

Abstract
Mental and neurological disorders are increasingly prevalent and constitute a major societal and economic burden worldwide. Many of these diseases and disorders are characterized by progressive deterioration over time, that ultimately results in identifiable symptoms that in turn dictate therapy. Disease-specific symptoms, however, often occur late in the degenerative process. A better understanding of presymptomatic events could allow for the development of new diagnostics and earlier interventions that could slow or stop the disease process. Such studies of progressive neurodegeneration require the use of animal models that are characterized by delayed or slowly developing disease phenotype(s). This brief review describes several examples of such animal models that have recently been developed with relevance to various neurological diseases and disorders, and delineates the potential of such models to aid in predictive diagnosis, early intervention and disease prevention.
Keywords: Preventative therapy, Epilepsy, Schizophrenia, Amyotrophic lateral sclerosis, Parkinson’s disease, Neurodevelopmental disorders
Introduction
Mental and neurological disorders are increasingly prevalent and constitute a major societal and economic burden worldwide. The impact of these disorders has traditionally been underestimated by epidemiological data that have relied on mortality rather than morbidity. According to the World Health Organization mental and neurological disorders are responsible for only about 1 % of deaths but account for almost 11% of the global disease burden [1]. Further, due to increased life expectancy and the aging of general populations in both developed and developing countries this number is expected to rise to 14.7% by 2020 [1]. Many of the most socially and economically devastating neurological diseases and disorders are characterized by progressive neurodegeneration. The prevalence of some of the most common of these diseases in the United States is depicted in Fig. 1. By extrapolation the prevalence worldwide is probably about 20x that of the USA.
Fig. 1.
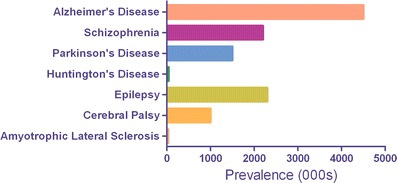
Prevalence in the USA of several important neurological diseases characterized by progressive neurodegeneration. Numbers are expressed in thousands (000 s) of persons affected. Data derived from [2]
Symptoms of many forms of progressive disease often appear in late adolescence or early adulthood and then become increasingly severe with increasing age. The neurodegenerative process however, usually begins long before the onset of clinically diagnosed symptoms. For example, it is estimated that up to 60% of the dopaminergic neurons in the substantia nigra need to be lost before the first clinical signs of Parkinson’s disease appear [3]. Thus, in many cases a precipitating event occurring much earlier in life, often around the time of birth or in early childhood, appears to initiate a degenerative cascade that proceeds undetected for some time prior to the onset of clinical signs. Presymptomatic detection of disease represents, therefore, a largely unexplored opportunity for therapeutic intervention. By detecting the disease process earlier, and initiating appropriate therapy to arrest the neurodegenerative process prior to the onset of symptoms, the disease process could be slowed or even stopped long before the patient becomes debilitated by both the primary disease process and secondary complications. This general concept is depicted in Fig. 2. While conceptually simple, such an approach to preventative therapy is by no means easy to implement. Even if pre-symptomatic biomarkers relevant to individual diseases could be identified, health budgets can not possibly support sustainable screening of all members of the population, and the incidence of these diseases is too large to permit accurate presymptomatic risk analysis (eg. Framingham). Further, most neurological diseases are presumed to not result from a single precipitating event, but rather arise from a complex interaction between initiating stimuli, genetic predisposition, environmental factors and lifestyle [4]. None-the-less, the overall goal of identifying disease earlier is both socially and economically responsible and should be vigourously pursued. Genetic predispositions and gene-environment interactions can be identified for individual diseases through careful study, and it is quite conceivable that the early indicators of neurodegeneration may be common or overlap between different diseases. This latter point gives rise to hope, and is supported by the knowledge that many neurodegenerative diseases demonstrate co-morbidity. In other words patients with a diagnosis of one disease often show signs of others. For example schizophrenia patients often suffer from epilepsy, Parkinson’s disease patients often develop dementia, and epilepsy patients often present with cognitive impairment. Thus, while the ultimate diagnosis of disease reflects the predominant symptomatology (which in turn dictates treatment), it is reasonable to assume that disease phenotypes overlap to some extent and may, therefore, have some common origin(s).
Fig. 2.
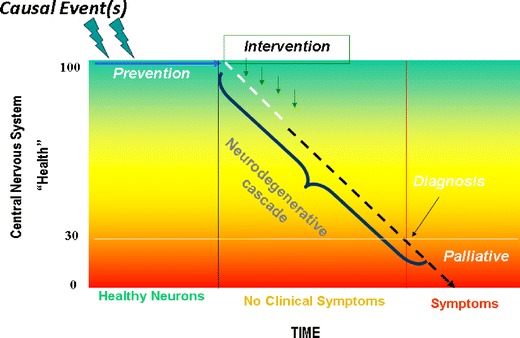
Representation of the concept of progressive neurodegeneration and presymptomatic therapeutic intervention. Events, often unidentified, initiate a progressive deterioration of brain health that continues for some time prior to the appearance of symptoms associated with a particular neurological disease. Traditionally therapy is initiated at the time of symptom onset and diagnosis, but earlier detection of deteriorating brain health would allow for presymptomatic intervention that could slow or prevent disease progression
Understanding the etiology, and initiation, of disease often relies on animal models, as does the development of new therapeutic strategies. But while there are a multitude of animal models that mimic specific features of end-stage disease for almost all neurological conditions, most of these models have been created with the aim of identifying new therapeutants to alleviate symptoms, and hence do not allow study of the development of disease. Understanding disease development is the key to identifying early intervention strategies consistent with the concept in Fig. 2. There exists, therefore, a largely unmet need to develop and exploit animal models designed to understand the origins and progression of neurological disease. This review will introduce several examples of animal models of progressive neurodegenerative disease. Because many diseases are neurodevelopmental in origin, those models that have been developed largely, but not exclusively, involve altering brain development in experimental animals. The descriptions below reflect that bias, and are not intended to be all-encompassing or even to be an exhaustive review of all such models. They are intended, rather, simply as examples to illustrate the concept of modeling disease progression as a means to identify predictive and/or preventative therapeutic strategies.
Progressive neurodevelopmental models of disease
Brain development
Brain development in mammals is a prolonged process beginning early in gestation and continuing into young adulthood. Abnormal development occurs as a result of changes in cellular processes that normally work in a delicate functional and temporal sequence. Whether these changes arise from physical damage or from neurotoxic insult, the impact of the injury depends upon the organism’s stage of growth [5, 6]. Generally, periods of rapid development are more vulnerable to damage and the extent of a given effect depends in large part on exposure timing. These critical periods, or “windows” of development have been divided by Vorhees [5] into four stages (although the final three have some overlap); namely, 1) implantation and all events preceding fertilization, 2) organogenesis, 3) differentiation and specialization of cells, and 4) functional organization, a period that encompasses both late fetal and early postnatal life and includes gene and receptor expression and post-translational modification of proteins that may be either programmed or be activity-dependent. It is this fourth developmental phase that manifests the greatest response to behavioural teratogens and is a prolonged period of vulnerability [5]. In the rat, the period from postnatal day (PND) 0 to PND 21 displays a particularly high rate of differentiation and acceleration of brain growth, with half of the total cell population forming during this time. In addition, processes such as synaptogenesis, developmental cell death (apoptosis), myelination, selective removal of excess synapses, and maturation of all major neurotransmitter systems occur at this time [6, 7]. This so-called “brain growth spurt” is, therefore, a period of particular vulnerability to neurological insult, and disruptions during this time may be manifested through a variety of behavioural and pathological abnormalities. In humans, the equivalent period occurs roughly between the third trimester of gestational development and the second year of life [8].
