complications. Biofactors. 1999;10:157–67. doi: 10.1002/biof.5520100211. [DOI] [PubMed] [Google Scholar]
- 50.Kowluru RA. Effect of advanced glycation end products on accelerated apoptosis of retinal capillary cells under in vitro conditions. Life Sci. 2005;76:1051–60. doi: 10.1016/j.lfs.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 51.Moore TC, Moore JE, Kaji Y, et al. The role of advanced glycation end products in retinal microvascular leukostasis. Invest Ophthalmol Vis Sci. 2003;44:4457–64. doi: 10.1167/iovs.02-1063. [DOI] [PubMed] [Google Scholar]
- 52.Tawfik A, Sanders T, Kahook K, et al. Suppression of retinal peroxisome proliferator-activated receptor gamma in experimental diabetes and oxygen-induced retinopathy: role of nadph oxidase. Invest Ophthalmol Vis Sci. 2009;50:878–84. doi: 10.1167/iovs.08-2005. [DOI] [PubMed] [Google Scholar]
- 53.Hammes HP, Hoerauf H, Alt A, et al. N(epsilon)(carboxymethyl)lysin and the age receptor rage colocalize in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:1855–9. [PubMed] [Google Scholar]
- 54.Spencer L, Mann C, Metcalfe M, et al. The effect of omega-3 fas on tumour angiogenesis and their therapeutic potential. Eur J Cancer. 2009;45:2077–86. doi: 10.1016/j.ejca.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 55.Koya D, King GL. Protein kinase c activation and the development of diabetic complications. Diabetes. 1998;47:859–66. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 56.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral pkc beta inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 57.Stauble B, Boscoboinik D, Tasinato A, et al. Modulation of activator protein-1 (ap-1) transcription factor and protein kinase c by hydrogen peroxide and d-alpha-tocopherol in vascular smooth muscle cells. Eur J Biochem. 1994;226:393–402. doi: 10.1111/j.1432-1033.1994.tb20064.x. [DOI] [PubMed] [Google Scholar]
- 58.Oikawa T, Shimamura M, Ashino H, et al. Inhibition of angiogenesis by staurosporine, a potent protein kinase inhibitor. J Antibiot (Tokyo) 1992;45:1155–60. doi: 10.7164/antibiotics.45.1155. [DOI] [PubMed] [Google Scholar]
- 59.Xia P, Aiello LP, Ishii H, et al. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase c, its isoforms, and endothelial cell growth. J Clin Invest. 1996;98:2018–26. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koya D, Jirousek MR, Lin YW, et al. Characterization of protein kinase c beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–26. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kowluru RA. Diabetes-induced elevations in retinal oxidative stress, protein kinase c and nitric oxide are interrelated. Acta Diabetol. 2001;38:179–85. doi: 10.1007/s592-001-8076-6. [DOI] [PubMed] [Google Scholar]
- 62.Wu Y, Wu G, Qi X, et al. Protein kinase c beta inhibitor ly333531 attenuates intercellular adhesion molecule-1 and monocyte chemotactic protein-1 expression in the kidney in diabetic rats. J Pharmacol Sci. 2006;101:335–43. doi: 10.1254/jphs.FP0050896. [DOI] [PubMed] [Google Scholar]
- 63.Ohshiro Y, Ma RC, Yasuda Y, et al. Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase cbeta-null mice. Diabetes. 2006;55:3112–20. doi: 10.2337/db06-0895. [DOI] [PubMed] [Google Scholar]
- 64.Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 65.Frank RN. The galactosemic dog. A valid model for both early and late stages of diabetic retinopathy. Arch Ophthalmol. 1995;113:275–6. doi: 10.1001/archopht.1995.01100030029016. [DOI] [PubMed] [Google Scholar]
- 66.Duh E, Aiello LP. Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes. 1999;48:1899–906. doi: 10.2337/diabetes.48.10.1899. [DOI] [PubMed] [Google Scholar]
- 67.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–5. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18:1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 69.Khan ZA, Chakrabarti S. Cellular signaling and potential new treatment targets in diabetic retinopathy. Exp Diabetes Res. 2007;2007:31867. doi: 10.1155/2007/31867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng TH, Shih NL, Chen SY, et al. Reactive oxygen species mediate cyclic strain-induced endothelin-1 gene expression via ras/raf/extracellular signal-regulated kinase pathway in endothelial cells. J Mol Cell Cardiol. 2001;33:1805–14. doi: 10.1006/jmcc.2001.1444. [DOI] [PubMed] [Google Scholar]
- 71.Chen S, Khan ZA, Barbin Y, et al. Pro-oxidant role of heme oxygenase in mediating glucose-induced endothelial cell damage. Free Radic Res. 2004;38:1301–10. doi: 10.1080/10715760400017228. [DOI] [PubMed] [Google Scholar]
- 72.Lutty GA, McLeod DS, Merges C, et al. Localization of vascular endothelial growth factor in human retina and choroid. Arch Ophthalmol. 1996;114:971–7. doi: 10.1001/archopht.1996.01100140179011. [DOI] [PubMed] [Google Scholar]
- 73.Aiello LP, Wong JS. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int Suppl. 2000;77:S113–9. doi: 10.1046/j.1523-1755.2000.07718.x. [DOI] [PubMed] [Google Scholar]
- 74.Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–24. doi: 10.1172/JCI1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caldwell RB, Bartoli M, Behzadian MA, et al. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6:511–24. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- 76.Tsai GY, Cui JZ, Syed H, et al. Effect of n-acetylcysteine on the early expression of inflammatory markers in the retina and plasma of diabetic rats. Clin Experiment Ophthalmol. 2009;37:223–31. doi: 10.1111/j.1442-9071.2009.02000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng Z, Chen H, Zhao H et al (2009) Pedf inhibits jak2/stat3-mediated vegf upregulation under high glucose condition through a mitochondrial ros pathway in vitro. Invest Ophthalmol Vis Sci (Epub ahead of print). [DOI] [PubMed]
