

Abstract
Cutaneous melanoma is a significant health problem worldwide. Available treatments can induce objective tumor regression in a small percent of patients, but these responses are not always associated with improved long-term survival. The resistance of melanoma to therapy and its predestined recurrence are related to the genetic heterogeneity and genomic instability of the tumor. For many years these genetic alterations were thought to be linked to the accumulation of random mutations in functionally differentiated cells which transform them into malignant cells that have lost their ability to differentiate and have acquired drug resistance. In the last few years it has been largely demonstrated that melanoma as other solid tumors contains a subpopulation of cells (CSCs) considered the source of the primary tumor mass, of new tumor nodules and responsible for drug resistance and cancer recurrence.
In this review, we provide an overview of findings and advances in CSCs research that are relevant to the initiation, natural history, and the response to treatment of malignant melanoma.
Keywords: Melanoma stem cells, Cancer stem cells
1. Introduction
Cutaneous melanoma is a significant health problem worldwide; its incidence has increased 3–7% on average over several decades. In US, the lifetime risk of melanoma in the year 2000 was estimated at 1 in 75 persons [1]. Patients with advanced disease have a poor prognosis with a reported median survival ranging between 3 and 11 months [2]. Therapies including immune therapy with systemic high dose interleukin-2 or interferon-α, antigen-specific immunization or chemotherapy with dacarbazine or temozolomide can induce objective tumor responses in only 5–20% of patients [2]. One type of adoptive immune therapy, the administration of tumor infiltrating lymphocyte following lympho-ablation, was reported to induced objective tumor regression in 60% of highly selected patients in a non-randomized study [3]. However, these responses do not result in overall survival benefit as the large majority of patients die with relapsing disease that is often resistant to further therapy. It has been suggested that the stubborn recurrence of cancer following a primary response to whatever treatment is applied, is due to the survival of a subset of cancer cells that display an intrinsic resistance to treatment-induced death [4]. The hypothesis that cancer stem cells (CSCs) lead the tumorigenic process has received several consents in the last years. The cancer stem cell hypothesis suggests that tumors are initiated and maintained by a subset of cancer cells capable of self-renewal and differentiation into bulk tumor cells [4] and [5]. It is important to note that the term cancer stem cells is more a functional definition that has been proposed to identify a subgroup of cancer cells able to initiate tumors and differentiate into a heterogeneous progeny, similar to the original tissue from which they have been obtained. The alternative terms tumor initiating cells can be inappropriate as well. Cancer stem cells can be genetically different from the cells that established the tumor [5]. Moreover, different subsets of cancer initiating cells as results of the genetic and epigenetic changes that characterize cancer cells, can be identified [6]. Even with several unanswered questions, the concept of cancer stem cells and the hierarchical model of tumorigenesis are considered a big challenge for the implications that they may have in the understanding of tumor biology and the development of more effective anti cancer treatments. As we know, the therapies currently available are modestly effective, and developed, according to the old model of carcinogenesis “The stochastic model”, to target and destroy a population of tumorigenic cells, undergone to transformation after random mutations targeting any cells in an organ and subsequent clonal selection [7].
This review will focus on aspects of CSCs that may be relevant to the natural history and the response to treatment of melanoma. The hypothesis that therapies capable of targeting such cells may be more likely to be successful than current therapies will also be discussed.
