

Abstract
Advanced drug delivery systems (DDS) present indubitable benefits for drug administration. Over the past three decades, new approaches have been suggested for the development of novel carriers for drug delivery. In this review, we describe general concepts and emerging research in this field based on multidisciplinary approaches aimed at creating personalized treatment for a broad range of highly prevalent diseases (e.g., cancer and diabetes). This review is composed of two parts. The first part provides an overview on currently available drug delivery technologies including a brief history on the development of these systems and some of the research strategies applied. The second part provides information about the most advanced drug delivery devices using stimuli-responsive polymers. Their synthesis using controlled-living radical polymerization strategy is described. In a near future it is predictable the appearance of new effective tailor-made DDS, resulting from knowledge of different interdisciplinary sciences, in a perspective of creating personalized medical solutions.
Keywords: Drug delivery system, Polymers, Stimuli-responsive polymers, Controlled/living radical polymerization
An overview in drug delivery systems
The introduction of drugs in human body may be accomplished by several anatomic routes [1]. In order to achieve the therapeutic purpose, the choice of the most suitable administration route is of unquestionable importance. Therefore, several factors must be taken into consideration when administrating a drug, namely its own properties, the disease to be treated and the desired therapeutic time. The drugs can be administrated directly to the target tissue or organ or can be delivered by systemic routes [2]. Systemic drug delivery routes are presented systematically in Table 1.
Table 1.
Classification of systemic drug delivery routes (adapted from [3])
| Anatomic routes for systemic drug delivery |
|---|
| Gastrointestinal systems |
| Oral |
| Rectal |
| Parenteral |
| Subcutaneous injection |
| Intramuscular injection |
| Intravenous injection |
| Intra-arterial injection |
| Transmucosal |
| Transnasal |
| Pulmonary (inhalation) |
| Transdermal |
| Intra-osseous |
Pharmaceutical treatments started plenty of decades, or even centuries ago (e.g., aspirin since 1828) either with the oral administration of solid pills (and liquids) [4], or with injectables active chemical drugs [5]. When either of these methods is applied, drug dose maintenance in the body is achieved by repeated administrations. Despite the effectiveness of these treatments, dose peaks at administration times alternated with sub-therapeutic drug levels are unavoidable. Therefore, the impossibility of controlling the drug level over a long period of time constituted an important drawback. During the past two decades, new approaches and strategies have been developed to control several parameters considered essential for enhancing the treatment performance such as the rate, period of time and targeting of delivery. This was the beginning of the so called drug delivery systems [3].
The main purpose of using a DDS is, as implied, not only to deliver a biologically active compound in a controlled manner (time period and releasing rate) but also to maintain drug level in the body within therapeutic window (Fig. 1). Besides, one can direct the drug towards a specific organ or tissue (targeted drug delivery) [6]. The first two features were addressed by using drug carriers, usually polymers (either biopolymers or synthetic polymers) which properties could be manipulated in order to improve DDS efficiency.
Fig. 1.
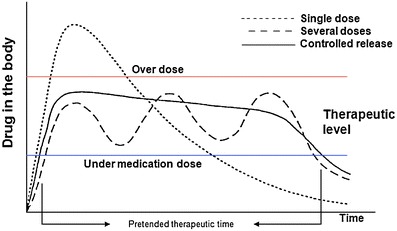
Scheme of the effect in drug concentration in the body when using different administration methods (adapted from [6])
Although both natural (Table 2) and synthetic polymers (Table 3) are being used in the preparation of DDS, there are some advantages that can be pointed to synthetic macromolecules [7].
Table 2.
Molecular structures of natural polymers used in drug delivery applications
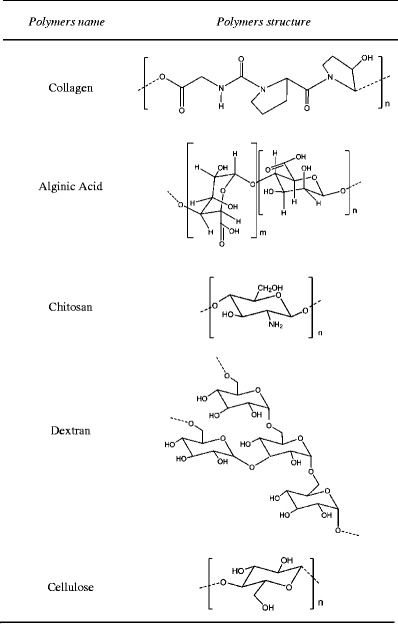
Table 3.
Molecular structures of synthetic polymers used in drug delivery applications
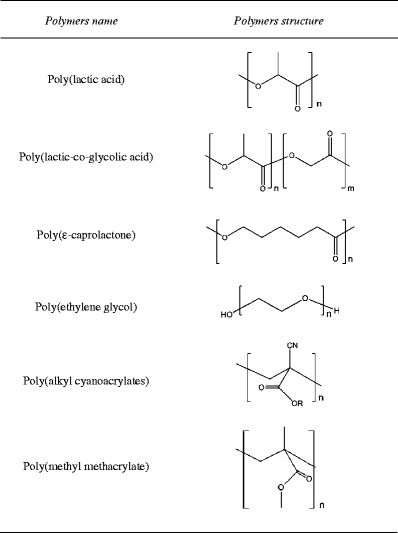
When the polymers are man-made, it becomes possible to control some aspects of polymer structure that allows producing tailor-made materials suitable to the desired biological application [8]. Also, three-dimensional structure as well as chemical composition can be controlled in order to adjust materials properties and orientation of specifics functional groups that can interact with the drug.
However, attention must be paid to molecular weight of synthetic polymers which are not biodegradable. Since biodegradation does not always occur, synthetic polymers must be eliminated through renal excretion [9]. Therefore, they should present a uniform molecular weight distribution that fits under the threshold of renal excretion. As further described in this paper, controlled/living radical polymerization is a very reliable and applied technology in order to obtain well defined macromolecular structures with narrow range molecular weights distributions [10].
DDS present several advantages. These include important factors from decrease of drug side-effects to increased patient compliance. However, DDS disadvantages are also well-known, e.g., DDS final price among others. DDS main advantages and disadvantages are summarized in Table 4.
Table 4.
DDS advantages and disadvantages
| DDS advantages |
| Extension of the duration of action and bioavailability of the drug |
| Minimization of drug degradation and loss |
| Prevention of drug’s adverse side-effects |
| Reduction of dosing frequency |
| Minimization of drug concentration fluctuations in plasma level |
| Improved drug utilization |
| Improved patient compliance |
| DDS disadvantages |
| Possibility of toxicity of the materials |
| Harmful degradation products |
| Necessity of surgical intervention either on systems application or removal |
| Patients discomfort with DDS device usage |
| High cost of final product |
Targeted drug delivery aroused the interest of the scientific community and consequently has witnessed tremendous developments over the last decade. The active compounds targeting involves the conjunction of different areas related to active compounds design, active compounds carriers, biological systems, genetic approaches and precise design of new molecules.
In order to improve the effectiveness of the existing methods for drug delivery, several steps need to be accomplished. The main goal is generically related to deliver suitable active compounds at a desired target without any sign of degradation during the whole process. The development of a controlled delivery system that can dose orally, being less expensive and less painful for the patients and at the same time extremely effective considering a specific disease represents a final target for the research community [11].
DDS must possess some features. The system should be recognized by the specific target tissues [12]. In fact, the delivering of the drug in a specific area of the body is extremely important, in terms of lowering possible side-effects of the active compounds, when enter non-targeted organs and tissues. The polymeric carrier itself, once in the targeting area should be able to control the drug administration by means of either a physiological or chemical trigger. The design of a delivery system should be done in such a way that it would be suitable for specific areas of the body, where it could be degraded by environmental conditions e.g., pH of the stomach or the presence of some enzymes [3].
Polymeric based DDS currently available can be classified as four different categories: diffusion-controlled systems, chemically controlled systems, solvent-activated systems, and magnetically controlled systems [13].
Diffusion controlled systems enclose both reservoir and matrix systems. The first type of system is based in a polymeric membrane that surrounds a core containing the drug, while the second type is based on a polymer matrix in which the drug is distributed homogeneously. Drug release is, in both cases, controlled by diffusion (Fig. 2). However, attention must be paid to the resistance of the polymeric membrane of the reservoir systems since its rupture would cause an abrupt drug release [14].
Fig. 2.
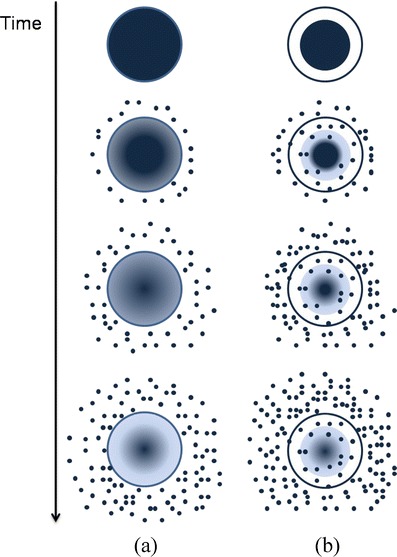
Drug diffusion profile for both matrix (a) and reservoir systems (b)
Chemically controlled systems include polymer-drug conjugates in which drug molecules are linked to a polymeric backbone often by means of a spacer molecule. Once inside the body the linkage between polymer carrier and the drug is cleaved either by hydrolysis or enzyme cleavage. Different types of biodegradable or hydrolysable chemical linkages are used to attach the drug to the polymer backbone (Fig. 3) [15].
Fig. 3.
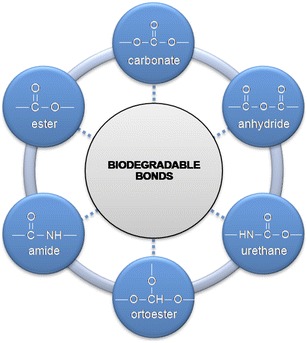
Biodegradable chemical linkages
These polymer-drug conjugates usually possess a transport system which is responsible for directing of the polymer to target organs or tissues (Fig. 4).
Fig. 4.
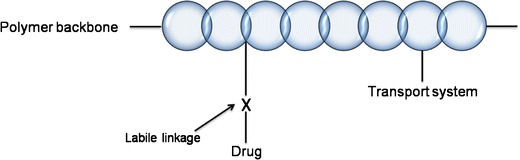
Schematization of general structure of polymer-drug conjugate
Another type of chemically controlled system is the one that involves the use of biodegradable/bioerodible polymers. The distinction between these two concepts is based on how degradation occurs. The term biodegradation is usually applied when polymer molecular weight decreases (by chain cleavage, Fig. 5) while bioerosion is used when the mass of the system diminishes in which case we can have surface or bulk eroding polymers (Fig. 6) [16]. In both cases the polymeric chains matrix disruption is the responsible for the drug release. By controlling polymer degradation rates it is possible to control drug delivery kinetics.
Fig. 5.
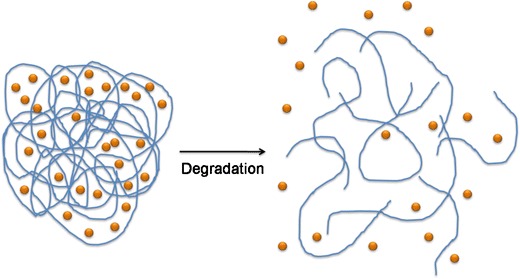
Biodegradation of polymer chains with consequent drug release
Fig. 6.
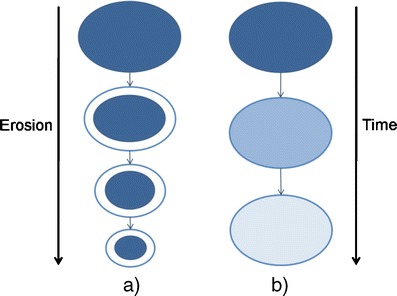
Scheme representing surface (a) and bulk (b) bioerosion
Solvent activated systems can be controlled either by swelling or by osmosis. Swelling controlled systems are based on a hydrophilic polymeric crosslinked chain that is able to absorb large amounts of water without dissolving. This water uptake allows the drug inside the system to diffuse outwards at a velocity that depends on the amount of water that enters the polymeric matrix (Fig. 7).
Fig. 7.
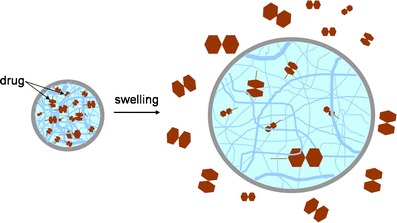
Drug release resulting from swelling of a polymeric matrix
Osmotically controlled systems relies on a device containing a semipermeable membrane through which a solvent without or with small amount of drug flows toward a chamber in which the drug is contained [17]. The solvent flow increases pressure inside the chamber containing the drug and forces the exit of the drug though an orifice present in the device (Fig. 8).
Fig. 8.
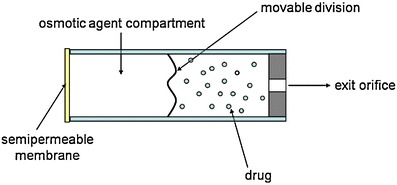
Scheme of an osmotically controlled DDS
Finally, magnetically controlled systems have been developed mostly by combining a polymer with magnetic microparticles. Due to these magnetic properties, the particles movement inside the body can be influenced by an externally applied magnetic field. This specific force combines with the hemodynamic force of the bloodstream resulting in a final motion force. However, in order to obtain an effective control over the particles movement, the external magnetic force has to overcome the hemodynamic force. Therefore, when in vivo application is desired, low values of magnetic fields must be applied. For this reason, materials with high magnetization at room temperature must be used. Among these, the most applied ones are iron, cobalt and nickel [18].
A quite used strategy in developing DDS using magnetic particles is their association with a smart polymer [19, 20]. This type of polymers will be further discussed in this paper, but one can advance that these polymers are sensitive and respond to some external stimuli, such as temperature and pH. The most studied of these smart polymers for drugs delivery applications are the thermo-responsive polymers since the temperature is an easily controlled parameter [21]. The most interesting point on using thermo-responsive hydrogels combined with magnetic particles results from the ability of these same particles to produce heat because of hysteresis energy loss when subjected to an external magnetic field [22]. Therefore, it is possible to produce a DDS that would be activated when a magnetic field is applied externally causing this energy release from the magnetic particles. This type of systems has been mainly applied in cancer treatment by attaching specific antibodies to their surface that allow a targeted delivery of the system (Fig. 9) [23, 24].
Fig. 9.
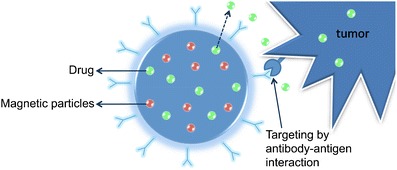
Drug loaded magnetic particle with specific antibodies attached to the surface applied in cancer treatment
In summary, advanced controlled DDS present indubitable advantages for pharmacologically active compounds administration. Owing to rapid advances in recent years, the application of polymers to drug delivery has grown noticeably. Different treatment methods aiming to control several diseases are currently available while some are still under development or even in researchers’ imagination.
Polymers in drug delivery systems
As already mentioned, DDS can be produced by using natural or synthetic polymers, which can be biodegradable or non-biodegradable (see Fig. 10).
Fig. 10.
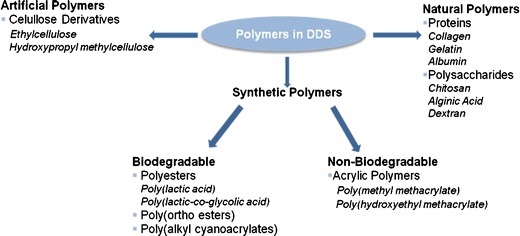
Overview of the polymers used in DDS
These polymeric systems can be used in the release of drugs, proteins and cells. The polymers used in DDS should present a set of properties that make them suitable materials to interact with the human body, as discussed in the previous section, being the biodegradability one of the most important features.
Biodegradable polymers are particularly attractive for application in DDS since, once introduced into the human body, they do not require removal or additional manipulation. Their degradation products are normal metabolites of the body or products that can be metabolized and easily cleared from the body [25, 26].
Some natural polymers, being biodegradable and with excellent biocompatibility, are very attractive materials for use in DDS. Besides, they are relatively inexpensive. However, some of them present some limitations, namely antigenicity, risk of viral infection and non-uniformity in the properties from batch-to-batch [25, 27].
Moreover, synthetic polymers offer a wide variety of compositions with adjustable properties. These materials open the possibility of developing new DDS with specific properties (chemical, interfacial, mechanical and biological) for a given application, simply by changing the building blocks or the preparation technique. Since their preparation is very reproducible, it is possible to prepare DDS with the same specifications quite easily [25–27]. Synthetic polymers can also be either biodegradable or non-biodegradable. Biodegradable synthetic polymers are those containing in their polymer backbone linkages such as ester, orthoester, amide, urea or urethane [26]. Thus, it is possible to increase biodegradability of non-biodegradable polymers by introducing such moieties in their backbone.
Another possible strategy is the design of DDS comprising natural and synthetic blocks. The final purpose is to gather in the same material the best properties of the natural polymer (biodegradability and biocompatibility) and the synthetic polymer (e.g., mechanical properties) [28, 29].
Natural polymers
Along this subsection, it is intended to give an overview of the most currently used natural biodegradable polymers in drug delivery devices. The source of the materials and some of the main application as DDS are presented.
Proteins
As it is well-known, proteins are high molecular weight compounds composed by amino acid residues, linked together by peptide (amide) linkages. They are the main structural components in human tissues. Among these, collagen, gelatin and albumin have been used in the design of DDS.
Collagen
Collagen is the most abundant protein in the human body, being the major component of skin, cartilage and bone. This natural polymer is non-toxic, biodegradable and low-immunogenic [30]. Collagen has been used in DDS, in a variety of shapes, namely microspheres, minirods and sponges [31].
Collagen microparticles have been tested as a carrier system for glucocorticoids, namely hydrocortisone. It was found that the release behaviour of this specific drug was not influenced by the pH of the surrounding medium [32].
Collagen minirods (minipellets) were tested as delivery systems for high-molecular-weight drugs [31]. As an example, the work of Metzmacher and co-authors is presented. They prepared an unsoluble and non-crosslinked collagen minirod delivery system, using fluorescein isothiocyanate (FTIC)-dextran as model drug. The results showed that the release profile of the drug from the matrix is a two step process being dependent on the molecular weight of the drug and length of mini-rod [33].
Collagenous matrices (sponges) have also been used in the sustained release of growth factors (e.g., vascular endothelial growth factor, VEGF and fibroblast growth factor, bFGF), as demonstrated by Kanematsu and co-authors [34].
Gelatin
Gelatin is a protein obtained by the partial hydrolysis of collagen. In this process, the collagen is converted to an unoriented water-soluble protein. In aqueous solution, gelatin undergoes a sol-gel transition, when temperature is lowered below 35°C. This compound being biodegradable, biocompatible and non-immunogenic is commonly used in biomedical field (e.g., drug delivery vehicles and wound dressings). Due to its high solubility in water and poor mechanical properties, crosslinking of gelatin with other materials may be necessary [35, 36].
Muvaffak and co-authors prepared gelatin crosslinked microspheres loaded with colchicine, an agent used in the cancer treatment. They obtained high drug entrapment efficiency and the release profiles were found to be dependent on the gelatin and crosslinking agent concentrations [37]. In the same field of research, microspheres of gelatin were used for the entrapment of some anti-cancer drugs, namely doxorubicin [38], 5-fluoruoracil (5-FU), bleomycin (BLM) and mitomycin C (MMC) [39].
Ofokansi and Adikwu used gelatin-mucin microspheres for the delivery of cefuroxime sodium (a bactericidal agent) in the rectal tract. The microspheres presented high drug entrapment efficiency. However, a fast drug release was observed [40].
Gelatin based hydrogels have also been used in the drug delivery of some active compounds like antibacterial proteins. As an example, Kuijpers and co-authors used gelatin and gelatin/chondroitin sulphate hydrogels, crosslinked via a carbodiimide mediated reaction, impregnated in Dracon® (poly(ethylene tereftalate) matrix), as delivery system for antibacterial proteins. Lysozyme was used as a model compound. The results showed an enhancement of both lysozyme loading and sustained release time, with the incorporation of chondroitin sulphate in gelatin hydrogels. An in vivo experiment showed that these materials are well tolerated by living tissues and are completely degraded in 18 days [41, 42].
Recently, Lin and co-authors proposed a novel drug delivery system composed of poly(ε-caprolactone) (PCL) particles embedded in a gelatin film, using ibuprofen as a model drug. This system showed a prolonged sustained drug release and an enhancement in the adhesion properties. This system can be applied, for instance, in wound healing [36].
Albumin
Albumin is the most abundant protein in the human blood plasma. It is hydrosoluble and presents a molecular weight of about 66 kDa [30]. Characteristics like biodegradability, non-toxicity and non-immunogenicity, makes albumin a very promising material for biomedical/pharmaceutical applications, including drug delivery purposes [43]. Albumin microspheres have proved to be a suitable carrier for drugs used in cancer treatment [44]. This is mainly due to the fact that albumin is used by cancer cells as a source of nitrogen and energy, being taken up by tumor cells by a mechanism of fluid phase endocytosis, followed by lysosomal breakdown. With this mechanism, the drugs in the albumin microspheres are delivery in the specific site of action, minimizing systemic toxicity [43, 44]. Albumin microspheres, loaded with anti-cancer drugs, have showed to be efficient in breast cancer treatment [45, 46].
Polysaccharides
Polysaccharides are high molecular weight compounds, composed by monosaccharide repeating units. They present a wide range of properties and structures. The presence of reactive lateral groups allows the modification of their structure, further increasing their possible applications. Among these, chitosan, alginate and dextran are widely used in DDS development.
Chitosan
Chitosan is a cationic polysaccharide obtained by the alkaline deacetylation of chitin (see Fig. 11), that is the main constituent of the shells of marine crustaceans.
Fig. 11.
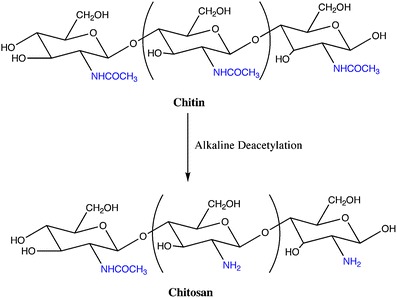
Schematic representation of the alkaline deacetylation of chitin to obtain chitosan
It is insoluble in water and in organic solvents, but it can be dissolved in mildly acidic solutions [47]. This polysaccharide possesses high biodegradability, low toxicity and good biocompatibility and, for that reason, it is widely used in biomedical/pharmaceutical applications, namely in drug delivery devices with different shapes and geometries [47, 48].
Chitosan micro/nanoparticles are a widely used drug delivery devices. Chitosan particles based on ionotropic gelation method (Fig. 12) between sodium tripolyphospate and chitosan were tested as drug carriers for proteins, using bovine serum albumin (BSA) as model compound [49, 50]. The same kind of particles was also investigated as drug carriers for ophthalmologic applications and the obtained results showed that the chitosan nanoparticles are well tolerated by the ocular surface tissues [51, 52]. This method of preparation is very attractive, since it does not require severe reaction conditions, thus maintaining the integrity of the drug [49].
Fig. 12.
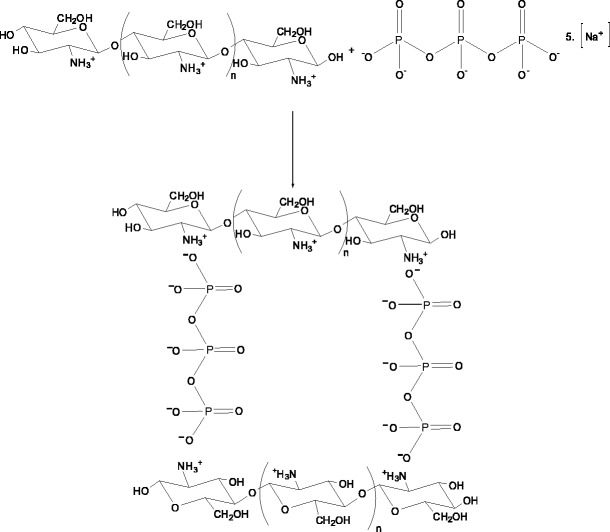
Schematic representation of the interaction of chitosan with sodium tripolyphospate giving micro/nanoparticles
Chitosan particles prepared by the complex coacervation method were used in the encapsulation of genetic material to be applied in gene therapy, as it will be further discussed in this paper. The system showed to be efficient in protecting the genetic material from nuclease attack. The transfection efficiency showed to be dependent on the molecular weight of chitosan, concentration of nucleotide and type of cells [53, 54].
Hydrogels based on chitosan have been used as DDS in the field of cancer treatment, as reviewed by Thu Ta and co-authors. Different methods of preparation and crosslinking agents were presented. Examples of entrapped drugs are paclitaxel, doxorubicin and camptothecin [55].
Alginic Acid
Alginic acid is a cationic polysaccharide extracted from brown algae. This polysaccharide is a block copolymer composed of two uronic acid units: β-D-mannuronic acid and α-L-glucoronic acid (Fig. 13). Alginate’s molecular weight can be higher than 500 kDa [27, 30]. Usually, this material is used in its sodium salt form.
Fig. 13.

Alginate molecular structure
Sodium alginate readily forms gels when in contact with divalent cations (e.g., Ca2+), at ambient temperature. This property is very important, since it opens the possibility of encapsulating some active compounds, under mild conditions, while maintaining their full biological activity [30]. Furthermore, sodium alginate is biocompatible and non-immunogenic. However, it presents the disadvantage of not being enzymatically degraded by mammals [30, 56].
Sodium alginate based hydrogels can be used for the sustained and localized release of low-molecular drugs and macromolecules. The release profile of the drug is dependent on the interaction between the drug and the biopolymer. The release profile of the active compounds can be adjusted by covalently crosslinking alginate with other materials [56].
Angiogenic growth factors, like VEGF and bFGF, have been entrapped in alginate microspheres. In these systems, a fast initial release of the molecules was observed, and to overcome the problem an alginate-heparin system, crosslinked with ethylenediamine was developed. This DDS was designed for specific application in tissue engineering field [57, 58]. Floating alginate beads have been also prepared and have shown to be useful in the delivery of drugs in the gastrointestinal tract [59].
Dextran
Dextran is a polysaccharide of bacterial origin being composed essentially by α-1,6-linked D-glucopyranose units. It may present side branches in the positions α-1,2-, α-1,3- or α-1,4 (Fig. 14) [60, 61].
Fig. 14.