- 78.Velazquez E, Winocour PH, Kesteven P, et al. Relation of lipid peroxides to macrovascular disease in type 2 diabetes. Diabet Med. 1991;8:752–8. doi: 10.1111/j.1464-5491.1991.tb01695.x. [DOI] [PubMed] [Google Scholar]
- 79.Kedziora-Kornatowska KZ, Luciak M, Blaszczyk J, Pawlak W. Lipid peroxidation and activities of antioxidant enzymes in erythrocytes of patients with non-insulin dependent diabetes with or without diabetic nephropathy. Nephrol Dial Transplant. 1998;13:2829–32. doi: 10.1093/ndt/13.11.2829. [DOI] [PubMed] [Google Scholar]
- 80.Liang XC, Guo SS, Wang XD. study on relationship of lipid peroxide in coronary heart disease with and without diabetes. Zhongguo Zhong Xi Yi Jie He Za Zhi. 1996;16:29–31. [PubMed] [Google Scholar]
- 81.Stringer MD, Gorog PG, Freeman A, et al. Lipid peroxides and atherosclerosis. BMJ. 1989;298:281–4. doi: 10.1136/bmj.298.6669.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Griesmacher A, Kindhauser M, Andert SE, et al. Enhanced serum levels of thiobarbituric-acid-reactive substances in diabetes mellitus. Am J Med. 1995;98:469–75. doi: 10.1016/S0002-9343(99)80347-7. [DOI] [PubMed] [Google Scholar]
- 83.Losada M, Alio JL. Malondialdehyde serum concentration in type 1 diabetic with and without retinopathy. Doc Ophthalmol. 1996;93:223–9. doi: 10.1007/BF02569062. [DOI] [PubMed] [Google Scholar]
- 84.Gallou G, Ruelland A, Campion L, et al. Increase in thiobarbituric acid-reactive substances and vascular complications in type 2 diabetes mellitus. Diabete Metab. 1994;20:258–64. [PubMed] [Google Scholar]
- 85.Kesavulu MM, Giri R, Kameswara Rao B, et al. Lipid peroxidation and antioxidant enzyme levels in type 2 diabetics with microvascular complications. Diabetes Metab. 2000;26:387–92. [PubMed] [Google Scholar]
- 86.Pan HZ, Zhang H, Chang D, et al. The change of oxidative stress products in diabetes mellitus and diabetic retinopathy. Br J Ophthalmol. 2008;92:548–51. doi: 10.1136/bjo.2007.130542. [DOI] [PubMed] [Google Scholar]
- 87.Nishigaki I, Hagihara M, Tsunekawa H, et al. Lipid peroxide levels of serum lipoprotein fractions of diabetic patients. Biochem Med. 1981;25:373–8. doi: 10.1016/0006-2944(81)90096-X. [DOI] [PubMed] [Google Scholar]
- 88.Dursun E, Dursun B, Suleymanlar G, et al. Effect of haemodialysis on the oxidative stress and antioxidants in diabetes mellitus. Acta Diabetol. 2005;42:123–8. doi: 10.1007/s00592-005-0191-1. [DOI] [PubMed] [Google Scholar]
- 89.Polak M, Zagorski Z. Lipid peroxidation in diabetic retinopathy. Ann Univ Mariae Curie Sklodowska Med. 2004;59:434–7. [PubMed] [Google Scholar]
- 90.Adaikalakoteswari A, Balasubramanyam M, Rema M, et al. Differential gene expression of nadph oxidase (p22phox) and hemoxygenase-1 in patients with type 2 diabetes and microangiopathy. Diabet Med. 2006;23:666–74. doi: 10.1111/j.1464-5491.2006.01879.x. [DOI] [PubMed] [Google Scholar]
- 91.Armstrong D, Abdella N, Salman A, et al. Relationship of lipid peroxides to diabetic complications. Comparison with conventional laboratory tests. J Diabetes Complicat. 1992;6:116–22. doi: 10.1016/1056-8727(92)90022-D. [DOI] [PubMed] [Google Scholar]
- 92.Armstrong D, al-Awadi F. Lipid peroxidation and retinopathy in streptozotocin-induced diabetes. Free Radic Biol Med. 1991;11:433–6. doi: 10.1016/0891-5849(91)90161-U. [DOI] [PubMed] [Google Scholar]
- 93.Augustin AJ, Breipohl W, Boker T, et al. Increased lipid peroxide levels and myeloperoxidase activity in the vitreous of patients suffering from proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 1993;231:647–50. doi: 10.1007/BF00921959. [DOI] [PubMed] [Google Scholar]
- 94.Augustin AJ, Spitznas M, Koch F, et al. Indicators of oxidative tissue damage and inflammatory activity in epiretinal membranes of proliferative diabetic retinopathy, proliferative vitreoretinopathy and macular pucker. Ger J Ophthalmol. 1995;4:47–51. [PubMed] [Google Scholar]
- 95.Diffley JM, Wu M, Sohn M, et al. Apoptosis induction by oxidized glycated ldl in human retinal capillary pericytes is independent of activation of mapk signaling pathways. Mol Vis. 2009;15:135–45. [PMC free article] [PubMed] [Google Scholar]
- 96.Wu M, Chen Y, Wilson K, et al. Intraretinal leakage and oxidation of ldl in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:2679–85. doi: 10.1167/iovs.07-1440. [DOI] [PubMed] [Google Scholar]
- 97.Song W, Barth JL, Yu Y, et al. Effects of oxidized and glycated ldl on gene expression in human retinal capillary pericytes. Invest Ophthalmol Vis Sci. 2005;46:2974–82. doi: 10.1167/iovs.04-1505. [DOI] [PubMed] [Google Scholar]
- 98.Fredrikson GN, Anand DV, Hopkins D, et al. Associations between autoantibodies against apolipoprotein b-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia. 2009;52:1426–33. doi: 10.1007/s00125-009-1377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morrow JD, Hill KE, Burk RF, et al. A series of prostaglandin f2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–7. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Morrow JD, Harris TM, Roberts LJ., 2nd Noncyclooxygenase oxidative formation of a series of novel prostaglandins: Analytical ramifications for measurement of eicosanoids. Anal Biochem. 1990;184:1–10. doi: 10.1016/0003-2697(90)90002-Q. [DOI] [PubMed] [Google Scholar]