2. Treatment failure in cancer: are we shooting at the wrong target?
Normal tissue homeostasis is guaranteed by the balance between cell loss and cell renewal, balance due to the capability of ASCs to undergo self-renewal and give rise to a more differentiated progeny to replenish cells in adult tissues [8]. To maintain indefinitely this aptitude, stem cells must perform asymmetric cell divisions; with every division, each ASC generates a cell with identical genetic and epigenetic content (self-renewal) and a genetically different cell programmed toward terminal differentiation. CSCs share with ASCs the capability for self-renewal, indefinite proliferation, and potential to differentiate into heterogeneous cancer cells that embody the tumor bulk. The genetic abnormalities present in CSCs, such as DNA repair deficiency, mutations, genetic imbalances and epigenetic alterations result in significant genomic instability [9] and [10]. This instability, in turn, is responsible for a rapidly evolving “express lane” process, in which the programmed asymmetrical division is frequently lost, leading to a large number of semi-differentiated cancer cells that randomly retain characteristics of the original tissue. Thus, only a small proportion of self-renewed and undifferentiated CSCs are maintained during cancer progression representing less than 1% of the bulk of the tumor. Because CSCs are undifferentiated they do not express functional proteins associated with tissue differentiation (differentiation antigens/biomarkers) and, therefore, are not susceptible to immune therapy targeting tumor differentiation antigens [11]. The self-renewed CSCs are also in a relatively quiescent mitotic state; they are characterized by high expression of ABC drug efflux transporter, growth factors and anti apoptotic signaling elements; all these characteristics can account for the resistance to current clinical therapies predominantly effective against rapidly dividing cells [12]. Thus, targeting molecules relevant to the survival of CSCs can lead to the identification of more effective therapies.
3. Recent advances in CSCs: lessons learned from leukemias
CSCs have been best characterized in hemopoietic neoplasia. The discovery of CSCs in leukemia was facilitated by the ease in which circulating cancer cells can be obtained in leukemias compared to solid tumors and the availability of technology for the extraction, purification and transplantation of haematopoietic stem cells [13].
It is well-accepted that normal hemopoietic cell development is organized according to a hierarchical model sustained by a small population of quiescent, pluripotent ASCs capable of self-renewal and differentiation into all blood cell types.
The haematopoietic system and related malignancies offered the first and strongest evidence of the existence of CSCs. In the 1970s several different groups observed that in hematological diseases only a subset of cells could grow in vitro giving rise to a clonal expansion of mutated stem-like cells [14], [15] and [16]. Subsequently, Lapidot et al. [17] identified an acute myeloid leukemia-initiating cell bearing a CD34+ CD38−phenotype that could be engrafted in SCID mice. Others [18] confirmed that cells bearing the same CD34+, CD38−, Lin–surface makers were able to initiate human acute myeloid leukemias in non-obese diabetic/severe combined immunodeficiency mice. Engrafted CD34+, CD38−, Lin–cells could differentiate and reconstitute the heterogeneous phenotype observed in the original tumor. Moreover, serial transplantations demonstrated that these cells possessed self-renewal capacity. This study represents the first evidence that cancer cells can be organized into a hierarchy that mirrors the ASCs model. The evidence of the existence of cells with stem cell-like properties in hematological malignancies opened a new era for cancer research that is focused on the identification of CSCs in solid tumors.
4. CSCs in solid tumors
It is known that, unlike haematopoietic malignancies, solid tumors are composed of a mixed population of cells at different stage of differentiation phenotypically and genetically heterogeneous. A recent study from our group characterized cell lines generated from the same melanoma metastasis and identified contrasting phenotypes represented by distinct cell morphology, level of pigmentation and genetic imbalances as indicated by karyotyping, chromosomal and array comparative genomic hybridization [19] and [20]. In spite of these differences, all the cell lines displayed an identical methylation pattern, according to the human androgen receptor assay, supporting the idea that all cell lines derived from a common ancestor. Moreover, mutational analysis of the β-catenin gene, identified a 1-bp mutation present in all the metastatic cells tested [19]. As we know the WNT/β-catenin pathway is involved in all of the stem cell defining characteristics [21]. In our model, the persistent mutation in β-catenin could indicate activation of stem cell-like signaling pathways that may contribute to the biological behavior of this patient tumor [21]. Our observations support the hypothesis that cancer cells can arise from a common ancestor and contain a subpopulation of cells with stem-like properties [22].