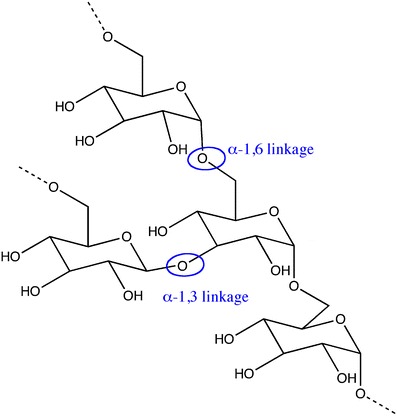
Molecular structure of dextran (adapted from [61])
It is an adequate material for biomedical applications due to its biodegradability, biocompatibility, non-immunogenicity and non-antigenicity [60].
Microspheres based on acrylated dextran, obtained by reaction of dextran with glycidyl methacrylate (Dex-MA) or hydroxyethyl methacrylate (Dex-HEMA), have been used for the controlled release of proteins. These microspheres present the advantage of being prepared in aqueous medium [62]. In vivo experiments, performed in rats, showed that these polysaccharide microspheres are well tolerated when injected subcutaneously [63].
Casadei and co-authors developed a DDS for ibuprofen, comprising solid lipid nanoparticles embedded in a Dex-MA hydrogel, crosslinked by ultraviolet (UV) radiation. This system permitted to obtain a percentage of drug retention nearly 60%, after 2 h in acidic medium, with a subsequent slow release in neutral medium. These results indicate that this system is adequated for modified delivery oral formulations of lipophilic drugs [64].
Recently, Raemdonck and co-authors evaluated the potential application of dextran-UV photopolymerized hydrogel nanoparticles as a carrier for genetic material. The hydrogel particles presented a high loading capacity. The citoxicity tests done with a human hepatoma cellular line HuH-7 demonstrated that these particles are slightly citotoxic. It was shown that the efficiency of gene silencing depends on the degradation profile of the nanoparticles. This can be modified by changing the derivatization degree of dextran [65].
Recently, Horning and co-authors, prepared a prodrug made from dextran and hydrophobic drugs (iboprufen and naproxen), in a N,N′-carbonyldiimidazole mediated reaction [60]. This prodrug is hydrophobic in nature and, when in contact with water or water miscible solvents, self-assembles into nanoparticles. These nanoparticles presented high load efficiency and showed to be stable under pHs in the range of 4 to 11, for several months. This system seems to be reliable for the sustained release of hydrophobic drugs.
Artificial polymers
Cellulose derivatives
Cellulose is the most abundant occurring biopolymer in the nature. It is a linear polymer composed of β(1→4) linked D-glucose units, each one presenting three hydroxyl groups. These hydroxyl groups are responsible for the strong intermolecular and intramolecular H-bonds that are established between the cellulosic chains, making it insoluble in water and organic solvents [66]. Thus, the chemical modification of cellulose is necessary to spread the fields of application of this polymer. Cellulose derivatives are also biocompatible polymers with application in biomedical field.
Hydroxypropylmethylcellulose (HPMC) (Fig. 15) is a cellulose ether widely used in the preparation of drug delivery devices. When in contact with water or biological fluids, this polymer becomes hydrated, leading to a ‘disentanglement’ of the polymeric matrix, forming a swelling gel layer. It is accepted that the drug release from a HPMC matrix comprises two steps: diffusion through the swelling gel layer and release due to the erosion of the swollen matrix [66, 67]. The drug release from this type of matrices can also be influenced by viscosity of the gel layer formed during the hydration of the polymer [66].
Fig. 15.
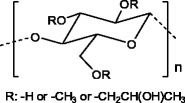
Hydroxypropylcellulose structure
Along the years, HPMC has been used as carrier for several drugs and the factors influencing the release behaviour have been studied, as documented by Kamel and co-authors [66]. Recently, a HPMC-indomethacin (an anti-inflammatory drug) composite was formulated by supercritical fluid (e.g., sc-CO2) assisted impregnation method [68]. The results indicated that hydrogen bonding is the primarily form of interaction between the polymer and the drug. Various processing conditions were used: the HPMC-indomethacin drug composite processed at 130°C and 17.2 MPa, presented a drug release behaviour that obeyed to a n-power law  , with n = 0.54. This strategy is very interesting and promising since it opens the possibility of preparing natural drug carriers in a ‘green’ way [68].
, with n = 0.54. This strategy is very interesting and promising since it opens the possibility of preparing natural drug carriers in a ‘green’ way [68].
Ethylcellulose (EC) (Fig. 16) is another cellulose derivative used in DDS. EC is a non-ionic cellulose ether, insoluble in water, but soluble in some polar organic solvents. In the last years, EC has been used for the controlled release of various types of drugs: diclofenac sodium [69], ketoprofen [70], betamethazone [71] and more recently, nimesulide [72]. In these contributions, some parameters like drug and polymer concentration or type of solvents were evaluated in order to get improved delivery systems based on this polymer.
Fig. 16.
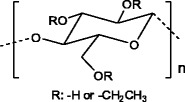
Ethylcellulose structure
Synthetic polymers
Along this section, the most common synthetic polymers used in drug delivery devices will be described. As stated above, this kind of polymers offer the great advantage of being synthesized with specific properties for a given application.
Biodegradable synthetic polymers
Polyesters
Poly(lactic acid) (PLA), and poly(lactic co-glycolic acid) (PLGA) (Fig. 17) are the most widely used polymers in drug delivery devices. This fact can be attributed to their biodegradability, biocompatibility, low-immunogenicity and low-toxicity [26]. A tailored degradation rate of these copolymers can be achieved only by varying the stereochemistry (D or L-lactic acid monomer) and the PLA/PGA (poly(glycolic acid)) ratios [27]. PLGA, due to its higher degradation rate comparatively to PLA, is sometimes the preferred polymer for drug delivery devices.
Fig. 17.
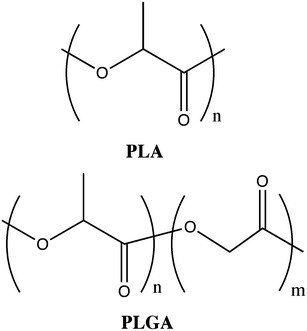
Structures of poly(lactic acid) and poly(lactic co-glycolic acid)
Micro/nanoparticles of PLGA have been used in the controlled delivery of proteins, vaccines, genes, antigens as well as growth factors. An excellent review on this matter was done by Mundargi and co-authors [73]. These particles are also suitable for the encapsulation of anti-cancer drugs [74]. Several studies have been done on the influence of certain parameters (presence or absence of stabilizers in the formulations [75], type of solvent [76] and molecular weight of the drug [77]) in the encapsulation efficiency or drug release profiles from these micro/nanoparticles.
This kind of micro/nanoparticles has demonstrated their potential for application in gene delivery. PLGA particles and a mixture of PLGA particles with polyoxyethylene derivatives were used in the encapsulation of genetic material. These systems have proved to be efficient in protecting the genetic material from the nuclease attack and high transfection efficiencies were obtained [78, 79]. A cationic complex of PLGA with polyethyleneimine was also used in the encapsulation of genetic material. It was shown that, in this particular case, the gene silencing mechanism is performed at the intracellular level [80].
A study done by Kompella and co-authors showed that PLA and PLGA nanoparticles have potentialities for the design of gene therapy strategies for ocular diseases of the posterior segment of the eye.
PCL (Fig. 18) is a semicrystalline polymer with low melting point (Tm = 55–60°C) and glass transition temperature (Tg = −60°C). It possesses a low degradation rate, reason why it has been mainly used for preparation of long-term drug delivery devices [25]. PCL is highly permeable to small drug molecules. Another important feature is related to the non-generation of acidic by-products when it is degraded (contrarily to what happens with PLA and PLGA). Additionally, PCL offers the possibility of being easily blended with other polymers [81].
Fig. 18.
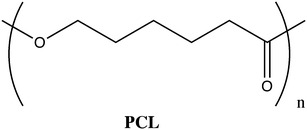
Structure of poly(ε-caprolactone)
PCL, in its native form or blended with other polymers has been used for the encapsulation of several drugs, as reviewed by Sinha and co-authors [81] and more recently by Kumari and co-authors [74].
Poly(ortho esters)
The development of poly(ortho esthers) (POE) is related with the necessity of having more hydrophobic polymers, containing hydrolytically labile chemical bonds, with a surface erosion degradation mechanism instead of a bulk degradation mechanism [25, 30]. Currently, four families of POE are known: POE I, POE II, POE III and POE IV. POE I is obtained by transesterification reaction between a diol and a diethoxytetrahydrofuran. POE II is synthesized from diols and diketene acetal 3,9-bis(ethylidene 2,4,8,10-tetraoxaspiro[5,5] undecane). This polymer is highly hydrophobic. Usually, it is necessary the addition of an acid excipient to make it appreciable degradable under physiological conditions. POE III can be obtained by a reaction between a triol and an ortho ester. The flexibility of the polymer backbone can be easily tailored by the selection of the triol. POE IV is a modification of POE II; in this specific case units of lactic acid or glycolic acid are incorporated in the polymer backbone, which enables the degradation of these polymers, without the addition of acidic excipients. Besides, the rate of degradation of these polymers can be tuned by the amount of lactic or glycolic acid present along the polymer chain [25, 30]. The structures of the mentioned POEs are presented in Fig. 19.
Fig. 19.
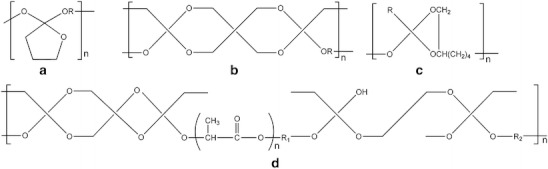
Structures of the different families of poly(ortho esters): a POE I; b POE II; c POE III and d POE IV
POE IV presents a number of advantages over the other POE families, namely the possibility of controlling the polymer properties and erosion rate, high stability at room temperature and drug release dependent on erosion mechanism [82]. Thus, POE IV seems to be the most adequate drug carrier for a variety of drugs, including proteins, in diverse applications, as well documented by Heller who described various types of DDS based on POE [82–84].
Poly(alkylcyanoacrylates)
Poly(alkyl cyanoacrylates) (PACA) (Fig. 20) are biodegradable acrylate polymers, with a wide range of applications in the biomedical/pharmaceutical field. Their C-C bonds are hydrolytic instable, which can be ascribed to the high inductive activation of methylene hydrogen atoms by the electron-withdrawing neighboring groups [25].
Fig. 20.
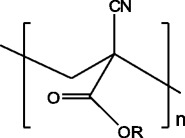
Structure of poly(alkyl cyanoacrylates); R is an alkyl chain of variable length
PACA exhibit high rates of degradation that can vary between hours and days, depending on the alkyl (R) chain length of the polymer. For instance, poly(methyl cyanoacrylate) can degrade within a few hours, but its degradation products (cyanoacetic acid and formaldehyde) are toxic to the organism. Therefore, the research has been directed towards PACA with longer alkyl chains [25, 30].
The development of PACA particles for drug delivery purposes has started two decades ago [85]. Almost all type of drugs have been successfully encapsulated in PACA particles (microparticles, nanoparticles or capsules). Among them, peptides, proteins, oligonucleotides, anti-cancer and anti-infectious compounds as well as anti-inflamatory compounds are included [74, 85]. A comprehensive review on the methods of preparation, potential applications, and drugs commonly incorporated was done by Vauthier and co-authors [85]. Recently, Graf and co-authors reviewed the methods of preparation, the factors influencing the encapsulation efficiency and the drug release profiles. They also presented results of some experiments done in vivo [86].
Non-biodegradable synthetic polymers
Acrylic Polymers
Poly(methyl methacrylate) (PMMA) (Fig. 21), a biocompatible and biostable polymer, was the first acrylic polymer used in a biomedical application.
Fig. 21.
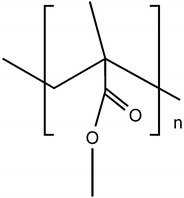
Structure of poly(methyl methacrylate)
PMMA is transparent, does not absorb water being dimensionally stable. Its first biomedical application was in intraocular lens (IOL), just after the Second World War. PMMA is still used in the fabrication of contact lenses [27].
Besides the use of PMMA based materials in ophthalmology, it can also be applied in the orthopedic field; PMMA has been used over 20 years in managing, for example, open fractures, total joint arthroplasty and chronic osteomyelitis. However, some of the applications have been impaired by its bio-inertness and, for this reason, it was proposed the addition of bioactive glasses/ceramics fillers. The work of Lin and co-authors is an example of that. They prepared a PMMA/silica composite via a sol-gel method and tested it as a drug delivery device for anti-inflammatory drugs, using acetylsalicylic acid as model drug. The obtained results showed that interface between polymeric matrix and silica particles plays a key role in drug release behaviour, that demonstrated to be well fitted by the Ficks’ law [87]. PMMA can also be used in orthopedic surgery as an efficient delivery device of anti-microbial agents, as shown by Anguita-Alonso and co-authors [88].
A PMMA microdevice (flat and thin, in order to maximize the area of contact), coated with lectins was successfully used in delivering of drugs to the gastrointestinal tract [89].
PMMA is a versatile biocompatible polymer with applications as DDS in various areas of the biomedical field.
Poly(2-hydroxyethyl methacrylate) (PHEMA) possesses a similar structure to PMMA (Fig. 22). The pendant methylester group in PMMA is substituted by a pendant hydroxyethyl ester group [27].
Fig. 22.
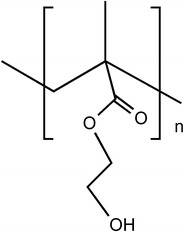
Structure of poly(2-hydroxyethyl methacrylate)
PHEMA is a biostable polymer, with the ability of forming hydrogels. It is a particularly interesting polymer due to its properties which can be easily manipulated, offering the possibility of having tailor-made materials for specific applications. The use of PHEMA based materials in controlled release applications is well known [90, 91]. In some cases, PHEMA delivery systems present an initial drug ‘burst release’: immediately after being hydrated. Therefore, sometimes, it is necessary to proceed to structural modifications. In a very recent work, Anderson and co-authors [92] developed a drug delivery device for the release of norfloxacin based on PHEMA, which surface was hydrophobized by the reaction with octadecyl isocyanate. This system showed to be adequate in the prevention of post-operative infection (endophthalmitis), after a cataract surgery.
Other areas of the biomedical field have been accessed by PHEMA drug carriers, namely cancer treatment [91, 93] and neurologic diseases treatment [94].
Acrylic polymers with pendant acidic (poly(acrylic acid)) and N-substituted acrylamide polymers (poly(N-isopropylacrylamide) (PNIPAAm)) are particularly interesting for drug delivery purposes, since they are stimuli-responsive materials [95]. The potential application of these polymers in DDS will be appropriately discussed in the following sections of this paper.
Responsive polymers
Introduction
Most of polymer-based DDS are hydrogels. Hydrogels are three-dimensional high-molecular weight networks composed of a polymer backbone, water and a crosslinking agent. They are polymeric materials that do not dissolve in water at physiological temperature and pH. Hydrogel are capable of absorbing water without undergoing dissolution due to their chemical structure which include hydrophilic functional groups such as –OH, –COOH, –CONH2, and –SO3H. Being insoluble, these three-dimensional hydrophilic networks can retain a large amount of water that not only contributes to their good blood compatibility but also maintains a certain degree of structural integrity and elasticity [96].
Hydrogels can be classified according to several different criteria depending on their preparation method and physicochemical properties. Table 5 shows some of these criteria.
Table 5.
| Responsive polymer materials | Type of stimulus |
|---|---|
| Bisacrylamide | pH |
| Poly(acrylic acid) | |
| Chitosan | |
| Poly(acetoacetoxyethylmethacrylate) | |
| Poly(acrylamides) | |
| Poly(butyl acrylate) | |
| Poly(2-(diethylamino)ethyl methacrylate) | |
| Poly(2-(diisopropylamino)ethyl methacrylate) | |
| Poly(ethylene oxide) | Temperature |
| Poly(N-isopropylacrylamide) | |
| Poly(propylene oxide) | |
| Poly(dimethylsiloxane) | Electrical, temperature |
| Poly(2-(methacryloyloxy)ethyl phosphorylcholine) | |
| Poly(N-isopropylacrylamide)-co-acrylamide containing ferromagnetic material | Magnetic |
| Poly(N-vinylcaprolactone) | Temperature, pH |
| Poly(N-acryloyl-N-propylpiperazine) |
Hydrogels can be prepared from natural or synthetic polymers [97]. On the other hand, they can be classified according to the nature of the crosslinks as chemical gels (when three-dimensional network is achieved by permanent covalent bonds usually achieved by using crosslinking agents) or as physical gels (formed by the growth of physically connected aggregates). Depending on the nature of the gelling system, in the physical gels, the connections can be achieved via hydrogen bonds, crystalline regions, ionic clusters, or phase-separated microdomains [98–100].
The synthesis and development of polymeric based materials that are able to respond to external conditions enhance even more the importance of polymers in DDS. The development of stimuli-responsive polymers is a broad area that has been attracting interest in the scientific community.
Responsive polymers are materials that can undergo abrupt changes that result from small variations in environmental conditions, such as: temperature, pH, electric charges, ionic strength, electromagnetic radiation, UV/visible light, ionic or metallic interactions or combinations of them. These stimuli can lead to different types of responses, such as degradation, drug release, dissolution/precipitation, swelling/collapsing. Figure 23 illustrates different types of stimuli and possible response from polymers. Table 6 presents some polymeric materials and the external stimulus that they are sensitive to.
Fig. 23.
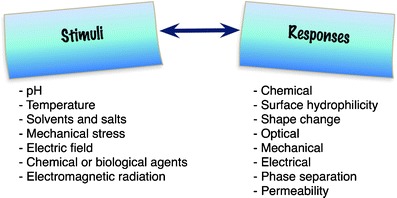
Table 6.
| Responsive polymer materials | Type of stimulus |
|---|---|
| Bisacrylamide | pH |
| Poly(acrylic acid) | |
| Chitosan | |
| Poly(acetoacetoxyethylmethacrylate) | |
| Poly(acrylamides) | |
| Poly(butyl acrylate) | |
| Poly(2-(diethylamino)ethyl methacrylate) | |
| Poly(2-(diisopropylamino)ethyl methacrylate) | |
| Poly(ethylene oxide) | Temperature |
| Poly(N-isopropylacrylamide) | |
| Poly(propylene oxide) | |
| Poly(dimethylsiloxane) | Electrical, temperature |
| Poly(2-(methacryloyloxy)ethyl phosphorylcholine) | |
| Poly(N-isopropylacrylamide)-co-acrylamide containing ferromagnetic material | Magnetic |
| Poly(N-vinylcaprolactone) | Temperature, pH |
| Poly(N-acryloyl-N-propylpiperazine) |
Although all of the previous stimulus has been studied in DDS, most of the works reported in the literature are related to temperature and pH stimulus [96, 103–105]. The main reason for that can be understood by the fact that variations in pH and temperature are easily found in the human body, e.g., fever, diseases or local infections. The deviations from normal values can work as a trigger for reversible phase transitions. The response of hydrogels to external stimulus is evaluated considering different aspects, such as: material change of shape, speed of response, viscoelastic behaviour and shape recovery.
Gels exhibiting a phase transition in response to change in external conditions such as pH, ionic strength, temperature and electric currents are known as “stimuli-responsive” or “smart” gels [6]. Thus, hydrogels have been developed as stimuli-responsive materials, which can undergo abrupt volume change in response to small changes in environmental parameters, as schematically shown in Fig. 24. Here, a drug is released when the material (hydrogel) is submitted to a specific stimulus.
Fig. 24.
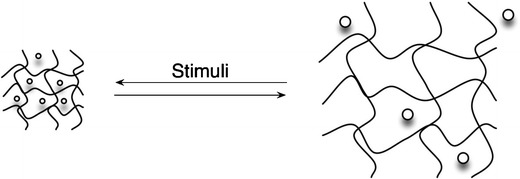
Schematic representation of a stimuli-responsive hydrogel releasing a drug. The predictive transition behaviour of responsive polymers is explained by the readjustment of interactions between polymer-polymer and polymer-solvent in small ranges of pH or temperature. Depending on the polymer structure the stimulus can lead to an abrupt volume change
Synthesis of stimuli-responsive polymers by conventional methods
Several methods can be used to synthesize hydrogels. However, most hydrogels are prepared by radical copolymerization [106], graft copolymerization [107], chemical or physical crosslinkage [108, 109] and ionizing radiation [110].
As represented in Fig. 25, chemical hydrogels are usually synthesized by polymerizing a water-soluble monomer (acrylic acid, acrylamide, hydroxypropylacrylate) in the presence of a bi- or multifunctional crosslinking agent. Another method to obtain a chemical hydrogel is, by chemical reaction, crosslink the functional groups of a water-soluble polymer. These functional groups can be either vinyl groups, hydroxyl groups, amine groups or carboxylic groups [101, 111].
Fig. 25.
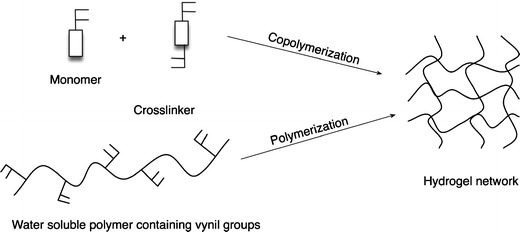
Methods for the synthesis of hydrogels (adapted from [101])
Physical gels are prepared by crosslinking without chemical reaction. They are formed by the growth of non-covalent bonds, which are formed through hydrogen bonding, electrostatic interactions, ionic clusters, antigen-antibody interactions and crystalline regions [100, 101]. These physically crosslinked gels can reversibly degrade into the corresponding precursors upon external stimuli [112].
Physical hydrogels present polymeric network composed by hydrophilic and hydrophobic domains, whereas chemical hydrogels present “clusters” or regions of high crosslinking density (which induce a high swelling structure) [113].
Smart polymeric materials respond to small changes in their environment with significant changes in their properties.
As previously mentioned, several stimuli have been exploited, although most of the works have been related with temperature or pH stimuli. Ideally, the response to the stimuli should be reversible. This fact makes chemical crosslinked hydrogels good candidates to be used in drug delivery applications due to their good mechanical stability [103, 105].
Temperature-responsive polymers
Temperature sensitive (or thermosensitive) hydrogels are among the most studied class of stimuli-responsive polymers for drug delivery systems.
Temperature sensitive polymers present an hydrophobic–hydrophilic balance in their structure and small temperature changes around a critical solution temperature (CST) make the chains collapse or extend, responding to adjustments of the hydrophobic and hydrophilic interactions between the polymer chains and the aqueous medium [114, 115]. A critical solution temperature can be defined as a temperature at which the polymer solution undergoes separation from one phase to two phases. Thus, temperature sensitive polymers undergo an abrupt change in volume as the temperature of the medium is varied above or below the CST [116].
Temperature is the most used triggering signal for DDS, which in principle, can be justified by the fact that the human body temperature frequently deviates from the normal value (37°C) in the presence of strange microorganisms. The idea to have a device able to recognize this deviation and at the same time release a therapeutic agent is particularly interesting. In terms of physical-chemical changes the thermoresponsive hydrogels can involve swelling effects, glass transitions, crystalline melting and thermally reversible transitions.
The temperature sensitive hydrogels are able swell/deswell as a result temperature changes in the environmental medium. According to Peppas and co-authors [117] these materials can be classified into negatively thermosensitive, positively thermosensitive and thermally reversible gels. Table 7 summarizes the most important features of each category.
Table 7.
Classification and characteristics of the different thermosensitive materials
| Classification | Characteristic | Transition | Example | T (°C) | Ref |
|---|---|---|---|---|---|
| Negatively thermosensitive | Lower critical solution temperature (LCST) | Below LCST the polymer swells, above the polymer contracts | NIPAAm | 32°C in pure water | [118] |
| Positively thermosensitive | Upper critical solution temperature (UCST) | Below UCST the polymer contracts | Poly(acrylamide-co-butyl methacrylate) | [119] | |
| Thermally reversible gels | Gelation temperature | Liquid to a gel | Poly(ethylene glycol-b-poly(lactic acid-co-glycolic acid)-b-poly(ethylene glycol) | [120] |
PNIPAAm hydrogels are typical examples among the temperature sensitive polymers. PNIPAAm gels swell when cooled below their lower critical solution temperature (LCST) around 31–34°C, and they collapse when heated above the LCST.
In earlier studies, Shibayama and co-authors [121–123] have shown that, in a swollen PNIPAAm gel, there are two types of water molecules association. About 10 to 15 water molecules per NIPAAm segment are associated with the phase transition, while about 1 to 3 water molecules per polymer segment may be considered as the lower limit for the hydrophobic hydration. The water molecules in the hydration layer are in an ordered state if the temperature is lower then the LCST. However, if the temperature is above the LCST, water molecules are dissociated.
The concept of drug delivery via temperature sensitive hydrogels can be illustrated according to Fig. 26. In Fig. 26a an hydrophilic drug is trapped in a swollen gel and once the temperature decreases below the LCST the drug is released due to the increase of diffusivity. In Fig. 26b, an hydrophobic drug is release from the matrix when temperature is above LCST. In Fig. 26c, the drug is trapped in the gel due to its heterogeneous nature which above the LCTS form a dense layer while the core remains swollen [105, 124].
Fig. 26.
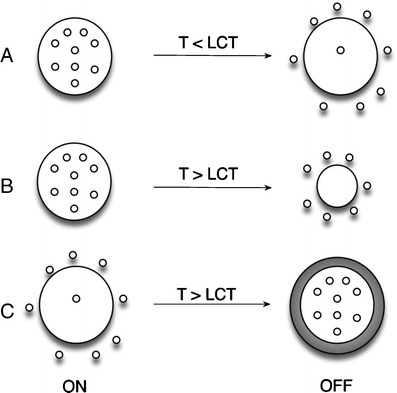
Drug delivery strategies from temperature-responsive hydrogels (adapted from [124])
Kikuchi and co-authors [125] grafted the thermo-responsive PNIPAAm arms onto an inert hydrogel matrix. This allowed a fast responding PNIPAAm hydrogel that can avoid the skin layer formation upon rapid temperature change.
Recently, highly deformable red blood cells were incorporated either between PNIPAAm gel and cover glass or patterned PNIPAAm gel by Pelah and co-authors [126]. When the temperature was increased above the LCST of PNIPAAm, the polymer gel shrinks, which causes the deformation of the embedded cells. The deformation of cells can be transformed into biochemical responses, which play critical roles in cell development, migration, and morphology [126].
pH-responsive materials
The pH changes within the body can be used to induce a response since different organs or tissues have different and specific pH. Table 8 shows the different pH of some organs or tissues within the human body.
Table 8.
pH in the different tissues (adapted from [13])
| Tissue/ organ | pH |
|---|---|
| Blood | 7.35–7.45 |
| Stomach | 1.0–3.0 |
| Duodenum | 4.8–8.2 |
| Colon | 7.0–7.5 |
| Early endosome | 6.0–6.5 |
| Late endosone | 5.0–6.0 |
| Lysosome | 4.5–5.0 |
| Tumor | 6.5–7.2 |
The presence of ionisable weak acid or basic moieties attached to a hydrophobic backbone of the material is the key element for a pH sensitive polymer. Once the side groups are ionized, an extension of the coiled chains occurs in response to electrostatic repulsion of the generated charges formed, which can be either anions or cations.
