

Abstract
Multiple sclerosis (MS) is a chronic autoimmune disease, where T-cells attack the myelin sheath in the central nervous system (CNS), characterized by relapsing-remitting episodes, or gradually increasing severity of symptoms and disability that accumulate over time. While current MS therapies have been proven in clinical trials to provide significant benefits, they cater only to subsets of patients. Moreover, there is an acute need to identify the most effective and safe treatment appropriate for each patient prospectively, since early intervention has been proven to prevent accumulation of irreversible dysfunction. In this review we discuss the current state-of-the-art in pharmacogenetic research as applied to the common marketed and in-development MS treatments, with respect to both efficacy and safety aspects. We conclude by discussing the relevance of pharmacogenetics and other biomarkers to the prediction, prevention and personalization of MS medications in the horizon.
Keywords: Multiple Sclerosis, Pharmacogenetics, Personalized medicine, Polymorphism, Predictive diagnostics, Preventive measures
Multiple sclerosis: clinical characteristics and pathoetiology
Multiple Sclerosis (MS) is the most common chronic neurological disease affecting young adults, with onset usually at the second to fourth decade of life [1]. Similarly to other autoimmune diseases, women are affected 3–4 times more often than men [1]. The pathoetiology of the disease is complex, with multiple factors influencing the mode of the disease, its severity and the progression rate. Epidemiological studies have clearly indicated that both genetic and environmental factors determine and modify the disease [1]. The two main clinical subtypes that characterize the onset of the disease are the relapsing-remitting (RRMS) and primary progressive (PPMS) disease courses, accounting for 85% and 10–15% of all MS cases, respectively [2]. It should be noted, that over two thirds of RR patients will succumb to a secondary progressive (SPMS) disease mode within 10–15 years. Other rarer disease subtypes include the extremely active form (malignant MS) and the mild type (benign MS). MS is characterized clinically by the manifestation of acute, as well as chronically accumulating symptoms, including numbness, motor weakness, optic neuritis, incoordination, diplopia, dizziness and vertigo. Other accompanying symptoms and signs may include fatigue, urinary urgency or retention, depression, heat intolerance, pain, cognitive dysfunction, spasticity, ataxia, nystagmus and others.
The underlying pathology that leads to clinical disability is comprised of intricate relationships between three disrupted processes involving the myelin sheath (mainly oligodendocytes) that wraps central nervous system (CNS) neurons: (a) the inflammatory axis involves the invasion of immune cells with aberrant activity into the CNS, where they attack the myelin components, a process termed demyelination and resulting in secondary neurodegeneration. This process subsequently leads to axonal and neuronal loss; (b) the primary neurodegeneration axis involves primary axonal and neuronal loss without triggers or signs of prominent inflammation; and (c) the impaired repair axis involves unsuccessful repair attempts by the CNS to mend the damage accumulated through insults from the other two processes. Unfortunately, this is but a partial and incomplete repair process, which is often the basis of residual deficits and disability. The dynamic interactions between these three processes are characterized by periods of acute attacks, interspaced by remissions during which chronic disability is accumulated. The attacks are manifested through paralysis, visual loss and other symptoms prohibiting the normal conduct of activities of daily living. These offensives occur due to an over-activation of immune cells in the inflammatory axis. The chronic phase of the disease, on the other hand, is considered to be dominated by the neurodegenerative processes. In this fashion, repair is mainly noted once the acute attack fades off and may give rise to spontaneous recovery that in the early phase of the disease does not necessitate pharmaceutical intervention. Triggers for initiation, duration, severity and rate of attacks can be both environmental and inherited and it is as yet unclear how to predict the disease course in any given patient.
The differential diagnoses of MS include, at early stages, cerebrovascular disease, epilepsy, vitamin-B12 deficiency and neuropathy. As disease progresses and neuroimaging techniques are employed (e.g. MRI), conditions that may appear similar to MS include vasculitis, congenital biochemical disorders, viral infection and lupus (SLE). Lastly, neuromyelitis optica (NMO) is a severe inflammation and astrocyte loss with profound demyelination in the optic nerves and the spinal cord. NMO symptoms are similar to MS (e.g., vision loss, muscle weakness, numbness and paralysis in the arms and legs, etc), and therefore in the past, NMO was considered a severe form of MS. However, research has shown that this condition results from the production of pathogenic autoantibodies against the water channel aquaporin-4 (AQP-4) on astrocytes [3]. This is therefore one important example where biomarkers serve as essential diagnosis tools. By screening NMO immunoglobulins (NMO-IgG) in serum it is now possible to accurately diagnose this condition and provide the required course of treatment. While acute attacks are treated similarly in MS and NMO (intravenous steroids), chronic treatment by IFN-β and GA does not seem to be beneficial in the case of NMO, but rather oral steroid medication and immunosuppressive drugs (e.g. azathioprine) may be used [4, 5]. In this case, early diagnosis is critical, since NMO immunosuppressive therapy is necessary to prevent attack-related disability. Indeed, this is one gateway to personalization facilitated by biomarkers in this therapeutic area.
Multiple sclerosis: nature versus nurture
Despite decades of research it is still debatable to what extent genetic factors, environmental variables, and interactions between them cause and modify MS disease susceptibility. A common method used to estimate the contribution of genetics employs heritability testing (h2) in kinships, and particularly in monozygotic (MZ) versus dizygotic (DZ) twins. A recent meta-analysis [6] suggests that commonly used methodology for estimating heritability has been largely non-informative and yielded mean estimates that are weak in North America and France (h2: 0.25–0.31); moderate in Italy (h2: 0.45); and high in the UK, Canada and Denmark (h2: 0.53–0.76). Moreover, confidence intervals literally span the entire gamut, ranging from zero to 0.94 [6]. The limited utility of these estimates stems both from flaws in heritability analysis [7], as well as the low disease prevalence and the challenges of collecting unbiased informative twin pairs. It is therefore important to employ other types of family-based data, such as cohorts of adoptee, half-sibling step-siblings and conjugal pedigrees in order to address questions about the extent to which MS susceptibility is heritable [7]. Overall, reported studies suggest that MS indeed has a substantial genetic basis (possibly with maternal parent-of-origin effect), although the number and size of studies published thus far are small.