In the last few years CSCs have been identified and described in several epithelial and other solid malignancies. Al-Hajii et al. [23] reported the isolation and characterization of a CD44+, CD24−/low cell population from metastatic pleural effusions in a patient with invasive breast cancer. Cells carrying these surface markers were infrequent but could sustain tumor initiation when injected in immune-deficient mice. Moreover, tumors derived from CD44+, CD24−/low cells recreated the phenotypic heterogeneity observed in the original sample. Thus, these cells demonstrated at the same time self-renewal capacity and the ability to differentiate into cells with low tumorigenic potential that represented the bulk of the tumor population. Hemmati HD et al. [24] observed brain tumor-derived progenitor cells that can form neurospheres and are capable of self-renewal. The neurospheres display an unusual ability to proliferate and sometimes differentiate into abnormal cells with multiple differentiation markers. Studies performed on glioblastoma multiforme and medulloblastoma have shown that tumorigenic cells are restricted to the CD133+ subpopulation, which usually represents 5–30% of total tumor cells. CD133 (prominin-1) is a membrane protein identified in both humans and mice and classified as a marker of primitive hemopoietic and neural ASCs [25]. Tumors arising from purified CD133+ cells demonstrated the ability to reproduce the phenotypic diversity and differentiation pattern of the parent tumors [26].
For brain tumors, the identification of self-renewing CSCs was facilitated by the availability of an in vitro method for the selection of “neurospheres” previously used for the isolation of ASCs. Using this method, Galli et al. [27] succeeded in the isolation and propagation of “cancer neurospheres” from human glioblastoma multiforme, which were highly enriched in long-term self-renewing, multi-lineage-differentiating, CSCs. Recently, it has been observed that CD133+ glioma stem-like cells are more resistant to ionizing radiation than bulk tumor cells in culture and in in vivo xenograft transplant model [28]. This observation attributes the irradiation resistance of brain tumors to a particular subset of CSCs. In addition to breast and brain tumors, progenitor/tumorigenic subpopulations of CSCs have been characterized in prostate and colorectal cancer that expressed the CSC-specific surface markers CD44 [29] and CD133, respectively [30]. In the last few years, a subpopulation of cancer stem cells has been described also in Melanoma.
5. Melanoma stem cells (MSCs)
Melanocytes are specialized cells derived from the neural crest during embryonic development that migrated to the hair follicle and the basal layer of the epidermis. Immature melanocytic cells have been identified in normal human epidermidis [31]. Nishimura et al. [32] reported the presence of ASCs of the melanocytes lineage in the lower portion of the mouse hair follicles. Melanocyte ASCs migrate from the hair follicle niche, divide and differentiate into pigmented melanocytes. Moreover, they migrate into vacant niches and replenish the reservoir of melanocytes ASCs. Thus, far no such melanocytic-lineage ASCs have been identified in humans. However, the observation that re-pigmentation can occur following the onset of vitiligo suggests that melanocytes can be replaced by a reservoir of ASCs [33] and [34].
The role of melanocyte ASCs in normal tissues may be critical also in humans and more important in melanoma development [35] and [36]. A recent genetic and transcriptional analysis of melanoma cell lines derived from metachronous metastases in a single patient during a decade of recurring disease demonstrated that each cell line was derived from the clonal expansion of a unique progenitor/stem-like cell that maintained a consistent genetic pattern throughout the patient’s life [19] and [20].
Experimental evidence supports the existence of a MSC phenotype; Fang et al. [37] described a subset of cells derived from freshly isolated or in vitro stabilized melanoma cell lines that was able to form “melanoma spheroids” when grown in a specific stem cell medium. The MSCs exhibited self-renewal properties after in vitro and in vivo cloning. The MSC-like population exhibited the ability to differentiate into astrocytes or cells of the mesenchymal-lineage and was extremely tumorigenic when injected into immune-deficient mice. The spheroids were enriched in CD20 expressing cells. In addition, they expressed CD133 and ABCG2. The expression of these two antigens was associated with higher tumorigenic potential and ability to create new and bigger spheres [37] and [38]. MSCs are similar to bone marrow-derived ASCs in that they are Hoechst dye-low [39] and [40], small in size and display a low proliferation rate [35]. However, the culture of single MSCs yielded large and morphologically heterogeneous melanoma cells that exhibited lineage and antigenic heterogeneity.