Schmaljohann [13] showed that the extent of ionization of the pH sensitive polymers depends on the concentration of the pendant acidic/basic groups. All the pH sensitive hydrogels contain pendant basic or acidic groups that are able either to accept or donate protons in response to the environmental pH.
To obtain a pH-responsive polymer, monomers like acrylic acid (AA), methacrylic acid (MAA), maleic anhydride (MA), and N,N-dimethylaminoethylmethacrylate (DMAEMA) are normally used [111]. Polymers containing phosphoric derivatives have been also reported [99].
The swelling and collapsing behaviour induced by a pH-responsive stimulus has been used in controlled release of compounds like caffeine, drugs like indomethacin, or cationic proteins like lysozome [13].
Poly(acrylic acid) has been widely used as a pH-responsive polymer. The carboxylic pendant groups of its chain, as shown in Fig. 27, accept protons at low pH, while releasing them above its pKa (4.28) [127]; therefore the corresponding hydrogels made with this system will exhibit a sudden increase in the hydrodynamic volume and in the swelling capability when the repeating units pass from a ionized to a deionized state. When a basic polymer is used, e.g., poly(N,N′-diethylaminoethylmethacrylate), the opposite effect is observed (Fig. 28).
Fig. 27.
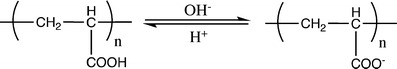
Poly(acrylic acid) behaviour in aqueous solution at low and high pH
Fig. 28.
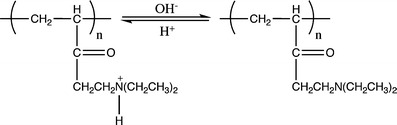
Poly(N,N′-diethylaminoethylmethacrylate) behaviour in aqueous solution at low and high pH
Polysaccharides can also be used as pH-responsive hydrogels. Examples of anionic natural macromolecules are alginate, hyaluronic acid and chondroitin sulphate. On the other hand, only chitosan is a cationic natural polysaccharide [105].
Other studies, which can again be used in drug delivery, have been also performed with micro/nanogels. Mainly, the immobilization of hydrolytically sensitive molecules like peptides and proteins has been accomplished, e.g., van derWeert and co-author [128] immobilized lysozyme in PLGA, and Peppas and co-authors [117] prepared anionic pH-sensitive hydrogels for calcitonin entrapment.
Temperature/pH-responsive materials
The combination of a thermo-responsive monomer (e.g., NIPAAm) and a pH-responsive monomer leads to a double-response copolymer. Temperature/pH-dual-responsive systems may have potential applications in the development of new anti-cancer drug delivery systems, since certain malignancies can alter simultaneously the two parameters around the tumor place, including a slight local increase of the temperature and a minor decrease in extracelular pH.
Ganorkar and co-authors [129] used temperature/pH-sensitive copolymers—Poly(NIPAAm-co-butyl methacrylate-co-AA)—to prepare insulin releasing. At acid pH and body temperature, the beads were insoluble, and thus no drug was released in the stomach. At pH 7.4 and body temperature, the low-molecular weight hydrophilic polymeric beads displayed a hump-like profile and dissolved within 2 h (inducing a controlled release mechanism), while the high-molecular weight hydrophilic polymeric beads swelled and released insulin slowly over a period of 8 h.
More recently, Asoh and co-authors [130] prepared gels with porosity by combining poly(acrylic acid) with porous PNIPAAm. These gels exhibited a faster deswelling in response to both pH and temperature, when compared with the corresponding nonporous gels.
UV and visible light sensitive materials
Light sensitive hydrogels include UV and visible light sensitive hydrogels. These polymer gels go through reversible photomechanical changes when exposed to UV or visible light.
UV sensitive hydrogels bearing triphenylmethane units swell in the presence of UV light and contract when the light is removed. However this volume transition is discontinuous.
In a study of Qiu and co-authors [131], visible light-sensitive hydrogels were prepared using copper chlorophyll bound to NIPAAm, which shrinks, in response to visible light and contracts when the light source is removed. This material may be used in artificial muscles, switches, and memory devices. However, response time is slow and chlorophyll can get leached out of the polymer matrix.
Electric-responsive materials
Kim and co-authors [132] synthesized an hydrogel composed of poly(vinyl alcohol) (PVA) and chitosan which exhibited electro-sensitive behaviour. They investigated the response of the hydrogel in electric fields. A swollen PVA/chitosan network was placed between a pair of electrodes and bending behaviour in response to the applied electric field was noted. The bending angle and the bending speed of the PVA/chitosan interpenetrated network (IPN) increased with increasing applied voltage and concentration of NaCl aqueous solution.
Synthetic as well as naturally occurring polymers either alone or in combination, have also been used. Examples of naturally occurring polymers include hyaluronic acid, chondroitin sulfate, agarose, xanthan gum and calcium alginate. The synthetic polymers are mostly methacrylate and acrylate derivatives such as partially hydrolyzed polyacrylamide, polydimethylaminopropylacrylamide, among others.
Tanaka and co-authors [134] were the first authors to explain the electrically induced anisotropic gel deswelling. They suggested that a force on both the mobile counter ions and the immobile charged groups of the gel’s polymeric network is generated by the electric field. When the gel is not fixed to either electrode, the attractive forces between the immobile negative charges of the polymer network and the anode can result in translational movement of the gels towards the anode.
In a recent study, Bajpai and co-authors [134] impregnated polyaniline into a macromolecular matrix of poly(vinyl alcohol)-g-poly(acrylic acid) and studied the electrical conductivity and electroactive behaviour of the resulting nanocomposite.
Magnetic-responsive materials
Living organisms are deeply influenced by magnetism. The iron-containing protein in our blood (hemoglobin) is magnetic. Blakemore and co-authors [135] found that magnetotactic bacteria were perhaps the first living organisms to orient themselves with the earth’s magnetic field. A work from Bahadur and co-authors [136] showed that all living organisms, including animals and humans, contain magnetic particles that act as magnetic receptors. Several researchers [137–140] have established that magnetism and magnetic materials have strong importance in healthcare and biological applications, such as gene and drug delivery, and magnetic intracellular hyperthermia treatment of cancer.
Cancer treatment by electromagnetically heating was studied by Rabin [141]. Cancer cells can be clinically heated either by ultrasound, radio frequency, thermal radiation, lasers or magnetic nanoparticles. These particles are subjected to an oscillating electromagnetic field so they can act like heaters. Thus, the application of a magnetic field produces a directional force on each magnetic particle. As the magnetic field oscillates at high frequency, the average force on the particle is zero. The energy of the oscillation is converted into heat, raising the temperature of the nanoparticles and their biological material.
Synthesis of stimuli-responsive polymers by controlled/living radical polymerization for DDS
Controlled/Living Radical Polymerization (CLRP) provides a powerful route for the preparation of macromolecules with controlled properties, such as: molecular weight, narrow molecular weight distribution, uniformity, topology, composition, architecture and functionality [142].
The precise synthesis opens unprecedented possibilities to synthesize targeted tailor-made polymers for DDS. The preparation of well defined copolymers based on stimulus-responsive polymers that can be pre-assembled to macrostructures with controlled morphologies is also extremely relevant to enhance the efficiency of drug releasing. In the same way, natural polymers and synthetic polymers can be covalently linked to afford new bioconjugates [143–145]. The covalent attachment of controlled synthetic polymers with well defined structures to biological entities such as nucleic acids, proteins, carbohydrates, virus and cells represents the combination of two fascinating worlds.
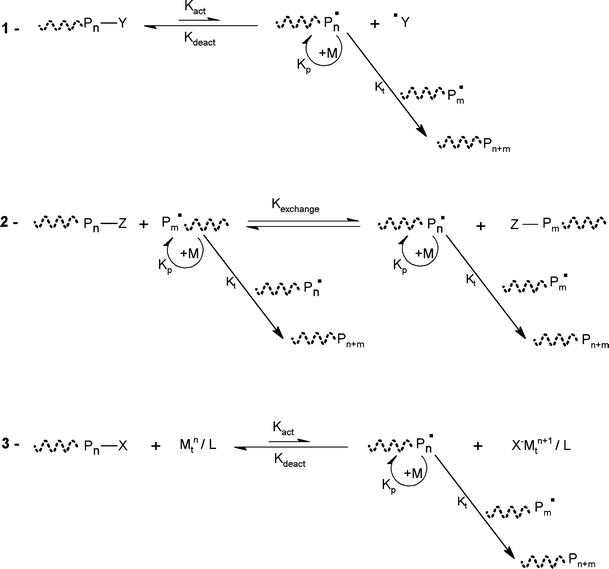
In the last decade, great progress has been made on the development of controlled/living radical polymerization methods [10, 142, 146–148]. The most successful CLRP methods are the stable free radical polymerization (SFRP), atom transfer radical polymerization (ATRP) and degenerative based methods such as reversible addition fragmentation transfer (RAFT) and iodine transfer (IT) (Fig. 29).
Fig. 29.
General schemes of the most used LRP methods: (1) SFRP; (2) RAFT/DT and (3) ATRP
The SFRP [142] uses stable radicals which reversibly react with active radicals together forming dormant covalent species. Several stable radicals have been successfully used to control the radical polymerization. The most used nitroxide is the (2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO)) [149].
RAFT [150] and Degenerative Chain Transfer (DCT) (also known as Degenerative Transfer (DT)) [151, 152] use a chain transfer agent that reacts reversibly with the propagation macro-radical. This reaction between the dormant species and the active radicals results in the transfer of an end-group from the transfer-agent to the radical. For the DT process, this transfer directly involves, for example, an iodine atom. In the RAFT process, an addition-fragmentation process is used to exchange a moiety (for example, a dithioester) between the two chains [153]. The macromolecular design via the interchange of xanthates (MADIX) process is similar to the RAFT process but uses xanthates as transfer-agents [154]. For the RAFT approach, generically the conventional initiators are used as radical source.
Among the various CLRP methods, the ATRP is most studied due to its simplicity, efficiency, high functional tolerance and the fact that most of the initiators and catalysts are available commercially.
The mechanism presented in Fig. 29 consists in the formation of active radicals through a redox process catalyzed by a transition metal complex. The Pn ● are known as propagation radicals, while the Pn−X are defined as dormant species. The transition-metal complex (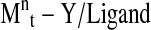 ) plays an indispensable role in this system, since it provides the activation and deactivation processes, which keep the concentration of radicals to be very low. These processes are related to a one-electron oxidation with concomitant abstraction of a (pseudo)halogen atom X from the dormant specie (Pn−X) (n = 0, initiator) [155]. The radicals (Pn ●) are able to react reversibly with the oxidized metal complex (X−Mtn+1 /ligand) to form again the dormant species and the activator. The ligand is essential to the solubilization of the transition metal salt in the organic medium and to the adjustment of the redox potential of the metal center, which defines the reactivity and the dynamics of the atom transfer process [155]. Once the radicals are active, the polymer chain growing process is similar to the free radical polymerization (FRP) process. The equilibrium between the active species and dormant species is shifted towards the dormant species via an excess of the higher oxidation state of the catalyst that is generated by a small amount of radical dimerization, during the initial steps of polymerization. This effect is known as the persistent radical effect (PRE) [156]. Several metal catalysts have been proposed as mediators of the ATRP process. Among these the copper based catalysts are extensively [157] studied due to their potential, low cost and large availability. New ligands for several transition metals have been developed with outstanding results achieved, related to the increase of the catalyst activity (10,000 fold when compared to the initial systems) [158]. In the last decade, the accomplished developments with respect to the capacity to polymerize different monomers and the smoothing of reaction conditions for CLRP methods are remarkable. The different strategies exploit the equilibria between growing radicals and dormant species and minimize the proportion of terminated chains in radical polymerization. However, the key point for the control is that the number of chains is much greater in CLRP than in FRP, therefore the rate of termination per chain is much lower in CLRP [158].
) plays an indispensable role in this system, since it provides the activation and deactivation processes, which keep the concentration of radicals to be very low. These processes are related to a one-electron oxidation with concomitant abstraction of a (pseudo)halogen atom X from the dormant specie (Pn−X) (n = 0, initiator) [155]. The radicals (Pn ●) are able to react reversibly with the oxidized metal complex (X−Mtn+1 /ligand) to form again the dormant species and the activator. The ligand is essential to the solubilization of the transition metal salt in the organic medium and to the adjustment of the redox potential of the metal center, which defines the reactivity and the dynamics of the atom transfer process [155]. Once the radicals are active, the polymer chain growing process is similar to the free radical polymerization (FRP) process. The equilibrium between the active species and dormant species is shifted towards the dormant species via an excess of the higher oxidation state of the catalyst that is generated by a small amount of radical dimerization, during the initial steps of polymerization. This effect is known as the persistent radical effect (PRE) [156]. Several metal catalysts have been proposed as mediators of the ATRP process. Among these the copper based catalysts are extensively [157] studied due to their potential, low cost and large availability. New ligands for several transition metals have been developed with outstanding results achieved, related to the increase of the catalyst activity (10,000 fold when compared to the initial systems) [158]. In the last decade, the accomplished developments with respect to the capacity to polymerize different monomers and the smoothing of reaction conditions for CLRP methods are remarkable. The different strategies exploit the equilibria between growing radicals and dormant species and minimize the proportion of terminated chains in radical polymerization. However, the key point for the control is that the number of chains is much greater in CLRP than in FRP, therefore the rate of termination per chain is much lower in CLRP [158].
Nevertheless, each CLRP technique has its advantages and disadvantages. A complete description of kinetics, controlling agents, kinetis, monomers, ligands, reactions conditions and synthetic approaches is far beyond the scope of this manuscript. There are a couple of comprehensive reviews about different aspects of CLRP methods [10, 142, 146, 147, 153, 155, 158–166].
The controlled synthesis of block copolymers that can self-assembled, leading to nanostructures, is of great potential for the conception of new drug delivery carriers. The possibility to introduce targeting residues (e.g., protein, peptides and antibodies) on the surface of the nanocarriers will make possible the delivery of the drug in specific regions and cells.
The control at the molecular level will allow tailoring relevant nanoscale features. It is possible to use block copolymers that undergo reversible conformational changes in response to external stimuli (pH, temperature, ionic strength among others). These block copolymers can form micelles and vesicles just by changing the environmental conditions. For that reason, the synthesis of diblock, triblock and star architecture has been focus of special attention due to their self-assembling potential in aqueous solution.
Under the scope of this manuscript, below it is presented some important and representative references for the development of new macromolecules with potential application as DDS.
The control over the structure is of prime importance to generate precise self-assembled nanostructures with controllable features. The block lengths affect a couple of parameters, like critical micelle concentration, stability, morphology, hydrodynamic size, chemical functionalities in the micelle corona and core. The available functionality at the micelle corona and core is particular important for further modification involving the crosslinking as route to stabilize the supramolecular structure via covalent linkage, and the conjugating with biological entities such as targeting molecules (e.g., antibodies, folic acid and so on) and therapeutics.
Self assembly (polymeric micelles and surfactants)
Stimuli responsive polymers are key elements in the design of controlled drug delivery systems. Frequently, the stimuli-responsive DDS are designed as “smart micelles”. These structures are formed through self-assembly of amphiphilic copolymers in a solvent that has no affinity for one of the moieties. When the micelles are prepared in water a hydrophobic core is shielded from the solvent by a hydrophilic shell [167–169]. The synthesis of amphiphilic block copolymers as building block of nanocarriers (micelles/vesicles/micro-nanoparticles/capsules) for potential drug delivery applications is being a topic of great attention by the scientific community.
Block copolymers composed of hydrophilic and hydrophobic segments, depending on their structures and compositions, will self-assemble in solution to form aggregates of different sizes and shapes. Another strategy involves the use of hydrophilic-hydrophilic segments in which one of the blocks changes its nature to hydrophobic in response to an external stimulus as temperature and pH [170, 171]. Due to remarkable development of knowledge in the area of CRLP the number of monomers used in the preparation of self-assembly structures is extremely vast.
Different strategies can be used to prepare micelle-like entities, typically the core is hydrophobic and is responsible for the phase transfer and sequestration of lipophilic molecules, while the outer “corona” is responsible for the stabilization of the structure in water [172].
There are several possibilities to incorporate therapeutic molecules into micelles and vesicles, such as: hydrophobic/hydrophilic interactions, electrostatic attractions, hydrogen bonding and/or covalent bonds.
Using, for instance pH, sensitive polymers, it is possible to synthesize block copolymers with precise smart polymers that will self-assemble, and can be used as nano-carriers for anti-cancer drugs, and therefore release the drug when triggered by the acidic nature of most tumors (5.8∼7.2), inflamed tumors, endosomal compartments or specific organs according with the characteristic pH (Table 8, in the previous section).
Recently, the CLRP methods turned possible the synthesis of a large number of stimuli-responsive polymeric systems, which led to the appearance of massive number of publications about this subject. It is extremely difficult to cover all contributions available for the different CRLP techniques and because of that only the most relevant publications will be considered, with the special focus on ATRP. Some important references on RAFT technology are also mentioned. Bioapplications of macromolecules prepared by RAFT polymerization has been recently reviewed in detail [173].
Temperature responsive polymers
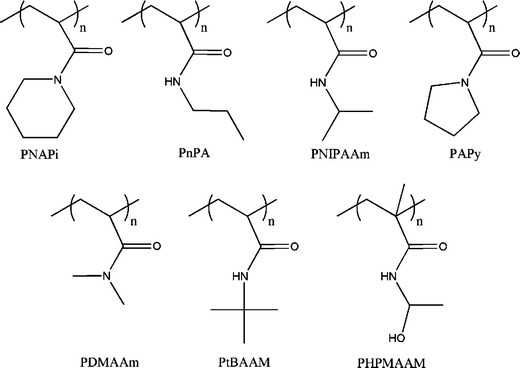
The N-substituted acrylamide polymers are the most commonly used thermo-responsive polymers. Some examples are presented in Fig. 30.
Fig. 30.
N-substituted acrylamide polymers used to synthesize thermo-responsive structures
The most studied N-substituted acrylamide is the PNIPAAm mainly because it displays a LCST value (32°C) very close to the human body temperature, and may therefore be applied in the biomedical applications, e.g., stimulus sensitive DDS. As mentioned before, the LCST value can be tunned by changing the molecular weight, end functionalities, adding hydrophilic and/or hydrophobic segments. On this matter, the CLRP methods are a powerful tool to synthesize precise NIPAAm based macrostructures with controlled molecular weight, low polydispersity polymers, complex architectures, and having at the same time stimuli-responsive properties.
The first report on the controlled ATRP of PNIPAAm was published in 2004 by Masci and co-authors [174]. The authors proposed the synthesis in a mixture of dimethylformamide (DMF)/water 50:50 (v/v) using a catalytic system of CuCl/tris(2-dimethyl aminoethyl) amine (Me6TREN). Following this work, several thermoresponsive copolymers had been synthesized via ATRP. Li and co-authors [175] reported the synthesis of biocompatible thermo-reponsive gelators based on ABA triblock (A-PNIPAAm and B—(poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC)) using a bifunctional initiator. Concerning ABC block copolymers, a doubly thermoresponsive poly(phenylene oxide)(PPO)-PMPC-PNIPAAm triblock copolymer gelators was synthesized by ATRP using a PPO-based macroinitiator [176]. The PPO exhibit an LCST near 15°C, and the authors demonstrated that for sufficiently long PPO blocks, the PPO-PMPC-PNIPAAm block copolymer presented two separate thermal transitions corresponding to micellization and gelation. The development of different block copolymers having PPO and PNIPAAm is particularly interesting, due to the presence of these two transitions. Above 15°C the PPO becomes hydrophobic leading to the formation of PPO-core micelles. Above 32°C the PNIPAAm becomes hydrophobic resulting in the formation of a micellar network (Fig. 31) [176].
Fig. 31.
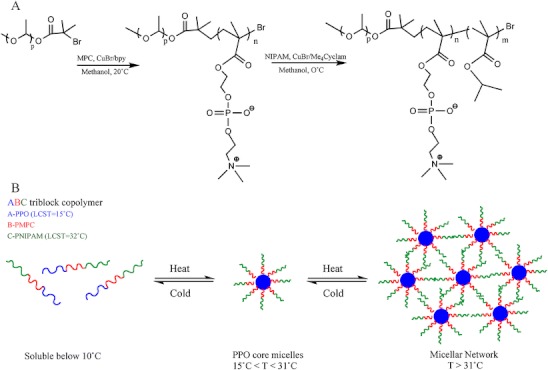
a Reaction scheme reported by Li and co-authors for the synthesis of PPO-PPMC-PNIPAAm triblock copolymer via ATRP; b Schematic representation of aqueous solution behaviour of the PPO-PMPC-PNIPAAm triblock copolymers: molecular dissolution at 5°C, formation of PPO-core micelles between 10 and 20°C, and formation of a micellar gel network above 31°C (adapted from [176])
In a very interesting contribution, Li and co-authors [177] prepared temperature/pH-responsive core-shell-corona micelles with different structures based on (poly(tert-butyl acrylate) (PtBA)-co-poly(acrylic acid) (PAA))-PNIPAAm. PtBA-b-PNIPAAm was firstly synthesized by sequential ATRP followed by partial hydrolysis of PtBA segments. At pH 5.8 and 25°C, the block copolymer self-assembled into spherical core-shell micelles with hydrophobic PtBA segments as the core and hydrophilic PAA/PNIPAAm segments as the mixed shell. Increasing temperature, core-shell micelles are converted into core–shell–corona micelles with PtBA as the core, collapsed PNIPAAm as the shell, and soluble PAA as the corona. Decreasing pH at 25°C, PAA chains collapsed onto the core resulting in core–shell–corona micelles with PtBA as the core [177].
Self-assembly of poly(t-butyl acrylate-co-acrylic acid)-b-poly(N-isopropylacrylamide) (P(rBA-co-AA)-b-PNIPAAm), which was obtained from part hydrolysis of PtBA-b-PNIPAAm synthesized by sequential atom transfer radical polymerization (ATRP) was studied. Thermo- and pH-responsive core-shell-corona (CSC) micelles with different structures were formed from (PtBA-co-PAA)-b-PNIPAAm in aqueous solution. At pH 5.8 and 25°C, the block copolymer self-assembled into spherical core-shell micelles with hydrophobic PtBA segments as the core, hydrophilic PAA/PNIPAAm segments as the mixed shell. Increasing temperatures, core-shell micelles converted into CSC micelles with PtBA as the core, collapsed PNIPAAm as the shell and soluble PAA as the corona. Moreover, decreasing pH at 25°C, PAA chains collapsed onto the core resulting in CSC micelles with PtBA as the core, PAA as the shell and PNIPAAm as the corona.
The PNIPAAm was successfully polymerized by RAFT using either benzyl dithiobenzoate or benzyl and cumyl dithiovarbamates as chain transfer agents (CTAs) [178] in 1,4-dioxane at 60°C. There are several contributions in the literature that describe the synthesis of PNIPAAm based materials via RAFT polymerization [178–182].
The controlled polymerization of a polymer from a biological structure is theoretically the more evident method for the synthesis of bioconjugates. In this approach the biomolecules are chemically modified with initiating groups for CLRP and are subsequently used as macroinitiators in the polymerization. Using modified biotin-moieties, Hong and co-authors [183] reported the synthesis of poly(NIPAAm-b-N-(2-hydroxypropyl) methacrylamide) via RAFT method. This block copolymer is able to form a coreshell nanostructure with biotin groups on the surface, by changing the temperature (Fig. 32).
Fig. 32.
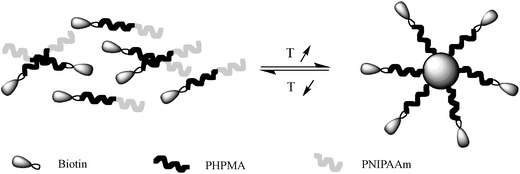
Schematic of the formation of nano core-shell structure from P(NIPAAm-b-HPMA) with biotin on the surface induced by temperature (adapted from [183])
In the same line of research, Kulkarni and co-authors [184] reported a very interesting example of post-modification of PNIPAAm with a biotin derivative. The reported block copolymer of biotin-terminated PNIPAAm-b-PAA was conjugated to streptavidin (SA) via the terminal biotin.
pH-responsive block copolymers
As referred before the pH-responsive polymers possess ionizable pendant weak acidic or basic moieties attached to the polymer backbone. Due to catalyst poisoning the controlled polymerization of acidic monomers was problematic via ATRP [185]. In 1999, Armes and co-authors [186] reported the “First example of the atom transfer radical polymerisation of an acidic monomer” by using methacrylic acid (MAA) in its sodium salt. This contribution represented a mark for the spread of the concept to other acid monomers, as the sodium 4-vinylbenzoic acid (VBA). Using the same approach, the synthesis of a wide range of pH-stimulus polymers was finally possible. Regarding basic monomers, Matyjaszewski and co-authors reported the CLRP of 2-(dimethylamino)ethyl methacrylate (DMAEMA) [187] using Cu(I)Br complexed with different amine ligands, ethyl-2-bromoisobutyrate (EBiB) or 2-bromopropionitrile (BPN) as the initiator in different solvents. Shortly before, the block copolymers of polystyrene-poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) were prepared by SFRP [188]. However, the molecular weight of the PDMAEMA segment was not controlled. The PDMAEMA is particularly interesting because it exhibits both temperature and pH-responsive behaviour. The pH-responsive copolymers containing polybase blocks, with pKa of 7.0–7.5, such as the tertiary amine-based methacrylate polymers, PDMAEMA [189], poly(N,N –diethyl aminoethyl methacrylate) (PDEAEMA) [189, 190] and poly(2-(diisopropylamino)ethyl methacrylate) (PDPAEMA) [190, 191] are among the most used polymers [192]. Their pendant amine groups gain protons under acidic conditions and release them under basic conditions.
Amphiphilic AB block copolymers of DMAEMA with methyl methacrylate (MMA), methyl acrylate (MA), or styrene (St) have been synthesized by ATRP [193] using well-defined poly(methyl methacrylate) (PMMA), poly(methyl acrylate) (PMA), and polystyrene (PS) macroinitiators. Different block copolymers containing different lengths of PDMAEMA segments were reported by varying the ration of DMAEMA and macroinitiators. ABA –triblock copolymers of PDMAEMA-PMMA-PDMAEMA were also reported by using difunctional PMMA macroinitiator [193].
The polybase-based micelles were amply used as pH-triggered drug carriers for delivery of several active substances, such as: chorambucil [194], anti-cancer taxomifen [195], antiparasitic miltephosine [196] and dipyridamole [189].