- 101.Morrow JD, Minton TA, Roberts LJ., 2nd The f2-isoprostane, 8-epi-prostaglandin f2 alpha, a potent agonist of the vascular thromboxane/endoperoxide receptor, is a platelet thromboxane/endoperoxide receptor antagonist. Prostaglandins. 1992;44:155–63. doi: 10.1016/0090-6980(92)90077-7. [DOI] [PubMed] [Google Scholar]
- 102.Morrow JD, Awad JA, Boss HJ, et al. Non-cyclooxygenase-derived prostanoids (f2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci U S A. 1992;89:10721–5. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang M, Cyrus T, Yao Y, et al. Involvement of thromboxane receptor in the proatherogenic effect of isoprostane f2alpha-iii: Evidence from apolipoprotein e- and ldl receptor-deficient mice. Circulation. 2005;112:2867–74. doi: 10.1161/CIRCULATIONAHA105.562223. [DOI] [PubMed] [Google Scholar]
- 104.Khasawneh FT, Huang JS, Mir F, et al. Characterization of isoprostane signaling: Evidence for a unique coordination profile of 8-iso-pgf(2alpha) with the thromboxane a(2) receptor, and activation of a separate camp-dependent inhibitory pathway in human platelets. Biochem Pharmacol. 2008;75:2301–15. doi: 10.1016/j.bcp.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Joy AP, Cowley EA. 8-iso-pge2 stimulates anion efflux from airway epithelial cells via the ep4 prostanoid receptor. Am J Respir Cell Mol Biol. 2008;38:143–52. doi: 10.1165/rcmb.2006-0295OC. [DOI] [PubMed] [Google Scholar]
- 106.Nourooz-Zadeh J. Key issues in f2-isoprostane analysis. Biochem Soc Trans. 2008;36:1060–5. doi: 10.1042/BST0361060. [DOI] [PubMed] [Google Scholar]
- 107.Nourooz-Zadeh J, Pereira P. F(2) isoprostanes, potential specific markers of oxidative damage in human retina. Ophthalmic Res. 2000;32:133–7. doi: 10.1159/000055603. [DOI] [PubMed] [Google Scholar]
- 108.Dandona P, Thusu K, Cook S, et al. Oxidative damage to DNA in diabetes mellitus. Lancet. 1996;347:444–5. doi: 10.1016/S0140-6736(96)90013-6. [DOI] [PubMed] [Google Scholar]
- 109.Kaneto H, Fujii J, Suzuki K, et al. DNA cleavage induced by glycation of cu, zn-superoxide dismutase. Biochem J. 1994;304(Pt 1):219–25. doi: 10.1042/bj3040219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Krapfenbauer K, Birnbacher R, Vierhapper H, et al. Glycoxidation, and protein and DNA oxidation in patients with diabetes mellitus. Clin Sci (Lond) 1998;95:331–7. doi: 10.1042/CS19970302. [DOI] [PubMed] [Google Scholar]
- 111.Ha H, Kim C, Son Y, et al. DNA damage in the kidneys of diabetic rats exhibiting microalbuminuria. Free Radic Biol Med. 1994;16:271–4. doi: 10.1016/0891-5849(94)90152-X. [DOI] [PubMed] [Google Scholar]
- 112.Nishikawa T, Sasahara T, Kiritoshi S, et al. Evaluation of urinary 8-hydroxydeoxy-guanosine as a novel biomarker of macrovascular complications in type 2 diabetes. Diabetes Care. 2003;26:1507–12. doi: 10.2337/diacare.26.5.1507. [DOI] [PubMed] [Google Scholar]
- 113.Kakimoto M, Inoguchi T, Sonta T, et al. Accumulation of 8-hydroxy-2′-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes. 2002;51:1588–95. doi: 10.2337/diabetes.51.5.1588. [DOI] [PubMed] [Google Scholar]
- 114.Suzuki S, Hinokio Y, Komatu K, et al. Oxidative damage to mitochondrial DNA and its relationship to diabetic complications. Diabetes Res Clin Pract. 1999;45:161–8. doi: 10.1016/S0168-8227(99)00046-7. [DOI] [PubMed] [Google Scholar]
- 115.Hinokio Y, Suzuki S, Hirai M, et al. Oxidative DNA damage in diabetes mellitus: its association with diabetic complications. Diabetologia. 1999;42:995–8. doi: 10.1007/s001250051258. [DOI] [PubMed] [Google Scholar]
- 116.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–6. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 117.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–94. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 118.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic Biol Med. 2002;32:790–6. doi: 10.1016/S0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 119.Dalle-Donne I, Giustarini D, Colombo R, et al. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–76. doi: 10.1016/S1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 120.Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–7. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cakatay U. Protein oxidation parameters in type 2 diabetic patients with good and poor glycaemic control. Diabetes Metab. 2005;31:551–7. doi: 10.1016/S1262-3636(07)70230-6. [DOI] [PubMed] [Google Scholar]
- 122.Ramakrishna V, Jailkhani R. Evaluation of oxidative stress in insulin dependent diabetes mellitus (iddm) patients. Diagn Pathol. 2007;2:22. doi: 10.1186/1746-1596-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Margetis PI, Antonelou MH, Petropoulos IK, et al. Increased protein carbonylation of red blood cell membrane in diabetic retinopathy. Exp Mol Pathol. 2009;87:76–82. doi: 10.1016/j.yexmp.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 124.El-Remessy AB, Al-Shabrawey M, Khalifa Y, et al. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–44. doi: 10.2353/ajpath.2006.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhan X, Du Y, Crabb JS, et al. Targets of tyrosine nitration in diabetic rat retina. Mol Cell Proteomics. 2008;7:864–74. doi: 10.1074/mcp.M700417-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med. 1998;25:392–403. doi: 10.1016/S0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 127.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–44. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 128.El-Remessy AB, Bartoli M, Platt DH, et al. Oxidative stress inactivates vegf survival signaling in retinal endothelial cells via pi 3-kinase tyrosine nitration. J Cell Sci. 2005;118:243–52. doi: 10.1242/jcs.01612. [DOI] [PubMed] [Google Scholar]