The MSC marker ABCB5 was first described by Frank et al. [41] and [42] as a regulator of cell–cell fusion in normal skin progenitor cells, and was considered as the most important mediator of resistance to doxorubicin in malignant melanoma cell lines. Moreover, he reported that this marker was expressed by a subgroup of melanoma cell lines coexpressing the CSCs marker CD133 [42]. Using gene expression profiling the authors demonstrated that ABCB5 expression was higher in melanoma lesions and metastases compared to benign skin nevi, thick melanomas compared to thin ones, and metastases compared to primary lesions, correlating with the clinical progression of malignant melanoma. They concluded that ABCB5 should be considered a marker of melanoma progression.
Based on these findings, Schatton et al. [43] hypothesized that ABCB5 could be a molecular marker defining tumorigenic malignant-melanoma-initiating cells. ABCB5 expression was found to be higher in primary or metastatic melanoma compared to benign melanocytic nevi. ABCB5 was expressed by 1.6–20.4% of melanoma cells. The expression of ABCB5 correlated with the expression of markers of primary melanoma phenotype such as TIE1, CD144 and BMPR1. These markers were expressed more frequently in ABCB5+ rather than ABCB5−cells. ABCB5− expressing MSCs displayed a significantly greater capability to initiate tumor growth in immune-deficient mice. Xenografts derived from ABCB5− expressing MSCs exhibited the same phenotypic heterogeneity of parental tumors and could in turn initiate secondary tumors when ABCB5− expressing cells were isolated and used for re-implantation. The tumor forming competence could be inhibited by anti-ABCB5 antibody-mediated cytotoxicity. Thus, this study provides a link between MSCs, cancer progression and chemotherapy resistance of melanoma [43].
The expression of MDR1 (ABCB1, PGY1 or GP170 gene product), one of the active transport pumps on the plasma membrane, has been described in many tumor types and is considered to be responsible for drug resistance. To test whether MDR1 represent a biomarker relevant to MSC biology, Keshet et al. [44] examined the expression of this gene by a melanoma self-renewing population obtained from primary melanoma culture. MDR1-expressing cells contributed 1.3–9.7% of the entire cell population in primary melanoma and co-expressed other MSC surface markers such as ABCB5, Nanog and hTERT, underlining the relationship between drug resistance and CSCs. Interestingly, CD133 expression was not detected in this population of MDR1-expressing MSCs.
We still lack a marker that can be used to reliably and consistently identify and isolate MSCs. Monzani et al. [38] found that CD133 is a useful marker for identifying MSC outlier cells with a high tumorigenic potential, but in contrast with Fang’s findings [37], these cells are primarily concentrated in adherent cell populations rather than in melanoma sphere characteristic of MSCs. Similarly, the expression of ABCG2 alone as a marker for MSCs could not reliably identify cells with a higher tumorigenic potential suggesting that it has low specificity as a melanoma initiating cell marker [45].
Although mounting evidence supports the existence of CSCs, their characterization is limited by difficulties related to the separation of these relatively rare cells. So far, markers used to distinguish CSCs from other tumor cells are predominantly based on our knowledge of embryonic stem cells (ESCs) surface markers or cancer drug resistance markers. Furthermore, cells studied as CSCs are cultured in vitro and selected according to their proliferation capacity. These cells favor sphere formation in culture and can differentiate into heterogenic populations and can self-renew. It is logical to postulate that the markers so far identified are not representative of stem-like cell subpopulation but rather represent an active phase of their biology.