Clearly, environmental factors play a significant role in the determination and modification of MS disease risk, progression and severity in addition and in combination with the effects of genetic factors. The unique geography of the disease, for instance, indicates a latitude effect that seems to alter risk estimates upon immigration [2]. It should be pointed, though, that the geographical features of the disease are complex and thus the irregular disease distribution pattern in secluded populations, such as Sardinia and the Inuit population in Canada, may support the hypothesis of a genetic etiology as well. Other lines of evidence support the contribution of environmental factors to MS etiology, including sunlight and ultraviolet radiation and vitamin D intake. Intensive investigation has been devoted to the influence of viral infections, primarily Epstein-Barr and Herpes simplex. These were recently reviewed by Milo and Kahan [2]. Another possible type of modifying factors relates to epigenetics, the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence [8]. The parent-of-origin and gender-specific effects noted in MS disease risk support the possibility of an epigenetic involvement, with the human leukocyte antigen (HLA) region effects published recently [9].
The main genetic association with MS disease susceptibility in northern Europeans has been shown with extended major histocompatibility complex (MHC) haplotypes, especially those containing HLADRB1*1501 (HLA DRb1 gene, 1501 allele), odds ratio (OR) = 7.5 for a dominant allele-dose effect [10]. The HLADBR5*0101 allele, which is strongly correlated with HLADRB1*1501, has been found to act as a modifying factor in interaction (epistasis) with the latter, affecting disease severity rather than susceptibility [11]. Other secondary signals to the DRB1*1501 allele in the HLA region have been found, with the top associated single nucleotide polymorphisms (SNPs) correlating with the HLA-B*4402 [12], HLADRB1*08 [10], HLA-DRB1*01 and HLA-DRB1*10 [13] alleles. A protective effect has been registered for the HLA-DRB1*14 allele [10]. Other complex epistatic interactions have been registered, where HLA-DQA1*0102 strongly interacts with HLADRB1*1501, so that in the presence of the latter MS risk is increased, while in its absence a protective effect has been registered [13]. In this context it should be noted that a recent publication suggests that the HLA*DRB profile of NMO patients is different from that observed for MS patients: DRB3 is over-represented in NMO patients, while DRB1 and DRB5 are over-abundant in MS patients [14]. This differential genetic signature serves both to support the progressively accepted claim that the two conditions are underlined by different etiologies, as well as assists in delineating the associated pathways and assessing novel therapies for each condition.
In addition to the HLA region, modest associations with MS susceptibility have been published and replicated for IL7R [15, 16], IL2R [17], EVI5 [18], CLEC16A and KIF1B [19]. The largest OR reported for a non-HLA variant was 1.34 in sporadic MS and 1.74 in multiplex families [20]. Indeed, functional studies have long shown a biological interaction between the HLA and KIR (killer immunoglobulin receptor) gene products (such as KIF1B) that seems to explain the reported genetic signatures [21]. Overall, most of the association data available to-date originated from candidate gene studies supplemented by functional analyses. These studies included admixture mapping strategies that were able to narrow down the association interval by studying populations with different linkage disequilibrium (LD) block structure (i.e. genomic sequences with dissimilar patterns of co-inheritance across populations with divergent geneology, such as European Caucasians versus African Americans) [22]. Recent years have seen the reports of genome wide association studies (GWAS), which screen the entire genome for SNPs using off-the-shelf chips. This methodology has been useful in highlighting common variations in regions of interest along the genome. A meta-analysis of a total of 2,624 MS cases and 7,220 controls, which was then replicated in additional 2,215 cases and 2,116 controls, pinpointed TNFRSF1A, IRF8, and CD6 with high significance level (combined p value <6 × 10(−6)) and ORs <1.6. It should be noted that in the same cohort the HLA-DRB1*1501 tagging SNP rs3135388 was associated with MS susceptibility at an OR of 2.75 and a combined p value of 3.8 × 10(−225) for a minor allele frequency (MAF) of 20%. The highest OR registered for a novel non-HLA association was found for a rare allele with an MAF of only 2%.
Genetic studies serve not only to highlight disease specific risk polymorphisms, but may also be designed to bring to light similarities between, and unique features of, a range of autoimmune diseases. It has been known for decades that autoimmune disorders tend to co-aggregate in specific individuals and nuclear families as reviewed by Baranzini [23]. As expected, the majority of genetic research has focused on the HLA region, which has been widely studied in the context of autoimmune diseases for several decades. For instance, it has been shown that HLADRB1 is associated with susceptibility to MS, but protection from type 1 diabetes (T1D) [10, 24]. Lately, comparison of GWAS data and gene-network analysis suggested that gene variant signatures in non-HLA genes are shared between MS and other autoimmune diseases, such as rheumatoid arthritis (RA), Type 1 Diabetes and possibly celiac disease [23]. Another study published by the Welcome Trust Case Control Consortium (WTCCC2) tested seven disease cohorts in parallel [25] and suggested novel MS-specific genes (TYK2, H6PD), which were further supported by follow-up genetic and functional studies [26, 27].
As whole, the most plausible explanation is that MS is caused by the combined effects of genetic, epigenetic and environmental factors, and is thus said to have a multifactorial etiology. It is likely that the interaction patterns between these factors gives rise to disease subtypes, and it is possible that eventually each subtype would be discreetly diagnosed with high sensitivity and specificity as was the case for NMO, reviewed above. Moreover, it is likely that subtypes branching from and refining the categorization into RR and SPMS/PPMS may delineate independent or differentially dominant pathological processes typical of each presentation, and thus susceptible and responsive to different medications. This heterogeneity could be the result of environmental factors (such as infectious agents, climate or diet), genetic determinants, or interactions between them. Understanding how such genetic polymorphisms may affect the response to interventional therapy is the focus of pharmacogenetic (PGx) research [28, 29].