Neonatal models of epileptogenesis
Epilepsy is a complex, symptomatically-defined cluster of disorders that affects 1–1.5% of the population worldwide [9]. Characterized by recurrent seizures manifesting either as motor convulsions or abnormalities in electroencephalographic (EEG) recordings, epilepsy can be either generalized (affecting the whole brain) or partial (affecting discrete brain regions such as the temporal lobe). Partial epilepsy may or may not progress to generalized seizures [10]. Epilepsy is also frequently neurodevelopmental in origin, whereby a strong correlation exists between developmental CNS trauma (eg. hypoxia, closed head trauma, febrile seizures) and the subsequent development of seizure disorders such as temporal lobe epilepsy (TLE) [11]. While childhood epilepsy is not uncommon, early trauma is often followed by a latent, or “silent”, period ranging from years to decades before the onset of epileptic seizures. This delayed onset is a time when the brain is undergoing progressive changes in structure and function that ultimately culminate in seizures, and is referred to as “epileptogenesis” (ie. the process of becoming epileptic) [10].
The process of epileptogenesis is poorly understood, but because it is presymptomatic, the epileptogenic period represents a window in which therapeutic intervention could slow or even stop the development of epilepsy. Understanding the process of epileptogenesis and identification of potential biomarkers for presymptomatic intervention, however, requires the development of appropriate animal models, and while there are a number of epilepsy models that involve chemical or electrical induction of spontaneous recurrent seizures in adult animals [for review see 11, 12], modeling epilepsy, and particularly epileptogenesis, in neonatal animals has proven more difficult. This is due, in part, to what superficially appears to be a paradox. It is well established that the immature brain is more prone than the adult brain to seizures arising from chemical or environmental intervention [13–16], probably because of a difference in the developmental maturation of excitatory and inhibitory systems [17]. Despite this increased susceptibility to seizure genesis, however, the neonatal brain has generally been regarded as largely resistant to the development of long-term consequences consistent with epilepsy (eg. spontaneous recurrent seizures, mossy fiber sprouting, hippocampal cell loss) [18–20], although others have reported long-lasting consequences of neonatal seizures particularly when multiple administrations of convulsants are administered during early development [21, 22].
Of particular relevance to understanding the neurodevelopmental origins of epilepsy and epileptogenesis with an eye on early intervention, however, are two recently described rodent models of epilepsy that produce slowly-developing or delayed onset changes in epilepsy-relevant morphology, neurochemistry and behaviour. These are the neonatal inflammation model and the neonatal domoic acid model; each of which is described in more detail below.
Neonatal inflammation
Recently an interesting model linking neonatal inflammation and seizure susceptibility has been described [23–25]. Sprague-Dawley rat pups injected intraperitoneally with the bacterial endotoxin lipopolysaccharide on PND14 demonstrate enhanced susceptibility to seizures induced by lithium-pilocarpine, kainic acid (KA) or the GABA antagonist pentylenetetrazol when challenged at 6–8 weeks of age. The effect is antagonized by concurrent administration of an antibody to tumor necrosis factor α (TNFα). The authors also investigated whether the same result was obtained following LPS injection at other stages of development, and reported that injections on PND 7 also reduced seizure threshold, but that LPS injections on PND 1 or PND 20 did not; strongly implicating the second postnatal week of life as a “critical window” for the lasting effects. A follow-up paper by the same group described how direct (icv) injections of the viral mimetic polyinosinic:polycytidylic acid into PND 14 rat pups produces central inflammation and also results in a reduced seizure threshold when tested at 7–8 weeks of age [24]. Thus both peripheral and central inflammation during the second postnatal week of life in the rat appears to initiate a chronic neurodegenerative condition that culminates in reduced seizure threshold.
Neonatal domoic acid
Another neurodevelopmental model of epileptogenesis that has been described is the neonatal domoic acid model. Domoic acid is a naturally occurring excitotoxin that is structurally similar to KA and an analogue of glutamate, and is recognized as a selective, but not specific, agonist at the kainate subclass of non-NMDA glutamate receptors [26, 27]. In the late 1980’s domoic acid was identified as the causative agent in an outbreak of human toxicity in which patients experienced dose-dependent neurotoxicity culminating in seizures and death [28, 29]. One of the patients who survived subsequently went on to develop temporal lobe epilepsy [30]. Building on this background of clinical data, Doucette and co-workers investigated the response of neonatal rats to very low doses of domoic acid [16, 31] and in 2004 they described a unique low-grade seizure response that occurred in rats treated neonatally but exposed to novel and/or stressful environments as adults [32]. In addition to manifesting seizures in adulthood, post-mortem analysis of these animals at 15 months of age revealed many hallmark features of clinical TLE including hippocampal cell loss, mossy fiber sprouting (MFS) in the dentate gyrus and area CA3, and regionally-selective elevations of the neurotrophin BDNF [32]. Of particular interest in the context of the current review, however, is that these changes do not occur at the time of drug administration (unpublished) but are progressive in nature whereby younger mature animals (PND75) display less severe MFS and no loss of hippocampal cells [33] and adolescent rats (PND 29) show no hippocampal pathology with the exception of mild atrogliosis [34]. Further confirming the relevance of this model to understanding epilepsy and epileptogenesis are two recent reports by Gill et al. [35, 36] demonstrating alterations in electroencephalographically (EEG) recorded sleep patterns and reductions in seizure threshold that accompany selective loss of inhibitory neurons in the hippocampus. A general overview of the current understanding of hippocampal epileptogenesis in this model is shown in Fig. 3.
Fig. 3.
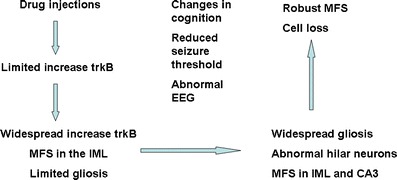
Preliminary schematic depiction of identified changes over time in a neurodevelopmental rat model of temporal lobe epilepsy. Low doses of domoic acid during the second postnatal week lead to a progressive series of changes in hippocampal neurotrophin signaling, astrogliosis, sprouting of dentate granule cell axons and cell loss, that are accompanied by changes in cognition, seizure threshold and EEG although the precise time of onset of each is currently unknown. Changes depicted are correlative only and not necessarily causally related. Data derived from [32–36]
One obvious deficiency in the neonatal domoate model at this time, however, is a lack of data on the ability to predict therapeutic efficacy for current or emerging drugs. Because the model does not exhibit spontaneous motor convulsions, typical anticonvulsants are not of immediate relevance, but other anti-epileptics such as channel modulators or ligands for pharmacologically defined receptors controlling excitability need to be tested to properly evaluate the predictive validity of the model in relation to more established protocols.