- 129.El-Remessy AB, Khalil IE, Matragoon S, et al. Neuroprotective effect of (-)delta9-tetrahydrocannabinol and cannabidiol in n-methyl-d-aspartate-induced retinal neurotoxicity: involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ali TK, Matragoon S, Pillai BA, et al. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes. 2008;57:889–98. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- 131.Ramakrishnan S, Rajesh M, Sulochana KN. Eales’ disease: oxidant stress and weak antioxidant defence. Indian J Ophthalmol. 2007;55:95–102. doi: 10.4103/0301-4738.30701. [DOI] [PubMed] [Google Scholar]
- 132.Julius U, Drel VR, Grassler J, et al. Nitrosylated proteins in monocytes as a new marker of oxidative-nitrosative stress in diabetic subjects with macroangiopathy. Exp Clin Endocrinol Diabetes. 2009;117:72–7. doi: 10.1055/s-2008-1078710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cakatay U, Kayali R. The evaluation of altered redox status in plasma and mitochondria of acute and chronic diabetic rats. Clin Biochem. 2006;39:907–12. doi: 10.1016/j.clinbiochem.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 134.Alderman CJ, Shah S, Foreman JC, et al. The role of advanced oxidation protein products in regulation of dendritic cell function. Free Radic Biol Med. 2002;32:377–85. doi: 10.1016/S0891-5849(01)00735-3. [DOI] [PubMed] [Google Scholar]
- 135.Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996;49:1304–13. doi: 10.1038/ki.1996.186. [DOI] [PubMed] [Google Scholar]
- 136.Baskol G, Gumus K, Oner A, et al. The role of advanced oxidation protein products and total thiols in diabetic retinopathy. Eur J Ophthalmol. 2008;18:792–8. doi: 10.1177/112067210801800521. [DOI] [PubMed] [Google Scholar]
- 137.Witko-Sarsat V, Nguyen Khoa T, Jungers P, et al. Advanced oxidation protein products: oxidative stress markers and mediators of inflammation in uremia. Adv Nephrol Necker Hosp. 1998;28:321–41. [PubMed] [Google Scholar]
- 138.Witko-Sarsat V, Friedlander M, Nguyen Khoa T, et al. Advanced oxidation protein products as novel mediators of inflammation and monocyte activation in chronic renal failure. J Immunol. 1998;161:2524–32. [PubMed] [Google Scholar]
- 139.Kalousova M, Skrha J, Zima T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol Res. 2002;51:597–604. [PubMed] [Google Scholar]
- 140.Vivekanadan-Giri A, Wang JH, Byun J, et al. Mass spectrometric quantification of amino acid oxidation products identifies oxidative mechanisms of diabetic end-organ damage. Rev Endocr Metab Disord. 2008;9:275–87. doi: 10.1007/s11154-008-9093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sampathkumar R, Balasubramanyam M, Sudarslal S, et al. Increased glutathionylated hemoglobin (hbssg) in type 2 diabetes subjects with microangiopathy. Clin Biochem. 2005;38:892–9. doi: 10.1016/j.clinbiochem.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 142.Selvam R, Anuradha CV. Lipid peroxidation and antiperoxidative enzyme changes in erythrocytes in diabetes mellitus. Indian J Biochem Biophys. 1988;25:268–72. [PubMed] [Google Scholar]
- 143.Kaji H, Kurasaki M, Ito K, et al. Increased lipoperoxide value and glutathione peroxidase activity in blood plasma of type 2 (non-insulin-dependent) diabetic women. Klin Wochenschr. 1985;63:765–8. doi: 10.1007/BF01733829. [DOI] [PubMed] [Google Scholar]
- 144.Hagglof B, Marklund SL, Holmgren G. Cuzn superoxide dismutase, mn superoxide dismutase, catalase and glutathione peroxidase in lymphocytes and erythrocytes in insulin-dependent diabetic children. Acta Endocrinol (Copenh) 1983;102:235–9. doi: 10.1530/acta.0.1020235. [DOI] [PubMed] [Google Scholar]
- 145.Hartnett ME, Stratton RD, Browne RW, et al. Serum markers of oxidative stress and severity of diabetic retinopathy. Diabetes Care. 2000;23:234–40. doi: 10.2337/diacare.23.2.234. [DOI] [PubMed] [Google Scholar]
- 146.Siemianowicz K, Gminski J, Telega A, et al. Blood antioxidant parameters in patients with diabetic retinopathy. Int J Mol Med. 2004;14:433–7. [PubMed] [Google Scholar]
- 147.Gupta MM, Chari S. Lipid peroxidation and antioxidant status in patients with diabetic retinopathy. Indian J Physiol Pharmacol. 2005;49:187–92. [PubMed] [Google Scholar]
- 148.Ford ES, Mokdad AH, Giles WH, et al. The metabolic syndrome and antioxidant concentrations: findings from the third national health and nutrition examination survey. Diabetes. 2003;52:2346–52. doi: 10.2337/diabetes.52.9.2346. [DOI] [PubMed] [Google Scholar]
- 149.Chowdhury TA, Dyer PH, Kumar S, et al. Genetic determinants of diabetic nephropathy. Clin Sci (Lond) 1999;96:221–30. doi: 10.1042/CS19980331. [DOI] [PubMed] [Google Scholar]
- 150.Chowdhury TA, Kumar S, Barnett AH, et al. Nephropathy in type 1 diabetes: the role of genetic factors. Diabet Med. 1995;12:1059–67. doi: 10.1111/j.1464-5491.1995.tb00422.x. [DOI] [PubMed] [Google Scholar]
- 151.Parving HH, Tarnow L, Rossing P. Genetics of diabetic nephropathy. J Am Soc Nephrol. 1996;7:2509–17. doi: 10.1681/ASN.V7122509. [DOI] [PubMed] [Google Scholar]
- 152.Tarnow L, Rossing P, Nielsen FS, et al. Cardiovascular morbidity and early mortality cluster in parents of type 1 diabetic patients with diabetic nephropathy. Diabetes Care. 2000;23:30–3. doi: 10.2337/diacare.23.1.30. [DOI] [PubMed] [Google Scholar]
- 153.Levy A, Asleh R, Blum S et al (2009) Haptoglobin: basic and clinical aspects. Antioxid Redox Signal (Epub ahead of print). [DOI] [PubMed]