Multiple sclerosis therapy in practice and in development
Currently there are no curative treatments for MS. Available therapies provide symptom relief and can be categorized into indications for acute, chronic and escalation therapy. Severe acute attacks are treated by in-hospital intravenous steroids administered for about 6 days at a time. Once diagnosis is made and frequency of attacks exhibited by RR patients reaches a certain threshold (determined by the expanded disability status score (EDSS) [30] and other physician driven input), treatment is commenced using one of four available injectible immunosuppressants in order to attenuate the disease (reduce the number of relapses per year): recombinant IFN-β1a agents (avonex and rebif); recombinant IFN-β1b (betaseron/betaferon) and glatiramer acetate (GA, copaxone). While these immunotherapies have been shown to be efficacious in clinical trials, they are beneficial in only ∼30% of the RR patients [31]. Escalation therapy includes two approved options at present: mitoxantrone (novatrone) and natalizumab (tysabri), both of which are associated with safety limitations.
The definition of positive response to MS intervention therapy is in itself a debatable subject. Response can be measured by clinical scales (most commonly the EDSS); prescribed based on neuroimaging scans; or be a composite of both. Still, the threshold of acceptable improvements and the implications and relevance of short-term improvement on long-term prognosis are largely unknown. In recent years MRI has been the primary tool used for confirming diagnosis and for establishing efficacy claims in late-stage pharmaceutical development. However, in the case of established MS, the correlation between clinical response and MRI measurements has been largely weak, suggesting MRI alone is a poor biomarker of predicted drug response [32]. Therefore, clinical end-points remain the main diagnostic criteria in drug R&D, with the recently published phase III trials of oral cladribine and figolimod both employing as primary end-point the rate of clinical relapses [33–35]. In both cases sustained progression of disability, relapse-free rate and MRI lesion burden improvement were used as supportive secondary end-points. However, the definition of response may still be heterogeneous even when considered in the terms of “relapse rate”: the duration of the study, the inclusion and exclusion criteria, the definition of relapse and acute treatment protocol, population demographics and ethnicity, are only some of the factors that show heterogeneity across cohorts [29]. Additionally, the optimized definition of response would incorporate elements of patient and care-giver reported quality of life in order to fully gage into personalized MS medicine [29].
Given the high non-response rates registered for MS therapy and the increasing number of drugs available for switching to as potentially beneficial alternatives, a vast amount of effort has been invested in recent years to identify appropriate biomarkers that indicate poor response early on. One such recent publication suggests that developing new T2-hyperintense lesions during IFN-β therapy predicts subsequent poor response with hazards ratio (HR) of 16.8 [36]. However, while useful as a research tool it is impractical and too expensive to incorporate routine MRI scans into clinical management of MS patients. The importance of these findings is in the identification of distinct and measurable pathological differences between responders and non-responders that are evident prior to clinical manifestation. It is therefore likely that other diagnostics, based on biological or genetic markers, would subsequently be found which would be useful as response predictors.
The focus of drug R&D in recent years has been devoted to oral therapies, with five such compounds currently in late-stage development [37]. The goal is to expand the therapeutic choice range, provide efficacious solutions for patients refractory to current medications, improve convenience and quality of life, increase patient compliance to treatment, enable earlier and possibly prophylactic therapy initiation and introduce novel mechanisms of actions to combat the disease, which may be useful for other modes of disease rather than merely RR. Biomarkers may have a seminal role in supporting the development of these new medications (via differentiation strategy and target population definition), provide helpful guidelines in regimen tailoring in the clinic, and provide pharmacovigilance indicators [38]. Further detail and discussion on these aspects is provided in the safety section under the “Pharmacogenetics in Multiple Sclerosis” heading below.
It should be noted that at the present time there are no approved treatments for patients with the progressive mode of disease (with the exception of mitoxantrone in worsening progressive MS [39]), and the limited available data suggest that RRMS available therapy is ineffective for this disease mode [40]. It is also unclear whether early and aggressive treatment of RR patients halts or delays the progression to SPMS. Ultimately, our lack of understanding of the pathophysiological processes that give rise to PPMS and SPMS underlines our failure in developing successful treatments for these phases. Research of the genetic, biological, clinical and epidemiological characteristics of patients with progressive disease modes as differentiated from the other forms of the disease would eventually yield both novel drug discovery and the development of diagnostics for treatment allocation and monitoring of progressive MS.
Biomarkers of multiple sclerosis disease and therapy
Currently there are no biomarkers available for MS to guide treatment selection, predict disease progression or monitor patients over time in the clinic. The only validated biomarkers in use for MS diagnosis confirmation are oligoclonal IgGs screened in CSF samples, and MRI scans. However, both are impractical in routine practice since the former requires an invasive procedure (i.e. lumbar puncture for acquirement of CSF fluid samples) and is not correlated with disease activity nor response to therapy; while the latter, which may have benefit in progression monitoring, is prohibitively expensive. A novel type of biomarkers has recently been studied intensively for the purpose of response prediction and monitoring of IFN-β therapy. Treatment by agents from the IFN-β class induces immunogenicity reactions in the form of binding and neutralizing antibodies (BABs and NABs, respectively) [41]. However, not all patients develop these antibodies and even when developed they may disappear spontaneously over time with poor correlation to response. Therefore the potential utility of these markers as diagnostics in the clinic is low unless coupled with additional markers that increase their specificity and sensitivity. Several other biomarkers are being investigated for their utility in MS disease management and monitoring, as reviewed in detail elsewhere [42].
One of the main stumbling blocks to the research of MS pathoetiology and subsequent development of prognostic biomarkers has been its neurodegenerative nature and the fact that most studies rely on animal models with limited applicability to the human disease. The leading animal model used for MS research has been the experimental autoimmune encephalomyelitis (EAE) model, which depicts only the immunological and inflammatory axis of the pathology. It is therefore necessary to study human subjects with each of the disease modes in great depth and develop biomarkers that delineate the processes that lead to clinical outcomes. These would then serve as mechanistic markers in the development of novel agents and will ultimately guide treatment regimen selection and disease monitoring in the clinic. One striking example to the limited utility of animal models for the development of MS drugs is the discordant efficacy and safety profiles registered for animals versus humans in the course of drug development [43].