Neonatal models of schizophrenia and related disorders
Schizophrenia is a complex and devastating mental disorder affecting approximately 1% of the general population. While the term schizophrenia was first introduced by Eugen Bleuler in 1911, it is believed that the disorder has been present in all cultures for centuries [37]. Characterized by impairments in the perception and expression of reality, the symptoms of schizophrenia are most often categorized into positive, negative and more recently, cognitive symptoms [38]. Schizophrenia is debilitating and results in great emotional cost to the person afflicted, as well as to friends and relatives. It also results in great financial cost to the economy worldwide [39], with the estimated annual cost of healthcare for those afflicted in Canada alone, being over $2 billion US [40]. While the outlook for those affected has improved somewhat, a diagnosis of schizophrenia is still likely to result in substantial disability and potentially, in reduced life expectancy due to suicide [41, 42].
Although schizophrenia has been present for centuries and progress has been made through research, much about the disorder is still unknown. This is probably due to the complexity of the proximal cause(s) of the disorder and the apparent delay of the onset of definitive symptoms. Although the cause(s) of schizophrenia is/are not yet known, a prominent hypothesis is that it is neurodevelopmental in origin and arises due to a combination of environmental factors and genetic susceptibility. According to this theory, factors which occur long before the formal onset of the illness (potentially during gestational or early developmental stages), disrupt normal CNS development in a way that may lead to severe and long-lasting changes in the functional integrity of the CNS [43]. Early life events that have been implicated in a higher than average risk of developing schizophrenia include maternal illness during gestation [44], obstetric complications [45, 46] and toxin exposure [47]. Such events can lead to subtle alterations in the functioning of the CNS which may result in an increased vulnerability to environmental triggers later in life [48]. However, while environmental factors likely cause an individual to be more susceptible to the illness or play a role in triggering the onset of symptoms, specific genetic vulnerabilities also appear to contribute to the manifestation of schizophrenia. Although the prevalence of schizophrenia may only be up to 1% in the general population, the prevalence of the disorder is much higher in those who have a schizophrenic relative. Additionally, though the disorder can occur in people who have no family history of its presence, the risk increases dramatically with the degree of genetic closeness to a person who is afflicted [37].
A variety of genes have been associated with schizophrenia including neuregulin (NRG1) [49], dysbindin (DTNBP1) [50], Catechol-O-methyl transferase (COMT) [51] and DISC1 [52], and consequently a number of genetically-based animal models exist for studying schizophrenia. A thorough discussion of these models and their use in schizophrenia research is beyond the scope of this review, but has been discussed in detail elsewhere [for review see 53].
Of more relevance to the current review are several well characterized and/or emerging rodent models of schizophrenia that involve experimental manipulation in the perinatal period that gives rise to schizophrenia-like symtomatology that develops over time. Such slowly progressing animal models provide good platforms for both understanding disease development and identifying potential early markers of disease progression.
Lesion models
First proposed by Lipska and co-workers in 1993 [54], the neonatal ventral hippocampal (NVH) lesion model is one of the most widely studied animal models of schizophrenia. By using ibotenic acid (a glutamate agonist) to lesion the ventral hippocampus of neonatal rats on PND 7 of development, many aspects of the disorder have been demonstrated. Compared to controls, rats with lesions demonstrated a higher degree of activity in response to a novel environment and/or amphetamine challenge when tested in early adulthood (PND 56), but not at earlier time points (PND 35) [54]. Deficits have also been observed in other behavioural measures including prepulse inhibition (PPI) [55], latent inhibition (LI) [56], measures of working memory [57] and social interaction measures, with rats who received NVH lesions spending less time in social interaction as compared to controls [58].
In addition, neurobiological factors such as an increase in hippocampal dopamine (DA) have also been found [59] and a number of studies have indicated that antipsychotic drugs normalize some of the lesion-induced behaviours [54, 60]. In addition to the NVH model, other lesion based models of schizophrenia have been described including the neonatal amygdalar lesion model [61] where ibotenic acid is used to lesion the amygdala on PND 7, and the prefrontal cortical lesion model [62] which uses ibotenic acid to lesion the prefrontal cortex in neonatal or adult animals.
Immunological models
Prenatal exposure to infection has also been implicated in an increased risk of developing schizophrenia in adulthood. Presently it is believed that the maternal immune response, particularly the pro-inflammatory cytokines that are released in such situations, can interfere with the normal development of the CNS. As such, immunological models seek to imitate this potential risk factor [63]. Seeking to elicit the maternal immune response by mimicking viral exposure, Zuckerman and Weiner created such a model by injecting pregnant rats with polyriboinosinic-polyribocytidilic acid (polyI:C). This procedure resulted in adult animals displaying a variety of schizophrenia-like anomalies including deficits in PPI [64] and LI [65], as well as increased sensitivity to the locomotor effects of amphetamine [66], impairments in novel object recognition [64] and reduced social interaction [67]. Neuroanatomical and neurochemical abnormalities have also been observed including alterations to hippocampal morphology and increases in striatal DA release [66].
Pharmacological models
Another animal model of schizophrenia proposed by Harris et al.[68], used the administration of MK-801 (a glutamate antagonist) to neonatal rats, to produce a schizophrenia-like phenotype in adulthood. The procedure involved the administration of two subcutaneous (s.c.) injections of 0.5 mg/kg of MK-801 to Sprague-Dawley rat pups on PND 7. As adults, the animals displayed reduced volume and neuronal numbers in the hippocampus, and female rats displayed PPI deficits and increased locomotor activity in the open field [68].
In another study, Kawabe et al. [69] administered 0.2 or 0.4 mg/kg of MK-801 by i.p. injection, twice daily to neonatal rats from PND 7–20. It was found that when they reached adulthood, these animals displayed significant deficits in radial arm maze (RAM) learning. Interestingly, when adult animals who had not received the neonatal MK-801 treatment neonatally were given the same treatment in adulthood, no changes to RAM performance were noted [69].
In addition to the aforementioned work on NMDA receptors, neonatal manipulation of non-NMDA receptors has also been shown to result in schizophrenia-like symptoms that develop as animals age. In a series of papers published over the last 10 years, the Csernansky research group at Washington University in St. Louis has described how intracerebroventricular (icv) injection of a sub-convulsive dose of kainic acid on PND 7 induces delayed histopathology consistent with schizophrenia. Looking at time points ranging from 8–75 days of age these authors have described progressive loss of neurons in the CA1 and CA3 hippocampal subfields [70, 71]. Interestingly, at a low dose of KA cell loss did not begin until around PND40; mimicking the delayed onset of schizophrenia, and appeared due to apoptosis rather than the necrosis typically associated with excitotoxicity [71]. Subsequent papers by the same group have also reported changes in hippocampal interneuron populations [72] and hippocampal neurogenesis [73] in this model.