- 154.Nakhoul FM, Miller-Lotan R, Awaad H, et al. Hypothesis-haptoglobin genotype and diabetic nephropathy. Nat Clin Pract Nephrol. 2007;3:339–44. doi: 10.1038/ncpneph0467. [DOI] [PubMed] [Google Scholar]
- 155.Petrovic MG, Cilensek I, Petrovic D. Manganese superoxide dismutase gene polymorphism (v16a) is associated with diabetic retinopathy in slovene (caucasians) type 2 diabetes patients. Dis Markers. 2008;24:59–64. doi: 10.1155/2008/940703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ye LX, Yang MP, Qiu H, et al. association of the polymorphism in manganese superoxide dismutase gene with diabetic retinopathy in chinese type 2 diabetic patients. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2008;25:452–4. [PubMed] [Google Scholar]
- 157.Hovnik T, Dolzan V, Natasa Ursic B, et al. Genetic polymorphisms in genes encoding antioxidant enzymes are associated with diabetic retinopathy in type 1 diabetes. Diabetes Care. 2009;32:2258–62. doi: 10.2337/dc09-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Katnik I, Jadach J. Haptoglobin concentration in serum and other body fluids measured by comparison of its reactivity with hemoglobin and concanavalin a. Arch Immunol Ther Exp (Warsz) 1996;44:45–50. [PubMed] [Google Scholar]
- 159.Dobryszycka W. Biological functions of haptoglobin-new pieces to an old puzzle. Eur J Clin Chem Clin Biochem. 1997;35:647–54. [PubMed] [Google Scholar]
- 160.Everse J, Hsia N. The toxicities of native and modified hemoglobins. Free Radic Biol Med. 1997;22:1075–99. doi: 10.1016/S0891-5849(96)00499-6. [DOI] [PubMed] [Google Scholar]
- 161.Miller YI, Altamentova SM, Shaklai N. Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry. 1997;36:12189–98. doi: 10.1021/bi970258a. [DOI] [PubMed] [Google Scholar]
- 162.Lim SK, Kim H, Bin Ali A, et al. Increased susceptibility in hp knockout mice during acute hemolysis. Blood. 1998;92:1870–7. [PubMed] [Google Scholar]
- 163.Nakhoul FM, Marsh S, Hochberg I, et al. Haptoglobin genotype as a risk factor for diabetic retinopathy. JAMA. 2000;284:1244–5. doi: 10.1001/jama.284.10.1244-a. [DOI] [PubMed] [Google Scholar]
- 164.Langlois MR, Delanghe JR, Buyzere ML, et al. Effect of haptoglobin on the metabolism of vitamin c. Am J Clin Nutr. 1997;66:606–10. doi: 10.1093/ajcn/66.3.606. [DOI] [PubMed] [Google Scholar]
- 165.Delanghe JR, Langlois MR. Haptoglobin polymorphism and body iron stores. Clin Chem Lab Med. 2002;40:212–6. doi: 10.1515/CCLM.2002.035. [DOI] [PubMed] [Google Scholar]
- 166.Brouwers A, Langlois M, Delanghe J, et al. Oxidized low-density lipoprotein, iron stores, and haptoglobin polymorphism. Atherosclerosis. 2004;176:189–95. doi: 10.1016/j.atherosclerosis.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 167.Miller-Lotan R, Miller B, Nakhoul F, et al. Retinal capillary basement membrane thickness in diabetic mice genetically modified at the haptoglobin locus. Diabetes Metab Res Rev. 2007;23:152–6. doi: 10.1002/dmrr.654. [DOI] [PubMed] [Google Scholar]
- 168.Chen Y, Huang H, Zhou J, et al. Polymorphism of the endothelial nitric oxide synthase gene is associated with diabetic retinopathy in a cohort of west africans. Mol Vis. 2007;13:2142–7. [PubMed] [Google Scholar]
- 169.Ezzidi I, Mtiraoui N, Mohamed MB, et al. Endothelial nitric oxide synthase glu298asp, 4b/a, and t-786c polymorphisms in type 2 diabetic retinopathy. Clin Endocrinol (Oxf) 2008;68:542–6. doi: 10.1111/j.1365-2265.2007.03089.x. [DOI] [PubMed] [Google Scholar]
- 170.Taverna MJ, Sola A, Guyot-Argenton C, et al. Enos4 polymorphism of the endothelial nitric oxide synthase predicts risk for severe diabetic retinopathy. Diabet Med. 2002;19:240–5. doi: 10.1046/j.1464-5491.2002.00681.x. [DOI] [PubMed] [Google Scholar]
- 171.Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via tnf-alpha suppression. Faseb J. 2002;16:438–40. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 172.Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–9. doi: 10.1016/S0002-9440(10)64869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Golubnitschaja O, Jaksche A, Moenkemann H, et al. Molecular imaging system for possible prediction of active retinopathy in patients with diabetes mellitus. Amino Acids. 2005;28:229–37. doi: 10.1007/s00726-005-0173-1. [DOI] [PubMed] [Google Scholar]
- 174.Oshitari T, Hata N, Yamamoto S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag. 2008;4:115–22. doi: 10.2147/vhrm.2008.04.01.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Imai H, Singh RS, Fort PE, et al. Neuroprotection for diabetic retinopathy. Dev Ophthalmol. 2009;44:56–68. doi: 10.1159/000223946. [DOI] [PubMed] [Google Scholar]
- 176.Daley ML, Watzke RC, Riddle MC. Early loss of blue-sensitive color vision in patients with type i diabetes. Diabetes Care. 1987;10:777–81. doi: 10.2337/diacare.10.6.777. [DOI] [PubMed] [Google Scholar]
- 177.Roy MS, Gunkel RD, Podgor MJ. Color vision defects in early diabetic retinopathy. Arch Ophthalmol. 1986;104:225–8. doi: 10.1001/archopht.1986.01050140079024. [DOI] [PubMed] [Google Scholar]
- 178.Sokol S, Moskowitz A, Skarf B, et al. Contrast sensitivity in diabetics with and without background retinopathy. Arch Ophthalmol. 1985;103:51–4. doi: 10.1001/archopht.1985.01050010055018. [DOI] [PubMed] [Google Scholar]
- 179.Hirsh J. Reduced contrast sensitivity may precede clinically observable retinopathy in type 1 diabetes. In: Henkind P, editor. Acta XXIV international congress of ophthalmology. New York: Lippincott; 1982. [Google Scholar]