Pharmacogenetics in multiple sclerosis: approved and experimental agents
Drug efficacy
Most of the PGx literature to-date has been dedicated to IFN-β treatments and predictors of positive versus partial or failed response (Table 1). 13 studies have been reported between 2001 and 2009 on investigation of IFN-β treatment efficacy, 4 of which in 2009 alone. In contrast, 2 studies were reported studying GA and only 1 studying mitoxantrone. The majority of studies conducted prior to 2009 are small candidate gene studies investigating one to several dozens of markers at a time. With the development of SNP detection technologies the research scope has expanded to investigation of whole pathways or genomes at a time. Most importantly, while many of the early studies lacked follow-up confirmation studies, the trend in recent years has been to incorporate discovery and replication cohorts into the initial publication in order to increase validity of the findings. However, even for the more recent publications several study design limitations exist which result in un-interpretable and non-replicated findings with little clinical utility information. A systematic examination of Table 1 highlights the main flaws: First, sample sizes are generally small although accurate phenotyping may compensate, to some degree, for small patient numbers; Second, rarely are markers studied in the context of healthy controls and placebo treated patients in order to decipher PGx-specific from disease progression and severity markers. Designs that do not explore the potential influence of prognostic markers on the PGx association are particularly vulnerable to false-positive findings; Third, the type of IFN-β used or the relative fraction of patients using each one of the three class drugs has been inconsistent across studies. This is mainly a result of the approved formularies in different countries for non-industry sponsored trials; Fourth, study populations vary by geographic residency and ethnic origin both within studies and across studies and population stratification control measurements have not been used in most cases; Fifth, while most studies focused on RR patients, some included a mixture of other MS disease modes with distinct clinical course and characteristics that may be particularly relevant to differential treatment response patterns; Sixth, the primary response definitions, for both positive and negative response span a wide range of possibilities and the incorporation of both clinical and MRI measurements is the exception rather than the rule. Choice of extreme phenotype investigation (i.e. “best” responders versus “worst” responders) is generally more powerful but comes at the cost of sample size and is critically dependent upon other factors, such as trial duration; Seventh, as mentioned above, the scope of studied genes and number and type of marker(s) studied within each genomic region limits the generalizability of reported findings. Moreover, in many instances, while overlapping genes may have been investigated, the selected markers within them were not identical. This is particularly relevant when studies were conducted in different populations that may be characterized by differential linkage disequilibrium properties; Eighth, not all studies screened for NABs as either an exclusion criterion or a potential confounder in statistical analyses; Lastly, the reported studies generally do not correct for multiple testing or multiple end-points studied. This is particularly concerning given the fact that in several instances the same study population (or partially overlapping populations) have been studied for different PGx markers and reported in separate publications, leading to misconception of independent findings. Overall, the power to detect PGx effects is objectively dependent upon factors beyond study design. Treatment response phenotypes are a composite outcome of many different factors in addition and complementary to genetic factors. Such environmental effects may include diet, concomitant treatments, viral infection and compliance. Moreover, low penetrance, gene-gene and gene-environment interactions, as well as limited statistical power are additional factors that may have hindered progress in MS PGx research thus far.
Table 1.
Pharmacogenetic studies published 2001–2009 studying multiple sclerosis therapies
| Drug | Reference | Responders [or ADR positive] | Non-responders [or ADR negative] | Healthy controls / placebo treated | Population and disease mode | Primary positive response [or ADR positive] definition | Non-responder definition | Follow-up (months) | Genetic regions and markers | Results |
|---|---|---|---|---|---|---|---|---|---|---|
| IFN-β | Fusco et al., 2001 | 22 | 17 | N/A | Italy; RR | No EDSS disability progression and no relapse or one mild relapse | ≥1 moderate or severe relapses and/or ≥1 point in the EDSS | 24 | HLA class II DRB1 and DQB1 genes | No association |
| Villoslada et al., 2002 | 77 | 57 | N/A | Spain; RR | No relapse and no EDSS increase sustained over 2 consecutive visits | ≥2 relapses and/or an increase in EDSS of ≥1 point sustained over 2 consecutive visits | 24 | HLA class I (A and B) & II (DRB1 and DQB1) genes | No association | |
| Sriram et al., 2003 | 57 | 48 | N/A | Spain; RR | No relapse and no EDSS increase sustained over 2 consecutive visits | ≥2 relapses and/or an increase in EDSS of ≥1 point sustained over 2 consecutive visits | 24 | IFNAR1 and IFNARA2 (8 SNPs) | No association | |
| Cunningham et al., 2005 | 94 | 68 | HC | Ireland; RR | Relapse rate reduced by a third and no sustained EDSS progressiona | Relapse rate unchanged or increases | 24 (response definition based on data after initial 6–9 months) | Candidate genes (32) with ISRE inthe promoter (54 SNPs) | IFNAR1, LMP7, CTSS, MXA | |
| Leyva et al., 2005 | 104 | 43 | HC | Spain; 100 RR & 47 SP | Unspecified | ≥1 relapses or an ≥0.5 points in the EDSS after the 1st year | 24 | IFNAR1 (2 SNPs) and IFNAR2 (18 SNP) | No association | |
| Fernandez et al., 2005 | 66 | 30 | N/A | Spain; 61 RR & 35 SP | Unspecified | ≥1 relapses or an ≥0.5 points in the EDSS after the 1st year (sustained over ≥6 months) | 12 | HLA class II DRB1, DQA1 and DQB1 genes | No association | |
| Martinez et al., 2006 | 39 | 71 | HC | Spain; RR | No relapses | ≥1 relapses | 24 | IFNG (CA repeat) | IFNG | |
| Weinstock-Guttman et al., 2007 | 19 | 18 | N/A | US; RR | No relapse and EDSS stable | ≥1 relapses in preceding 12 months | 24 | MXA (2 promoter SNPs) | No association | |
| Byun et al., 2008 | 99 | 107 | N/A | Spain (3 sites); France (1 Site); RR | No relapse and no EDSS increase | ≥2 relapses or an increase in EDSS of ≥1 point | 24 | GWAS (100K SNPs) using pooled and top 35 SNP individual DNA genotyping | Nominally significant SNPs in various genes, including GPC5, HAPLN1, PTGER3, FLJ32978, COL25A1 | |
| 242; including original 99 | 249; including original cohort | |||||||||
| Comabella et al., 2009 | 53 | 53 | N/A | Spain; RR | No EDSS score increase and no relapses | ≥1 relapses and an ≥1 point in the EDSS | 24 | GWAS (500K SNPs) on pooled DNA | After validation cohort analysis GRIA3 SNP associted in female only, 6 more nominally significant SNPs in various genes | |
| 49 | 45 | Aim to replicate of top 383 SNPs (individual DNAs) | ||||||||