Closely related to these findings are the recent data emerging from two independent groups that have administered domoic acid (DOM) during perinatal development. As described previously in this review, DOM is a naturally occurring environmental toxin produced by various marine organisms such as red algae [74] and has been associated with human intoxication resulting in neurological damage as well as being used to induce seizure states in experimental animals. A 2005 study by Levin and co-workers [75] revealed that a single prenatal exposure to DOM during midgestational development (with doses ranging from 0.3–1.2 mg/kg) produced dose related changes which included an increase in response latency in a spontaneous alternation task in the T-maze, as well as an increase in initial activity and a larger habituation score in the Figure-8 maze. Additionally, a later study by the same group [76], considered the effect of administering DOM (0.025–0.1 mg/kg) to rat pups twice per day, on PND 1 and 2. While the higher doses proved to be lethal, they did report some effects at lower doses, but these were less frequently observed than those seen following DOM exposure during mid-gestation.
In a related series of experiments Doucette et al. [31] demonstrated that when tested during the neonatal period, administration of very low doses of DOM to Sprague-Dawley rats did not alter a number of developmental milestones but treated rats did display increased conditioned place preference in an olfactory conditioning paradigm; confirming that this low dose of DOM (20 μg/kg) was centrally active at doses that were not overtly toxic. The same group then went on to administer low-dose DOM throughout the second postnatal week (PND 8–14) and reported a number of schizophrenia-like changes in behaviour when the animals were tested as adults. Treated animals have been shown to have abnormal responses to novel spatial environments [32] as well as impairments in prepulse inhibition [77], spatial cognition [78] and working memory [79]. Alterations in emotionality (elevated plus maze) as well as changes in response to novelty and increased sensitivity to nicotine have also been found in this paradigm [79–81]; as have increased sensitivity to reward (in a choice task) and psychomotor agitation (in a novelty conditioning paradigm) and social withdrawal (unpublished). Interestingly, and potentially of great importance, the same experimental manipulation (ie. low dose neonatal domoic acid) also produces a behavioural phenotype with relevance to partial complex epilepsy originating in the temporal lobe as described previously in this review (see above). Because the same drug, administered during the same neonatal window, results in features of both epilepsy and schizophrenia, there may be common molecular origins relevant to both disorders, although in the absence of further mechanistic data on both models such a conjecture is hypothetical at this time.
Thus, there are a number of different rodent models that utilize changes in perinatal brain development resulting in slowly progressing deterioration of central function relevant to schizophrenia and related psychiatric disorders.
Prenatal/Neonatal models of movement disorders
Amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD) are two of society’s most prevalent diseases of progressive motor dysfunction (Fig. 1). Most in vivo studies of ALS and Parkinson’s disease have examined associated movement disorders primarily in adult animals. There are good theoretical reasons and practical reasons for this emphasis. First, most cases of both diseases in humans are of the late onset form and are usually sporadic in nature. Gene-based models in fact have not been able to duplicate early onset human neurological diseases. For example, the most common ALS models of the familial (genetic) forms of the disease, mutant superoxide dismutase (mSOD) and the alsin protein deletion associated with juvenile ALS 2, do not show clear behavioural or pathological outcomes until well into postnatal life, if at all. For mSOD, the rapidly progressive G93A mutation only begins so show pathological features at about 50 days of postnatal life [82]. The G37R variant takes up to a year to demonstrate behavioural or pathological deficits [83]. ALS2/alsin -/- mice show almost no disease phenotype at any age [84]. Similarly, genetic models of PD usually show few early behavoural or neuronal outcomes [4].
Studies of environmental factors typically use adult animals (post sexual maturity and older) for a variety of factors such as ease of handling and testing and toxin administration. These factors may be one reason why no good age-dependence animal studies exist for either disease. In spite of these difficulties, for both ALS and Parkinson’s disease, there have been some attempts to probe the impact of some of the toxins studied in adulthood in prenatal mice. Steryl glucosides, notably those isolated from the seeds of the cycad palm [85, 86] have been implicated in a form of ALS that is sometimes linked to the co-expression of a form of parkinsonism (parkinsonism dementia complex), thereby forming a spectrum of neurological disorders termed ALS-PDC [87–89] (see below). A recent series of experiments by Shaw and Banjo [90] began to examine the neonatal and postnatal outcomes of steryl glucoside neurotoxins related to those previously implicated in ALS-PDC [86, 91] and shown to cause neurodegeneration following adult exposure [92]. This study administered these toxins to pregnant dams during the period of greatest differentiation of the nigro-striatal system, and also sought to determine if the impact of a second toxin exposure in adulthood was influence by the prenatal one. Finally, the experiments separated male from female mice to determine the role that sex might play.
Prenatal exposure to steryl glucoside toxins showed that mice exposed in utero had greater body weights that persisted throughout life and showed behavioural deficits associated with damage in the nigro-striatal system. Male mice were more profoundly impacted than females. A second adult toxin exposure had more negative impacts on the nigro-striatal system than in the spinal cord. These data mirror the earlier findings of Barlow et al. [93] that exposure of pregnant dams to the pesticides maneb and paraquat caused damage to the nigro-striatal system, that the effects were sex specific as above, and that an adult toxin exposure was more likely to trigger a Parkinson’s phenotype.
The results of these experiments reaffirm the notion that prenatal and likely neonatal life are potentially crucial periods that might set the conditions for later neurodegenerative disorders.
Slowly progressing adult models of movement disorders
There are a variety of adult models of ALS and Parkinson’s disease, based generally on genetic or environmental models.
For ALS, the primary genetic models are based on various mutations of SOD that produce, with different time courses, many of the features of ALS, including progressive motor deficits and the loss of spinal cord motor neurons. Alsin mutations have not produced particularly profound ALS phenotypes to date. The mSOD model has been used with great success in isolating the putative stages of neurodegeneration which is now believed to have a partial origin in the loss of neuromuscular junction endplates in mutant animals [94]. A number of studies with the mSOD model using various therapeutic drugs have shown modest delays in symptom onset and life expectancy [95–99]. However, no single agent or “drug cocktail” to date has provided lasting protection or actually delayed the eventual outcomes of motor neuron death [100].
Environmental links to ALS have arisen from a variety of sources, with the most well studied cases related to the cluster of ALS-PDC found on Guam and elsewhere in the Western Pacific during the 1950s and 1960s. As described previously, the strongest epidemiological correlate for ALS-PDC was the consumption of a local food product, the seeds of the cycad palm [88, 89]. Cycad seed has been shown to contain a variety of neurotoxic compounds, some of them water insoluble. Mice fed washed cycad seed flour exhibit a progressive form of motor neuron loss accompanied by motor behavioural deficits. These are later followed by gait disorders associated with pathological outcomes in the nigro-striatal system, the latter including the loss of tyrosine hydroxylase and apoptosis in the striatum. Increases in reactive astrocytes and microglia were components of both areas. In marked contrast, rats fed cycad seed flour showed profound parkinsonism outcomes of gait disturbances that initially presented unilaterally, later freezing, and highly significant losses of neurons in substantia nigra. Treated rats did not show any losses of motor neurons in the spinal cord [101] (Table 1).
Table 1.