- 180.Bresnick GH. Diabetic retinopathy viewed as a neurosensory disorder. Arch Ophthalmol. 1986;104:989–90. doi: 10.1001/archopht.1986.01050190047037. [DOI] [PubMed] [Google Scholar]
- 181.Wolter JR. Diabetic retinopathy. Am J Ophthalmol. 1961;51:1123–41. doi: 10.1016/0002-9394(61)91802-5. [DOI] [PubMed] [Google Scholar]
- 182.Bloodworth JM., Jr Diabetic retinopathy. Diabetes. 1962;11:1–22. [PubMed] [Google Scholar]
- 183.Fortune B, Schneck ME, Adams AJ. Multifocal electroretinogram delays reveal local retinal dysfunction in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 1999;40:2638–51. [PubMed] [Google Scholar]
- 184.Greco AV, Leo MA, Caputo S, et al. Early selective neuroretinal disorder in prepubertal type 1 (insulin-dependent) diabetic children without microvascular abnormalities. Acta Diabetol. 1994;31:98–102. doi: 10.1007/BF00570544. [DOI] [PubMed] [Google Scholar]
- 185.Leo MA, Caputo S, Falsini B, et al. Presence and further development of retinal dysfunction after 3-year follow up in iddm patients without angiographically documented vasculopathy. Diabetologia. 1994;37:911–6. doi: 10.1007/BF00400947. [DOI] [PubMed] [Google Scholar]
- 186.Ghirlanda G, Leo MA, Caputo S, et al. Detection of inner retina dysfunction by steady-state focal electroretinogram pattern and flicker in early iddm. Diabetes. 1991;40:1122–7. doi: 10.2337/diabetes.40.9.1122. [DOI] [PubMed] [Google Scholar]
- 187.Parisi V, Uccioli L. Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev. 2001;17:12–8. doi: 10.1002/dmrr.177. [DOI] [PubMed] [Google Scholar]
- 188.Ghirlanda G, Leo MA, Caputo S, et al. From functional to microvascular abnormalities in early diabetic retinopathy. Diabetes Metab Rev. 1997;13:15–35. doi: 10.1002/(SICI)1099-0895(199703)13:1<15::AID-DMR176>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 189.Greenstein VC, Shapiro A, Zaidi Q, et al. Psychophysical evidence for post-receptoral sensitivity loss in diabetics. Invest Ophthalmol Vis Sci. 1992;33:2781–90. [PubMed] [Google Scholar]
- 190.Bearse MA, Jr, Han Y, Schneck ME, et al. Local multifocal oscillatory potential abnormalities in diabetes and early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2004;45:3259–65. doi: 10.1167/iovs.04-0308. [DOI] [PubMed] [Google Scholar]
- 191.Bresnick GH, Palta M. Predicting progression to severe proliferative diabetic retinopathy. Arch Ophthalmol. 1987;105:810–4. doi: 10.1001/archopht.1987.01060060096041. [DOI] [PubMed] [Google Scholar]
- 192.Bengtsson B, Heijl A, Agardh E. Visual fields correlate better than visual acuity to severity of diabetic retinopathy. Diabetologia. 2005;48:2494–500. doi: 10.1007/s00125-005-0001-x. [DOI] [PubMed] [Google Scholar]
- 193.Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–91. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Abu El-Asrar AM, Dralands L, Missotten L, et al. Expression of apoptosis markers in the retinas of human subjects with diabetes. Invest Ophthalmol Vis Sci. 2004;45:2760–6. doi: 10.1167/iovs.03-1392. [DOI] [PubMed] [Google Scholar]
- 195.Abu El-Asrar AM, Dralands L, Missotten L, et al. Expression of antiapoptotic and proapoptotic molecules in diabetic retinas. Eye (Lond) 2007;21:238–45. doi: 10.1038/sj.eye.6702225. [DOI] [PubMed] [Google Scholar]
- 196.Abu El-Asrar AM, Steen PE, Al-Amro SA, et al. Expression of angiogenic and fibrogenic factors in proliferative vitreoretinal disorders. Int Ophthalmol. 2007;27:11–22. doi: 10.1007/s10792-007-9053-x. [DOI] [PubMed] [Google Scholar]
- 197.Oshitari T, Yamamoto S, Hata N, et al. Mitochondria- and caspase-dependent cell death pathway involved in neuronal degeneration in diabetic retinopathy. Br J Ophthalmol. 2008;92:552–6. doi: 10.1136/bjo.2007.132308. [DOI] [PubMed] [Google Scholar]
- 198.Schmidt KG, Bergert H, Funk RH. Neurodegenerative diseases of the retina and potential for protection and recovery. Curr Neuropharmacol. 2008;6:164–78. doi: 10.2174/157015908784533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Oshitari T, Roy S. Diabetes: a potential enhancer of retinal injury in rat retinas. Neurosci Lett. 2005;390:25–30. doi: 10.1016/j.neulet.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 200.Yang LP, Sun HL, Wu LM, et al. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:2319–27. doi: 10.1167/iovs.08-2642. [DOI] [PubMed] [Google Scholar]
- 201.Fox TE, Han X, Kelly S, et al. Diabetes alters sphingolipid metabolism in the retina: a potential mechanism of cell death in diabetic retinopathy. Diabetes. 2006;55:3573–80. doi: 10.2337/db06-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nakazawa T, Takahashi H, Nishijima K, et al. Pitavastatin prevents nmda-induced retinal ganglion cell death by suppressing leukocyte recruitment. J Neurochem. 2007;100:1018–31. doi: 10.1111/j.1471-4159.2006.04274.x. [DOI] [PubMed] [Google Scholar]
- 203.Zhang J, Wu Y, Jin Y, et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci. 2008;49:732–42. doi: 10.1167/iovs.07-0721. [DOI] [PubMed] [Google Scholar]
- 204.Zhang B, Ma JX. Serpina3k prevents oxidative stress induced necrotic cell death by inhibiting calcium overload. PLoS One. 2008;3:e4077. doi: 10.1371/journal.pone.0004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Reiter CE, Wu X, Sandirasegarane L, et al. Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes. 2006;55:1148–56. doi: 10.2337/diabetes.55.04.06.db05-0744. [DOI] [PubMed] [Google Scholar]
- 206.Fort PE, Freeman WM, Losiewicz MK, et al. The retinal proteome in experimental diabetic retinopathy: up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics. 2009;8:767–79. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schlingemann RO, Witmer AN. Treatment of retinal diseases with vegf antagonists. Prog Brain Res. 2009;175:253–67. doi: 10.1016/S0079-6123(09)17517-9. [DOI] [PubMed] [Google Scholar]