Di- and tri-block copolymers of MAA and MMA were reported via ATRP [197]. In aqueous solutions, the polymers are water-soluble at high pH and the core-shell micelle consists in the hydrophobic MMA core surrounded by hydrophilic MAA shell. P(MMA-b-MAA-b-MMA) with longer MMA segment self-assembles via the closed association mechanism through stronger self-entanglement of MMA chains, whereas P(MMA-b-MAA-b-MMA) with shorter MMA chain self-assembles via the open association mechanism [198].
Dai and co-authors [199] studied the block copolymerization of PMAA-b-PDEA through ATRP. At high pH micelles were formed consisting of poly(N,N-diethylacrylamide) (PDEA) core and PMMA shell. However, at low pH, unimers co-existed with micelles comprising PMMA core and PDEA core.
The possibility to easily tailor the segment chain and composition is of outstanding importance to the stringent control over the self-assembly process. Ma and co-authors [175] described the synthesis of biocompatible pH- responsive ABA triblock copolymer based on A= 2-(diisopropylaminoethyl methacrylate) (DPA) and B = 2-methacryloyloxyethyl phosphorylcholine (MPC). The MPC can be used to produce highly biocompatible surface coatings that exhibit remarkably resistance to protein adsorption and bacterial adhesion [200].
Teoh and co-authors [201] reported the synthesis of fullerene (C60) containing ampholytic block copolymer of poly((methacrylic acid)-block-(2-(dimethylamino)ethyl methacrylate))-block-C60 (P(MAA-b-DMAEMA)-b-C60) by ATRP (Fig. 33).
Fig. 33.
Synthetic scheme reported by Teoh and co-authors for synthesis of P(MAA102-b-DMAEMA67)-b-C60 (adapted from [201])
The authors found that the amphiphilic mono-C60 end-capped block copolymer showed enhanced solubility in aqueous medium at room and elevated temperatures and at low and high pH, but phase separates at intermediate pH between 5.4 and 8.8. At pH 11 and 55°C the polymer formed micelles and aggregates, whereas at the same temperature but pH 3 the polymer formed unimers and aggregates.
Armes and co-authors reported a novel zwitterionic poly(4-vinylbenzoic acid-block-2-N-(morpholino)ethyl methacrylate) (P(VBA(63)-b-MEMA(123))) [202] diblock copolymer synthesized via ATRP using protecting group chemistry for the acidic residues (Fig. 34). VBA core micelles were formed at pH below 6, while above pH 6 the block copolymer dissolved unimers. Very interesting was the fact that under alkakine media and in the presence of salt (Na2S2O4) or at elevated temperature, well-defined MEMA-core micelles were formed. The MEMA is a very interesting polymer since it displays both pH and temperature sensitive behaviour.
Fig. 34.
Schematic representation of the synthesis of P(VAB63-b-MEMA123) reported by Armes and co-authors (adapted from [202])
The core-shell micellar structures based on acidic-containing copolymers were used as pH-triggered drug carriers for delivery several drugs with different natures. Star-shaped polymer containing ethyl methacrylate (hydrophilic), tert-butyl methacrylate, poly(ethylene glycol)methacrylate (hydrophilic) were synthesized by ATRP using a four-armed multifunctional initiator [203]. The results suggest that star-shaped with high molar ratio of hydrophobic monomers tend to form high molecular weight aggregates in water. The system was used as potential delivery system for the oral administration of hydrophobic drugs (progesterone (PRG) was used as drug model) [203]. Sant and co-authors prepared diblock copolymers of PEG, tert-butyl methacrylate, ethyl acrylate or n-butyl acrylate by ATRP [204]. After the hydrolysis of the t-butyl groups all the polymers exhibited pH-dependent aggregation behaviour and their critical aggregation concentration decreased with the increase in the hydrophobic block length. The stimuli-responsive system was designed for poorly water-soluble model drugs (indomethacin (IND), fenofibrate (FNB) and PRG). The results presented by the authors indicate that supramolecular assemblies with high drug loadings and pH-dependent release kinetics can potentially enhance the oral bioavailability of poorly water-soluble drugs [204]. Satturwar and co-authors synthesized block copolymer poly(ethylene glycol) and t-butyl methacrylate, iso-butyl acrylate, n-butyl acrylate or propyl methacrylate by ATRP. Again, pH-sensibility was obtained by hydrolysis of t-butyl groups. The model drug used was candesartan cilexetil (CDN) and the results suggest that the pH-sensitive micelles was triggered upon an increase in pH from 1.2 to 7.2 [205]. Tian and co-authors [206] attached PAA at the ends of Pluronic P85 copolymer via ATRP to produce a novel block copolymer PAA-b-P85-b-PAA.
The complex formation and drug loading were found to be strongly depend on the PAA segment and pH. The protonation of carboxyl groups in the PAA segment at pH < 7.2 reduced the binding sites of doxorubicin (DOX, water soluble cationic drug) onto P85PAA chains, resulted in diminished DOX uptake at low pH. The block copolymer PAA-b-P85-b-PAA proved to be efficient in the complexation of DOX and could be pH-triggered in an acidic environment at pH 5.0. (Fig. 35)
Fig. 35.
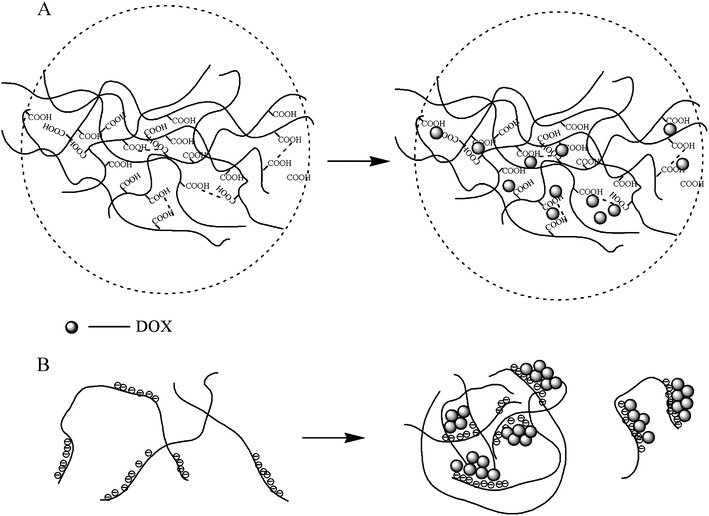
a Schematic representation of the complexation of P85PAA60/DOX behaviour at pH 3.87; b Schematic representation of the complexation of P85PAA60/DOX behaviour at pH 7.2 (adapted from [206])
The same research group reported a similar study with Pluronic® F87 [207].
Using RAFT methods, McCormick and co-authors [208] were the first to report RAFT stimuli responsive block copolymers that undergo reversible micellization in aqueous solutions. The authors used dithioester-capped sodium 4-styrenesulfonate homopolymer as a macro-CTA to prepare the block copolymer with sodium 4-vinylbenzoate in aqueous media. The micelles hydrodynamic diameter ranged from 18–38 nm. Other block copolymers with pH-responsive behaviour were prepared by using RAFT agents, such as: copolymers of sodium 2-acrylamido-2-methylpropanesulfonate (AMPS) and sodium 3-acrylamido-3-methylbutanoate (AMBA) [209]; AB diblock and AB statistical block copolymer of AMPS and AMBA [210]; poly(sodium 2-(acrylamido)-2-methylpropanesulfonate-block-sodium 6-acrylamidohexanoate) (pNaAMPS-AaH) [209]; acryloamido-styrenic diblock copolymers AB (A-poly(N,N-dimethylacrylamide) and B- N,Ndimethylvinylbenzylamine (DMVBA)) [211]. Double responsive block copolymers of PNIPAAm-b-PAA were synthesized in methanol using 1-cyanoethyl-2-pyrrolidone-1-carbodithioate as RAFT agent [212]. The authors demonstrated that the solution behaviour was strongly dependent on the hydrogen-bond interactions between NIPAAm and AA blocks. The block copolymers were sensitive to pH, temperature, block length and solvents (Fig. 36) [212].
Fig. 36.
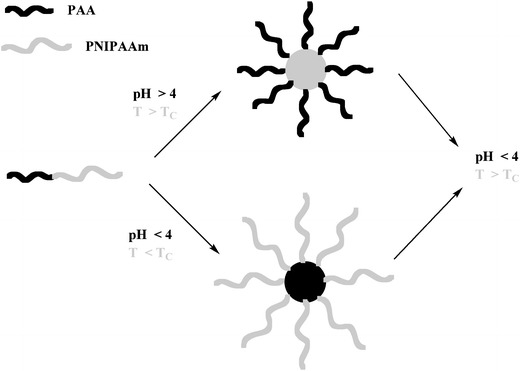
Modes of aggregate formation for block copolymers PNIPAAm-b-PAA in aqueous solution in dependence of pH and temperature (adapted from [212])
As it will be presented later, liposomes possess several features that make these structures very interesting for drug delivery purposes. Modified liposomes have shown to have excellent pharmokinetic profiles for the delivery of several structures such as nucleic acids, proteins and chemotherapeutic agents [213]. However, these nanocarriers have low stability and the lack of tunable triggers for drug release. In order to overcome both problems, Lee and co-authors [213] have proposed a very elegant strategy via a simple drop in procedure using a cholesterol-functionalized PAA. The active carboxylate groups are cross-linked with telechelic 2,2′-(ethylenedioxy)bis(ethylamine) linkers, resulting in polymer-caged liposomes that are highly stable and have tunable pH-sensitive responses (Fig. 37). The cholesterol-terminated PAA was synthesized via nitroxide mediated polymerization (NMP) of tert-butyl acrylate followed by acidolysis.
Fig. 37.
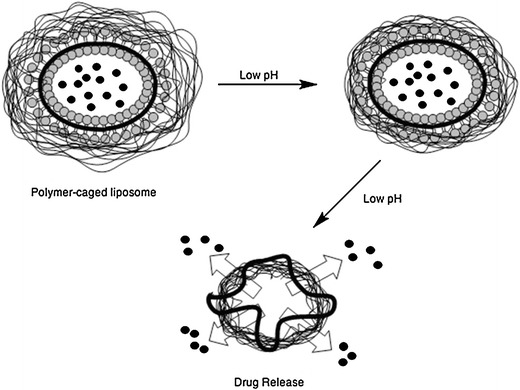
Drug release from a pH-responsive liposome with a PAA crosslinked shell (adapted from [213])
CLRP and “click chemistry”
The application of CLRP and “click chemistry” offers a robust method to produce new polymers with tailor-made features aiming to improve the biocompatibility and biodegradability of the DDS. “Click chemistry” represents a philosophy inspired by the simplicity and efficiency of the chemistry of the mother Nature. The concept was introduced by Sharpless and co-authors [214]. The main objective is to establish straightforward and highly selective reaction systems to synthesize precise structures. The most representative reaction of “click chemistry” is the copper catalyzed Huisgen 1,3 –dipolar cycloaddition of azides and terminal alkynes [215, 216] (Fig. 38).
Fig. 38.
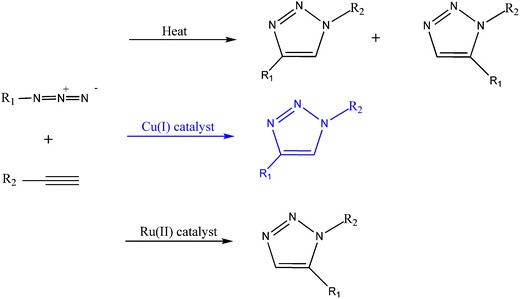
1,3-Dipolar cycloaddition between azides and alkynes
Since alkynes are poor 1,3-dipole acceptors in the absence of a proper catalyst the reaction is fairly slow. In the presence of copper (I) that can bind to terminal alkynes, the reaction becomes extremely fast, regioselective and efficient. Another important feature is the fact that the cooper catalyzed Huisgen cycloaddition can be performed at room temperature in various solvents and is tolerant to several functional groups [216]. The combination of ATRP with “click chemistry” opens the door to the creation of new range of materials through polymer functionalization and macromolecular engineering. In these works, most frequently, the ATRP polymers are transformed into azides by nucleophilic substitution and subsequently reaction with alkyne-functionalized molecules (Fig. 39).
Fig. 39.
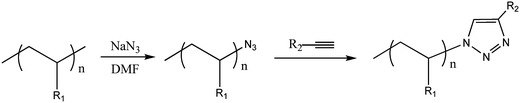
Schematic representation of the transformation of a bromide chain end into azide and subsequent reaction alkyne-functionalized molecules
Polypeptide-based rod-coil diblock copolymers (poly(gamma-benzyl-L-glutamate) (PBLG)-PDMAEMA) were synthesized via combination of ATRP and “click chemistry”. PDMAEMA was synthesized by ATRP from alkyne- or azide-functionalized initiators and coupled with the corresponding azide-or alkyne- modified synthetic polypeptide prepared from ring-opening polymerization [217].
Degradable brushes of PHEMA-PDMAEMA were synthesized via ATRP and “click chemistry” by Jiang and co-authors [218]. The brushes were composed of backbone of PHEMA onto which PDMAEMA was grafted via a hydrolysable linker. Both PDMAEMA and PHEMA were synthesized by ATRP with controlled molecular weight and narrow distributions. Click reaction between PHEMA with alkyne side groups and the azide end group in the PDMAEMA resulted in a high-molecular-weight polymer composed of low-molecular-weight constituents via an easily degradable carbonate ester linker (Fig. 40). The degradable brushes were able to transfect cells efficiently in the presence of the endosomal-disrupting INF-7 peptide.
Fig. 40.
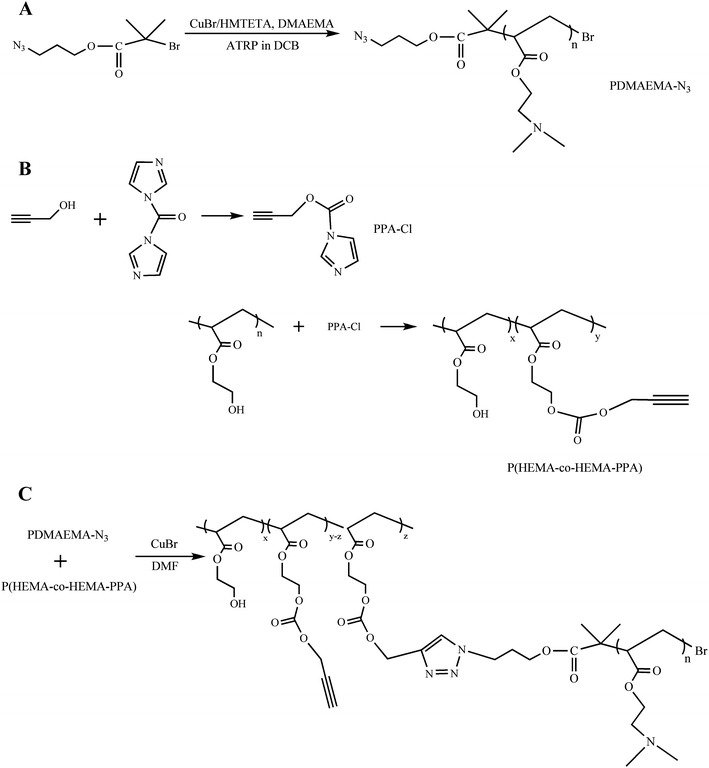
Schematic representation of the strategy reported by [218] to prepare degradable brushes of PHEMA-PDMAEMA. A ATRP of DMAEMA from 2-bromo-isobutyric acid 3-azidopropylester (BiBAP in Dichlorobenzene (DCB); b PHEMA with side chain of alkynes; c “click chemistry” to form degradable brushed PHEMA–PDMAEMA
Miniaturization of DDS
Micro- and nanoparticles
Drug delivery systems often use polymeric carriers that act as “drug transporters”. The use of a carrier allows overcoming some problems associated with the development and application of new drugs. Despite the new fact that new biologically active molecules are being discovered constantly, some of them present serious limitations to their use such as: rapid drug elimination; poor solubility and biodistribution which can compromise interaction of drug and site of action; low solubility at physiological pH [219] and insufficient cellular uptake [220]. The use of a DDS can be a very effective way of overcoming these barriers. However, when DDS are concerned, both design and engineering are to be considered in order to optimize drug efficiency as well as reduce their side-effects [221].
Particulate systems as DDS can be prepared using different types of materials like polymers, lipids or peptides. Particles prepared from either natural or synthetic polymers have been extensively investigated to be applied as DDS. Their synthesis can be achieved from different techniques [222]. Among them, are emulsion polymerization, solvent evaporation, ionic gelation, self-assembly, nanoprecipitation and supercritical fluid technology [223].
Within particles, two different categories can be distinguished: spheres and capsules. The first type refers to spherical particles composed of homogeneous mixtures of active agent and polymer while the second is applied when the particle presents a core (where the active agent is placed) that is delimited by a different material (usually the polymer). The core may present itself in distinct forms and states from solid to even gas. Several active agent domains may be found inside the capsule core. Some potential particles morphologies are presented in Fig. 41.
Fig. 41.
Potential particles morphologies (adapted from [224])
Another parameter used in particles classification refers to their size. Therefore, two categories are considered: micro- and nanoparticles. The distinction between both classes is often confusing and even polemic. Lately, the most accepted classification reports that particles up to 100 nm are considered nanoparticles while the ones from 1 µm up to 1,000 µm are classified as microparticles [225]. However, because of their small size, both can be injected directly into the systemic circulation or a certain compartment of the body. This process allows a high local drug concentration and can also be adapted to alternative administration routes such as inhalation or topical administration if properly sustained. They are extremely stable allowing the encapsulation of both hydrophobic and hydrophilic drugs. However, the most explored ability of the particles during last years is the possibility of directing them to specific places where drug is to be delivered by using targeting strategies. Several factors can influence drug release from both micro- and nanoparticles, namely the nature of the carried drug (including charge and even size), particle size and composition and even surrounding conditions such as pH and temperature [226].
Size difference between micro- and nanoparticles, implies differences in their performance. This means that each system presents its own advantages as well as limitations. Mathematically speaking, the surface area/volume ratio is inversely proportional to the radius of the sphere. This means that, when smaller particles are formed, drug diffusion from the particles surface occurs in higher proportions [227]. Due to the increased surface area, water diffuses easier towards inside smaller particles causing higher drug release kinetics. This enlarged drug release rate does not always consist on a system advantage since is often accompanied by initial burst release.
Micro- and nanoparticles performance also differ in terms of their fate after injection, their ability to cross barriers and enter cells as well as tissue reaction [227].
While microparticles are likely to stay in place after injection in tissues, nanoparticles tend to clear from injection place after some time. Kohane and co-authors injected several concentrations of both micro- and nanoparticles in the abdominal cavity of mice [228]. They observed that while microparticles could still be seen in injection site after 2 weeks, the same did not happen to the nanoparticles.
The problem associated with size is even more important when intravenous injection is used. When microparticles are too large, they may block blood flow through vessels with the same caliber as their diameter. This can result in strokes at end organs. However, microparticles up to 20 µm are widely applied in DDS design for inhalation applications [229].
Because of its very small size (lower than cells diameter, Fig. 42) nanoparticles can easily cross biologic barriers as well as easily enter all type of cells. Contrarily, microparticles, can only be incorporated by cells with phagocytic capacity [230].
Fig. 42.
Sizes of nanoparticles compared with other biological entities
Nanoparticle-based DDS were primarily developed as carriers for vaccines and cancer chemotherapy agents [231] but have so far proved to comprise considerable potential for treatment of several diseases [232]. In fact, nano-DDS can be applied in the treatment of different diseases such as cancer [233], AIDS (acquired immune deficiency syndrome) [234], diabetes [235], malaria [236], tuberculosis [237] and even prion disease [238]. This broad range of application is directly related to the possibility of targeting nanoparticles and therefore directing them to a specific organ.
Liposomes
Liposomes were reported, for the first time, during the 1960s and their clinical use was approved in the mid-1990s. These self-assembled vesicles consist of an aqueous core domain entrapped by a lipidic bilayer [239].
The formation of liposomes is related to hydrophilic/hydrophobic interaction between lipid/lipid and lipid/water molecules. In aqueous medium, lipids rearrange themselves, first, as bilayer sheets, with the headgroup of the lipid (hydrophilic) facing the water phase while the hydrocarbon chains (hydrophobic) are forced to face each other. Immediately afterwards, the formation of liposomes occurs (Fig. 43) [240, 241].
Fig. 43.
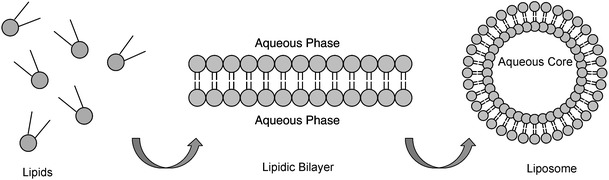
Schematic representation of the liposomes’ formation
Liposomes can be classified according to their lamellarity (number of bilayer membranes) and size (Fig. 44) in [239, 240]: small unilamellar vesicle (SUVs) (∼0.02 μm to ∼0.2 μm), large unilamellar vesicles (LUVs) (∼0.2 μm to ∼1 μm), giant unilamellar vesicles (GUVs) (>1 μm) and multilamellar vesicles (MLVs) (> 0.1 μm).
Fig. 44.
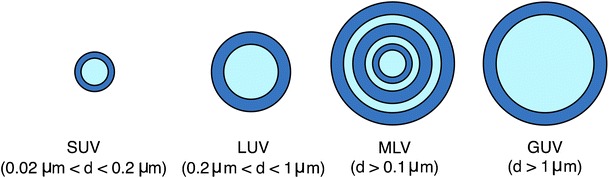
Schematic representation of the different types of liposomes, depending on their size and number of lamellae
The simplest method for the preparation of liposomes is the thin-film hydration method, which yields mostly MLVs. Other methods, like reverse-phase evaporation technique and solvent injection are also used and produce mostly LUVs. Preparations containing MLVs or LUVs can, subsequently, be sonicated or extruded in order to give SUVs [239, 240].
In the thin-film hydration method, a dried lipidic film is hydrated in an aqueous buffer solution, at a temperature above the transition temperature (temperature at which a lipid passes from the gel state to a liquid state) of the lipids. As stated above, this method originates preferentially MLVs, with high heterogeneity in size, size distribution and lamellarity. In solvent injection method, an ethanolic or etheric solution of lipids is slowly injected into an aqueous phase, forming unilamellar liposomes. The vesicles obtained by this method present a well-defined size distribution and high encapsulation efficiency. Removal of residual solvent is done by dialysis against distilled water [240]. Figure 45 presents the different methods that can be used in the liposomes preparation.
Fig. 45.
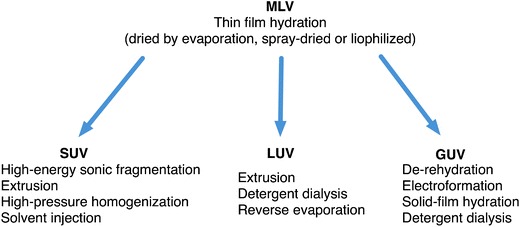
Different methods used in the liposomes preparation (adapted from [240])
Recently, due to their biocompatibility, non-immunogenicity, non-toxicity and ability of entrapping both hydrophilic and hydrophobic compounds, liposomes emerged as attractive materials for the development of DDS (Fig. 46) [239, 242]. Furthermore, a wide spectrum of properties for liposomes can be obtained by just varying the lipid (natural or synthetic) or lipid composition used in their preparation. Chemical surface modification of liposomes is also possible. The incorporation of specific molecules or macromolecules can enhance the interaction of liposome with a target site [243].
Fig. 46.
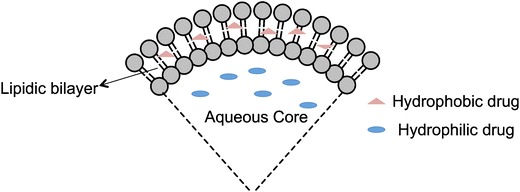
Localization of hydrophilic and hydrophobic drugs within the liposome
Although liposomes are promising carriers for drug delivery, they have shown low transfection efficiency and low stability after intravenous injection, due to absorption of plasma lipoproteins at the liposome surface. They also tend to be rapidly cleared from systemic circulation through reticuloendothelial system (RES) in living cells [244, 245]. In order to increase long-term stability of the liposome in the blood stream, it is necessary to alter some of the vesicles properties, namely particle size, surface charge and fluidity. Sterical stabilization of liposomes can be achieved by incorporating compounds bearing hydrophilic groups in lipidic bilayer [239, 245]. One of the most widely used method for liposome stabilization, with consequent enhancement of circulation time, is the incorporation of a poly(ethylene glycol) conjugated phosphatidylethanolamine (PEG-PE) into its structure [246–248].
An additional stabilizing effect of the liposome bilayer can be achieved by including cholesterol in the formulation. Cholesterol is used as a membrane sealer, minimizing bilayers permeability and providing mechanical and cohesive strength [244, 249].
A possible strategy to increase the usefulness of liposomes in the drug delivery field is to prepare functional liposomes that can response to changes, for example, in pH or temperature of surrounding medium. These are useful for site-specific and/or cytoplasmatic drug delivery [250]. The most effective method for that purpose is the incorporation of functional polymers in the liposome. These polymers should be amphiphilic, with the polymers hydrophobic part acting as an anchor to the liposome surface. The stimulus-induced conformational changes of the polymers induce a partial or total arrangement of the liposome structure, which is believed to be responsible for the release of drugs from the liposome [251].
Temperature sensitive liposomes are prepared from polymers that exhibit a LCST in aqueous medium. These kinds of polymers are highly hydrophilic below the LCST, but become hydrophobic above this temperature value. Therefore, incorporation of the polymer in the liposome is expected to destabilize the liposomal structure when temperature is raised above the LCST. Their copolymerization with other monomers, with varying hydrophilicity/hydrophobicity, enables the adjustment of the LCST to a desired temperature [252, 253]. Poly(N-isopropylacrylamide), that exhibits a LCST around 32°C, is the most widely used polymer for the preparation of temperature sensitive liposomes.
Takagishi and co-authors prepared phosphatidylcholine (PC) liposomes thermal-responsive, by coating them with a NIPAAm-octadecylacrylate (ODA) copolymer [254]. In other study, Takagishi and co-authors used a copolymer of NIPAAm and N,N-didodecylacrylamide (NDDAAm) to coat a PE based liposome. NDDAAm presents along acyl chain, which allows the copolymer to be tightly bound to the liposome. In addition to hydrophobic interactions, the interaction between the membrane and the copolymer can be enhanced by the establishment of hydrogen bonds. During this work, it was shown that the incorporation of PE in liposome formulation is important for the development of more efficient drug delivery systems [252]. Han and co-authors studied the release of doxorubicin from liposomes modified with poly(NIPAAm-co-acrylamide) P(NIPAAm-AAm) and poly(ethylene glycol). They showed that incorporation of the copolymer and poly(ethylene glycol) enhanced drug release from liposomes and reduced protein adsorption in serum [255].