Comparison of features of ALS-PDC of Guam with animal models of the disorder. The various features of the Guamanian spectrum of disorders are compared to two rodent models fed flour made from seeds of the local cycad palm, Cycas micronesica. Cycad consumption is still considered to be the strongest epidemiological link to ALS-PDC [88, 89]. Both model systems have also examined dietary exposure to isolated cycad neurotoxins, notably two variant steryl glucoside, B-sitosterol B-D glucoside (BSSG) and stigmasterol glucoside (SG). Combined, the two models faithfully reproduce the salient behavioural and pathological features of the ALS-PDC spectrum
| Disease outcomes | ALS-parkinsonism dementia complex | Rodent models of ALS-PDC (cycad or steryl glucoside) |
|---|---|---|
| Behavioural changes motor function | Expresses as ALS, PD, or both for motor function losses | Mice: ALS-phenotype is dominant; Rats: parkinsonism only |
| Cognitive function | Dementia accompanies parkinsonism | Cognitive decline on various memory tests both models |
| Olfactory function | ALS-PDC shows early olfactory disturbance | Mice fail to discriminate odours |
| Motor neuron loss | Yes, all levels of motor system | Mice, yes; Rats, no. |
| Nigro-striatal neuron loss | PDC | Mice: significant but not dominant feature; Rats: significant losses of cells in SNpc and striatum |
| Loss of neurons in hippocampus/cortex | Features of PDC | Mice at late stages |
| Expression of hyperphosphorylated tau | Both ALS and PDC | Mice at late stages in lumbar spinal cord motor neurons with steryl glucoside feeding |
| Α-synuclein expression | Approx. 20% PDC cases | Rats with cycad |
| TDP-43 translocation | Noted | Mice at late stages in lumbar cord motor neurons with steryl glucoside feeding |
| Dopamine loss | PDC | Mice and rats |
| D2 receptor upregulation; DAT down regulation | unknown | Mice; Rats unknown |
| Increases in reactive astrocytes | ALS-PDC | Both mice and rats in various affected regions of CNS |
| Increases in microglial proliferation | ALS-PDC | Both mice and rats in various affected regions of the CNS |
Mice fed the putative neurotoxic factor in cycad, variant steryl glucosides, showed many of the same features as those fed cycad [92]. Preliminary studies using steryl glucosides in rats suggests that they exhibit only parkinsonism phenotypes (unpublished).
Attempts have been made to duplicate ALS-PDC in its various forms in monkeys [85] using the free plant amino acid, beta-methyl amino alanine (BMAA) and earlier by Dastur and colleagues using washed and unwashed cycad seed flour [102]. The Spencer results [85] have not been repeated and the original data could not be duplicated in mice fed BMAA [103, 104].
In regard to Parkinson's disease, a great variety of studies have focused on either genetic links to the disease, including mutations in alpha-synuclein and others [105, 106], or to various toxins linked to some forms of parkinsonism. These models rarely produce full blown Parkinson's disease phenotypes. The latter have included toxins such as MPTP, linked originally to rapid parkinsonism onset following the use of a synthetic opioid, 6 hydroxydopamine, proteasome inhibitors [107–109], and various pesticides as well as cycad and steryl glucosides as cited above. Continuous delivery of MPTP via a pump or slowing MPTP excretion by concomitant administration of probenecid has been used to produce a slower neurodegenerative process [110–112].
One of the more interesting animal models of Parkinson's disease is based on the cluster on the island of Guadeloupe linked to annonacin neurotoxins contained in the fruit of the soursop (summarized in Shaw and Hoglinger [4]. Here, as in ALS-PDC, an environmental neurotoxin epidemiologically linked to a human neurodegenerative disease condition can duplicate many of the disease phenotypes in in vivo models.
Outlook
Animal models that allow examination of the developmental processes underlying neurodegenerative disease represent an important opportunity to develop therapies directed at prevention or amelioration of the disease phenotype. As stated at the beginning of this review, the task of identifying pre-symptomatic markers of disease in human populations and then implementing appropriate screening and treatment options is daunting, but potentially of enormous value. One obvious opportunity is identification of early biomarkers of disease that could translate rapidly into diagnostic and clinical settings. Another is identification of early targets common to multiple disease processes. While seeming fanciful to some, some of the examples described above give credence to this notion. In some of the models described herein, both neonatal inflammation and neonatal glutamatergic stimulation have been used to model both epilepsy and schizophrenia; two conditions known to exhibit co-morbidity but currently treated with widely differing classes of drugs. Administration of steryl glucosides results in symptoms common to both ALS and Parkinson’s disease with the relative proportion of phenotype influenced by the species of choice (mice versus rats). At this stage it is too early to know to what extent the relevant disease processes overlap and which precise molecular events result in each disease phenotype, but better understanding of early disease progression, combined with improved epidemiological data on appropriate risk factors, represents an avenue for improving predictive and personalized therapy.
Conclusion
Further development and analysis of slowly progressing animal models of neurological disease is an area of increasing interest and considerable potential. Understanding the origins, molecular targets, and internal and external modifiers of disease progression through the use of such models constitutes a promising new approach to new therapy development, and if factors common to multiple diseases can be discovered the value, and practicality, of pre-symptomatic intervention will be greatly enhanced. Thus, continued study and development of slowly progressing animal models of disease appears to be a viable option for advancing the agenda of predicting and preventing, rather than simply treating, some of society’s most devastating neurological conditions.
Acknowledgements
Amber Adams-Marriott is supported by a Canada Graduate Scholarship awarded through the Natural Sciences and Engineering Research Council of Canada.
References
- 1.World Health Organization (2006) Neurology and Public Health. http://www.who.int/mental_health/neurology/en. Cited 15 March 2010.
- 2.OHSU Brain Institute (2010) Disease Statistics. Oregon Health & Science University. http://www/ohsu.edu/xd/education/schools/research-institutes/brain-institute/Cited 15 March 2010.
- 3.Schulz JB, Falkenburger BH. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2004;318:135–137. doi: 10.1007/s00441-004-0954-y. [DOI] [PubMed] [Google Scholar]
- 4.Shaw CA, Hoglinger GU. Neurodegenerative diseases: neurotoxins as sufficient etiological agents ? Neuromolecular Med. 2008;10:1–9. doi: 10.1007/s12017-007-8016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vorhees CV. Principles of behavioural teratology. In: Riley EP, Vorhees CV, editors. Handbook of Behavioural Teratology. New York: Plenum Press; 1986. [Google Scholar]
- 6.Kaufmann W. Developmental neurotoxicity. In: Krinkle GJ, editor. The Laboratory Rat. London: Academic Press; 2000. [Google Scholar]
- 7.Ben-Ari Y. Developing networks play a similar melody. Trends Neurosci. 2001;24:353–360. doi: 10.1016/s0166-2236(00)01813-0. [DOI] [PubMed] [Google Scholar]
- 8.Dobbing J, Smart JL. Vulnerability of developing brain and behaviour. Brit Med Bull. 1974;30:164–168. doi: 10.1093/oxfordjournals.bmb.a071188. [DOI] [PubMed] [Google Scholar]
- 9.Murray CJL, Lopez AD (Eds)(1994) Global comparative assessment in the health sector: disease burden, expenditures, and intervention packages. World Health Organization, Geneva.
- 10.Engel J, Jr, Schwartzkroin PA. What should be modeled ? In: Pitkanen A, Schwartzkroin PA, Moshe SL, editors. Models of Seizures and Epilepsy. London: Academic Press; 2006. [Google Scholar]
- 11.Jefferys JGR. Models and mechanisms of experimental epilepsies. Epilepsia. 2003;44:44–50. doi: 10.1111/j.0013-9580.2003.12004.x. [DOI] [PubMed] [Google Scholar]
- 12.Pitkanen A, Schwartzkroin PA, Moshe SL (Eds) Models of Seizures and Epilepsy. Academic Press, London.