Thus, CSCs can be defined as progenitor cells unlikely to be exposed by current experimental protocols while their active phase could be best called cancer stemloid cells (or CSCs-like) that can proliferate, undergo clonal selection, and accumulate mutations. Since they do proliferate to a certain extent they can also be the target of cancer therapies [46]. Depending on the accumulation of acquired mutations, CSCs and their stemloid progeny at different stages of differentiation may display fluctuations of CSC-marker expression as exemplified by varying levels of CD133 expression [47].
It is clear that novel strategies should be sought for the identification of CSCs at different stages of differentiation beyond the validation of ASCs markers or markers of drug resistance. Genomic instability is the hallmark of malignant disease including melanoma [48], [49] and [50]. Melanomas contain high number of chromosomal rearrangements, such as translocations, chromosomal amplifications, or deletions, and, in addition, germline or somatic point mutations that can affect cancer development and progression [51] and [52]. Comparative analysis of genetic and epigenetic alterations could further illuminate the genetic make up and stability of CSCs compared to their progeny [20]. Different chromosomal aberration patterns were identified by comparative genomic hybridization between vertical and radial growth phase of primary melanomas [53] and between primary and metastatic melanomas [54]. We compared melanoma cell lines derived from metachronous melanoma metastases in a patient who experienced several recurrences and clinical remissions over more than a decade. In spite of marked phenotypic variations, a conserved core of genetic alterations was maintained throughout the natural history of the disease that could have been inherited from the progenitor stem cell [19] and [20]. A rare point mutation of the β-catenin gene was identified in each of the metastatic samples suggesting that all cell lines were derived from the same progenitor. Although some genetic changes were shared in this series of patient biopsies, not all of the aberrations were perpetuated. Thus, while extensive genetic instability could be observed at the 12th different stages of this patient’s disease, alterations were not sequential or cumulative. This suggests that each metastasis evolved independently from the other from a common ancestor MSC whose vestiges were reverberated in each by a consistent and invariable genetic core of chromosomal aberrations.
Taken together, these findings suggest that melanomas arise from mutations and chromosomal rearrangements that accumulate in a common precursor cell, which subsequently drives the establishment of primary and metastatic lesions (Fig. 1). This cancer stem-like precursor is likely responsible for the metastatic process while the less stable progeny are unable to spread locally or distantly [35] and [4].
Fig 1.
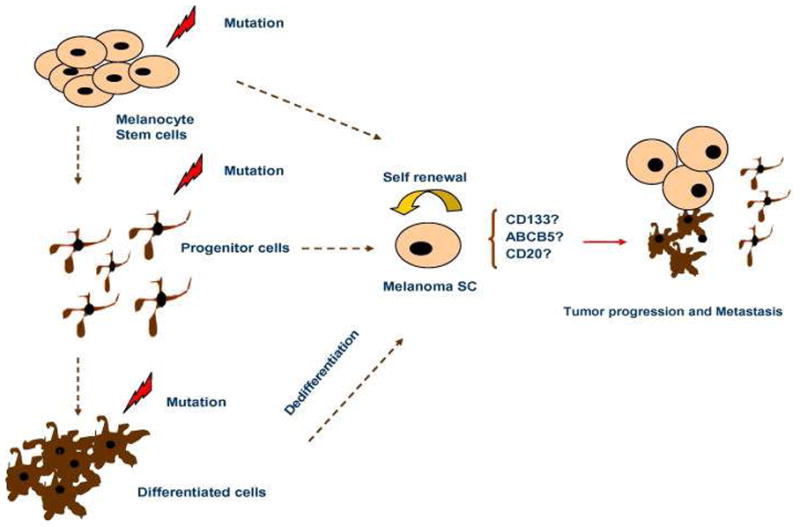
Melanoma stem-like cells may appear after mutations occurred in melanocytes stem cells, or progenitor cells. They may also be the results of transformation in differentiated cells. They are responsible for tumor growth and metastasis.