| Cenit et al., 2009 | 55 | 79 | HC | Spain; RR | No EDSS score increase and no relapses | ≥2 relapses or an increase in EDSS of ≥1 point | 24 | Aim to replicate Byun GWAS in HAPLN1 (3 SNPs) and GPC5 (3 SNPs) | Significant association and dose response for 2 out of 3 SNPs in GPC5 | |
| O’Doherty et al., 2009 | 155 | 100 | N/A | Ireland; RR | Relapse rate reduced by a third and no sustained EDSS progressiona | Relapse rate unchanged or increases | 24 (response definition based on data after initial 6–9 months) | 34 candidate genes (61 SNPs) | Allelic combinations, particularly of JAK2-IL10RB-GBP1-PIAS1 and JAK2-IL10-CASP3 | |
| Comabella et al., 2009 | 74 | 75 | N/A | Spain; RR | No EDSS score increase and no relapses | ≥1 relapses and an ≥1 point in the EDSS persistant for ≥2 consecutive visits separated by 6 month | 24 | HLA class I and II genes | No association | |
| Anthracenedione Mitoxantrone | Cotte et al., 2009 | Monotherapy: 121; Combination: 91 | Monotherapy: 34; Combination: 63 | HC | Germany (3 sites), Spain (1 site); RR, SP and atypical | At least one of: EDSS or MSFC or MRI stability/improvement; relapse rate improvement; lack of physician determined disease progression | EDSS deterioration of 1 point (EDSS <6.0) or 0.5 points (EDSS >6.0) | 12 (89.3%) or 9 (10%) or >21 (0.7%) | ABCB1 (2 SNPs); ABCG2 (2 SNPs); experimental in vitro and in vivo validation done as well | Monotherapy reached nominal statistical significance for an additive effect of the ABCB1-ABCG2 haplotype (but site effect pronounced); in vitro human MX efflux nominally significant between the extreme haplotype groups; EAE clinical response significantly associated with ABCG2 null vs. wt status |
| 36 (11%), half on monotherapy | 283 | Severe cardiac or hematological ADRs | Treated patients not meeting ADR definition | No association (a rare haplotype was found in one acute case) | ||||||
| Glatiramer Acetate | Fusco et al., 2001 | 22 | 22 | N/A | Italy; RR | No EDSS disability progressionb and no relapse or one mild relapse | ≥1 moderate or severe relapses and/or ≥1 point in the EDSS | 24 | HLA class II DRB1 and DQB1 genes | No association |
| Grossman et al., 2007 | 17 | 32 | Placebo and HC | Europe, Canada; RR | (1) No relapse and ≤1 T1-enhancing lesion in the third trimester (2) no T1-enhancing lesions in the third trimester | Treated patients not meeting positive response definition | 9 | 27 candidate genes (63 SNPs) and HLA DRB1*1501 | TCRB@, CTSS, MBP, CD86, IL1R1 | |
| PBO 10 | PBO 42 | |||||||||
| 14 | 22 | Placebo and HC | US: RR | No EDSS disability progressiona and no relapse | 24 | TCRB@, IL1R12RB2 | ||||
| PBO 11 | PBO 26 |
ABC ATP-binding cassette; MSFC multiple sclerosis functional composite; EDSS expanded disability status scale; ADR adverse drug reaction; RR relapsing remitting; SP secondary progressive; N/A not applicable; ISRE IFN stimulated response elements; HC healthy controls; wt wild type; PBO placebo
This table is an extension of Table 2 from Miller et al. [29]
aincrease of at least one point in the EDSS score sustained at least over 3 months
bmore than one point if baseline EDSS < 5.5, or more than 0.5 points if baseline EDSS at least 5.5
In the context of novel drug development the need to prove efficacy in comparison to existing medications presents new challenges to drug developers. Drugs that show superiority to placebo but not gold-standard comparator (such as IFN or GA) are unlikely to be approved by regulators and have low chances of gaining reimbursement status by health insurers. For this purpose biomarkers have the opportunity to pinpoint those subpopulations that may benefit from novel treatments while having little chance of benefiting from current approved therapy. Given the low percentage of individuals responding favorably to existing treatments, it is likely that new compounds with novel mechanisms of action may provide superior efficacy to substantial proportions of the MS population. For instance, fingolimod is a novel agent in late-stage development that is based on a novel mode-of-action. It is a sphingosine-1-phosphate receptor agonist that alters lymphocyte trafficking. The drug has shown efficacy in a 2-year, placebo controlled trial (FREEDOMS), but did not show clinical superiority to IFN-β1a in a head-to-head trial (TRANSFORMS) in the overall study population. However, the hypothesis that a subset of patients with a particular genetic, genomic or biomarker profile show clear benefit when treated with fingolimod rather than avonex has not been reported to have been explored.
Drug safety
Adverse drug reactions (ADRs) present particular concerns in the field of MS since most novel therapies are immunosuppressive in nature. Therefore, side effects tend to be potentially life-threatening and are often not visible unless exposure time and dosage are extended beyond the customary 12–18 months clinical trial duration. Some of these agents deplete immune cells for sustained periods of times which may result in life-threatening vulnerability in the face of infection or malignancy. This safety concern has been demonstrated through the experience with the drugs natalizumab (a humanized monoclonal antibody against α4β1 integrin) and mitoxantrone (a DNA-intercalating agent and topoisomerase II inhibitor [39, 44]). In fact, natalizumab was withdrawn from the market for 18 months in 2005, but has gained increased popularity since its re-introduction, exceeding $1B in 2009. 24 ADR cases of progressive multifocal leukoencephalopathy (PML) were reported prior to withdrawal and 11 more (including 4 deaths) since reintroduction. Mitoxantrone, on the other hand, is associated with severe immunosuppressive and cardiovascular side effects. The rare ADRs associated with both natalizumab and mitoxantrone have deemed their indication only to severe and non-tolerant cases, since there are currently no validated, specific and sensitive markers that would pinpoint individuals prone to develop these events. In the case of mitoxantrone a recent study suggested PGx markers in the ATP-binding cassette transporters (ABC), which carry and eliminate the drug from its target cells [45] (Table 1). While small and heterogeneous with respect to nature of ADR (i.e. severe cardiovascular or hematological adverse events were lumped together) as well as treatment regimen (patients received either mono- or combination therapy), the study presents a first attempt to identify genetic markers of mitxantrone ADRs. Such a marker could serve as a screening tool prior to therapy prescription and counter-indicate therapy for individuals at high risk for ADR development. In this fashion the majority of patients that are not at risk of developing the event would benefit from the option of starting treatment with one of these agents earlier in the disease course due to a “personalized” favorable benefit-risk profile. Such a study case has been proven efficacious and safe in the treatment of HIV with abacavir, which is associated with a hyper sensitivity ADR. The development of a genetic marker, HLA-B*5701, has recently been adopted into clinical guidelines as indicator of treatment allocation and has reduced the incidence of hyper sensitivity reactions to miniscule levels [46, 47]. Furthermore, various ADRs of common therapies have by now been confirmed to be highly associated with specific genetic markers that demonstrate high predictive power [48–51].