- 13.Cavalheiro EA, Silva DF, Turski WA, et al. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Dev Brain Res. 1987;37:43–58. doi: 10.1016/0165-3806(87)90227-6. [DOI] [PubMed] [Google Scholar]
- 14.Stafstrom CE, Thompson JL, Holmes GL. Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Brain Res Dev Brain Res. 1992;65:227–236. doi: 10.1016/0165-3806(92)90184-x. [DOI] [PubMed] [Google Scholar]
- 15.Stafstrom CE, Chronopoulos A, Thurber S, et al. Age-dependent cognitive and behavioral deficits after kainic acid-induced seizures. Epilepsia. 1993;34:420–432. doi: 10.1111/j.1528-1157.1993.tb02582.x. [DOI] [PubMed] [Google Scholar]
- 16.Doucette TA, Strain SM, Allen GV, et al. Comparative behavioural toxicity of domoic acid and kainic acid in neonatal rats. Neurotoxicol Teratol. 2000;22:863–869. doi: 10.1016/s0892-0362(00)00110-0. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Ari Y, Khazipov R, Leinekugel X, et al. GABA-A, NMDA and AMPA receptors: a developmentally regulated ‘ménage a trois’. Trends Neurosci. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- 18.Nitecka L, Tremblay E, Charton G, et al. Maturation of kainic acid seizure-brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience. 1984;13:1073–1094. doi: 10.1016/0306-4522(84)90289-6. [DOI] [PubMed] [Google Scholar]
- 19.Sperber EF, Haas KZ, Stanton PK, et al. Resistance of the immature hippocampus to seizure-induced synaptic reorganization. Brain Res Dev Brain Res. 1991;60:88–93. doi: 10.1016/0165-3806(91)90158-f. [DOI] [PubMed] [Google Scholar]
- 20.Haas KZ, Sperber EF, Opanashuk LA, et al. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- 21.Holmes GL, Sarkisian M, Ben-Ari Y, et al. Mossy fiber sprouting after recurrent seizures during early development in rats. J Comp Neurol. 1999;22:537–553. doi: 10.1002/(sici)1096-9861(19990222)404:4<537::aid-cne9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Yang Y, Silveira DC, et al. Consequences of recurrent seizures during early brain development. Neuroscience. 1999;92:1443–1454. doi: 10.1016/s0306-4522(99)00064-0. [DOI] [PubMed] [Google Scholar]
- 23.Galic MA, Riazi K, Heida JG, et al. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–6913. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galic MA, Riazi K, Henderson AK, et al. Viral-like brain inflammation during development causes increased seizure susceptibility in adult rats. Neurobiol Dis. 2009;36:343–351. doi: 10.1016/j.nbd.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riazi K, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89:34–42. doi: 10.1016/j.eplepsyres.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Verdoorn TA, Johansen TH, Drejer J, et al. Selective block of recombinant GluR6 receptors by NS-102; a novel non-NMDA receptor antagonist. Eur J Pharmacol. 1994;269:43–49. doi: 10.1016/0922-4106(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 27.Tasker RA, Strain SM, Drejer J. Selective reduction in domoic acid toxicity in vivo by a novel non-N-methyl-D-aspartate receptor antagonist. Can J Physiol Pharmacol. 1996;74:1047–1054. [PubMed] [Google Scholar]
- 28.Perl TM, Bedard L, Kosatsky T, et al. An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N Engl J Med. 1990;322:1775–1780. doi: 10.1056/NEJM199006213222504. [DOI] [PubMed] [Google Scholar]
- 29.Teitelbaum J, Zatorre R, Carpenter S, et al. Neurologic sequelae of domoic acid intoxication due to ingestion of contaminated mussels. N Engl J Med. 1990;322:1781–1787. doi: 10.1056/NEJM199006213222505. [DOI] [PubMed] [Google Scholar]
- 30.Cendes F, Andermann F, Carpenter S, et al. Temporal lobe epilepsy caused by domoic acid intoxication: evidence for glutamate receptor-mediated excitotoxicity in humans. Ann Neurol. 1995;37:123–126. doi: 10.1002/ana.410370125. [DOI] [PubMed] [Google Scholar]
- 31.Doucette TA, Bernard PB, Yuill PC, et al. Low doses of non-NMDA glutamate receptor agonists alter neurobehavioural development in the rat. Neurotox Teratol. 2003;25:473–479. doi: 10.1016/s0892-0362(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 32.Doucette TA, Bernard PB, Husum H, et al. Low doses of domoic acid during postnatal development produce permanent changes in rat behaviour and hippocampal morphology. Neurotox Res. 2004;6:555–563. doi: 10.1007/BF03033451. [DOI] [PubMed] [Google Scholar]
- 33.Bernard PB, Macdonald DS, Gill DA, et al. Hippocampal mossy fiber sprouting and elevated trkB receptor expression following systemic administration of low dose domoic acid during neonatal development. Hippocampus. 2007;17:1121–1133. doi: 10.1002/hipo.20342. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald DS, Bernard PB, Ramsay LA, et al. Progressive changes in hippocampal cytoarchitecture in a non-convulsive developmental model of TLE. Proc. Soc Neurosci. 2008;449:8. [Google Scholar]
- 35.Gill DA, Bastlund JF, Anderson NC, et al. Reductions in paradoxical sleep time in adult rats treated neonatally with low dose domoic acid. Behav Brain Res. 2009;205:564–569. doi: 10.1016/j.bbr.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Gill DA, Ramsay SL, Tasker RA. Selective reductions in subpopulations of GABAergic neurons in a developmental rat model of epilepsy. Brain Res. 2010;1331:114–123. doi: 10.1016/j.brainres.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 37.Tamminga CA, Holcomb HH. Phenotype of schizophrenia: A review and formulation. Mol Psychiatr. 2005;10:27–39. doi: 10.1038/sj.mp.4001563. [DOI] [PubMed] [Google Scholar]
- 38.Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia “just the facts” 4: Clinical features and conceptualization. Schizophr Res. 2009;110:1–23. doi: 10.1016/j.schres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Rössler W, Salize HJ, Os J, et al. Size of burden of schizophrenia and psychotic disorders. Eur Neuropsychopharmacol. 2005;15:399–409. doi: 10.1016/j.euroneuro.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Goeree R, Farahati F, Burke N, et al. The economic burden of schizophrenia in Canada in 2004. Curr Med Res Opin. 2005;21:2017–2028. doi: 10.1185/030079905X75087. [DOI] [PubMed] [Google Scholar]
- 41.Caldwell CB, Gottesman II. Schizophrenics kill themselves too: A review of risk factors for suicide. Schizophr Bull. 1990;16:571–589. doi: 10.1093/schbul/16.4.571. [DOI] [PubMed] [Google Scholar]
- 42.Radomsky ED, Haas GL, Mann JJ, et al. Suicidal behavior in patients with schizophrenia and other psychotic disorders. Am J Psychiatry. 1999;156:1590–1595. doi: 10.1176/ajp.156.10.1590. [DOI] [PubMed] [Google Scholar]
- 43.Rapoport JL, Addington AM, Frangou S. The neurodevelopmental model of schizophrenia: Update 2005. Mol Psychiatry. 2005;10:434–449. doi: 10.1038/sj.mp.4001642. [DOI] [PubMed] [Google Scholar]