Recently, Paasonen and co-authors prepared a temperature sensitive PC and PE based liposomes using poly(N-(2-hydroxypropyl) methacrylamide) mono/dilactate. The copolymer was functionalized with a cholesterol moiety that acts as an anchor to the lipidic vesicle. When compared with the NIPAAm based copolymers, this kind of copolymer presents the advantage of being biodegradable, with a LCST ranging from 13 to 65°C [242].
The sensization of liposomes to pH by incorporation of a pH-sensitive polymer was reported by Tirrel and co-authors, in the early 80’s. They showed that the attachment of poly(2-ethyl acrylic acid) (PEAA) onto liposome surface gave pH-sensitive content release properties. This polymer interacts with lipidic membrane in a pH dependent manner. At mildly acidic conditions, in high concentrations, PEAA induces micelization of the membranes, whereas at low concentrations, it permeabilizes them [256]. The pH at which permeabilization occurs can be modulated by adjusting the polymer molecular weight or substituting PEAA by another acrylic acid derivative [257].
Succinylated poly(glycidol) (SucPG), that present –OH and –COOH groups on the side chain, can also be used to prepare pH-sensitive liposomes [257]. Kono and co-authors have incorporated (SucPG) bearing long alkyl chains in the structure of PC vesicles [258]. This complex is stable at neutral pH, being destabilized under mildly acidic conditions. It was shown that it could deliver calcein into the cytosol through fusion with endosome/lysosome membranes. It is believed that the mechanism responsible for the destabilization of liposome, when in contact with SucPG, is related to two phenomena: the hydrophobic interaction of SucPG backbone with lipid membrane and hydrogen bond formation between polymer carboxyl groups and phosphate groups of the phospholipid membrane [257]. More recently, Sakaguchi and co-authors demonstrated that pH sensization of eggPC was enhanced with increasing side-chain hydrophobicity of poly(glycidol) derivatives [259]. The introduction of a titratable comonomer in the structure of hydrophobically-modified PNIPAAm can make the polymer pH-responsive. Leroux and co-authors have prepared a pH-sensitive liposome from eggPC/cholesterol, by incorporating a copolymer of NIPAAm, methacrylic acid and ODA into the liposomes lipidic layer [260].
Recently, Cho and co-authors prepared a pH-sensitive system composed of poly(methacrylic acid-co-stearyl methacrylate), PC and cholesterol. The complex was stable at pH above 6, becoming slightly disorganized when pH was 5 [251].
All the systems above referred are commonly known as polymer incorporated liposomes (PIL). Although they are able to respond to temperature or pH changes of the medium, it is known that they can dissociate from the liposome surface, returning them to the unstable state. This is related with the chain length and/or saturation degree of the anchor.
Cell and gene therapy
Cell therapy
Due to the need of increasing the level of drugs safety, capable of reaching the target with minimal side-effects emerged a great interest of cell therapy in the area of DDS. The bio-distribution of pharmaceuticals throughout the body has been reported as one of the main problems associated with systemic drug delivery administration. This leads to indiscriminated distribution requiring the use of a excessive concentration of drug to achieve the desired therapeutic level. This means that the drug is wasted in normal tissues and has a subsequently limited therapeutic efficacy. Thus, the ideal drug is the one that has pharmacological activity only at target site, using concentrations as low as possible and without negative effects on non-target compartments [261–263].
Cell-based delivery systems have been referred as the nearer to ideal system for delivery of drugs. Cells are capable of delivering drugs in response to an external stimulus, which is highly advantageous to maintain homeostasis for patients suffering from chronic diseases [263]. These systems can be divided into two categories: cells capable of expressing pharmaceutically relevant agents (e.g., cytosine deaminase-expressing neural stem/progenitor cells upon systemic 5-fluorocytosine administration) [264] and cell carriers that can be loaded with drugs or therapeutics. They can release the drug content in circulation or at selected sites or even could target the drug to other relevant cells in the body. Cell carriers investigated include so far animal and bacterial cells [261].
Stem cells
Stem cells are characterized by having a prolonged self-renewal capacity and the ability to differentiate into mature stages and different tissue types by asymmetric replication [265–267]. Therefore, it is essential to deeply know, understand and identify cells differentiation and growth in order to safely direct this knowledge towards clinical applications [268].
Stem cells can be isolated from various sources, such as a bone marrow, umbilical cord blood, peripheral blood, adipose tissue, skin and hair follicules [267, 269–274].
Stem cells can be delivered through intravenous infusion, intra-arterial infusion, direct implantation, intrathecal infusion and also biomaterial implantation (engineered scaffolding constructs) [263]. Despite the diversity of delivery mechanisms, it has been reported that cells directly injected into the body experienced less than desired therapeutic efficacies for many reasons, including rapid decrease in cell viability and immune rejection. Entrapped cells into biomaterials has shown promising results for reducing immune response and increasing efficacy and viability of transplanted cells [262].
Biomaterials support for stem cells
Stem cells are implanted in the host organism seeded in polymeric matrix that is design to work as a scaffold. Scaffolds can be modified to mimic extracellular matrix (ECM) and promote the attachment, growth and differentiation of cells. After implantation, they may remain locally obtaining long-term delivery or migrate to surrounding tissues [275].
Scaffold design and materials must present certain characteristics including: capacity for delivery without loss of function, adequate cellular adhesion and influence cell differentiation down desired cell lineages via mechanical or chemical interaction with support matrix [263]. Table 9 presents some polymers used in supports for stem cells applications.
Table 9.
Examples of biomaterials used to improve cell-based drug delivery
| Biomaterial | Cell type | Cell-secreted protein | Reference |
|---|---|---|---|
| Poly(ethylene glycol) | Islet cells | Insulin | [276–278] |
| Hyaluronic Acid | Fibroblasts and embryonic stem cells | – | [279] |
| Sodium Alginate | Osteoblasts | – | [280] |
| Fibroblasts | – | [281] | |
| Fibroblasts | Vascular endothelial growth factor | [282] | |
| Islet cells | Insulin | [283, 284] | |
| Polyethersulfone | Engineered kidney cells | Ciliary neurotrophic factor | [285] |
The interaction between polymer scaffolds and cells is crucial. It can be regulated by controlling specific ligand–receptor interactions, physical properties of the scaffolds (e.g., mechanical properties and degradation rate), and release of incorporated molecules such as growth factors or deoxyribonucleic acid (DNA) from the scaffolds [275]. Analogous compounds or identical to adhesion molecules (CAMs), such as integrins and cadherins, naturally present at ECM must be added to the scaffold. These molecules interact with proteins such as fibronectin and vitronectin and enhance cell adhesion [263, 275].
The influence of the entrapment process as well as the material immunogenicity, porosity, and stiffness are important factors in improving cell viability, cell function and subsequent therapeutic efficacy, both in vitro and in vivo [262].
Differentiated cells
Although most of research in DDS has been focused on stem cells, mature cells (leukocytes, hepatocytes, platelets, erythrocytes, islet cells, among others) can also be used [286]. For example, many cells, such as ovary cells, kidney cells, myoblasts, and fibroblasts, have been genetically engineered to secrete specific therapeutic proteins [262]. The most extensively studied are macrophages and erythrocytes.
Macrophages
Macrophages are differentiated cells of the immune system that are able to phagocytize microorganisms, nanoparticulate materials and soluble compounds. Mainly because of the phagocytosis phenomenon, macrophages have been considered as cell targets for selective delivery of drugs using nanoparticles as well as cell carriers for the delivery of therapeutic agents. This approach requires that macrophages should be adoptively transferred to the recipient after ex vivo loading with nanoparticles formulated drugs and/or contrasting agents [287, 288]. The majority of these macrophages (74-81%) are immediately sequestered in the liver and in the spleen (13–18%) [287], suggesting that they are more appropriate as a targeting system than as a drug delivery system [261].
Erythrocytes
Human red blood cells (RBCs) or erythrocytes, the most common cells of blood, are responsible for oxygen transport and have a typical biconcave shape. RBCs compared to other delivery systems have some unique features making them not only natural, safe and abundant carriers but, being endowed with enzymes involved in bioconversion reactions, also active bioreactors. RBCs are safe carriers that persist in circulation for months and can release active pharmacological agents in circulation for an equivalent period. RBCs permit to have a low and constant drug concentration in blood that once specifically selected can be therapeutically efficient without side-effects [261, 286].
The erythrocytes constitute potential biocompatible carriers for different bioactive substances, including protein drugs, as well as conventional therapeutics. The main features of using erythrocytes as a drug delivery system are the following [261, 286, 289]:
complete biodegradability without generation of toxic products;
high biocompatibility (especially when autologous erythrocytes are employed);
easily handled ex vivo by means of several techniques for the encapsulation of different molecules, after which one can obtain loaded erythrocytes with morphological, immunological and biochemical properties similar to those native cells;
large volume available for the encapsulation drugs due to lack of nucleus and other organelles;
protection of encapsulated substance from premature inactivation and degradation by endogenous factors and, at same time, the organism against the toxic effects of the drugs thus avoiding immunological reactions;
wide variety of compounds can be entrapped, even peptides of high molecular weight;
longer life-span in circulation (approximately 120 days);
could act as bioreactors due to the presence of several enzymatic activities that can turn a loaded prodrug into the active drug itself;
can use as a selective targeting of drugs directly to macrophages without affecting the non-targeted compartments. After their natural life-span in systemic circulation, the senescent RBCs are recognized by the cells of the reticuloendothelial system and removed from circulation to be destroyed.
The first clinical applications of RBCs as carriers were in enzyme replacement therapy by Beutler and co-authors who entrapped placental glucocerebrosidase into RBCs for a selective delivery to macrophages [290].
Some other active substances that have been encapsulated into red bloods cells include, toxins, peptides, glutathione, antisense PNA, glucocorticoid analogues and nucleoside/nucleotide analogues [261].
More recently several methods have been developed to couple molecules of interest to RBCs for their in vivo applications. The biotinylation of RBCs can be used to encapsulate drugs or contrasting agents, normally, fluorescent agents and/or superparamagnetic nanoparticles [261, 286].
RBCs have been used for therapeutic applications: in amino acid-based biopharmaceuticals (peptides and peptidomimetics (enalaprilat, glutathione, dermaseptin S4, listeriolysin, ubiquitin analogue), proteins (insulin, erythropoeietin, interleukin-2, interleukin-3, interferon α-2b), therapeutic enzymes (for example, β-galactosidase, β-glucosidase, fumarase, glyceraldehydes 3-phosphate dehydrogenase, etc); nucleic acid-based biopharmaceuticals (nucleosides, nucleotides and their analogues); and immunogenic biopharmaceuticals (model antigens, bacterial toxoids, viral subunit vaccines, cancer vaccines) [286].
Gene therapy
Gene therapy can be defined as the transfer of genetic material, a functional gene or DNA/ribonucleic acid (RNA) fragment into specific cells to elicit a desired therapeutic phenotype in order to reduce symptoms or treat human diseases [291]. There are two categories of somatic cell gene therapy, according to genetic material delivery method, ex vivo or in vivo. Ex vivo approach involves tissue biopsy, followed by cells. Finally, modified cells are returned to the body. In vivo approach consists in direct application of genetic material into cells, e.g., direct tissue injection or modification of culture cells for posterior implantation [292, 293]. These therapies have an ample potential and have been heavily investigated during the past 30 years. The first clinical trial with gene therapy was initiated in 1990, and involved two infants suffering from a form of severe combined immunodeficiency (SCID) resulting from adenosine deaminase (ADA) deficiency. However, it took until April 2000 before the first clinical success was reported [293, 294].
The success of gene therapy is highly dependent on the development of a vector or vehicle that can selectively and efficiently deliver the genetic material to target cells with minimal toxicity [295, 296]. An ideal gene delivery vector should be effective, specific, long lasting and safe. Vectors can be generally categorized into viral and non-viral vectors. Viruses carry and express modificated genes into host cells. They are used in the majority of clinical trials and are often the most efficient vectors. However they often initiate immune responses, are limited in size of genetic material they can carry, are difficult to produce and purify and exhibit limited target-cell specificity (or often nonspecifity). Moreover non-viral vectors as cationic polymers have the potential to be non toxic and nonimmunogenic, are chemically and physically stable, are relatively easy to produce in large quantities and can be targeted to desired cell types [267, 291].
There are two main types of viral vectors that transfer foreign genetic information into cells: simple capsid virions (the genetic material is inside a proteinaceous shell) like recombinant adenovirus or adeno-associated virus (AAV) and viruses with enveloped virions (the capsid is surrounded by a lipid bilayer envelope) which include retrovirus, lentivirus, alphavirus and herpes virus (Fig. 47). Viral vectors are distinguished essentially by their size and transgene capacity, target (efficiency and specifity with which the viral vector infects the target cells), performance, duration of required expression, immunitary response and safety [297, 298]. All of these characteristics must be taken into account when a viral vector is chosen in order to achieve better results.
Fig. 47.
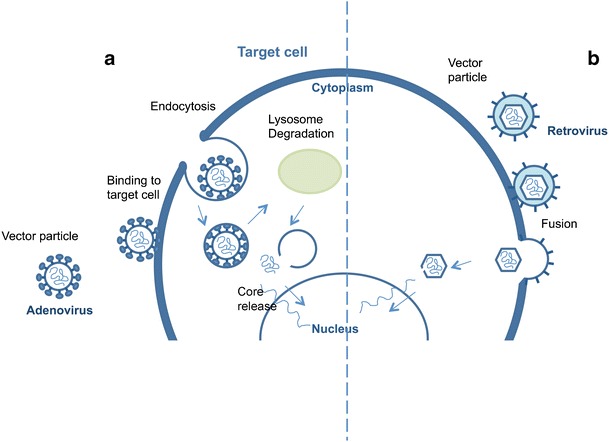
Two different approaches of virus entering the cell, simple capsid (a) and enveloped virions (b) (adapted from [293, 297])
Viral vectors are in a prominent position in strategies for gene therapy essentially because of their high delivery and transfection efficiency’s. Adenovirus (a double-stranded DNA virus) is the vector most widely used in gene therapy (24%), followed by retrovirus (20.9%) (single-stranded RNA viruses that replicate through a DNA intermediate) [299]. Adeno-associated viruses (single-stranded, non-enveloped DNA viruses) represent only 4.3% of vectors used in gene therapy clinical trials. Although viruses are efficient in transducing cells, its production however, is time and cost consuming and it is important to take into account the risk of local or systemic infections [267]. Due to adverse events in past clinical trials, the safety issue of gene vector became a major concern that has hindered the advance of gene therapy. Consequently the use of non-viral vectors in clinical trials in recent years is increasing mainly due to their reduced immunotoxicity and side-effects [296, 300, 301].
The simplest approach to non-viral delivery systems is inducing gene expression by direct gene transfer (intra tissue injection) with naked plasmid DNA (‘naked’ DNA). ‘Naked’ DNA are very specific and can promote long lasting gene expression [296]. It is the third most used vector in gene therapy clinical trials [301]. Recent advances improved the precision and reproducibility of this approach using a computer-controlled catheter guided injection device [302]. Moreover, intratracheal and intravenous administrations normally requires the use of a delivery vector or vehicle. A variety of non viral delivery methods for gene therapy with potential for clinical trials are currently available. The gene transfer systems most extensively studied are the cationic-lipid and polymer-based systems [295, 303–306]. These include cationic lipids or polymers, liposomes [307–310], and nanoparticles [311–313]. In most cases, these vectors have net positive charge, which contributes to both complex formation with polyanionic nucleic acid and interaction with negatively charged cell membrane [314, 315].
As mentioned before, liposomes consist of a phospholipid bilayer vesicle entrapping an aqueous domain. They are particles with stable physicochemical properties. Lipoplexes, moreover, are cationic lipid/DNA complexes, spontaneously formed via interaction of positively charged lipids and negatively charged nucleic acids and therefore are unstable particles and should be prepared immediately before use [316, 317]. The first description of a successful in vitro transfection with cationic lipid was reported by Felgner and co-authors in 1987 [318]. In following years, many progress were made in this area. Surface modified lipid-DNA nanoparticles are recent strategies for delivering siRNA to specific tissues in vivo [319]. There are some cationic lipids for genetic material delivery commercial available, like Lipofectin®, RNAiFect™, Oligofectamine™, Lipofectamine™ and TransIT® [314].
Nanoparticles have great potential as a strategy for gene therapy. They meet the majority of discussed characteristics for a successful vector. As mentioned before there are many different formulations of nanoparticles. Nanoparticles and complexes polymer-genetic material should be <100 nm to reduce renal excretion and to be captured by cells [314, 320]. Typically, in gene therapy, a segment of DNA or RNA (circular or linear) is compacted with a policationic polymer. These particles enter the cell via endocitosis and/or macropinocytosis and rapidly transfficked to the nucleolus. In many cases expression levels are superior that those observed after treatment with naked plasmid DNA [305]. The success of this vector is directly associated with nanoparticle formulation, size or electric charge. Nanoparticles can be stably stored under a variety of conditions and concentrations (up to 12 mg/ml of DNA). They are tolerant of a wide range of temperatures, salt concentrations and pH and they tend to protect their genetic material from enzyme degradation (deoxyribonuclease or ribonuclease). Polyethyleneimine (PEI) has been widely tested for DNA, oligonucleotide and siRNA delivery [314, 321, 322].
Non-viral vectors generally are superior to viral vectors in safety, due to low risk of infection [296]. The simplicity of use and the possibility of large-scale production is also advantageous [295]. Different strategies are now being developed to improve the non viral vector efficiency.
RNAi: Novel gene therapy approach
RNA interference (RNAi) was discovered by Fire and co-authors in 1998 [323]. This discovery revolutionized biological research. In 2006, the Nobel Prize in Physiology or Medicine was attributed to Andrew Z. Fire and Craig C. Mello for their discovery of RNA interference-gene silencing by double-stranded RNA. RNAi is a natural process through which expression of a targeted gene can be knocked down with high specificity and selectivity [324]. It plays an important role in the regulation of gene expression. With this potential therapeutic application, it is now possible to create small inhibitory RNA molecules, introduce them into specific cells and inhibit expression of a disease gene or block a destructive pathway [325, 326].
RNAi is a potent and specific gene silencing event in which small interfering RNA degrades target mRNA which has a complementary sequence to the siRNA [327]. siRNA has, as convenient, specific and potent gene silencing effect compared with conventional techniques such as antisense oligodeoxynucleotides and homologous recombination-based knockout strategies [324, 328, 329].
Gene therapy can be applied in many different diseases including cancer, neurodegenerative diseases, muscular dystrophy, immunologic diseases, blood diseases, among others [297, 324, 330–333]. However, the easier choices for gene therapy are the monogenic loss of function diseases, associated with one gene mutation that results in loss of gene function in a specific tissue. Gene therapy with RNAi is a potential therapy for human diseases including human genetic and acquired disorders such as cancer and viral infections [334, 335]. Actually, cancer is the disease with more developments in gene therapy, about 64,6% of all research worldwide activity, followed by cardiovascular diseases (8.9%), monogenic diseases (8.1%) and infectious diseases (7.9%) [300, 301]. The future challenges are those that optimize the individualized vector according to each clinical setting.
A recent clinical possibility is to provide long-term phenotypic correction by genetic modification of stem cells in vitro before transplantation. Different cells have been transduced with particular genes using different types of vectors [261, 336]. The main characteristics of stem cells for use in therapeutic cell transplants are schematically shown in Table 10.
Table 10.
Main characteristics of stem cells for use in therapeutic cell transplants and as vehicle for gene therapy
| Cell transplantation | Gene therapy |
|---|---|
| They can grow in culture indefinitely (they are easy to maintain) | They can be deployed directly on the site (e.g., the brain can pass the blood brain barrier) |
| They are multipotent (forming various types of cells) | They are easily transfected with therapeutic genes, may express more than one gene of interest |
| The presence of viruses/diseases can be analyzed in culture | |
| They assume the phenotype of neighboring cells after transplantation | They have a correct implementation in the host tissue, allowing gene transcription (proteic expression) |
| They have the capacity to migrate after transplantation | |
| They produce factors that contribute to tissue repair |
Gene and cell therapies have been widely developed in recent years. Much has been done, but much still remains to be done. Table 11 presents some other pathologies that may profit in a near future from this top of the art technology.
Table 11.
Some examples of gene and cell therapy applications
Conclusion and outlook
As the pathological and physiological mechanisms of disease come more in focuse, a design of more effective controlled release delivery systems becomes a greater challenge. The development of new polymers more suitable as biomaterials allows their extensive application in delivery device construction, drug targeting as well as gene and protein delivery. Also, the possibility of designing more compact systems, such as micro- and nanoparticles makes polymers a highly attractive field of research. More recently, the advances in CLRP of several monomers that lead to stimuli-responsive polymers and hydrogels open several options that, in years to come, will allow a new and more effective generation of DDS to arise.
Biodistribution, stability, solubility, targeting, immunogenicity can be altered rationally designing macromolecules synthesized by CLRP methods. The combination of these methods and “click chemistry” present significant opportunities for the design and synthesis of advanced macromolecules for DDS. It is predictable the appearance of new high efficient macrostructures for DDS in a near future.
DDS technology has emerged as an unquestionable interdisciplinary science aimed at advanced healthcare.
Recent technological breakthroughs in drug discovery and development have resulted in novel therapeutics for targeted prevention and individualized therapies potentially leading to ultimate improvement of life quality of patients treated. These innovations have been accomplished by increasing interest (and consequently investments) in the research field by pharmaceutical companies. In fact, drug delivery technologies have become one of the major players in biopharmaceutical industry. Multidisciplinary interaction between drug delivery systems, pharmaceutical industry and biotechnologies is summarized as represented in Fig. 48.
Fig. 48.
Interrelationship of DDS, pharmaceutical industry and biotechnology (adapted from [3])
However, and despite the understandable interest from pharmaceutical industries in these devices, some new advances are still to come. Most of recent research in DDS is focused on developing new materials and preparation of responsive polymers with specific macro/micro-structure and chemical profile. Special attention is being paid to the design of copolymers with hydrophilic/hydrophobic segments, the synthesis of star shaped polymers and dendrimes as nanocarriers for bioactive compounds as well as the synthesis of bioconjugates. The medicine of future is treated in a personalized way in accordance with the individual parameters. By this concept it is predictable the appearance of new effective tailor-made DDS, resulting from knowledge of interdisciplinary research creating personalized medical solutions.
Glossary
- AA
Acrylic acid
- AAm
Acrylamide
- AAV
Adeno-associated virus
- ADA
Adenosine deaminase
- AIDS
Acquired immunodeficiency syndrome
- AMBA
Sodium 3-acrylamido-3-methylbutanoate
- AMPS
Sodium 2-acrylamido-2-methylpropanesulfonate
- ATRP
Atom transfer radical polymerization
- bFGF
Fibroblast growth factor
- BLM
Bleomycin
- BPN
2-bromopropionitrile
- BSA
Bovine serum albumin
- C60
Fullerene
- CAM
Compounds to adhesion molecules
- CDN
Candesartan cilexetil
- CLRP
Controlled/living radical polymerization
- CST
Critical solution temperature
- CTA
Chain transfer agents
- DCT
Degenerative chain transfer
- DDS
Drug delivery systems
- Dex-HEMA
Dextran with Hydroxyethyl methacrylate
- Dex-MA
Dextran with glycidyl methacrylate
- DMAEMA
N,N-dimethylaminoethylmethacrylate
- DMF
Dimethylformamide
- DMVBA
B-N,N-dimethylvinylbenzylamine
- DNA
Desoxyribonucleic acid
- DOX
Doxorubicin
- DPA
2-(diisopropylamido)ethyl methacrylate
- DT
Degenerative transfer
- EBiB
Ethyl-2-bromoisobutyrate
- EC
Ethylcellulose
- ECM
Extracellular matrix
- FITC
Fluorescein isothiocyanate
- FNB
Fenofibrate
- FRP
Free radical polymerization
- FU
Fluorouracil
- GUV
Giant unilamellar vesicle
- HPMC
Hydroxypropylmethylcellulose
- HuH-7
Human hepatoma cellular line
- IND
Indomethacin
- IOL
Intraocular lens
- IPN
Interpenetrated network
- IT
Iodine transfer
- LCST
Low critical solution temperature
- LUV
Large unilamellar vesicle
- MA
Maleic anhydride
- MAA
Methacrylic acid
- MADIX
Macromolecular design via the interchange of xanthates
- Me6TREN
tris(2-dimethyl aminoethyl) amine
- MEMA
2-(N-morpholino)ethyl methacrylate
- MLV
Multilamellar vesicle
- MMA
Methacrylic acid
- MMC
Mitomycin C
- MPC
Methacryloyloxyethyl phosphorylcholine
- NDDAAm
N,N-didodecylacrylamide
- NIPAAm
N-isopropylacrylamide
- NMP
Nitroxide-mediated polymerization
- ODA
Octadecylacrylate
- P(MAA-b-DMAEMA)-b-C60
Poly((methacrylic acid)-block-(2-(dimethylamino)ethyl methacrylate))-block-C60
- PAA
Poly(acrylic acid)
- PACA
Poly(alkyl cyanoacrylates)
- PBLG
Poly(gamma-benzyl-L-glutamate)
- PC
Phosphatidylcholine
- PCL
Poly(ε-caprolactone)
- PDEA
Poly(N,N-diethylacrylamide)
- PDMAEMA
Poly(2-(dimethylamino)ethyl methacrylate)
- PE
Phosphatidylethanolamine
- PEAA
Poly(2-ethyl acrylic acid)
- PEG
Poly(ethylene glycol)
- PEI
Poly(ethyleneimine)
- PGA
Poly(glycolic acid)
- PHEMA
Poly(2-hydroxyethyl methacrylate)
- PLA
Poly(lactic acid)
- PLGA
Poly(lactic-co-glycolic acid)
- PMA
Poly(methyl acrylate)
- PMMA
Poly(methyl methacrylate)
- PMPC
Poly(2-methacryloyloxyethyl phosphorylcholine)
- pNaAMPS-AaH
Poly(sodium 2-(acrylamido)-2-methylpropanesulfonate-block-sodium 6-acrylamidohexanoate)
- PNIPAAm
Poly(N-isopropylacrylamide)
- POE
Poly(ortho esters)
- PPO
Poly(phenylene oxide)
- PRE
Persistent radical effect
- PRG
Progesterone
- PS
Polystyrene
- PtBA
Poly(tert-butyl acrylate)
- PVA
Poly(vinyl alcohol)
- PVBA(63)-b-MEMA(123)
Poly(4-vinylbenzoic acid-block-2-N-(morphohno)ethyl methacrylate)
- RAFT
Reversible addition fragmentation transfer
- RBC
Red blood cells
- RES
Reticuloendothelial system
- RNA
Ribonucleic acid
- RNAi
RNA interference
- SA
Streptavidin
- SCID
Severe combined immunodeficiency
- SFRP
Stable free radical polymerization
- siRNA
Small interfering ribonucleic acid
- St
Styrene
- SucPG
Succinylated poly(glycidol)
- SUV
Small unilamellar vesicle
- TEMPO
2,2,6,6-tetramethylpiperidinyl-1-oxyl
- UCST
Upper critical solution temperature
- UV
Ultraviolet
- VBA
Sodium 4-vinylbenzoic acid
- VEGF
Vascular endothelial growth factor
References
- 1.Zempsky WT. Alternative routes of drug administration—advantages and disadvantages (subject review) Pediatrics. 1998;101:730–1. doi: 10.1542/peds.101.4.730. [DOI] [PubMed] [Google Scholar]
- 2.Golan DE, Armen H, Tashjian J, Armstrong EJ, et al. Principles of pharmacology: the pathophysiological basis of drug therapy. Philadelphia: Lippincott Williams&Wilkins; 2008. [Google Scholar]
- 3.Jain KK. Drug delivery systems—an overview. In: Jain KK, editor. Drug delivery systems. New York: Humana; 2008. [Google Scholar]
- 4.Jeffreys D. Aspirin: the remarkable story of a wonder drug. New York: Bloomsbury; 2005. [Google Scholar]
- 5.Magalhães PO, Lopes AM, Mazzola PG, et al. Methods of endotoxin removal from biological preparations: a review. J Pharm Sci. 2006;10:388–404. [PubMed] [Google Scholar]
- 6.Bajpai AK, Shukla SK, Bhanu S, et al. Responsive polymers in controlled drug delivery. Prog Polym Sci. 2008;33:1088–118. doi: 10.1016/j.progpolymsci.2008.07.005. [DOI] [Google Scholar]
- 7.Gilhotra RM, Mishra DN. Polymeric systems for ocular inserts. Latest Reviews 7. 2009.