6. Conclusions
The CSCs hypothesis is not totally new as a similar concept was suggested more than 100 years ago [55] and [56]. However, it is still at its infancy and much study needs to be done to confirm it. Accumulating evidence supported by an increasing number of publications in the last few years suggests that cancers are originated from a subpopulation of cells responsible for tumorigenesis, tumor maintenance, growth and metastasis. Assuming that CSCs have dramatically different biological properties compared with the rest of the cancer cells, it is possible to explain the poor effectiveness of current therapies by the fact that most were developed by testing their activity against the bulk of cancer cells independent of functional subsets. For melanoma, immune therapy should consider alternate target antigens unrelated to tissue differentiation such as cancer testis antigens [57] whose expression is increasingly stabilized in the later stages of cancer progression or mutated neo antigens associated with the oncogenic process and most likely expressed by CSCs [58] and [59]. Chemotherapy should target pathways less strictly associated with cell division but more closely related to the metabolism and, most importantly, relatively resting CSCs [60].
Despite the experimental evidence described in the mini-review, the CSC theory applied to solid tumors need to be validated and many questions remain. Most of the characterization of CSCs, among them MSCs, has been based on the expression of stem cell markers; the ability to form spheres; and the capacity to self-renew, proliferate, differentiate and initiate tumors when injected in immunodeficient mice. These arbitrarily selected criteria may suffer some limitations. The markers used to isolate cancer stem cells are not unique to these cells, they are often expressed in normal tissues from different organs [61]. Their expression can be modulated by different experimental and environmental conditions; for example hypoxia can induce increased expression of stem cell-like surface markers and interfere with the gene expression machinery of cancer cells [62]. The surface markers may be not identify a pure population of cancer stem cells, but enrich the specific population, help to sort for further evaluation such as the ability to initiate tumors in animal models. The in vivo assay and their results can be difficult to interpret because extremely variable according to the experimental conditions and the host microenvironment [63]. The ability of tumor cells to survive and regenerate in xenografts may be unrelated to stem cell-like features but instead may be due to random alterations in the regulation of apoptotic pathways, cell cycle regulation or altered methylation patterns. Genetic and epigenetic may allow stochastic changes for better adaptation to the microenvironment independent of a programmed asymmetric process.
In physiologic conditions ASCs require a specific environment to maintain their “stemness” and de-differentiated status. An environmental “niche” that fosters ASC survival has been described in bone marrow and in mouse hair follicles in physiological condition. CSC-niches that may play a role in maintaining tumorigenesis and determining cell behavior have not yet been characterized. It is possible that CSCs do not co-localize with their daughter cells but instead hide in a special micro-environmental niche. This may explain why cancer reoccurrence after complete regression rarely occur at the same site.
In a transgenic transforming rat Her-2/neu breast cancer BALB-neuT mouse model and in a MMTV-polyomavirus-middle T transgenic mice model, disseminated tumor cells (CK+ and HER-2+) become detectable in bone marrow at as early as 4–9 weeks of age when even the most meticulous analysis of the mammary gland can only detect areas of atypical ductal hyperplasia. This suggests that an early spread of cancer occurs through the migration of CSC-like progenitors in the bone marrow [64]. Moreover, these disseminated tumors in bone marrow do not significantly increase in number during tumor growth and progression suggesting that they retain a quiescent phenotype and asymmetrical self-renew in a niche. Similar disseminated tumor cells have also been identified in the bone marrow of patients with breast cancer at different stages and carrying both normal or abnormal karyotypes [64]. Thus, much still needs to be learned about the true biology of CSCs, their genetic and phenotypic evolution in response to environmental stimuli, the hierarchy governing their differentiation, and their salient survival strategies.