- 208.Kilic U, Kilic E, Jarve A, et al. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating erk-1/2 and akt pathways. J Neurosci. 2006;26:12439–46. doi: 10.1523/JNEUROSCI.0434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nishijima K, Ng YS, Zhong L, et al. Vascular endothelial growth factor-a is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–8. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Barber AJ, Antonetti DA, Kern TS, et al. The ins2akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–8. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 212.Gastinger MJ, Kunselman AR, Conboy EE, et al. Dendrite remodeling and other abnormalities in the retinal ganglion cells of ins2 akita diabetic mice. Invest Ophthalmol Vis Sci. 2008;49:2635–42. doi: 10.1167/iovs.07-0683. [DOI] [PubMed] [Google Scholar]
- 213.Gastinger MJ, Singh RS, Barber AJ. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and ins2akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci. 2006;47:3143–50. doi: 10.1167/iovs.05-1376. [DOI] [PubMed] [Google Scholar]
- 214.Smith SB, Duplantier J, Dun Y, et al. In vivo protection against retinal neurodegeneration by sigma receptor 1 ligand (+)-pentazocine. Invest Ophthalmol Vis Sci. 2008;49:4154–61. doi: 10.1167/iovs.08-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yoshioka M, Kayo T, Ikeda T, et al. A novel locus, mody4, distal to d7mit189 on chromosome 7 determines early-onset niddm in nonobese c57bl/6 (akita) mutant mice. Diabetes. 1997;46:887–94. doi: 10.2337/diabetes.46.5.887. [DOI] [PubMed] [Google Scholar]
- 216.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 217.Barile GR, Pachydaki SI, Tari SR, et al. The rage axis in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:2916–24. doi: 10.1167/iovs.04-1409. [DOI] [PubMed] [Google Scholar]
- 218.Sohn EJ, Kim YS, Kim CS, et al. Kiom-79 prevents apoptotic cell death and ages accumulation in retinas of diabetic db/db mice. J Ethnopharmacol. 2009;121:171–4. doi: 10.1016/j.jep.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 219.Ning X, Baoyu Q, Yuzhen L, et al. Neuro-optic cell apoptosis and microangiopathy in kkay mouse retina. Int J Mol Med. 2004;13:87–92. [PubMed] [Google Scholar]
- 220.Martin PM, Roon P, Ells TK, et al. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004;45:3330–6. doi: 10.1167/iovs.04-0247. [DOI] [PubMed] [Google Scholar]
- 221.Sanderson TH, Kumar R, Murariu-Dobrin AC, et al. Insulin activates the pi3k-akt survival pathway in vulnerable neurons following global brain ischemia. Neurol Res. 2009;31:947–58. doi: 10.1179/174313209X382449. [DOI] [PubMed] [Google Scholar]
- 222.Felice FG, Vieira MN, Bomfim TR, et al. Protection of synapses against alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–6. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Watanabe M, Osada J, Aratani Y, et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–9. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Aydemir O, Turkcuoglu P, Guler M, et al. Plasma and vitreous homocysteine concentrations in patients with proliferative diabetic retinopathy. Retina. 2008;28:741–3. doi: 10.1097/IAE.0b013e31816079fb. [DOI] [PubMed] [Google Scholar]
- 225.Wright AD, Martin N, Dodson PM. Homocysteine, folates, and the eye. Eye (Lond) 2008;22:989–93. doi: 10.1038/sj.eye.6703061. [DOI] [PubMed] [Google Scholar]
- 226.Brazionis L, Rowley K, Sr, Itsiopoulos C, et al. Homocysteine and diabetic retinopathy. Diabetes Care. 2008;31:50–6. doi: 10.2337/dc07-0632. [DOI] [PubMed] [Google Scholar]
- 227.Roedl JB, Bleich S, Reulbach U, et al. Vitamin deficiency and hyperhomocysteinemia in pseudoexfoliation glaucoma. J Neural Transm. 2007;114:571–5. doi: 10.1007/s00702-006-0598-z. [DOI] [PubMed] [Google Scholar]
- 228.Roedl JB, Bleich S, Reulbach U, et al. Homocysteine in tear fluid of patients with pseudoexfoliation glaucoma. J Glaucoma. 2007;16:234–9. doi: 10.1097/IJG.0b013e31802d6942. [DOI] [PubMed] [Google Scholar]
- 229.Ganapathy PS, Moister B, Roon P, et al. Endogenous elevation of homocysteine induces retinal neuron death in the cystathionine-beta-synthase mutant mouse. Invest Ophthalmol Vis Sci. 2009;50:4460–70. doi: 10.1167/iovs.09-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ganapathy PS, Roon P, Moister TK et al (2009) Diabetes accelerates retinal neuronal cell death in a mouse model of endogenous hyperhomocysteinemia. Ophthalmology and eye Diseases. In press. [DOI] [PMC free article] [PubMed]
- 231.Seigel GM. Establishment of an e1a-immortalized retinal cell culture. In Vitro Cell Dev Biol Anim. 1996;32:66–8. doi: 10.1007/BF02723034. [DOI] [PubMed] [Google Scholar]
- 232.Nakanishi Y, Nakamura M, Mukuno H, et al. Latanoprost rescues retinal neuro-glial cells from apoptosis by inhibiting caspase-3, which is mediated by p44/p42 mitogen-activated protein kinase. Exp Eye Res. 2006;83:1108–17. doi: 10.1016/j.exer.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 233.Kline CL, Schrufer TL, Jefferson LS, et al. Glucosamine-induced phosphorylation of the alpha-subunit of eukaryotic initiation factor 2 is mediated by the protein kinase r-like endoplasmic-reticulum associated kinase. Int J Biochem Cell Biol. 2006;38:1004–14. doi: 10.1016/j.biocel.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 234.Shen J, Wu Y, Xu JY et al (2009) Erk- and akt-dependent neuroprotection by erythropoietin (epo) against glyoxal-ages via modulation of bcl-xl, bax and bad. Invest Ophthalmol Vis Sci (Epub ahead of print). [DOI] [PubMed]