Overall, pharmacogenetic research of the ADRs associated with approved MS treatments has been limited thus far. This is mostly due to the fact that chronic treatments are relatively safe, and the experience with escalation therapy has been limited with little availability of genetic material for the purpose of marker research and diagnostic development. IFN-β therapy in general is associated with ADRs such as flu-like symptoms (experienced by about a third of treated patients), depression and transient laboratory abnormalities [52], while for GA the majority of ADRs are limited to local brief reaction at the site of injection. These ADRs are known to reduce patients’ adherence to medication, but since they are rarely severe there has been no research of diagnostic markers.
Novel oral treatments in development present a significant quality-of-life improvement to patients who are now limited to injectable options, but these new agents are also associated with a concerning safety profile. For instance, the recently concluded phase III trials for fingolimod reported increased infections, liver-function test abnormalities, cardiovascular disorders, as well as the mechanism-based expected leucopenia and lymphopenia [33, 35]. Similarly, cladribine treatment was associated with ADRs of infections and neoplasms [34]. Lymphocytopenia was again markedly more frequent in cladribine treated patients (27%) as expected given its selective induced apoptosis effects in T-lymphocytes and CD19+ B-lymphocytes. ADRs of lymphopenia increased with dose of both cladribine and fingolimod. Genetic or genomic variants may exist which predispose individual to react more strongly upon treatment and may therefore benefit from tailored decreased dosing regimen. It remains to be seen whether biological samples have been collected and studied to identify such markers of ADRs.
Implications of pharmacogenetics for prediction and personalization in multiple sclerosis
Healthcare systems worldwide are gradually adopting models of comparative-effectiveness, where demonstrated favorable benefit-cost profiles serve as the basis for inclusion in national formularies (e.g. UK) or health insurance reimbursement programs (e.g. US). It is becoming clear that “blockbuster drugs” will be few and far between in years to come. It is expected that demonstrated superior efficacy in subpopulations diagnostically identified via PGx markers will serve to qualify newly approved medicines, as well as serve as means for economic health management in choosing generic versus branded therapeutics [53]. PGx markers are already becoming important factors in regulatory approval of new medicines and are being incorporated into existing drug labels [54–56]. MS clinical management is a case in point. Traditional therapy with IFN-β and GA has proven to be disappointing and selection of first or subsequent switch therapy is done currently by trial-and-error [57]. On the other hand, novel treatments are about to enter the market. These innovative treatments are associated with concerning side effects and the lack of longitudinal medical experience of treating neurologists poises a considerable entry to market barrier. It is therefore a pressing priority to identify treatment response predictive diagnostics (Fig. 1).
Fig. 1.
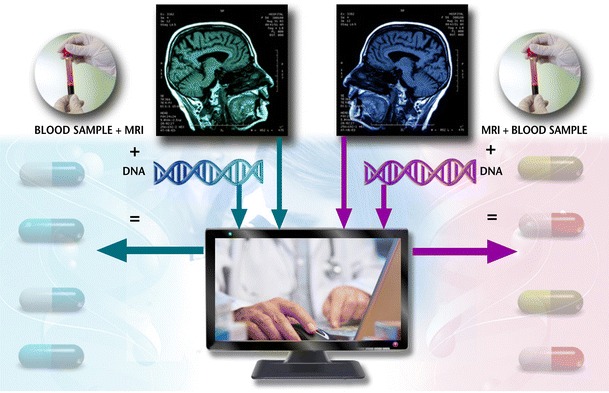
Scheme of Tailored Therapeutics in Multiple Sclerosis Management by Integrated Pharmacogenetic and Other Clinical and Diagnostic Measures. The framework of Personalized, Predictive and Preventative Multiple Sclerosis Management relies on integrative analysis of input from diagnostic tests with proven utility in selection of treatment regimen for the individual patient. In this fashion, blood samples (or cheek swabs / spit samples) are collected from the patient upon early signs of disease development. DNA is extracted and key, validated genetic markers of disease progression /subtype and response to marketed drugs (safety and efficacy indicators) are genotyped. The genetic data is then analyzed in combination with other validated disease and treatment response variables, including indicators of clinical characteristics, MRI imaging, and / or other biomarkers in body fluids. Specific algorithms provide the physician with predictive pharmacogenetics- and bioinformatics-based tools to allow informed medical decision prior to initiation of pharmacological intervention. Hospitalizations, treatment switching and inadequate efficacy results are thus considerably reduced, leading to improved patients’ medical care management and quality of life, as well as a decrease in healthcare costs
Overall, PGx research has failed to identify any major genetic contributors to phenotypes of MS drug efficacy and/or safety thus far. However, we propose that the lion’s share of research conducted thus far has been based on inconsistent study designs, small population sizes, limited scientific scope, heterogeneous response and non-response definitions and little functional experimentation follow-up (Table 1). The substantial improvement in all of these categories seen through 2009 and the expansion of research into methxantrone, in addition to IFN- β and GA PGx, marks a trend for change in the quality and utility of results coming out of contemporary research. While it is possible that genetic variants do not serve as significant and useful predictors of any MS treatment response, the poor level of evidence available today contributes little to support or refute this hypothesis. To this end, the recent establishment of a European network of research groups that promotes MS PGx research (the United Europeans for development of pharmacogenetics in MS network- UEPHA*MS) [58] denotes an international effort toward standardization and harmonization of methodology in order to maximize the usefulness of study results and to allow directly interpretable replication analyses. Expansion of the network to include non-European groups will further allow the generalizability of outcomes to other world populations [59].