- 8.Kelner A, Schacht EH. Tailor-made polymers for local drug delivery: release of macromolecular model drugs from biodegradable hydrogels based on poly(ethylene oxide) J Control Release. 2005;101:13–20. doi: 10.1016/j.jconrel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Kabanov A, Okano T. Challenges in polymer therapeutics, state of the art and prospects of polymer drugs. In: Maeda H, Kabanov A, editors. Polymer drugs in the clinical stage: advantages and prospects. New York: Kluwer Academic/ Plenum; 2001. [Google Scholar]
- 10.Braunecker WA, Matyjaszewski K. Controlled/living radical polymerization: features, developments, and perspectives. Prog Polym Sci. 2007;32:93–146. doi: 10.1016/j.progpolymsci.2006.11.002. [DOI] [Google Scholar]
- 11.Robinson JR, Lee VHL. Controlled drug delivery: fundamentals and applications. New York: Dekker; 1987. [Google Scholar]
- 12.Koo OM, Rubinstein I, Onyuksel H. Role of nanotechnology in targeted drug delivery and imaging: a concise review. Nanomed Nanotechnol Biol Med. 2005;1:193–212. doi: 10.1016/j.nano.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Schmaljohann D. Thermo- and pH-responsive polymers in drug delivery. Adv Drug Deliv Rev. 2006;58:1655–70. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Brannon-Peppas L. Polymers in controlled drug delivery. In: Medical Plastics and Biomaterials Available via. http://www.devicelink.com/mpb/archive/97/11/003.html. Cited 14 Nov 2009; 1997.
- 15.Chasin M, Langer RS. Biodegradable polymers as drug delivery systems. New York: Dekker; 1990. [Google Scholar]
- 16.Burkersroda FV, Schedl L, Göpferich A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials. 2002;23:4221–31. doi: 10.1016/S0142-9612(02)00170-9. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S. Osmotic controlled drug delivery system. Latest Reviews 6. 2008.
- 18.Urbina MC, Zinoveva S, Miller T, et al. Investigation of magnetic nanoparticle-polymer composites for multiple-controlled drug delivery. J Phys Chem C. 2008;112:11102–8. doi: 10.1021/jp711517d. [DOI] [Google Scholar]
- 19.Liu T-Y, Hu S-H, Liu K-H, et al. Preparation and characterization of smart magnetic hydrogels and its use for drug release. J Magn Magn Mater. 2006;304:397–9. doi: 10.1016/j.jmmm.2006.01.203. [DOI] [Google Scholar]
- 20.Craciunescu I, Nan A, Turcu R, et al. Synthesis, characterization and drug delivery application of the temperature responsive pNIPA hydrogel. J Phys Conf Ser. 2009;182:1–4. doi: 10.1088/1742-6596/182/1/012060. [DOI] [Google Scholar]
- 21.Huang J, Huang Z, Bao Y, et al. Thermosensitive poly(N-isopropylacrylamide-co-acrylonitrile) hydrogels with rapid response. Chin J Chem Eng. 2006;14:87–92. doi: 10.1016/S1004-9541(06)60042-6. [DOI] [Google Scholar]
- 22.Hergt R, Dutz S. Magnetic particle hyperthermia-biophysical limitations of a visionary tumour therapy. J Magn Magn Mater. 2007;311:187–92. doi: 10.1016/j.jmmm.2006.10.1156. [DOI] [Google Scholar]
- 23.Hergt R, Dutz S, Muller R, et al. Magnetic particle hyperthermia: nanoparticle magnetism and materials development for cancer therapy. J Phys Condens Matter. 2006;18:S2919–34. doi: 10.1088/0953-8984/18/38/S26. [DOI] [Google Scholar]
- 24.Shinkai M, Ito A. Functional magnetic particles for medical application. In: Kobayashi T, editor. Recent progress of biochemical and biomedical engineering in Japan II. Berlin: Springer Berlin / Heidelberg; 2004. [Google Scholar]
- 25.Nair LS, Laurencin CT. Polymers as biomaterials for tissue engineering and controlled drug delivery. Adv Biochem Eng Biotechnol. 2006;102:47–90. doi: 10.1007/b137240. [DOI] [PubMed] [Google Scholar]
- 26.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5:447–51. doi: 10.1016/S1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 27.Uchegbu IF, Schatzlein AG. Polymers in drug delivery. New York: CRC; 2006. [Google Scholar]
- 28.Sahoo S, Sasmala A, Nanda R, et al. Synthesis of chitosan–polycaprolactone blend for control delivery of ofloxacin drug. Carbohydr Polym. 2010;79:106–13. doi: 10.1016/j.carbpol.2009.07.042. [DOI] [Google Scholar]
- 29.Cascone MG, Sim B, Downes S. Blends of synthetic and natural polymers as drug delivery systems for growth hormone. Biomaterials. 1995;16:569–74. doi: 10.1016/0142-9612(95)91131-H. [DOI] [PubMed] [Google Scholar]
- 30.Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32:762–98. doi: 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- 31.MaHam A, Tang ZW, Wu H, et al. Protein-based nanomedicine platforms for drug delivery. Small. 2009;5:1706–21. doi: 10.1002/smll.200801602. [DOI] [PubMed] [Google Scholar]
- 32.Berthold A, Cremer K, Kreuter J. Collagen microparticles: carriers for glucocorticosteroids. Eur J Pharm Biopharm. 1998;45:23–9. doi: 10.1016/S0939-6411(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 33.Metzmacher I, Radu F, Bause M, et al. A model describing the effect of enzymatic degradation on drug release from collagen minirods. Eur J Pharm Biopharm. 2007;67:349–60. doi: 10.1016/j.ejpb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Kanematsu A, Yamamoto S, Ozeki M, et al. Collagenous matrices as release carriers of exogenous growth factors. Biomaterials. 2004;25:4513–20. doi: 10.1016/j.biomaterials.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 35.Kuijpers AJ, Engbers GHM, Feijen J, et al. Characterization of the network structure of carbodiimide cross-linked gelatin gels. Macromolecules. 1999;32:3325–33. doi: 10.1021/ma981929v. [DOI] [Google Scholar]
- 36.Lin M, Meng S, Zhong W, et al. Novel drug-loaded gelatin films and their sustained-release performance. J Biomed Mater Res B. 2009;90B:939–44. doi: 10.1002/jbm.b.31366. [DOI] [PubMed] [Google Scholar]
- 37.Muvaffak A, Gurhan I, Hasirci N. Prolonged cytotoxic effect of colchicine released from biodegradable microspheres. J Biomed Mater Res B. 2004;71B:295–304. doi: 10.1002/jbm.b.30092. [DOI] [PubMed] [Google Scholar]
- 38.Leo E, Vandelli MA, Cameroni R, Forni F. Doxorubicin-loaded gelatin nanoparticles stabilized by glutaraldehyde: Involvement of the drug in the crosslinking process. Int J Pharm. 1997;155:75–82. doi: 10.1016/S0378-5173(97)00149-X. [DOI] [Google Scholar]
- 39.Jeyanthi R, Panduranga RK. Release characteristics of bleomycin, mitomycin C, and 5-fluorouracil from gelatin microspheres. Int J Pharm. 1989;55:31–37. doi: 10.1016/0378-5173(89)90273-1. [DOI] [Google Scholar]
- 40.Ofokansi KC, Adikwu MU. Formulation and evaluation of microspheres based on gelatin-mucin admixtures for the rectal delivery of cefuroxime sodium. Trop J Pharmaceut Res. 2007;6:825–32. [Google Scholar]
- 41.Kuijpers AJ, Wachem PB, Luyn MJA, et al. In vitro and in vivo evaluation of gelatin-chondroitin sulphate hydrogels for controlled release of antibacterial proteins. Biomaterials. 2000;21:1763–72. doi: 10.1016/S0142-9612(00)00064-8. [DOI] [PubMed] [Google Scholar]
- 42.Kuijpers AJ, Wachem PB, Luyn MJA, et al. In vivo and in vitro release of lysozyme from cross-linked gelatin hydrogels: a model system for the delivery of antibacterial proteins from prosthetic heart valves. J Control Release. 2000;67:323–36. doi: 10.1016/S0168-3659(00)00221-2. [DOI] [PubMed] [Google Scholar]
- 43.Chuang VTG, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm Res. 2002;19:569–77. doi: 10.1023/A:1015396825274. [DOI] [PubMed] [Google Scholar]
- 44.Wunder A, Muller-Ladner U, Stelzer EHK, et al. Albumin-based drug delivery as novel therapeutic approach for rheumatoid arthritis. J Immunol. 2003;170:4793–801. doi: 10.4049/jimmunol.170.9.4793. [DOI] [PubMed] [Google Scholar]
- 45.Almond BA, Hadba AR, Freeman ST, et al. Efficacy of mitoxantrone-loaded albumin microspheres for intratumoral chemotherapy of breast cancer. J Control Release. 2003;91:147–55. doi: 10.1016/S0168-3659(03)00214-1. [DOI] [PubMed] [Google Scholar]
- 46.Doughty JC, Anderson JH, Willmott N, et al. Intra-arterial administration of adriamycin-loaded albumin microspheres for locally advanced breast cancer. Postgrad Med J. 1995;71:47–9. doi: 10.1136/pgmj.71.831.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha VR, Singla AK, Wadhawan S, et al. Chitosan microspheres as a potential carrier for drugs. Int J Pharm. 2004;274:1–33. doi: 10.1016/j.ijpharm.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 48.Denkbas EB. Perspectives on: chitosan drug delivery systems based on their geometries. J Bioact Compat Polym. 2006;21:351–68. doi: 10.1177/0883911506066930. [DOI] [Google Scholar]
- 49.Xu Y, Du Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int J Pharm. 2003;250:215–26. doi: 10.1016/S0378-5173(02)00548-3. [DOI] [PubMed] [Google Scholar]
- 50.Gan Q, Wang T. Chitosan nanoparticle as protein delivery carrier-systematic examination of fabrication conditions for efficient loading and release. Colloids Surf, B. 2007;59:24–34. doi: 10.1016/j.colsurfb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 51.Campos AMD, Diebold Y, Carvalho ELS, et al. Chitosan nanoparticles as new ocular drug delivery systems: in vitro stability, in vivo fate, and cellular toxicity. Pharm Res. 2004;21:803–10. doi: 10.1023/B:PHAM.0000026432.75781.cb. [DOI] [PubMed] [Google Scholar]
- 52.Salamanca AED, Diebold Y, Calonge M, et al. Chitosan nanoparticles as a potential drug delivery system for the ocular surface: toxicity, uptake mechanism and in vivo tolerance. Investig Ophthalmol Vis Sci. 2006;47:1416–25. doi: 10.1167/iovs.05-0495. [DOI] [PubMed] [Google Scholar]
- 53.Yuan Y, Tan J, Wang Y, et al. Chitosan nanoparticles as non-viral gene delivery vehicles based on atomic force microscopy study. Acta Biochim Biophys Sin. 2009;41:515–26. doi: 10.1093/abbs/gmp038. [DOI] [PubMed] [Google Scholar]
- 54.Mao H-Q, Roy K, Troung-Le VL, et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70:399–421. doi: 10.1016/S0168-3659(00)00361-8. [DOI] [PubMed] [Google Scholar]
- 55.Ta HT, Dass CR, Dunstan DE. Injectable chitosan hydrogels for localised cancer therapy. J Control Release. 2008;126:205–16. doi: 10.1016/j.jconrel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 56.Augst AD, Kong HJ, Mooney DJ. Alginate hydrogels as biomaterials. Macromol Biosci. 2006;6:623–33. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 57.Ohta M, Suzuki Y, Chou H, et al. Novel heparin/alginate gel combined with basic fibroblast growth factor promotes nerve regeneration in rat sciatic nerve. J Biomed Mater Res A. 2004;71A:661–8. doi: 10.1002/jbm.a.30194. [DOI] [PubMed] [Google Scholar]
- 58.Tanihara M, Suzuki Y, Yamamoto E, et al. Sustained release of basic fibroblast growth factor and angiogenesis in a novel covalently crosslinked gel of heparin and alginate. J Biomed Mater Res. 2001;56:216–21. doi: 10.1002/1097-4636(200108)56:2<216::AID-JBM1086>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 59.Gadad AP, Patil MB, Naduvinamani SN, et al. Sodium alginate polymeric floating beads for the delivery of cefpodoxime proxetil. J Appl Polym Sci. 2009;114:1921–6. doi: 10.1002/app.30617. [DOI] [Google Scholar]
- 60.Hornig S, Bunjes H, Heinze T. Preparation and characterization of nanoparticles based on dextran-drug conjugates. J Colloid Interface Sci. 2009;338:56–62. doi: 10.1016/j.jcis.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 61.Chiellini E, Sunamoto J, Migliaresi C, et al. Biomedical polymers and polymer therapeutics. New York: Kluwer Academic/Plenum; 2001. [Google Scholar]
- 62.Vlugt-Wensink KDF, Vlugt TJH, Jiskoot W, et al. Modeling the release of proteins from degrading crosslinked dextran microspheres using kinetic Monte Carlo simulations. J Control Release. 2006;111:117–27. doi: 10.1016/j.jconrel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 63.Cadee JA, Brouwer LA, Otter W, et al. A comparative biocompatibility study of microspheres based on crosslinked dextran or poly(lactic-co-glycolic)acid after subcutaneous injection in rats. J Biomed Mater Res. 2001;56:600–9. doi: 10.1002/1097-4636(20010915)56:4<600::AID-JBM1133>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 64.Casadei MA, Cerreto F, Cesa S, et al. Solid lipid nanoparticles incorporated in dextran hydrogels: a new drug delivery system for oral formulations. Int J Pharm. 2006;325:140–6. doi: 10.1016/j.ijpharm.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Raemdonck K, Naeye B, Buyens K, et al. Biodegradable dextran nanogels for RNA interference: focusing on endosomal escape and intracellular siRNA delivery. Adv Funct Mater. 2009;19:1406–15. doi: 10.1002/adfm.200801795. [DOI] [Google Scholar]
- 66.Kamel S, Ali N, Jahangir K, et al. Pharmaceutical significance of cellulose: a review. Express Polymer Lett. 2008;2:758–78. doi: 10.3144/expresspolymlett.2008.90. [DOI] [Google Scholar]
- 67.Kavanagh N, Corrigan OI. Swelling and erosion properties of hydroxypropylmethylcellulose (Hypromellose) matrices-influence of agitation rate and dissolution medium composition. Int J Pharm. 2004;279:141–52. doi: 10.1016/j.ijpharm.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 68.Gong K, Rehman IU, Darr JA. Characterization and drug release investigation of amorphous drug-hydroxypropyl methylcellulose composites made via supercritical carbon dioxide assisted impregnation. J Pharm Biomed Anal. 2008;48:1112–9. doi: 10.1016/j.jpba.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 69.Murthy TEGK, Chowdary KPR. Formulation and evaluation of ethyl cellulose-coated diclofenac sodium microcapsules: Influence of solvents. Indian J Pharm Sci. 2005;67:216–9. [Google Scholar]
- 70.Yamada T, Onishi H, Machida Y. Sustained release ketoprofen microparticles with ethylcellulose and carboxymethylethylcellulose. J Control Release. 2001;75:271–82. doi: 10.1016/S0168-3659(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 71.Santhi K, Venkatesh D, Dhanaraj S, et al. Development and in-vitro evaluation of a tropical drug delivery system containing betamethazone loading ethyl cellulose nanospheres. Trop J Pharmaceut Res. 2005;4:495–500. [Google Scholar]
- 72.Ravikumara NR, Madhusudhan B, Nagaraj TS, et al. Preparation and evaluation of nimesulide-loaded ethylcellulose and methylcellulose nanoparticles and microparticles for oral delivery. J Biomater Appl. 2009;24:47–64. doi: 10.1177/0885328209103406. [DOI] [PubMed] [Google Scholar]
- 73.Mundargi RC, Babu VR, Rangaswamy V, et al. Nano/micro technologies for delivering macromolecular therapeutics using poly(D, L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125:193–209. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 74.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B. 2009;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 75.Kumar PS, Saini TR, Chandrasekar D, et al. Novel approach for delivery of insulin loaded poly(lactide-co-glycolide) nanoparticles using a combination of stabilizers. Drug Deliv. 2007;14:517–23. doi: 10.1080/10717540701606467. [DOI] [PubMed] [Google Scholar]
- 76.Bilati U, Allémann E, Doelker E. Poly(D, L-lactide-co-glycolide) protein-loaded nanoparticles prepared by the double emulsion method-processing and formulation issues for enhanced entrapment efficiency. J Microencapsul. 2005;22:205–14. doi: 10.1080/02652040400026442. [DOI] [PubMed] [Google Scholar]
- 77.Sandor M, Enscore D, Weston P, et al. Effect of protein molecular weight on release from micron-sized PLGA microspheres. J Control Release. 2001;76:297–311. doi: 10.1016/S0168-3659(01)00446-1. [DOI] [PubMed] [Google Scholar]
- 78.Csaba N, Caamano P, Sanchez A, et al. PLGA: poloxamer and PLGA: poloxamine blend nanoparticles: new carriers for gene delivery. Biomacromolecules. 2005;6:271–8. doi: 10.1021/bm049577p. [DOI] [PubMed] [Google Scholar]
- 79.He Q, Liu J, Sun X, et al. Preparation and characteristics of DNA-nanoparticles targeting to hepatocarcinoma cells. World J Gastroenterol. 2004;10:660–3. doi: 10.3748/wjg.v10.i5.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patil Y, Panyam J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm. 2009;367:195–203. doi: 10.1016/j.ijpharm.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinha VR, Bansal K, Kaushik R, et al. Poly-[epsilon]-caprolactone microspheres and nanospheres: an overview. Int J Pharm. 2004;278:1–23. doi: 10.1016/j.ijpharm.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 82.Heller J, Barr J, Ng SY, et al. Poly(ortho esters)—their development and some recent applications. Eur J Pharm Biopharm. 2000;50:121–8. doi: 10.1016/S0939-6411(00)00085-0. [DOI] [PubMed] [Google Scholar]
- 83.Heller J, Barr J. Poly(ortho esters)—From concept to reality. Biomacromolecules. 2004;5:1625–32. doi: 10.1021/bm040049n. [DOI] [PubMed] [Google Scholar]
- 84.Heller J, Barr J, Ng SY, et al. Poly(ortho esters): synthesis, characterization, properties and uses. Adv Drug Deliv Rev. 2002;54:1015–39. doi: 10.1016/S0169-409X(02)00055-8. [DOI] [PubMed] [Google Scholar]
- 85.Vauthier C, Labarre D, Ponchel G. Design aspects of poly(alkylcyanoacrylate) nanoparticles for drug delivery. J Drug Target. 2007;15:641–63. doi: 10.1080/10611860701603372. [DOI] [PubMed] [Google Scholar]
- 86.Graf A, McDowell A, Rades T. Poly(alkycyanoacrylate) nanoparticles for enhanced delivery of therapeutics—is there real potential? Expert Opin Drug Deliv. 2009;6:371–84. doi: 10.1517/17425240902870413. [DOI] [PubMed] [Google Scholar]
- 87.Lin M, Wang H, Meng S, et al. Structure and release behavior of PMMA/silica composite drug delivery system. J Pharm Sci. 2007;96:1518–26. doi: 10.1002/jps.20809. [DOI] [PubMed] [Google Scholar]
- 88.Anguita-Alonso P, Giacometti A, Cirioni O, et al. RNAIII-inhibiting-peptide-loaded in vivo Polymethylmethacrylate prevents in vivo Staphylococcus aureus biofilm formation. Antimicrob Agents Chemother. 2007;51:2594–6. doi: 10.1128/AAC.00580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tao SL, Lubeley MW, Desai TA. Bioadhesive poly(methyl methacrylate) microdevices for controlled drug delivery. J Control Release. 2003;88:215–28. doi: 10.1016/S0168-3659(03)00005-1. [DOI] [PubMed] [Google Scholar]
- 90.Singhal R, Datta M. Studies on the development of biodegradable poly(HEMA)/cloisite nanocomposites. Polym Compos. 2009;30:887–90. doi: 10.1002/pc.20627. [DOI] [Google Scholar]
- 91.Chouhan R, Bajpai AK. An in vitro release study of 5-fluoro-uracil (5-FU) from swellable poly-(2-hydroxyethyl methacrylate) (PHEMA) nanoparticles. J Mater Sci, Mater Med. 2009;20:1103–14. doi: 10.1007/s10856-008-3677-x. [DOI] [PubMed] [Google Scholar]
- 92.Anderson EM, Noble ML, Garty S, et al. Sustained release of antibiotic from poly(2-hydroxyethyl methacrylate) to prevent blinding infections after cataract surgery. Biomaterials. 2009;30:5675–81. doi: 10.1016/j.biomaterials.2009.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nyangoga H, Zecheru T, Filmon R, et al. Synthesis and use of pHEMA microbeads with human EA.hy 926 endothelial cells. J Biomed Mater Res B. 2009;89B:501–7. doi: 10.1002/jbm.b.31240. [DOI] [PubMed] [Google Scholar]
- 94.Jhaveri SJ, Hynd MR, Dowell-Mesfin N, et al. Release of nerve growth factor from HEMA hydrogel-coated substrates and its effect on the differentiation of neural cells. Biomacromolecules. 2009;10:174–83. doi: 10.1021/bm801101e. [DOI] [PubMed] [Google Scholar]
- 95.Hobabi M-R, Hassanzadeh D, Azarmi S, et al. Effect of synthesis method and buffer composition on the LCST of a smart copolymer of N-isopropylacrylamide and acrylic acid. Polym Adv Technol. 2007;18:986–92. doi: 10.1002/pat.951. [DOI] [Google Scholar]
- 96.Li Q, Wang J, Shahani S, et al. Biodegradable and photocrosslinkable polyphosphoester hydrogel. Biomaterials. 2006;27:1027–34. doi: 10.1016/j.biomaterials.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chang C, Duan B, Cai J, et al. Superabsorbent hydrogels based on cellulose for smart swelling and controllable delivery. Eur Polym J. 2010;46:92–100. doi: 10.1016/j.eurpolymj.2009.04.033. [DOI] [Google Scholar]
- 98.Ahn SK, Kasi RM, Kim SC, et al. Stimuli-responsive polymer gels. Soft Matter. 2008;4:1151–7. doi: 10.1039/b714376a. [DOI] [PubMed] [Google Scholar]
- 99.Li L, Aoki Y. Rheological images of poly(vinyl chloride) gels. 1. The dependence of sol-gel transition on concentration. Macromolecules. 1997;30:7835–41. doi: 10.1021/ma971045w. [DOI] [Google Scholar]
- 100.Knoben W, Besseling NAM, Stuart MAC. Rheology of a reversible supramolecular polymer studied by comparison of the effects of temperature and chain stoppers. J Chem Phys. 2007;126:024907. doi: 10.1063/1.2409296. [DOI] [PubMed] [Google Scholar]
- 101.Jeong HS, Huh KM, Park K. Hydrogel drug delivery systems. In: Uchegbu IF, Schatzlein AG, editors. Polymers in drug delivery. Boca Raton: Taylor&Francis; 2006. [Google Scholar]
- 102.Roy I, Gupta MN. Smart polymeric materials: emerging biochemical applications. Chem Biol. 2003;10:1161–71. doi: 10.1016/j.chembiol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 103.Geever LM, Minguez CM, Devine DM, et al. The synthesis, swelling behaviour and rheological properties of chemically crosslinked thermosensitive copolymers based on N-isopropylacrylamide. J Mater Sci. 2007;42:4136–48. doi: 10.1007/s10853-006-0912-z. [DOI] [Google Scholar]
- 104.Geever LM, Nugent MJD, Higginbotham CL. The effect of salts and pH buffered solutions on the phase transition temperature and swelling of thermoresponsive pseudogels based on N-isopropylacrylamide. J Mater Sci. 2007;42:9845–54. doi: 10.1007/s10853-007-1814-4. [DOI] [Google Scholar]
- 105.Mano JF. Stimuli-responsive polymeric systems for biomedical applications. Adv Eng Mater. 2008;10:515–27. doi: 10.1002/adem.200700355. [DOI] [Google Scholar]
- 106.Yue YM, Sheng X, Wang PX. Fabrication and characterization of microstructured and pH sensitive interpenetrating networks hydrogel films and application in drug delivery field. Eur Polym J. 2009;45:309–15. doi: 10.1016/j.eurpolymj.2008.10.038. [DOI] [Google Scholar]
- 107.Zhang JP, Wang L, Wang AQ. Preparation and swelling behavior of fast-swelling superabsorbent hydrogels based on starch-g-poly (acrylic acid-co-sodium acrylate) Macromol Mater Eng. 2006;291:612–20. doi: 10.1002/mame.200500387. [DOI] [Google Scholar]
- 108.D’Erricot G, Lellis M, Mangiapia G, et al. Structural and mechanical properties of UV-photo-cross-linked poly(N-vinyl-2-pyrrolidone) hydrogels. Biomacromolecules. 2008;9:231–40. doi: 10.1021/bm7008137. [DOI] [PubMed] [Google Scholar]
- 109.Marandi GB, Esfandiari K, Biranvand F, et al. pH sensitivity and swelling behavior of partially hydrolyzed formaldehyde-crosslinked poly(acrylamide) superabsorbent hydrogels. J Appl Polym Sci. 2008;109:1083–92. doi: 10.1002/app.28205. [DOI] [Google Scholar]
- 110.El-Rehim HAA, Hegazy ES, El-Mohdy HLA. Effect of various environmental conditions on the swelling property of PAAm/PAAcK superabsorbent hydrogel prepared by ionizing radiation. J Appl Polym Sci. 2006;101:3955–62. doi: 10.1002/app.22904. [DOI] [Google Scholar]
- 111.Kopecek J, Yang JY. Revie—hydrogels as smart biomaterials. Polym Int. 2007;56:1078–98. doi: 10.1002/pi.2253. [DOI] [Google Scholar]
- 112.Oh JK, Drumright R, Siegwart DJ, et al. The development of microgels/nanogels for drug delivery applications. Prog Polym Sci. 2008;33:448–77. doi: 10.1016/j.progpolymsci.2008.01.002. [DOI] [Google Scholar]
- 113.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Delivery Rev. 2002;54:3–12. doi: 10.1016/S0169-409X(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 114.Ebara M, Yamato M, Hirose M, et al. Copolymerization of 2-carboxyisopropylacrylamide with N-isopropylacrylamide accelerates cell detachment from grafted surfaces by reducing temperature. Biomacromolecules. 2003;4:344–9. doi: 10.1021/bm025692t. [DOI] [PubMed] [Google Scholar]
- 115.Schmaljohann D, Oswald J, Jorgensen B, et al. Thermo-responsive PNiAAm-g-PEG films for controlled cell detachment. Biomacromolecules. 2003;4:1733–9. doi: 10.1021/bm034160p. [DOI] [PubMed] [Google Scholar]
- 116.Xu FJ, Kang ET, Neoh KG. pH- and temperature-responsive hydrogels from crosslinked triblock copolymers prepared via consecutive atom transfer radical polymerizations. Biomaterials. 2006;27:2787–97. doi: 10.1016/j.biomaterials.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 117.Peppas NA, Bures P, Leobandung W, et al. Hydrogels in pharmaceutical formulations. Eur J Pharm Biopharm. 2000;50:27–46. doi: 10.1016/S0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 118.Schild HG. Poly (N-Isopropylacrylamide)—experiment, theory and application. Prog Polym Sci. 1992;17:163–249. doi: 10.1016/0079-6700(92)90023-R. [DOI] [Google Scholar]
- 119.Gil ES, Hudson SA. Stimuli-reponsive polymers and their bioconjugates. Prog Polym Sci. 2004;29:1173–222. doi: 10.1016/j.progpolymsci.2004.08.003. [DOI] [Google Scholar]
- 120.Jeong B, Choi YK, Bae YH et al. New biodegradable polymers for injectable drug delivery systems. In: Conference on challenges for drug delivery and pharmaceutical technology. Tokyo: Elsevier Science Bv; 1998.