- 235.Abcouwer SF, Shanmugam S, Gomez PF, et al. Effect of il-1beta on survival and energy metabolism of r28 and rgc-5 retinal neurons. Invest Ophthalmol Vis Sci. 2008;49:5581–92. doi: 10.1167/iovs.07-1032. [DOI] [PubMed] [Google Scholar]
- 236.Santiago AR, Rosa SC, Santos PF, et al. Elevated glucose changes the expression of ionotropic glutamate receptor subunits and impairs calcium homeostasis in retinal neural cells. Invest Ophthalmol Vis Sci. 2006;47:4130–7. doi: 10.1167/iovs.06-0085. [DOI] [PubMed] [Google Scholar]
- 237.Krishnamoorthy RR, Agarwal P, Prasanna G, et al. Characterization of a transformed rat retinal ganglion cell line. Brain Res Mol Brain Res. 2001;86:1–12. doi: 10.1016/S0169-328X(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 238.Bergen NJ, Wood JP, Chidlow G, et al. Recharacterization of the rgc-5 retinal ganglion cell line. Invest Ophthalmol Vis Sci. 2009;50:4267–72. doi: 10.1167/iovs.09-3484. [DOI] [PubMed] [Google Scholar]
- 239.Ganapathy PS, Dun Y, Ha Y, et al. Sensitivity of staurosporine-induced differentiated rgc-5 cells to homocysteine. Curr Eye Res. 2010;35:80–90. doi: 10.3109/02713680903421194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Osborne NN, Ji D, Majid A et al (2009) Acs67, a hydrogen sulphide-releasing derivative of latanoprost acid, attenuates retinal ischemia and oxidative stress to rgc-5 cells in culture. Invest Ophthalmol Vis Sci (Epub ahead of print). [DOI] [PubMed]
- 241.Biswas SK, Zhao Y, Sandirasegarane L. Imatinib induces apoptosis by inhibiting pdgf- but not insulin-induced pi 3-kinase/akt survival signaling in rgc-5 retinal ganglion cells. Mol Vis. 2009;15:1599–610. [PMC free article] [PubMed] [Google Scholar]
- 242.Koriyama Y, Ohno M, Kimura T, Kato S. Neuroprotective effects of 5-s-gad against oxidative stress-induced apoptosis in rgc-5 cells. Brain Res. 2009;1296:187–95. doi: 10.1016/j.brainres.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 243.Sharma RK, Chalam KV. In vitro evaluation of bevacizumab toxicity on a retinal ganglion cell line. Acta Ophthalmol. 2009;87:618–22. doi: 10.1111/j.1755-3768.2008.01410.x. [DOI] [PubMed] [Google Scholar]
- 244.Schallenberg M, Charalambous P, Thanos S. Gm-csf regulates the erk1/2 pathways and protects injured retinal ganglion cells from induced death. Exp Eye Res. 2009;89:665–77. doi: 10.1016/j.exer.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 245.Kanamori A, Naka M, Fukuda M, et al. Tafluprost protects rat retinal ganglion cells from apoptosis in vitro and in vivo. Graefes Arch Clin Exp Ophthalmol. 2009;247:1353–60. doi: 10.1007/s00417-009-1122-6. [DOI] [PubMed] [Google Scholar]
- 246.Frassetto LJ, Schlieve CR, Lieven CJ, et al. Kinase-dependent differentiation of a retinal ganglion cell precursor. Invest Ophthalmol Vis Sci. 2006;47:427–38. doi: 10.1167/iovs.05-0340. [DOI] [PubMed] [Google Scholar]
- 247.Lieven CJ, Millet LE, Hoegger MJ, et al. Induction of axon and dendrite formation during early rgc-5 cell differentiation. Exp Eye Res. 2007;85:678–83. doi: 10.1016/j.exer.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Iizuka Y, Hong S, Kim CY, et al. Agmatine pretreatment protects retinal ganglion cells (rgc-5 cell line) from oxidative stress in vitro. Biocell. 2008;32:245–50. [PubMed] [Google Scholar]
- 249.Harvey R, Chintala SK. Inhibition of plasminogen activators attenuates the death of differentiated retinal ganglion cells and stabilizes their neurite network in vitro. Invest Ophthalmol Vis Sci. 2007;48:1884–91. doi: 10.1167/iovs.06-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Barres BA, Silverstein BE, Corey DP, et al. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- 251.Dun Y, Mysona B, Ells T, et al. Expression of the cystine-glutamate exchanger (xc-) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Dun Y, Thangaraju M, Prasad P, et al. Prevention of excitotoxicity in primary retinal ganglion cells by (+)-pentazocine, a sigma receptor-1 specific ligand. Invest Ophthalmol Vis Sci. 2007;48:4785–94. doi: 10.1167/iovs.07-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Desco MC, Asensi M, Marquez R, et al. Xanthine oxidase is involved in free radical production in type 1 diabetes: Protection by allopurinol. Diabetes. 2002;51:1118–24. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 254.Kutty RK, Samuel W, Chen S, et al. Immunofluorescence analysis of the expression of norpeg (rai14) in retinal muller and ganglion cells. Neurosci Lett. 2006;404:294–8. doi: 10.1016/j.neulet.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 255.Umapathy NS, Gnana-Prakasam JP, Martin PM, et al. Cloning and functional characterization of the proton-coupled electrogenic folate transporter and analysis of its expression in retinal cell types. Invest Ophthalmol Vis Sci. 2007;48:5299–305. doi: 10.1167/iovs.07-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dun Y, Duplantier J, Roon P, et al. Serine racemase expression and d-serine content are developmentally regulated in neuronal ganglion cells of the retina. J Neurochem. 2008;104:970–8. doi: 10.1111/j.1471-4159.2007.05015.x. [DOI] [PubMed] [Google Scholar]
- 257.Umapathy NS, Dun Y, Martin PM, et al. Expression and function of system n glutamine transporters (sn1/sn2 or snat3/snat5) in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008;49:5151–60. doi: 10.1167/iovs.08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]