- 44.Mednick SA, Machon RA, Huttunen MO. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1998;45:189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- 45.Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: Historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- 46.Geddes JR, Verdoux H, Takei N, et al. Schizophrenia and complications of pregnancy and labor: An individual patient data meta-analysis. Schizophr Bull. 1999;25:413–423. doi: 10.1093/oxfordjournals.schbul.a033389. [DOI] [PubMed] [Google Scholar]
- 47.Fiore M, Grace AA, Korf J, et al. Impaired brain development in the rat following prenatal exposure to methylazoxymethanol acetate at gestational day 17 and neurotrophin distribution. NeuroReport. 2004;15:1791–1795. doi: 10.1097/01.wnr.0000135934.03635.6a. [DOI] [PubMed] [Google Scholar]
- 48.Lieberman JA, Perkins D, Belger A, et al. The early stages of schizophrenia: Speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol Psychiatry. 2001;50:884–897. doi: 10.1016/s0006-3223(01)01303-8. [DOI] [PubMed] [Google Scholar]
- 49.Stefansson H, Sigurdsson E, Steinthorsdottir V, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–348. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Pro Natl Acad Sci U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 53.O'Tuathaigh CM, Babovic D, O'Meara G, et al. Susceptibility genes for schizophrenia: characterisation of mutant mouse models at the level of phenotypic behaviour. Neurosci Behav Rev. 2007;31:60–78. doi: 10.1016/j.neubiorev.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: A potential animal model of schizophrenia. Neuropsychopharmacol. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- 55.Lipska BK, Swerdlow NR, Geyer MA, et al. Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacol. (Berl) 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- 56.Grecksch G, Bernstein HG, Becker A, et al. Disruption of latent inhibition in rats with postnatal hippocampal lesions. Neuropsychopharmacol. 1999;20:525–532. doi: 10.1016/S0893-133X(98)00081-5. [DOI] [PubMed] [Google Scholar]
- 57.Lipska BK, Aultman JM, Verma A, et al. Neonatal damage of the ventral hippocampus impairs working memory in the rat. Neuropsychopharmacol. 2002;27:47–54. doi: 10.1016/S0893-133X(02)00282-8. [DOI] [PubMed] [Google Scholar]
- 58.Becker A, Grecksch G, Bernstein HG, et al. Social behaviour in rats lesioned withibotenic acid in the hippocampus: Quantitative and qualitative analysis. Psychopharmacol. (Berl) 1999;144:333–338. doi: 10.1007/s002130051015. [DOI] [PubMed] [Google Scholar]
- 59.Alquicer G, Silva-Gómez AB, Peralta F, et al. Neonatal ventral hippocampus lesion alters the dopamine content in the limbic regions in postpubertal rats. Int J Dev Neurosci. 2004;22:103–111. doi: 10.1016/j.ijdevneu.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 60.Becker A, Grecksch G. Haloperidol and clozapine affect social behaviour in rats postnatally lesioned in the ventral hippocampus. Pharmacol Biochem Behav. 2003;76:1–8. doi: 10.1016/s0091-3057(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 61.Daenen EW, Wolterink G, Ree JM. Hyperresponsiveness to phencyclidine in animals lesioned in the amygdala on day 7 of life. Implications for an animal model of schizophrenia. Eur Neuropsychopharmacol. 2003;13:273–279. doi: 10.1016/s0924-977x(03)00029-4. [DOI] [PubMed] [Google Scholar]
- 62.Miner LA, Ostrander M, Sarter M. Effects of ibotenic acid-induced loss of neurons in the medial prefrontal cortex of rats on behavioral vigilance: Evidence for executive dysfunction. J Psychopharmacol. 1997;11:169–178. doi: 10.1177/026988119701100210. [DOI] [PubMed] [Google Scholar]
- 63.Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 64.Ozawa K, Hashimoto K, Kishimoto T, et al. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 65.Zuckerman L, Weiner I. Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacol. (Berl) 2003;169:308–313. doi: 10.1007/s00213-003-1461-7. [DOI] [PubMed] [Google Scholar]
- 66.Zuckerman L, Rehavi M, Nachman R, et al. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: A novel neurodevelopmental model of schizophrenia. Neuropsychopharmacol. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]
- 67.Shi L, Fatemi SH, Sidwell RW, et al. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harris LW, Sharp T, Gartlon J, et al. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- 69.Kawabe K, Iwasaki T, Ichitani Y. Repeated treatment with N-methyl-d-aspartate antagonists in neonatal, but not adult, rats causes long-term deficits of radial-arm maze learning. Brain Res. 2007;1169:77–86. doi: 10.1016/j.brainres.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 70.Montgomery EME, Bardgett ME, Lall B, et al. Delayed neuronal loss after administration of intracerebroventricular kainic acid to preweanling rats. Dev Brain Res. 1999;112:107–116. doi: 10.1016/s0165-3806(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 71.Humphrey WM, Dong H, Csernansky CA, et al. Immediate and delayed hippocampal neuronal loss induced by kainic acid during early postnatal development in the rat. Dev Brain Res. 2002;137:1–12. doi: 10.1016/s0165-3806(02)00344-9. [DOI] [PubMed] [Google Scholar]
- 72.Dong H, Csernansky CA, Chu Y, et al. Intracerebroventricular kainic acid administration to neonatal rats alters interneuron development in the hippocampus. Dev Brain Res. 2003;145:81–92. doi: 10.1016/s0165-3806(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 73.Dong H, Csernansky CA, Goico B, et al. Hippocampal neurogenesis follows kainic acid-induced apoptosis in neonatal rats. J Neurosci. 2003;23:1742–1749. doi: 10.1523/JNEUROSCI.23-05-01742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pulido OM. Domoic acid toxicologic pathology: A review. Mar Drugs. 2008;6:180–219. doi: 10.3390/md20080010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levin ED, Pizarro K, Pang WG, et al. Persisting behavioral consequences of prenatal domoic acid exposure in rats. Neurotoxicol Teratol. 2005;27:719–725. doi: 10.1016/j.ntt.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 76.Levin ED, Pang WG, Harrison J, et al. Persistent neurobehavioral effects of early postnatal domoic acid exposure in rats. Neurotoxicol Teratol. 2006;28:673–680. doi: 10.1016/j.ntt.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 77.Adams1 AL, Doucette TA, Ryan CL. Altered pre-pulse inhibition in adult rats treated neonatally with domoic acid. Amino Acids. 2008;35:157–60. 35. [DOI] [PubMed]