- 121.Shibayama M, Mizutani S, Nomura S. Thermal properties of copolymer gels containing N-isopropylacrylamide. Macromolecules. 1996;29:2019–24. doi: 10.1021/ma951390q. [DOI] [Google Scholar]
- 122.Shibayama M, Suetoh Y, Nomura S. Structure relaxation of hydrophobically aggregated poly(N-isopropylacrylamide) in water. Macromolecules. 1996;29:6966–8. doi: 10.1021/ma960866i. [DOI] [Google Scholar]
- 123.Shibayama M, Morimoto M, Nomura S. Phase-separation induced mechanical transition of poly(N-isopropylacrylamide) water isochore gels. Macromolecules. 1994;27:5060–6. doi: 10.1021/ma00096a031. [DOI] [Google Scholar]
- 124.Bromberg LE, Ron ES. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv Drug Delivery Rev. 1998;31:197–221. doi: 10.1016/S0169-409X(97)00121-X. [DOI] [PubMed] [Google Scholar]
- 125.Kikuchi A, Okano T. Stimuli-sensitive hydrogels. In: Kwon GS, editor. Polymeric drug delivery systems. Boca Raton: Taylor&Francis; 2005. [Google Scholar]
- 126.Pelah A, Seemann R, Jovin TM. Reversible cell deformation by a polymeric actuator. J Am Chem Soc. 2007;129:468–9. doi: 10.1021/ja067171+. [DOI] [PubMed] [Google Scholar]
- 127.Kurkuri MD, Aminabhavi TM. Poly(vinyl alcohol) and poly(acrylic acid) sequential interpenetrating network pH-sensitive microspheres for the delivery of diclofenac sodium to the intestine. J Control Release. 2004;96:9–20. doi: 10.1016/j.jconrel.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 128.Kim B, Flamme K, Peppas NA. Dynamic swelling behavior of pH-sensitive anionic hydrogels used for protein delivery. J Appl Polym Sci. 2003;89:1606–13. doi: 10.1002/app.12337. [DOI] [Google Scholar]
- 129.Ramkissoon-Ganorkar C, Liu F, Baudys M, et al. Modulating insulin-release profile from pH thermosensitive polymeric beads through polymer molecular weight. J Control Release. 1999;59:287–98. doi: 10.1016/S0168-3659(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 130.Asoh T, Kaneko T, Matsusaki M, et al. Rapid deswelling of semi-IPNs with nanosized tracts in response to pH and temperature. J Control Release. 2006;110:387–94. doi: 10.1016/j.jconrel.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 131.Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev. 2001;53:321–39. doi: 10.1016/S0169-409X(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 132.Kim SJ, Park SJ, Kim IY, et al. Electric stimuli responses to poly(vinyl alcohol)/chitosan interpenetrating polymer network hydrogel in NaCl solutions. J Appl Polym Sci. 2002;86:2285–9. doi: 10.1002/app.11215. [DOI] [Google Scholar]
- 133.Tanaka T, Nishio I, Sun ST, et al. Collapse of gels in a electric-field. Science. 1982;218:467–9. doi: 10.1126/science.218.4571.467. [DOI] [PubMed] [Google Scholar]
- 134.Bajpai AK, Bajpai J, Soni SN. Preparation and characterization of electrically conductive composites of poly(vinyl alcohol)-g-poly(acrylic acid) hydrogels impregnated with polyaniline (PANI) Express Polymer Lett. 2008;2:26–39. doi: 10.3144/expresspolymlett.2008.5. [DOI] [Google Scholar]
- 135.Blakemore RP, Frankel RB. Magnetic navigation in bacteria. Sci Am. 1981;245:58–65. doi: 10.1038/scientificamerican1281-58. [DOI] [Google Scholar]
- 136.Bahadur D, Giri J. Biomaterials and magnetism. Sadhana-Acad P Eng S. 2003;28:639–56. [Google Scholar]
- 137.Jordan A, Scholz R, Wust P, et al. Magnetic fluid hyperthermia (MFH): Cancer treatment with AC magnetic field induced excitation of biocompatible superparamagnetic nanoparticles. J Magn Magn Mater. 1999;201:413–9. doi: 10.1016/S0304-8853(99)00088-8. [DOI] [Google Scholar]
- 138.Shinkai M, Yanase M, Suzuki M, et al. Intracellular hyperthermia for cancer using magnetite cationic liposomes. J Magn Magn Mater. 1999;194:176–84. doi: 10.1016/S0304-8853(98)00586-1. [DOI] [Google Scholar]
- 139.Kim DK, Zhang Y, Kehr J, et al. Characterization and MRI study of surfactant-coated superparamagnetic nanoparticles administered into the rat brain. J Magn Magn Mater. 2001;225:256–61. doi: 10.1016/S0304-8853(00)01255-5. [DOI] [Google Scholar]
- 140.Mykhaylyk O, Cherchenko A, Ilkin A, et al. Glial brain tumor targeting of magnetite nanoparticles in rats. J Magn Magn Mater. 2001;225:241–7. doi: 10.1016/S0304-8853(00)01264-6. [DOI] [Google Scholar]
- 141.Rabin Y. Cancer treatment by electromagnetic activator nanoheaters. In: Eureka alert! Nanotechnology in context. Available via. http://www.me.cmu.edu/faculty1/rabin/Rabin_Pub/Rabin_Pub501.pdf. Cited 14 Nov 2009; 2002.
- 142.Qiu J, Charleux B, Matyjaszewski K. Controlled/living radical polymerization in aqueous media: homogeneous and heterogeneous systems. Prog Polym Sci. 2001;26:2083–134. doi: 10.1016/S0079-6700(01)00033-8. [DOI] [Google Scholar]
- 143.Cunliffe D, Pennadam S, Alexander C. Synthetic and biological polymers-merging the interface. Eur Polym J. 2004;40:5–25. doi: 10.1016/j.eurpolymj.2003.10.020. [DOI] [Google Scholar]
- 144.Klok HA. Biological-synthetic hybrid block copolymers: combining the best from two worlds. J Polym Sci, A Polym Chem. 2005;43:1–17. doi: 10.1002/pola.20527. [DOI] [Google Scholar]
- 145.Lutz JF, Borner HG. Modern trends in polymer bioconjugates design. Prog Polym Sci. 2008;33:1–39. doi: 10.1016/j.progpolymsci.2007.07.005. [DOI] [Google Scholar]
- 146.Matyjaszewski K, Tsarevsky NV. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat Chem. 2009;1:276–88. doi: 10.1038/nchem.257. [DOI] [PubMed] [Google Scholar]
- 147.Cunningham MF. Controlled/living radical polymerization in aqueous dispersed systems. Prog Polym Sci. 2008;33:365–98. doi: 10.1016/j.progpolymsci.2007.11.002. [DOI] [Google Scholar]
- 148.Hadjichristidis N, Iatrou H, Pitsikalis M, et al. Macromolecular architectures by living and controlled/living polymerizations. Prog Polym Sci. 2006;31:1068–132. doi: 10.1016/j.progpolymsci.2006.07.002. [DOI] [Google Scholar]
- 149.Georges MK, Veregin RPN, Kazmaier PM, et al. Narrow molecular-weight resins by a free radical polymerization process. Macromolecules. 1993;26:2987–8. doi: 10.1021/ma00063a054. [DOI] [Google Scholar]
- 150.Chiefari J, Chong YK, Ercole F, et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: the RAFT process. Macromolecules. 1998;31:5559–62. doi: 10.1021/ma9804951. [DOI] [Google Scholar]
- 151.Matyjaszewski K, Gaynor S, Wang JS. Controlled radical polymerizations—the use of akyl iodides in degenerative transfer. Macromolecules. 1995;28:2093–5. doi: 10.1021/ma00110a050. [DOI] [Google Scholar]
- 152.Percec V, Popov AV, Ramirez-Castillo E, et al. Non-transition metal-catalyzed living radical polymerization of vinyl chloride initiated with iodoform in water at 25 degrees C. J Polym Sci, A Polym Chem. 2004;42:6267–82. doi: 10.1002/pola.20481. [DOI] [Google Scholar]
- 153.Cunningham MF. Living/controlled radical polymerizations in dispersed phase systems. Prog Polym Sci. 2002;27:1039–67. doi: 10.1016/S0079-6700(02)00008-4. [DOI] [Google Scholar]
- 154.Perrier S, Takolpuckdee P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/Xanthates (MADIX) polymerization. J Polym Sci, A Polym Chem. 2005;43:5347–93. doi: 10.1002/pola.20986. [DOI] [Google Scholar]
- 155.Matyjaszewski K, Xia JH. Atom transfer radical polymerization. Chem Rev. 2001;101:2921–90. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 156.Fischer H. The persistent radical effect: a principle for selective radical reactions and living radical polymerizations. Chem Rev. 2001;101:3581–610. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]
- 157.Kamigaito M, Ando T, Sawamoto M. Metal-catalyzed living radical polymerization. Chem Rev. 2001;101:3689–745. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 158.Zetterlund PB, Kagawa Y, Okubo M. Controlled/living radical polymerization in dispersed systems. Chem Rev. 2008;108:3747–94. doi: 10.1021/cr800242x. [DOI] [PubMed] [Google Scholar]
- 159.Save M, Guillaneuf Y, Gilbert RG. Controlled radical polymerization in aqueous dispersed media. Aust J Chem. 2006;59:693–711. doi: 10.1071/CH06308. [DOI] [Google Scholar]
- 160.Cunningham MF. Recent progress in nitroxide-mediated polymerizations in miniemulsion. C R Chim. 2003;6:1351–74. [Google Scholar]
- 161.McLeary JB, Klumperman B. RAFT mediated polymerisation in heterogeneous media. Soft Matter. 2006;2:45–53. doi: 10.1039/b513575c. [DOI] [PubMed] [Google Scholar]
- 162.Schork FJ, Luo YW, Smulders W, et al. Miniemulsion polymerization. In: Okubo M, et al., editors. Polymer particles. Berlin: Springer-Verlag Berlin; 2005. [Google Scholar]
- 163.Hawker CJ, Bosman AW, Harth E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem Rev. 2001;101:3661–88. doi: 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- 164.Golas PL, Matyjaszewski K. Click chemistry and ATRP: a beneficial union for the preparation of functional materials. QSAR Comb Sci. 2007;26:1116–34. doi: 10.1002/qsar.200740059. [DOI] [Google Scholar]
- 165.Min K, Matyjaszewski K. Atom transfer radical polymerization in aqueous dispersed media. Cent Eur J Chem. 2009;7:657–74. doi: 10.2478/s11532-009-0092-1. [DOI] [Google Scholar]
- 166.Tang W, Matyjaszewski K. Kinetic Modeling of Normal ATRP, Normal ATRP with [Cu-II](o), Reverse ATRP and SR&NI ATRP. Macromol Theory Simul. 2008;17:359–75. doi: 10.1002/mats.200800050. [DOI] [Google Scholar]
- 167.Riess G. Micellization of block copolymers. Prog Polym Sci. 2003;28:1107–70. doi: 10.1016/S0079-6700(03)00015-7. [DOI] [Google Scholar]
- 168.Gohy JF. Block copolymer micelles. In: Abetz V, editor. Block copolymers II. Berlin: Springer-Verlag Berlin; 2005. [Google Scholar]
- 169.Read ES, Armes SP. Recent advances in shell cross-linked micelles. Chem Commun 3021–3035. 2007. [DOI] [PubMed]
- 170.McCormack CL, Lowe AB. Aqueous RAFT polymerization: recent developments in synthesis of functional water-soluble (Co)polymers with controlled structures. Acc Chem Res. 2004;37:312–25. doi: 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- 171.Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloid Surf, B Biointerfaces. 1999;16:3–27. doi: 10.1016/S0927-7765(99)00058-2. [DOI] [Google Scholar]
- 172.McCormick CL, Kirkland SE, York AW. Synthetic routes to stimuli-responsive micelles, vesicles, and surfaces via controlled/living radical polymerization. Polym Rev. 2006;46:421–43. doi: 10.1080/15583720600945428. [DOI] [Google Scholar]
- 173.Boyer C, Bulmus V, Davis T, et al. Bioapplications of RAFT polymerization. Chem Rev. 2009;1009:5402–36. doi: 10.1021/cr9001403. [DOI] [PubMed] [Google Scholar]
- 174.Masci G, Giacomelli L, Crescenzi V. Atom transfer radical polymerization of N-isopropylacrylamide. Macromol Rapid Comm. 2004;25:559–64. doi: 10.1002/marc.200300140. [DOI] [Google Scholar]
- 175.Ma YH, Tang YQ, Billingham NC, et al. Synthesis of biocompatible, stimuli-responsive, physical gels based on ABA triblock copolymers. Biomacromolecules. 2003;4:864–8. doi: 10.1021/bm034118u. [DOI] [PubMed] [Google Scholar]
- 176.Li CM, Buurma NJ, Haq I, et al. Synthesis and characterization of biocompatible, thermoresponsive ABC and ABA triblock copolymer gelators. Langmuir. 2005;21:11026–33. doi: 10.1021/la0515672. [DOI] [PubMed] [Google Scholar]
- 177.Li GY, Shi LQ, An YL, et al. Double-responsive core-shell-corona micelles from self-assembly of diblock copolymer of poly(t-butyl acrylate-co-acrylic acid)-b-poly (N-isopropylacrylamide) Polymer. 2006;47:4581–7. doi: 10.1016/j.polymer.2006.04.041. [DOI] [Google Scholar]
- 178.Schilli C, Lanzendorfer MG, Muller AHE. Benzyl and cumyl dithiocarbamates as chain transfer agent in the RAFT polymerization of N-isopropylacrylamide. In situ FT-NIR and MALDI-TOF MS investigation. Macromolecules. 2002;35:6819–27. doi: 10.1021/ma0121159. [DOI] [Google Scholar]
- 179.Ganachaud F, Monteiro MJ, Gilbert RG, et al. Molecular weight characterization of poly(N-isopropylacrylamide) prepared by living free-radical polymerization. Macromolecules. 2000;33:6738–45. doi: 10.1021/ma0003102. [DOI] [Google Scholar]
- 180.Kujawa P, Segui F, Shaban S, et al. Impact of end-group association and main-chain hydration on the thermosensitive properties of hydrophobically modified telechelic poly(N-isopropylacrylamides) in water. Macromolecules. 2006;39:341–8. doi: 10.1021/ma051876z. [DOI] [Google Scholar]
- 181.Ray B, Isobe Y, Morioka K, et al. Synthesis of isotactic poly(N-isopropylacrylamide) by RAFT polymerization in the presence of Lewis acid. Macromolecules. 2003;36:543–5. doi: 10.1021/ma0257595. [DOI] [Google Scholar]
- 182.Li YT, Lokitz BS, McCormick CL. RAFT synthesis of a thermally responsive ABC triblock copolymer incorporating N-acryloxysuccinimide for facile in situ formation of shell cross-linked micelles in aqueous media. Macromolecules. 2006;39:81–9. doi: 10.1021/ma052116r. [DOI] [Google Scholar]
- 183.Hong CY, Pan CY. Direct synthesis of biotinylated stimuli-responsive polymer and diblock copolymer by RAFT polymerization using biotinylated trithiocarbonate as RAFT agent. Macromolecules. 2006;39:3517–24. doi: 10.1021/ma052593+. [DOI] [Google Scholar]
- 184.Kulkarni S, Schilli C, Grin B, et al. Controlling the aggregation of conjugates of streptavidin with smart block copolymers prepared via the RAFT copolymerization technique. Biomacromolecules. 2006;7:2736–41. doi: 10.1021/bm060186f. [DOI] [PubMed] [Google Scholar]
- 185.Patten TE, Matyjaszewski K. Atom transfer radical polymerization and the synthesis of polymeric materials. Adv Mater. 1998;10:901–15. doi: 10.1002/(SICI)1521-4095(199808)10:12<901::AID-ADMA901>3.0.CO;2-B. [DOI] [Google Scholar]
- 186.Ashford EJ, Naldi V, O’Dell R et al. First example of the atom transfer radical polymerisation of an acidic monomer: direct synthesis of methacrylic acid copolymers in aqueous media. Chem Commun 1285–1286. 1999.
- 187.Zhang X, Xia JH, Matyjaszewski K. Controlled/“living” radical polymerization of 2-(dimethylamino)ethyl methacrylate. Macromolecules. 1998;31:5167–9. doi: 10.1021/ma980477j. [DOI] [PubMed] [Google Scholar]
- 188.Lokaj J, Vlcek P, Kriz J. Synthesis of polystyrene-poly(2-(dimethylamino)ethyl methacrylate) block copolymers by stable free-radical polymerization. Macromolecules. 1997;30:7644–6. doi: 10.1021/ma971192u. [DOI] [Google Scholar]
- 189.Tang YQ, Liu SY, Armes SP, et al. Solubilization and controlled release of a hydrophobic drug using novel micelle-forming ABC triblock copolymers. Biomacromolecules. 2003;4:1636–45. doi: 10.1021/bm030026t. [DOI] [PubMed] [Google Scholar]
- 190.Ma YH, Tang YQ, Billingham NC, et al. Well-defined biocompatible block copolymers via atom transfer radical polymerization of 2-methacryloyloxyethyl phosphorylcholine in protic media. Macromolecules. 2003;36:3475–84. doi: 10.1021/ma021762c. [DOI] [Google Scholar]
- 191.Liu H, Jiang XZ, Fan J, et al. Aldehyde surface-functionalized shell cross-linked micelles with pH-tunable core swellability and their bioconjugation with lysozyme. Macromolecules. 2007;40:9074–83. doi: 10.1021/ma071494o. [DOI] [Google Scholar]
- 192.Xu FJ, Neoh KG, Kang ET. Bioactive surfaces and biomaterials via atom transfer radical polymerization. Prog Polym Sci. 2009;34:719–61. doi: 10.1016/j.progpolymsci.2009.04.005. [DOI] [Google Scholar]
- 193.Zhang X, Matyjaszewski K. Synthesis of well-defined amphiphilic block copolymers with 2-(dimethylamino)ethyl methacrylate by controlled radical polymerization. Macromolecules. 1999;32:1763–6. doi: 10.1021/ma981332f. [DOI] [Google Scholar]
- 194.Yuan WZ, Yuan JY, Zheng SX, et al. Synthesis, characterization, and controllable drug release of dendritic star-block copolymer by ring-opening polymerization and atom transfer radical polymerization. Polymer. 2007;48:2585–94. doi: 10.1016/j.polymer.2007.03.039. [DOI] [Google Scholar]
- 195.Licciardi M, Tang Y, Billingham NC, et al. Synthesis of novel folic acid-functionalized biocompatible block copolymers by atom transfer radical polymerization for gene delivery and encapsulation of hydrophobic drugs. Biomacromolecules. 2005;6:1085–96. doi: 10.1021/bm049271i. [DOI] [PubMed] [Google Scholar]
- 196.Karanikolopoulos N, Pitsikalis M, Hadjichristidis N, et al. pH-responsive aggregates from double hydrophilic block copolymers carrying zwitterionic groups. Encapsulation of antiparasitic compounds for the treatment of leishmaniasis. Langmuir. 2007;23:4214–24. doi: 10.1021/la0628827. [DOI] [PubMed] [Google Scholar]
- 197.Ravi P, Wang C, Tam KC, et al. Association behavior of poly(methacrylic acid)-block-poly(methyl methacrylate) in aqueous medium: potentiometric and laser light scattering studies. Macromolecules. 2003;36:173–9. doi: 10.1021/ma021302j. [DOI] [Google Scholar]
- 198.Yao H, Ravi P, Tam KC, et al. Association behavior of poly(methyl methacrylate-b-methacrylic acid-b-methyl methacrylate) in aqueous medium. Polymer. 2004;45:2781–91. doi: 10.1016/j.polymer.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 199.Dai S, Ravi P, Tam KC, et al. Novel pH-responsive amphiphilic diblock copolymers with reversible micellization properties. Langmuir. 2003;19:5175–7. doi: 10.1021/la0340652. [DOI] [Google Scholar]
- 200.Lewis AL. Phosphorylcholine-based polymers and their use in the prevention of biofouling. Colloids Surf, B. 2000;18:261–75. doi: 10.1016/S0927-7765(99)00152-6. [DOI] [PubMed] [Google Scholar]
- 201.Teoh SK, Ravi P, Dai S, et al. Self-assembly of stimuli-responsive water-soluble [60]fullerene end-capped ampholytic block copolymer. J Phys Chem B. 2005;109:4431–8. doi: 10.1021/jp045413r. [DOI] [PubMed] [Google Scholar]
- 202.Liu SY, Armes SP. Synthesis and aqueous solution behavior of a pH-responsive schizophrenic diblock copolymer. Langmuir. 2003;19:4432–38. doi: 10.1021/la020951l. [DOI] [Google Scholar]
- 203.Jones MC, Ranger M, Leroux JC. pH-sensitive unimolecular polymeric micelles: synthesis of a novel drug carrier. Bioconjug Chem. 2003;14:774–81. doi: 10.1021/bc020041f. [DOI] [PubMed] [Google Scholar]
- 204.Sant VP, Smith D, Leroux JC. Novel pH-sensitive supramolecular assemblies for oral delivery of poorly water soluble drugs: preparation and characterization. J Control Release. 2004;97:301–12. doi: 10.1016/j.jconrel.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 205.Satturwar P, Eddine MN, Ravenelle F, Leroux JC. pH-responsive polymeric micelles of poly(ethylene glycol)-b-poly(alkyl(meth)acrylate-co-methacrylic acid): Influence of the copolymer composition on self-assembling properties and release of candesartan cilexetil. Eur J Pharm Biopharm. 2007;65:379–387. doi: 10.1016/j.ejpb.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 206.Tian Y, Bromberg L, Lin SN, et al. Complexation and release of doxorubicin from its complexes with pluronic P85-b-poly(acrylic acid) block copolymers. J Control Release. 2007;121:137–45. doi: 10.1016/j.jconrel.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 207.Tian Y, Ravi P, Bromberg L, et al. Synthesis and aggregation behavior of Pluronic F87/poly(acrylic acid) block copolymer in the presence of doxorubicin. Langmuir. 2007;23:2638–46. doi: 10.1021/la060780a. [DOI] [PubMed] [Google Scholar]
- 208.Mitsukami Y, Donovan MS, Lowe AB, et al. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34:2248–56. doi: 10.1021/ma0018087. [DOI] [Google Scholar]
- 209.Yusa S, Shimada Y, Mitsukami Y, et al. pH-responsive micellization of amphiphilic diblock copolymers synthesized via reversible addition-fragmentation chain transfer polymerization. Macromolecules. 2003;36:4208–15. doi: 10.1021/ma030065x. [DOI] [Google Scholar]
- 210.Sumerlin BS, Lowe AB, Thomas DB, et al. Aqueous solution properties of pH-responsive AB diblock acrylamido copolymers synthesized via aqueous RAFT. Macromolecules. 2003;36:5982–7. doi: 10.1021/ma034088n. [DOI] [Google Scholar]
- 211.Sumerlin BS, Lowe AB, Thomas DB, et al. Aqueous solution properties of pH-responsive AB diblock acrylamido-styrenic copolymers synthesized via aqueous reversible addition-fragmentation chain transfer. J Polym Sci, A Polym Chem. 2004;42:1724–34. doi: 10.1002/pola.11069. [DOI] [Google Scholar]
- 212.Schilli CM, Zhang MF, Rizzardo E, et al. A new double-responsive block copolymer synthesized via RAFT polymerization: poly(N-isopropylacrylamide)-block-poly(acrylic acid) Macromolecules. 2004;37:7861–6. doi: 10.1021/ma035838w. [DOI] [Google Scholar]
- 213.Lee SM, Chen H, Dettmer CM, et al. Polymer-caged liposomes: a pH-Responsive delivery system with high stability. J Am Chem Soc. 2007;129:15096–7. doi: 10.1021/ja070748i. [DOI] [PubMed] [Google Scholar]
- 214.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40:2004–21. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 215.Huisgen R. 1.3-dipolare cycloadditionen—ruckschau und ausblick. Angew Chem Int Ed. 1963;75:604–37. doi: 10.1002/ange.19630751304. [DOI] [Google Scholar]
- 216.Rostovtsev VV, Green LG, Fokin VV, et al. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed. 2002;41:2596–9. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 217.Agut W, Taton D, Lecommandoux S. A versatile synthetic approach to polypeptide based rod-coil block copolymers by click chemistry. Macromolecules. 2007;40:5653–61. doi: 10.1021/ma070579m. [DOI] [Google Scholar]
- 218.Jiang X, Lok MC, Hennink WE. Degradable-brushed pHEMA-pDMAEMA synthesized via ATRP and click chemistry for gene delivery. Bioconjug Chem. 2007;18:2077–84. doi: 10.1021/bc0701186. [DOI] [PubMed] [Google Scholar]
- 219.Persson EM, Gustafsson AS, Carlsson AS, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm Res. 2005;22:2141–51. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 220.Lysik MA, Wu-Pong S. Innovations in oligonucleotide drug delivery. J Pharm Sci. 2003;92:1559–73. doi: 10.1002/jps.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kingsley JD, Dou HY, Morehead J, et al. Nanotechnology: a focus on nanoparticles as a drug delivery system. J Neuroimmune Pharm. 2006;1:340–50. doi: 10.1007/s11481-006-9032-4. [DOI] [PubMed] [Google Scholar]
- 222.Pinto Reis C, Neufeld RJ, Ribeiro AJ, et al. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed Nanotechnol Biol Med. 2006;2:8–21. doi: 10.1016/j.nano.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 223.Lee LY, Wang CH, Smith KA. Supercritical antisolvent production of biodegradable micro- and nanoparticles for controlled delivery of paclitaxel. J Control Release. 2008;125:96–106. doi: 10.1016/j.jconrel.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 224.Birnbaum DT, Brannon-Peppas L. Microparticle drug delivery systems. In: Brown DM, editor. Drug delivery systems in cancer therapy. Totowa: Humana; 2004. [Google Scholar]
- 225.PAS 71:2005, Vocabulary—nanoparticles.