Abbreviations
- ASC(s)
adult stem cell(s)
- CSC(s)
cancer stem cell(s)
- EB
embryonic bodies
- ESC(s)
embryonic stem cell(s)
- MSC(s)
melanoma stem cell(s)
References
- 1.Beddingfield FC., III The melanoma epidemic: res ipsa loquitur. Oncologist. 2003;8:459–465. doi: 10.1634/theoncologist.8-5-459. [DOI] [PubMed] [Google Scholar]
- 2.Rietschel P, Wolchok JD, Krown S, Gerst S, Jungbluth AA, Busam K, Smith K, Orlow I, Panageas K, Chapman PB. Phase II study of extended-dose temozolomide in patients with melanoma. J Clin Oncol. 2008;26:2299–2304. doi: 10.1200/JCO.2007.14.5292. [DOI] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68:6932–6941. doi: 10.1158/0008-5472.CAN-07-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lagasse E. Cancer stem cells with genetic instability: the best vehicle with the best engine for cancer. Gene Ther. 2008;15:136–142. doi: 10.1038/sj.gt.3303068. [DOI] [PubMed] [Google Scholar]
- 7.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 8.Seaberg RM, van der KD. Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci. 2003;26:125–131. doi: 10.1016/S0166-2236(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 9.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 10.Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43–49. doi: 10.1016/j.semcancer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–182. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 12.Kvinlaug BT, Huntly BJ. Targeting cancer stem cells. Expert Opin Ther Targets. 2007;11:915–927. doi: 10.1517/14728222.11.7.915. [DOI] [PubMed] [Google Scholar]
- 13.Guo W, Lasky JL, III, Wu H. Cancer stem cells. Pediatr Res. 2006;59:59R–64R. doi: 10.1203/01.pdr.0000203592.04530.06. [DOI] [PubMed] [Google Scholar]
- 14.Minden MD, Buick RN, McCulloch EA. Separation of blast cell and T-lymphocyte progenitors in the blood of patients with acute myeloblastic leukemia. Blood. 1979;54:186–195. [PubMed] [Google Scholar]
- 15.Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976;295:913–916. doi: 10.1056/NEJM197610212951702. [DOI] [PubMed] [Google Scholar]
- 16.Fialkow PJ, Najfeld V, Reddy AL, Singer J, Steinmann L. Chronic lymphocytic leukaemia: clonal origin in a committed B-lymphocyte progenitor. Lancet. 1978;2:444–446. doi: 10.1016/s0140-6736(78)91444-7. [DOI] [PubMed] [Google Scholar]
- 17.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 19.Wang E, Voiculescu S, Le Poole IC, el Gamil M, Li X, Sabatino M, Robbins PF, Nickoloff BJ, Marincola FM. Clonal persistence and evolution during a decade of recurrent melanoma. J Invest Dermatol. 2006;126:1372–1377. doi: 10.1038/sj.jid.5700193. [DOI] [PubMed] [Google Scholar]
- 20.Sabatino M, Zhao Y, Voiculescu S, Monaco A, Robbins PF, Nickoloff BJ, Karai L, Selleri S, Maio M, Selleri S, Marincola FM, Wang E. Conservation of a core of genetic alterations over a decade of recurrent melanoma supports the melanoma stem cell hypothesis. Cancer Res. 2008;68:222–231. doi: 10.1158/0008-5472.CAN-07-1939. [DOI] [PubMed] [Google Scholar]
- 21.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 22.Pathak S. Organ- and tissue-specific stem cells and carcinogenesis. Anticancer Res. 2002;22:1353–1356. [PubMed] [Google Scholar]
- 23.Al-Hajj M, Wicha MS, ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizrak D, Brittan M, Alison MR. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 27.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De VS, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 28.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 29.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De MR. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 31.Grichnik JM, Ali WN, Burch JA, Byers JD, Garcia CA, Clark RE, Shea CR. KIT expression reveals a population of precursor melanocytes in human skin. J Invest Dermatol. 1996;106 doi: 10.1111/1523-1747.ep12338471. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura EK, Jordan SA, Oshima H, Yoshida H, Osawa M, Moriyama M, Jackson IJ, Barrandon Y, Miyachi Y, Nishikawa S. Dominant role of the niche in melanocyte stem-cell fate determination. Nature. 2002;416:854–860. doi: 10.1038/416854a. [DOI] [PubMed] [Google Scholar]