It is interesting to mention in the context of MS PGx research, that treatment response to another interferon therapy, peginterferon-alpha-2 in combination with ribavirin, used for the treatment of hepatitis C viral infection has been recently tightly associated with a genetic marker near the IL28B gene, encoding IFN-λ-3 [60]. This report further supports the notion that adequately powered and well-designed MS PGx studies may be able to identify markers that will be useful in future clinical management of immunomodulatory and other MS treatments. It is our belief that with the establishment of the above international consortia, dedicated to standardized and harmonized study designs and sample handling procedures, MS PGx research will unravel treatment response mechanisms and promote to the development of predictive diagnostics integrated with tailored therapeutics.
Contributor Information
Iris Grossman, Phone: +919-699-9967, Email: irisg@isragene.com, Email: irisg@cabernetpharma.com.
Ariel Miller, Phone: +972-4-8250851, FAX: +972-4-8250909, Email: millera@tx.technion.ac.il.
References
- 1.Compston A, Confavreux C, Lassmann H, et al. McAlpine’s multiple sclerosis. Philadelphia: Churchill Livingstone/Elsevier; 2006. [Google Scholar]
- 2.Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev. 2010;9(5):A387–94. PMID: 19932200. [DOI] [PubMed]
- 3.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66(5):630–43. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM. Neuromyelitis optica. Int MS J. 2006;13(2):42–50. [PubMed] [Google Scholar]
- 5.Uzawa A, Mori M, Hayakawa S, et al. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol. 2010;17(5):672–6. PMID: 20039942. [DOI] [PubMed]
- 6.Hawkes CH, Macgregor AJ. Twin studies and the heritability of MS: a conclusion. Mult Scler. 2009;15(6):661–7. doi: 10.1177/1352458509104592. [DOI] [PubMed] [Google Scholar]
- 7.Ramagopalan SV, Dyment D, AEbers GC. Genetic epidemiology: the use of old and new tools for multiple sclerosis. Trends Neurosci. 2008;31(12):645–52. doi: 10.1016/j.tins.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Wu C, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293(5532):1103–5. doi: 10.1126/science.293.5532.1103. [DOI] [PubMed] [Google Scholar]
- 9.Chao MJ, Ramagopalan SV, Herrera BM, et al. Epigenetics in multiple sclerosis susceptibility: difference in transgenerational risk localizes to the major histocompatibility complex. Hum Mol Genet. 2009;18(2):261–6. doi: 10.1093/hmg/ddn353. [DOI] [PubMed] [Google Scholar]
- 10.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006;15(18):2813–24. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 11.Gregersen JW, Kranc KR, Ke X, et al. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature. 2006;443(7111):574–7. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 12.Rioux JD, Goyette P, Vyse TJ, et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci USA. 2009;106(44):18680–5. doi: 10.1073/pnas.0909307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lincoln MR, Ramagopalan SV, Chao MJ, et al. Epistasis among HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci USA. 2009;106(18):7542–7. doi: 10.1073/pnas.0812664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brum DG, Barreira AA, Santos AC, et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler. 2010;16(1):21–9. doi: 10.1177/1352458509350741. [DOI] [PubMed] [Google Scholar]
- 15.Lundmark F, Duvefelt K, Iacobaeus E, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39(9):1108–13. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- 16.Peltonen L. Old suspects found guilty-the first genome profile of multiple sclerosis. N Engl J Med. 2007;357(9):927–9. doi: 10.1056/NEJMe078147. [DOI] [PubMed] [Google Scholar]
- 17.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 18.Hoppenbrouwers IA, Aulchenko YS, Ebers GC, et al. EVI5 is a risk gene for multiple sclerosis. Genes Immun. 2008;9(4):334–7. doi: 10.1038/gene.2008.22. [DOI] [PubMed] [Google Scholar]
- 19.Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV, et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet. 2008;40(12):1402–3. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- 20.D’Netto MJ, Ward H, Morrison KM, et al. Risk alleles for multiple sclerosis in multiplex families. Neurology. 2009;72(23):1984–8. doi: 10.1212/WNL.0b013e3181a92c25. [DOI] [PubMed] [Google Scholar]
- 21.Traherne JA. Human MHC architecture and evolution: implications for disease association studies. Int J Immunogenet. 2008;35(3):179–92. doi: 10.1111/j.1744-313X.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reich D, Patterson N, Jager PL, et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat Genet. 2005;37(10):1113–8. doi: 10.1038/ng1646. [DOI] [PubMed] [Google Scholar]
- 23.Baranzini SE. The genetics of autoimmune diseases: a networked perspective. Curr Opin Immunol. 2009;21(6):596–605. doi: 10.1016/j.coi.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Nejentsev S, Howson JM, Walker NM, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450(7171):887–92. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burton PR, Clayton DG, Cardon LR, et al. Association scan of 14, 500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39(11):1329–37. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ban M, Goris A, Lorentzen AR, et al. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet. 2009;17(10):1309–13. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcina A, Ramagopalan SV, Fernandez O, et al. Hexose-6-phosphate dehydrogenase: a new risk gene for multiple sclerosis. Eur J Hum Genet. 2010;18(5):618–20. PMID: 19935835. [DOI] [PMC free article] [PubMed]
- 28.Grossman I. Routine pharmacogenetic testing in clinical practice: dream or reality? Pharmacogenomics. 2007;8(10):1449–59. doi: 10.2217/14622416.8.10.1449. [DOI] [PubMed] [Google Scholar]