- 78.Adams AL, Doucette TA, James R, et al. Persistent changes in learning and memory in rats following neonatal treatment with domoic acid. Physiol Behav. 2009;96:505–512. doi: 10.1016/j.physbeh.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 79.Doucette TA, Ryan CL, Tasker RA. Gender-based changes in cognition and emotionality in a new rat model of epilepsy. Amino Acids. 2007;32:317–322. doi: 10.1007/s00726-006-0418-7. [DOI] [PubMed] [Google Scholar]
- 80.Burt MA, Ryan CL, Doucette TA. Altered responses to novelty and drug reinforcement in adult rats treated neonatally with domoic acid. Physiol Behav. 2008;93:327–336. doi: 10.1016/j.physbeh.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 81.Burt MA, Ryan CL, Doucette TA. Low dose domoic acid in neonatal rats abolishes nicotine induced place preference during late adolescence. Amino Acids. 2008;35:247–249. doi: 10.1007/s00726-007-0584-2. [DOI] [PubMed] [Google Scholar]
- 82.Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 83.Wong PC, Pardo CA, Borchelt DR, et al. An adverse property of familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 84.Devon RS, Orban PC, Gerrow K, et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. PNAS. 2006;103:9595–9600. doi: 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spencer PS, Hugon J, Ludolph A, et al. Discovery and partial characterization of primate motor-system toxins. Ciba Found Symp. 1987;126:221–238. doi: 10.1002/9780470513422.ch14. [DOI] [PubMed] [Google Scholar]
- 86.Khabazian I, Bains JS, Williams DE, et al. Isolation of various forms of sterol β-d-glucoside from the seed of cycas circinalis: neurotoxicity and implications for ALS-PDC. J Neurochem. 2002;83:1–13. doi: 10.1046/j.1471-4159.2002.00976.x. [DOI] [PubMed] [Google Scholar]
- 87.Kurland LT. Amyotrophic lateral sclerosis and Parkinson’s disease complex on Guam linked to an environmental neurotoxin. Trends Neurol. Sci. 1988;11:51–54. doi: 10.1016/0166-2236(88)90163-4. [DOI] [PubMed] [Google Scholar]
- 88.Whiting MG (1964) Food practices in ALS foci in Japan, the Marianas, and New Guinea. Federation Proceedings, Third Conference on Toxicity of Cycads 23: 1343–5. [PubMed]
- 89.Borenstein AR, Mortimer JA, Schofield E, et al. Cycad exposure and risk of dementia, MCI, and PDC in the Chamorro population of Guam. Neurology. 2007;68:1764–1767. doi: 10.1212/01.wnl.0000262027.31623.b2. [DOI] [PubMed] [Google Scholar]
- 90.Shaw CA, Banjo OC. Fetal exposure of mice to cycad neurotoxins impacts postnatal behaviour. Abstracts: Proc. Soc Neurosci; 2008. [Google Scholar]
- 91.Wilson JMB, Khabazian I, Wong MC, et al. Behavioral and neurological correlates of ALS-parkinsonism dementia complex in adult mice fed washed cycad flour. J Neuromol. Med. 2002;1:207–222. doi: 10.1385/NMM:1:3:207. [DOI] [PubMed] [Google Scholar]
- 92.Tabata RC, Wilson JMB, Ly P, et al. Chronic exposure to dietary sterol glucosides is neurotoxic to motor neurons and induces an ASL-PDC phenotype. Neuromol. Med. 2008;10:24–39. doi: 10.1007/s12017-007-8020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barlow BK, Richfield EK, Cory-Slechta DA, et al. A fetal risk factor for Parkinson’s disease. Dev Neurosci. 2004;26:11–23. doi: 10.1159/000080707. [DOI] [PubMed] [Google Scholar]
- 94.Fisher LR, Culver DG, Tennat P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 95.Feeney SJ, Austin L, Bennett TM, et al. The effect of leukaemia inhibitory factor on SOD1 G93A murine amyotrophic lateral sclerosis. Cytokine. 2003;23:108–118. doi: 10.1016/s1043-4666(03)00217-5. [DOI] [PubMed] [Google Scholar]
- 96.Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26:1–13. doi: 10.1016/j.nbd.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 97.Feng HL, Leng Y, Ma CH, et al. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155:567–572. doi: 10.1016/j.neuroscience.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nicaise C, Coupier J, Dabadie MP, et al. Gemals, a new drug candidate, extends lifespan and improves electromyographic parameters in a rat model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:85–90. doi: 10.1080/17482960701788180. [DOI] [PubMed] [Google Scholar]
- 99.Lee J, Ryu H, Kowall NW. Motor neuronal protection by L-arginine prolongs survival of mutant SOD1 (G93A) ALS mice. Biochem Biophys Res Commun. 2009;384:524–529. doi: 10.1016/j.bbrc.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scott S, Kranz JE, Cole J, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 101.Shen W-B, McDowell KA, Siebert AA, et al. (2010, in press) Environmental neurotoxin-induced progressive model of parkinsonism in rats. Annals Neurol. [DOI] [PMC free article] [PubMed]
- 102.Dastur DK. Cycad toxicity in monkeys: clinical, pathological, and biochemical aspects. Fed Proc. 1964;23:1368–1369. [PubMed] [Google Scholar]
- 103.Perry TL, Bergeron C, Biro AJ, et al. Beta-N-Methylamino-l-alanine. Chronic oral administration is not neurotoxic to mice. J Neurol Sci. 1989;94:173–180. doi: 10.1016/0022-510x(89)90227-x. [DOI] [PubMed] [Google Scholar]
- 104.Cruz-Aguado R, Winkler D, Shaw CA. Lack of behavioral and neuropathological effects of dietary b-methylaminoalanine (BMAA) in mice. Pharmacol Biochem Behav. 2006;84:294–299. doi: 10.1016/j.pbb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 105.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 106.Wider C, Wszolek ZK. Clinical genetics of Parkinson’s disease are related disorders. Parkinsonism Relat Disord. 2007;13(Suppl 3):S229–S232. doi: 10.1016/S1353-8020(08)70007-5. [DOI] [PubMed] [Google Scholar]
- 107.McNaught KS, Olanow CW. Proteasome inhibitor-induced model of Parkinson’s disease. Ann Neurol. 2006;60:243–247. doi: 10.1002/ana.20936. [DOI] [PubMed] [Google Scholar]
- 108.Nair VD, McNaught KS, González-Maeso J, et al. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem. 2006;281:39550–39560. doi: 10.1074/jbc.M603950200. [DOI] [PubMed] [Google Scholar]
- 109.Fishman-Jacob T, Reznichenko L, Youdim MB, et al. A sporadic Parkinson disease model via silencing of the ubiquitin-proteasome/E3 ligase component SKP1A. J Biol Chem. 2009;284:32835–32845. doi: 10.1074/jbc.M109.034223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petroske E, Merdith GE, Callen S, et al. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/s0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 111.Alvarez-Fischer D, Guerreiro S, Hunot S, et al. Modelling Parkinson-like neurodegeneration via osmotic minipump delivery of MPTP and probenecid. J Neurochem. 2008;107:701–711. doi: 10.1111/j.1471-4159.2008.05651.x. [DOI] [PubMed] [Google Scholar]
- 112.Schintu N, Frau L, Ibba M, et al. Progressive dopaminergic degeneration in the chronic MPTPp mouse model of Parkinson’s disease. Neurotox Res. 2009;16:127–139. doi: 10.1007/s12640-009-9061-x. [DOI] [PubMed] [Google Scholar]