- 33.Yu HS. Melanocyte destruction and repigmentation in vitiligo: a model for nerve cell damage and regrowth. J Biomed Sci. 2002;9:564–573. doi: 10.1159/000067283. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307:720–724. doi: 10.1126/science.1099593. [DOI] [PubMed] [Google Scholar]
- 35.Grichnik JM. Genomic instability and tumor stem cells. J Invest Dermatol. 2006;126:1214–1216. doi: 10.1038/sj.jid.5700240. [DOI] [PubMed] [Google Scholar]
- 36.Grichnik JM, Burch JA, Schulteis RD, Shan S, Liu J, Darrow TL, Vervaert CE, Seigler HF. Melanoma, a tumor based on a mutant stem cell? J Invest Dermatol. 2006;126:142–153. doi: 10.1038/sj.jid.5700017. [DOI] [PubMed] [Google Scholar]
- 37.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 38.Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, Invernici G, Parati E, Alessandri G, La Porta CA. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank NY, Pendse SS, Lapchak PH, Margaryan A, Shlain D, Doeing C, Sayegh MH, Frank MH. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem. 2003;278:47156–47165. doi: 10.1074/jbc.M308700200. [DOI] [PubMed] [Google Scholar]
- 42.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 43.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keshet GI, Goldstein I, Itzhaki O, Cesarkas K, Shenhav L, Yakirevitch A, Treves AJ, Schachter J, Amariglio N, Rechavi G. MDR1 expression identifies human melanoma stem cells. Biochem Biophys Res Commun. 2008;368:930–936. doi: 10.1016/j.bbrc.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 45.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and A ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 46.Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007;6:1684–1690. doi: 10.4161/cbt.6.11.5167. [DOI] [PubMed] [Google Scholar]
- 47.Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol. 2007;20:102–107. doi: 10.1038/modpathol.3800720. [DOI] [PubMed] [Google Scholar]
- 48.Kwong J, Lo KW, Chow LS, To KF, Choy KW, Chan FL, Mok SC, Huang DP. Epigenetic silencing of cellular retinol-binding proteins in nasopharyngeal carcinoma. Neoplasia. 2005;7:67–74. doi: 10.1593/neo.04370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang J, Wei W, Zhang J, Liu G, Bignell GR, Stratton MR, Futreal PA, Wooster R, Jones KW, Shapero MH. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 51.Bevilacqua RA, Nunes DN, Stroun M, Anker P. The use of genetic instability as a clinical tool for cancer diagnosis. Semin Cancer Biol. 1998;8:447–453. doi: 10.1006/scbi.1998.0122. [DOI] [PubMed] [Google Scholar]
- 52.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 53.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- 54.Balazs M, Adam Z, Treszl A, Begany A, Hunyadi J, Adany R. Chromosomal imbalances in primary and metastatic melanomas revealed by comparative genomic hybridization. Cytometry. 2001;46:222–232. doi: 10.1002/cyto.1131. [DOI] [PubMed] [Google Scholar]
- 55.Durante F. Nesso phisio-patologico tra la struttura dei nei materni e la genesi di alcuni tumori maligni. Arch Memor Observ Chir Pract. 1874;11:217–226. [Google Scholar]
- 56.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea –a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 57.Costa FF, Le BK, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells. 2007;25:707–711. doi: 10.1634/stemcells.2006-0469. [DOI] [PubMed] [Google Scholar]
- 58.Robbins PF, el-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185–1192. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA. 2005;102:16013–16018. doi: 10.1073/pnas.0500090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007;19:61–64. doi: 10.1097/CCO.0b013e328011a8d6. [DOI] [PubMed] [Google Scholar]
- 61.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells –perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 62.Greijer AE, van der GP, Kemming D, Shvarts A, Semenza GL, Meijer GA, van de Wiel MA, Belien JA, van Diest PJ, van der WE. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1) J Pathol. 2005;206:291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 63.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 64.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]