- 226.Tirelli N. (Bio)Responsive nanoparticles. Curr Opin Colloid Interface Sci. 2006;11:210–6. doi: 10.1016/j.cocis.2006.09.002. [DOI] [Google Scholar]
- 227.Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2007;96:203–9. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- 228.Kohane DS, Tse JY, Yeo Y, et al. Biodegradable polymeric microspheres and nanospheres for drug delivery in the peritoneum. J Biomed Mater Res A. 2006;77A:351–61. doi: 10.1002/jbm.a.30654. [DOI] [PubMed] [Google Scholar]
- 229.Yang Y, Bajaj N, Xu P, et al. Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials. 2009;30:1947–53. doi: 10.1016/j.biomaterials.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 230.Thiele L, Diederichs JE, Reszka R, et al. Competitive adsorption of serum proteins at microparticles affects phagocytosis by dendritic cells. Biomaterials. 2003;24:1409–18. doi: 10.1016/S0142-9612(02)00525-2. [DOI] [PubMed] [Google Scholar]
- 231.Kumar MNVR. Nano and microparticles as controlled drug delivery devices. J Pharm Pharm Sci. 2000;3:234–58. [PubMed] [Google Scholar]
- 232.Jong WHD, Borm PJA. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3:133–49. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mu L, Feng SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 2003;86:33–48. doi: 10.1016/S0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]
- 234.Coester C, Kreutera J, Briesen H, Langer K. Preparation of avidin-labelled gelatin nanoparticles as carriers for biotinylated peptide nucleic acid (PNA) Int J Pharm. 2000;196:147–9. doi: 10.1016/S0378-5173(99)00409-3. [DOI] [PubMed] [Google Scholar]
- 235.Damgé C, Maincent P, Ubrich N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Control Release. 2007;117:163–70. doi: 10.1016/j.jconrel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 236.Date AA, Joshi MD, Patravale VB. Parasitic diseases: liposomes and polymeric nanoparticles versus lipid nanoparticles. Adv Drug Deliv Rev. 2007;59:505–21. doi: 10.1016/j.addr.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 237.Ahmad Z, Pandey R, Sharma S, et al. Alginate nanoparticles as antituberculosis drug carriers: formulation development, pharmacokinetics and therapeutic potential. Indian J Chest Dis Allied Sci. 2006;48:171–6. [PubMed] [Google Scholar]
- 238.Calvo P, Gouritin B, Brigger I, et al. PEGylated polycyanoacrylate nanoparticles as vector for drug delivery in prion diseases. J Neurosci Methods. 2001;111:151–5. doi: 10.1016/S0165-0270(01)00450-2. [DOI] [PubMed] [Google Scholar]
- 239.Sharma A, Sharma US. Liposomes in drug delivery: progress and limitations. Int J Pharm. 1997;154:123–40. doi: 10.1016/S0378-5173(97)00135-X. [DOI] [Google Scholar]
- 240.Jesorka A, Orwar O. Liposomes: technologies and analytical applications. Annu Rev Anal Chem. 2008;1:801–32. doi: 10.1146/annurev.anchem.1.031207.112747. [DOI] [PubMed] [Google Scholar]
- 241.Mozafari MR. Liposomes: an overview of manufacturing techniques. Cell Mol Biol Lett. 2005;10:711–9. [PubMed] [Google Scholar]
- 242.Paasonen L, Romberg B, Storm G, et al. Temperature-sensitive poly(N-(2-hydroxypropyl)methacrylamide mono/dilactate)-coated liposomes for triggered contents release. Bioconjug Chem. 2007;18:2131–6. doi: 10.1021/bc700245p. [DOI] [PubMed] [Google Scholar]
- 243.Jones MN. The surface-properties of phospholipid liposome systems and their characterization. Adv Colloid Interface Sci. 1995;54:93–128. doi: 10.1016/0001-8686(94)00223-Y. [DOI] [PubMed] [Google Scholar]
- 244.Sulkowski WW, Pentak D, Nowak K, et al. The influence of temperature, cholesterol content and pH on liposome stability. J Mol Struct. 2005;744:737–47. doi: 10.1016/j.molstruc.2004.11.075. [DOI] [Google Scholar]
- 245.Takeuchi H, Kojima H, Yamamoto H, et al. Polymer coating of liposomes with a modified polyvinyl alcohol and their systemic circulation and RES uptake in rats. J Control Release. 2000;68:195–205. doi: 10.1016/S0168-3659(00)00260-1. [DOI] [PubMed] [Google Scholar]
- 246.Ulrich AS. Biophysical aspects of using liposomes as delivery vehicles. Biosci Rep. 2002;22:129–50. doi: 10.1023/A:1020178304031. [DOI] [PubMed] [Google Scholar]
- 247.Semple SC, Chonn A, Cullis PR. Interactions of liposomes and lipid-based carrier systems with blood proteins: relation to clearance behaviour in vivo. Adv Drug Deliv Rev. 1998;32:3–17. doi: 10.1016/S0169-409X(97)00128-2. [DOI] [PubMed] [Google Scholar]
- 248.Bondurant B, Mueller A, O’Brien DF. Photoinitiated destabilization of sterically stabilized liposomes. Biochim Biophys Acta Biomembr. 2001;1511:113–22. doi: 10.1016/S0005-2736(00)00388-6. [DOI] [PubMed] [Google Scholar]
- 249.Coderch L, Fonollosa J, Estelrich J, et al. Influence of cholesterol on liposome fluidity by EPR—relationship with percutaneous absorption. J Control Release. 2000;68:85–95. doi: 10.1016/S0168-3659(00)00240-6. [DOI] [PubMed] [Google Scholar]
- 250.Hayashi H, Kono K, Takagishi T. Temperature sensitization of liposomes using copolymers of N-ispopropylacrylamide. Bioconjug Chem. 1999;10:412–8. doi: 10.1021/bc980111b. [DOI] [PubMed] [Google Scholar]
- 251.Cho EC, Lim HJ, Kim HJ, et al. Role of pH-sensitive polymer-liposome complex in enhancing cellular uptake of biologically active drugs. Mat Sci Eng C-Biomimetic and Supramolecular Systems. 2009;29:774–8. [Google Scholar]
- 252.Kono K, Henmi A, Yamashita H, et al. Improvement of temperature-sensitivity of poly(N-isopropylacrylamide)-modified liposomes. J Control Release. 1999;59:63–75. doi: 10.1016/S0168-3659(98)00180-1. [DOI] [PubMed] [Google Scholar]
- 253.Kono K. Thermosensitive polymer-modified liposomes. Adv Drug Deliv Rev. 2001;53:307–19. doi: 10.1016/S0169-409X(01)00204-6. [DOI] [PubMed] [Google Scholar]
- 254.Hayashi H, Kono K, Takagishi T. Temperature-controlled release property of phospholipid vesicles bearing a thermo-sensitive polymer. Biochim Biophys Acta Biomembr. 1996;1280:127–34. doi: 10.1016/0005-2736(95)00273-1. [DOI] [PubMed] [Google Scholar]
- 255.Han HD, Shin BC, Choi HS. Doxorubicin-encapsulated thermosensitive liposomes modified with poly(N-isopropylacrylamide-co-acrylamide): drug release behavior and stability in the presence of serum. Eur J Pharm Biopharm. 2006;62:110–6. doi: 10.1016/j.ejpb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 256.Seki K, Tirrell DA. Interactions of synthetic-polymers with cell-membrane and model membrane systems. 5. pH-dependent complexation of poly(acrylic acid) derivatives with phospholipid vesicle membranes. Macromolecules. 1984;17:1692–8. doi: 10.1021/ma00139a009. [DOI] [Google Scholar]
- 257.Drummond DC, Zignani M, Leroux JC. Current status of pH-sensitive liposomes in drug delivery. Progr Lipid Res. 2000;39:409–60. doi: 10.1016/S0163-7827(00)00011-4. [DOI] [PubMed] [Google Scholar]
- 258.Kono K, Igawa T, Takagishi T. Cytoplasmic delivery of calcein mediated by liposomes modified with a pH-sensitive poly(ethylene glycol) derivative. Biochim Biophys Acta Biomembr. 1997;1325:143–54. doi: 10.1016/S0005-2736(96)00244-1. [DOI] [PubMed] [Google Scholar]
- 259.Sakaguchi N, Kojima C, Harada A, et al. Preparation of pH-sensitive poly(glycidol) derivatives with varying hydrophobicities: their ability to sensitize stable liposomes to pH. Bioconjug Chem. 2008;19:1040–8. doi: 10.1021/bc7004736. [DOI] [PubMed] [Google Scholar]
- 260.Leroux JC, Roux E, Garrec D, et al. N-isopropylacrylamide copolymers for the preparation of pH-sensitive liposomes and polymeric micelles. J Control Release. 2001;72:71–84. doi: 10.1016/S0168-3659(01)00263-2. [DOI] [PubMed] [Google Scholar]
- 261.Pierige F, Serafini S, Rossi L, et al. Cell-based drug delivery. Adv Drug Deliv Rev. 2008;60:286–95. doi: 10.1016/j.addr.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 262.Schmidt JJ, Rowley J, Kong HJ. Hydrogels used for cell-based drug delivery. J Biomed Mater Res A. 2008;87A:1113–22. doi: 10.1002/jbm.a.32287. [DOI] [PubMed] [Google Scholar]
- 263.Walker PA, Aroom KR, Jimenez F, et al. Advances in progenitor cell therapy using scaffolding constructs for central nervous system injury. Stem Cell Rev Rep. 2009;5:283–300. doi: 10.1007/s12015-009-9081-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Aboody KS, Najbauer J, Schmidt NO, et al. Targeting of melanoma brain metastases using engineered neural stem/progenitor cells. Neuro-Oncology. 2006;8:119–26. doi: 10.1215/15228517-2005-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100:157–68. doi: 10.1016/S0092-8674(00)81692-X. [DOI] [PubMed] [Google Scholar]
- 266.Cha JS, Falanga V. Stem cells in cutaneous wound healing. Clin Dermatol. 2007;25:73–8. doi: 10.1016/j.clindermatol.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 267.Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing. Burns. 2009;35:171–80. doi: 10.1016/j.burns.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Quattrocelli M, Cassano M, Crippa S et al. Cell therapy strategies and improvements for muscular dystrophy. Cell Death Differ. (in press). 2009. doi:10.1038/cdd.2009.160. [DOI] [PubMed]
- 269.Nakagawa H, Akita S, Fukui M, et al. Human mesenchymal stem cells successfully improve skin-substitute wound healing. Br J Dermatol. 2005;153:29–36. doi: 10.1111/j.1365-2133.2005.06554.x. [DOI] [PubMed] [Google Scholar]
- 270.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77. doi: 10.1016/S0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 271.Huss R. Isolation of primary and immortalized CD34(-) hematopoietic and mesenchymal stem cells from various sources. Stem Cells. 2000;18:1–9. doi: 10.1634/stemcells.18-1-1. [DOI] [PubMed] [Google Scholar]
- 272.Hows JM. Status of umbilical cord blood transplantation in the year 2001. J Clin Pathol. 2001;54:428–34. doi: 10.1136/jcp.54.6.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kim WS, Park BS, Sung JH, et al. Wound heating effect of adipose-derived stem cells: a critical role of secretory factors on human dermal fibroblasts. J Dermatol Sci. 2007;48:15–24. doi: 10.1016/j.jdermsci.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 274.Lu W, Zhang YJ, Jin Y. Potential of stem cells for skin regeneration following burns. Expert Rev Dermatol. 2009;4:97–9. doi: 10.1586/edm.09.9. [DOI] [Google Scholar]
- 275.Orive G, Anitua E, Pedraz JL, et al. Biomaterials for promoting brain protection, repair and regeneration. Nat Rev Neurosci. 2009;10:682–92. doi: 10.1038/nrn2685. [DOI] [PubMed] [Google Scholar]
- 276.Weber LM, Hayda KN, Haskins K, et al. The effects of cell-matrix interactions on encapsulated beta-cell function within hydrogels functionalized with matrix-derived adhesive peptides. Biomaterials. 2007;28:3004–11. doi: 10.1016/j.biomaterials.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 277.Cruise GM, Hegre OD, Lamberti FV, et al. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly(ethylene glycol) diacrylate membranes. Cell Transplant. 1999;8:293–306. doi: 10.1177/096368979900800310. [DOI] [PubMed] [Google Scholar]
- 278.Cruise GM, Scharp DS, Hubbell JA. Characterization of permeability and network structure of interfacially photopolymerized poly(ethylene glycol) diacrylate hydrogels. Biomaterials. 1998;19:1287–94. doi: 10.1016/S0142-9612(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 279.Khademhosseini A, Eng G, Yeh J, et al. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. J Biomed Mater Res A. 2006;79A:522–32. doi: 10.1002/jbm.a.30821. [DOI] [PubMed] [Google Scholar]
- 280.Kong HJ, Smith MK, Mooney DJ. Designing alginate hydrogels to maintain viability of immobilized cells. Biomaterials. 2003;24:4023–9. doi: 10.1016/S0142-9612(03)00295-3. [DOI] [PubMed] [Google Scholar]
- 281.Sawhney AS, Hubbell JA. Poly(ethylene oxide)-graft-poly(L-lysine) copolymers to enhance the biocompatibility of poly(L-lysine)-alginate microcapsules membranes. Biomaterials. 1992;13:863–70. doi: 10.1016/0142-9612(92)90180-V. [DOI] [PubMed] [Google Scholar]
- 282.Keshaw H, Forbes A, Day RM. Release of angiogenic growth factors from cells encapsulated in alginate beads with bioactive glass. Biomaterials. 2005;26:4171–9. doi: 10.1016/j.biomaterials.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 283.Darrabie MD, Kendall WF, Opara EC. Characteristics of poly-L-ornithine-coated alginate microcapsules. Biomaterials. 2005;26:6846–52. doi: 10.1016/j.biomaterials.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 284.Chae SY, Lee M, Kim SW, et al. Protection of insulin secreting cells from nitric oxide induced cellular damage by crosslinked hemoglobin. Biomaterials. 2004;25:843–50. doi: 10.1016/S0142-9612(03)00605-7. [DOI] [PubMed] [Google Scholar]
- 285.Bloch J, Bachoud-Levi AC, Deglon N, et al. Neuroprotective gene therapy for Huntington’s disease, using polymer-encapsulated cells engineered to secrete human ciliary neurotrophic factor: results of a phase I study. Hum Gene Ther. 2004;15:968–75. doi: 10.1089/hum.2004.15.968. [DOI] [PubMed] [Google Scholar]
- 286.Hamidi M, Zarrin A, Foroozesh M, et al. Applications of carrier erythrocytes in delivery of biopharmaceuticals. J Control Release. 2007;118:145–60. doi: 10.1016/j.jconrel.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 287.Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006;108:2827–35. doi: 10.1182/blood-2006-03-012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Gorantla S, Dou HY, Boska M, et al. Quantitative magnetic resonance and SPECT imaging for macrophage tissue migration and nanoformulated drug delivery. J Leukoc Biol. 2006;80:1165–74. doi: 10.1189/jlb.0206110. [DOI] [PubMed] [Google Scholar]
- 289.Rossi L, Serafini S, Pierigé F, et al. Erythrocyte-based drug delivery. Expert Opin Drug Deliv. 2005;2:311–22. doi: 10.1517/17425247.2.2.311. [DOI] [PubMed] [Google Scholar]
- 290.Beutler E, Dale GL, Guinto E, et al. Enzyme replacement therapy in gauchers-disease—preliminary clinical-trial of a new enzyme preparation. Proc Nat Acad Sci USA. 1977;74:4620–3. doi: 10.1073/pnas.74.10.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Frontiers of engineering: reports on leading-edge engineering from the 2008 symposium. Washington, DC: National Academies; 2009. [Google Scholar]
- 292.Mulligan RC. The basic science of gene-therapy. Science. 1993;260:926–32. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 293.Anderson WF. Human gene therapy. Nature. 1998;392:25–30. doi: 10.1038/32801. [DOI] [PubMed] [Google Scholar]
- 294.Cavazzana-Calvo M, Hacein-Bey S, Basile CD, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–72. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 295.Li S, Huang L. Nonviral gene therapy: promises and challenges. Gene Ther. 2000;7:31–4. doi: 10.1038/sj.gt.3301110. [DOI] [PubMed] [Google Scholar]
- 296.Li SD, Huang L. Non-viral is superior to viral gene delivery. J Control Release. 2007;123:181–3. doi: 10.1016/j.jconrel.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 297.Davidson BL, Breakefield XO. Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci. 2003;4:353–64. doi: 10.1038/nrn1104. [DOI] [PubMed] [Google Scholar]
- 298.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 299.Gardlik R, Palffy R, Hodosy J, et al. Vectors and delivery systems in gene therapy. Med Sci Monit. 2005;11:RA110–21. [PubMed] [Google Scholar]
- 300.Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007—an update. J Gene Med. 2007;9:833–42. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 301.Edelstein M, Abedi M, Wixon J. Gene therapy clinical trials worldwide In: J. Gene Med. Available via. http://www.wiley.co.uk/genetherapy/clinical/. Cited 13 Nov 2009; 2009.
- 302.Suda T, Suda K, Liu DX. Computer-assisted hydrodynamic gene delivery. Mol Ther. 2008;16:1098–104. doi: 10.1038/mt.2008.66. [DOI] [PubMed] [Google Scholar]
- 303.Nguyen DN, Green JJ, Chan JM, et al. Polymeric materials for gene delivery and DNA vaccination. Adv Mat. 2009;21:847–67. doi: 10.1002/adma.200801478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Seow WY, Yang YY. Functional polycarbonates and their self-assemblies as promising non-viral vectors. J Control Release. 2009;139:40–7. doi: 10.1016/j.jconrel.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 305.Cai X, Conley S, Naash M. Nanoparticle applications in ocular gene therapy. Vis Res. 2008;48:319–24. doi: 10.1016/j.visres.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Andrieu-Soler C, Bejjani RA, Bizemont T, et al. Ocular gene therapy: a review of nonviral strategies. Mol Vis. 2006;12:1334–47. [PubMed] [Google Scholar]
- 307.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–31. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Chen JL, Wang H, Gao JQ, et al. Liposomes modified with polycation used for gene delivery: preparation, characterization and transfection in vitro. Int J Pharm. 2007;343:255–61. doi: 10.1016/j.ijpharm.2007.05.045. [DOI] [PubMed] [Google Scholar]
- 309.McNeil SE, Perrie Y. Gene delivery using cationic liposomes. Expert Opin Ther Pat. 2006;16:1371–82. doi: 10.1517/13543776.16.10.1371. [DOI] [Google Scholar]
- 310.Oliveira AC, Ferraz MP, Monteiro FJ, et al. Cationic liposome-DNA complexes as gene delivery vectors: development and behaviour towards bone-like cells. Acta Biomater. 2009;5:2142–51. doi: 10.1016/j.actbio.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 311.Juliano R, Alam MR, Dixit V, et al. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158–71. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Yoo HS, Lee JE, Chung H, et al. Self-assembled nanoparticles containing hydrophobically modified glycol chitosan for gene delivery. J Control Release. 2005;103:235–43. doi: 10.1016/j.jconrel.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 313.Zhao J, Gou ML, Dai M, et al. Preparation, characterization, and in vitro cytotoxicity study of cationic PCL-Pluronic-PCL (PCFC) nanoparticles for gene delivery. J Biomed Mater Res A. 2009;90A:506–13. doi: 10.1002/jbm.a.31950. [DOI] [PubMed] [Google Scholar]
- 314.Gao K, Huang L. Nonviral methods for siRNA Delivery. Mol Pharmacol. 2009;6:651–8. doi: 10.1021/mp800134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173–84. doi: 10.1038/nrg2006. [DOI] [PubMed] [Google Scholar]
- 316.Fougerolles AR. Delivery vehicles for small interfering RNA in vivo. Hum Gene Ther. 2008;19:125–32. doi: 10.1089/hum.2008.928. [DOI] [PubMed] [Google Scholar]
- 317.Ziady AG, Davis PB, Konstan NW. Non-viral gene transfer therapy for cystic fibrosis. Expert Opin Biol Ther. 2003;3:449–58. doi: 10.1517/14712598.3.3.449. [DOI] [PubMed] [Google Scholar]
- 318.Felgner PL, Gadek TR, Holm M, et al. Lipofection—a highly efficient, lipid-mediated DNA-transfection procedure. Proc Nat Acad Sci USA. 1987;84:7413–7. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Moreira JN, Santos A, Moura V, et al. Non-viral lipid-based nanoparticles for targeted cancer systemic gene silencing. J Nanosci Nanotechnol. 2008;8:2187–204. doi: 10.1166/jnn.2008.319. [DOI] [PubMed] [Google Scholar]
- 320.Li WJ, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24:438–49. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 321.Aigner A. Delivery systems for the direct application of siRNAs to induce RNA interference (RNAi) in vivo. J Biomed Biotechnol. 2006;2006:1–15. doi: 10.1155/JBB/2006/71659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kawakami S, Hashida M. Targeted delivery systems of small interfering RNA by systemic administration. Drug Metabol Pharmacokinet. 2007;22:142–51. doi: 10.2133/dmpk.22.142. [DOI] [PubMed] [Google Scholar]
- 323.Fire A, Xu SQ, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 324.Rao DD, Vorhies JS, Senzer N, et al. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61:746–59. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 325.Hannon GJ. RNA interference. Nature. 2002;418:244–51. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 326.Silva JM, Hammond SM, Hannon GJ. RNA interference: a promising approach to antiviral therapy? Trends Mol Med. 2002;8:505–8. doi: 10.1016/S1471-4914(02)02421-8. [DOI] [PubMed] [Google Scholar]
- 327.Takahashi Y, Nishikawa M, Takakura Y. Nonviral vector-mediated RNA interference: its gene silencing characteristics and important factors to achieve RNAi-based gene therapy. Adv Drug Deliv Rev. 2009;61:760–6. doi: 10.1016/j.addr.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 328.Miyagishi M, Hayashi M, Taira K. Comparison of the suppressive effects of antisense oligonucleotides and siRNAs directed against the same targets in mammalian cells. Antisense Nucleic Acid Drug Dev. 2003;13:1–7. doi: 10.1089/108729003764097296. [DOI] [PubMed] [Google Scholar]
- 329.Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004;3:318–29. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- 330.Pushparaj PN, Aarthi JJ, Manikandan J, et al. siRNA, miRNA, and shRNA: in vivo applications. J Dent Res. 2008;87:992–1003. doi: 10.1177/154405910808701109. [DOI] [PubMed] [Google Scholar]
- 331.Rao DD, Senzer N, Cleary MA, et al. Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 2009;16:807–9. doi: 10.1038/cgt.2009.53. [DOI] [PubMed] [Google Scholar]
- 332.Manfredsson FP, Okun MS, Mandel RJ. Gene therapy for neurological disorders: challenges and future prospects for the use of growth factors for the treatment of Parkinson’s disease. Curr Gene Ther. 2009;9:375–88. doi: 10.2174/156652309789753400. [DOI] [PubMed] [Google Scholar]
- 333.Candolfi M, Kroeger KM, Muhammad A, et al. Gene therapy for brain cancer: combination therapies provide enhanced efficacy and safety. Curr Gene Ther. 2009;9:409–21. doi: 10.2174/156652309789753301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Davidson BL, Paulson HL. Molecular medicine for the brain: silencing of disease genes with RNA interference. Lancet Neurol. 2004;3:145–9. doi: 10.1016/S1474-4422(04)00678-7. [DOI] [PubMed] [Google Scholar]
- 335.He S, Zhang DC, Cheng F, et al. Applications of RNA interference in cancer therapeutics as a powerful tool for suppressing gene expression. Mol Biol Rep. 2009;36:2153–63. doi: 10.1007/s11033-008-9429-7. [DOI] [PubMed] [Google Scholar]
- 336.Kumar S, Nagy TR, Ponnazhagan S. Therapeutic potential of genetically modified adult stem cells for osteopenia. Gene Ther. 2010;17:105–16. doi: 10.1038/gt.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Fink D, Mata M, Glorioso JC. Cell and gene therapy in the treatment of pain. Adv Drug Deliv Rev. 2003;55:1055–64. doi: 10.1016/S0169-409X(03)00103-0. [DOI] [PubMed] [Google Scholar]
- 338.Soldner F, Hockemeyer D, Beard C, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–77. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Lindvall O, Kokaia Z. Prospects of stem cell therapy for replacing dopamine neurons in Parkinson’s disease. Trends Pharmacol Sci. 2009;30:260–7. doi: 10.1016/j.tips.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 340.Willmon C, Harrington K, Kottke T, et al. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17:1667–76. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Power AT, Bell JC. Cell-based delivery of oncolytic viruses: a new strategic alliance for a cancer. Mol Ther. 2007;15:660–5. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 342.Josiah DT, Zhu D, Dreher F, et al. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol Ther. 2010;18:337–85. doi: 10.1038/mt.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Menasche P. Cell-based therapy for heart disease: a clinically oriented perspective. Mol Ther. 2009;17:758–66. doi: 10.1038/mt.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]