- 29.Miller A, Avidan N, Tzunz-Henig N, et al. Translation towards personalized medicine in Multiple Sclerosis. J Neurol Sci. 2008;274(1–2):68–75. doi: 10.1016/j.jns.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 30.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33(11):1444–52. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Diego RS, Weiner HL. Novel therapeutic strategies for multiple sclerosis—a multifaceted adversary. Nat Rev Drug Discov. 2008;7(11):909–25. doi: 10.1038/nrd2358. [DOI] [PubMed] [Google Scholar]
- 32.Rovaris M. The definition of non-responder to multiple sclerosis treatment: neuroimaging markers. Neurol Sci. 2008;29(Suppl 2):S222–4. doi: 10.1007/s10072-008-0943-0. [DOI] [PubMed] [Google Scholar]
- 33.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15. PMID: 20089954. [DOI] [PubMed]
- 34.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416-26. PMID: 20089960. [DOI] [PubMed]
- 35.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. PMID: 20089952. [DOI] [PubMed]
- 36.Prosperini L, Gallo V, Petsas N, et al. One-year MRI scan predicts clinical response to interferon beta in multiple sclerosis. Eur J Neurol. 2009;16(11):1202–9. doi: 10.1111/j.1468-1331.2009.02708.x. [DOI] [PubMed] [Google Scholar]
- 37.Hughes B. First oral therapies for multiple sclerosis anticipated. Nat Rev Drug Discov. 2009;8(11):831–3. doi: 10.1038/nrd3043. [DOI] [PubMed] [Google Scholar]
- 38.Akkari PA, Swanson TW, Crenshaw DG, et al. Pipeline pharmacogenetics: a novel approach to integrating pharmaco-genetics into drug development. Curr Pharm Des. 2009;15(32):3754–63. doi: 10.2174/138161209789649538. [DOI] [PubMed] [Google Scholar]
- 39.Martinelli V, Radaelli M, Straffi L, et al. Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci. 2009;30(Suppl 2):S167–70. doi: 10.1007/s10072-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 40.Rojas JI, Romano M, Ciapponi A, et al. Interferon Beta for primary progressive multiple sclerosis. Cochrane Database Syst Rev. 2010;(1): CD006643. PMID: 20091602. [DOI] [PubMed]
- 41.Vartanian TK, Zamvil SS, Fox E, et al. Neutralizing antibodies to disease-modifying agents in the treatment of multiple sclerosis. Neurology. 2004;63(11 Suppl 5):S42–9. doi: 10.1212/wnl.63.11_suppl_5.s42. [DOI] [PubMed] [Google Scholar]
- 42.Miller A, Paperna T, Caspi O, et al. Theranostics and Translation towards Personalized Medicine for Multiple Sclerosis. In: Koslow EGSH, editor. Personalized Medicine, Healthcare and Integrative Neuroscience: Oxford University Press; 2010 (in press).
- 43.Friese MA, Montalban X, Willcox N, et al. The value of animal models for drug development in multiple sclerosis. Brain. 2006;129(Pt 8):1940–52. doi: 10.1093/brain/awl083. [DOI] [PubMed] [Google Scholar]
- 44.Fox EJ. Management of worsening multiple sclerosis with mitoxantrone: a review. Clin Ther. 2006;28(4):461–74. doi: 10.1016/j.clinthera.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 45.Cotte S, Ahsen N, Kruse N, et al. ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain. 2009;132(Pt 9):2517–30. doi: 10.1093/brain/awp164. [DOI] [PubMed] [Google Scholar]
- 46.Lalonde RG, Thomas R, Rachlis A, et al. Successful implementation of a national HLA-B*5701 genetic testing service in Canada. Tissue Antigens. 2010;75(1):12–8. PMID: 19843279. [DOI] [PubMed]
- 47.Hughes AR, Brothers CH, Mosteller M, et al. Genetic association studies to detect adverse drug reactions: abacavir hypersensitivity as an example. Pharmacogenomics. 2009;10(2):225–33. doi: 10.2217/14622416.10.2.225. [DOI] [PubMed] [Google Scholar]
- 48.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816–9. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 49.Nelson MR, Bacanu SA, Mosteller M, et al. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 2009;9(1):23–33. doi: 10.1038/tpj.2008.4. [DOI] [PubMed] [Google Scholar]
- 50.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008;359(8):789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 51.Fellay J, Thompson AJ, Ge D, et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010. PMID: 20173735. [DOI] [PubMed]
- 52.Vandenbroeck K, Matute C. Pharmacogenomics of the response to IFN-beta in multiple sclerosis: ramifications from the first genome-wide screen. Pharmacogenomics. 2008;9(5):639–45. doi: 10.2217/14622416.9.5.639. [DOI] [PubMed] [Google Scholar]
- 53.Epstein RS, Frueh FW, Geren D, et al. Payer perspectives on pharmacogenomics testing and drug development. Pharmacogenomics. 2009;10(1):149–51. doi: 10.2217/14622416.10.1.149. [DOI] [PubMed] [Google Scholar]
- 54.Frueh FW, Amur S, Mummaneni P, et al. Pharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug use. Pharmacotherapy. 2008;28(8):992–8. doi: 10.1592/phco.28.8.992. [DOI] [PubMed] [Google Scholar]
- 55.Conmmittee for medical products for human use (CHMP) E. Reflection paper on the use of pharmacogenetics in the pharmacogenetic evaluation of medical products. PMID. 2007.
- 56.Food and Drug Administration. Guidance for industry and FDA staff pharmacogenetic tests and genetic tests for heritable markers. 2007.
- 57.Kirstein-Grossman I, Beckmann JS, Lancet D, et al. Pharmacogenetic development of personalized medicine: multiple sclerosis treatment as a model. Drug News Perspect. 2002;15(9):558–67. doi: 10.1358/dnp.2002.15.9.740230. [DOI] [PubMed] [Google Scholar]
- 58.Vandenbroeck K, Comabella M, Tolosa E, et al. United Europeans for development of pharmacogenomics in multiple sclerosis network. Pharmacogenomics. 2009;10(5):885–94. doi: 10.2217/pgs.09.33. [DOI] [PubMed] [Google Scholar]
- 59.Benedek G, Paperna T, Avidan N, et al. Opposing effects of the HLA-DRB1*0301-DQB1*0201 haplotype on the risk of developing multiple sclerosis in diverse Arab populations in Israel. Genes & Immunity 2010; doi:10.1038/gene.2010.20. [DOI] [PubMed]
- 60.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]