

Abstract
Aortic stenosis (AS) is the most frequent valvular heart disease. Severe AS results in concentric left ventricular hypertrophy, and ultimately, the heart dilates and fails. During a long period of time patients remain asymptomatic. In this period a pathology progression should be monitored and effectively thwarted by targeted measures. A cascade of cellular and molecular events leads to chronic degeneration of aortic valves. There are some molecular attributes characteristic for the process of valvular degeneration with clear functional link between shifted cell-cycle control, calcification and tissue remodelling of aortic valves. Bioactivity of implanted bioprosthesis is assumed to result in its dysfunction. Age, gender (females), smoking, Diabetes mellitus, and high cholesterol level dramatically shorten the re-operation time. Therefore, predictive and preventive measures would be highly beneficial, in particular for young female diabetes-predisposed patients. Molecular signature of valvular degeneration is reviewed here with emphases on clinical meaning, risk-assessment, predictive diagnosis, individualised treatments.
Keywords: Degenerative valve disease, Bioprostheses, Risk assessment, Diabetes, Predictive diagnosis, Personalised medicine
Degenerative valve disease: clinical aspects and molecular signature
Clinical assessment of aortic stenosis
Aortic stenosis (AS) is the most frequent valvular heart disease. Its prevalence increases with age, and has been reported between 2–4% in a population ≥65 years old [1, 2]. Aortic sclerosis is the precursor of AS and has been found in 25–30% [3]. Calcific AS refers to a narrowing of the aortic valve lumen as a result of the deposition of calcium in the cusps and valve ring. Severe AS results in concentric left ventricular hypertrophy, and ultimately, the heart dilates and fails. During a long period with increasing outflow tract obstruction, which results in increasing left ventricular pressure load, patients remain asymptomatic, acute complications are rare. Therefore, these asymptomatic patients with AS should be monitored closely for the development of symptoms and progression of disease, especially by Doppler-echocardiography, an accurate non-invasive measurement of the stenosis severity (Fig. 1).
Fig. 1.
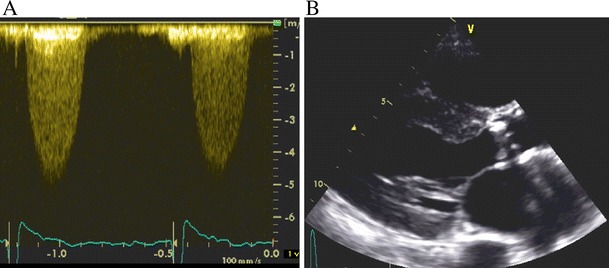
Clinical assessment by (a) Doppler echocardiography; (b) two dimensional echocardiography (parasternal short axis view)
However, as soon as symptoms occur, such as exertional dyspnoea, angina, and syncope, outcome becomes poor. Average survival after the onset of symptoms has been reported to be less than 2–3 years [4]. In this situation, valve replacement does not only result in dramatic symptomatic improvement but also in good long term survival [5]. This holds true even for patients with already reduced left ventricular function, as long as functional impairment is, indeed, caused by AS. Thus, there is general agreement that urgent surgery must be strongly recommended in symptomatic patients [5–7].
A cascade of cellular and molecular events leads to chronic degeneration of aortic valves
Mechanical stress is currently considered as the main cause that triggers degenerative processes. This is accompanied by a thickening of the valve cusps, and remodelling of the left ventricular geometry. Clinical-pathological studies of aortic stenosis have demonstrated an abundant deposition of extracellular matrix (ECM) proteins physiologically present in bones [6], and cuspal calcific deposits associated with mineralisation of devitalised cells [8]. Moreover, bone-marrow derived endothelial progenitor and dendritic cells have been identified in both native degenerative aortic valves and degenerative prostheses; the co-localisation of those cells with inflammatory infiltrates has been demonstrated [9]. A cascade of cellular and molecular events leading to the degeneration of aortic valves is summarised in Fig. 2.
Fig. 2.
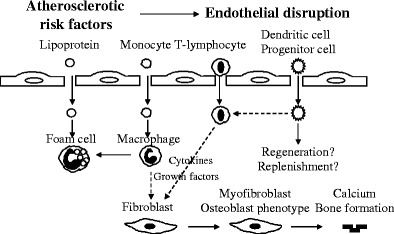
Cascade of cellular and molecular events leading to the degeneration of aortic valves: Inflammatory cells release cytokines and growth factors that act on valve fibroblasts. A subset of myofibroblasts may differentiate into the osteoblast cell-phenotype that secretes bone matrix proteins involved in the valve calcification process. However, several question remain open, such as - whether there is a differential role of the multiple subset of immune / inflammatory cells in the depicted cascade of events followed by the question, - whether the above demonstrated cellular events can serve as indicators for predictive diagnostics at pre-stages of valvular calcification
Mineralisation of skeletal and dental tissue is genetically programmed and physiologically well-regulated. In contrast, non-physiological calcification occurs in numerous pathological cardiovascular conditions including atherosclerosis, valvular stenosis, and reperfused ischemic myocardium. This is proposed to be an undesired common feature of degenerative or / and inflammatory tissue changes throughout the body. Pathomechanisms leading to the calcification of heart valves are still largely unknown. Contrary to physiological formation of bones, cuspal calcific deposits in the heart are non-physiological and normally not found in healthy cardiovascular tissues [6, 8, 10–12]. Numerous clinical-pathological studies of calcified valves have demonstrated cuspal calcific deposits tightly associated with mineralisation of devitalised cells, indicating a cascade of (programmed?) molecular events leading to chronic degeneration of myocardial tissue [6]. Tissue homeostasis strictly depends on a balance between cell growth and death. These aspects have been investigated at the level of gene transcription as reported earlier [7]: Table 1 summarises the list of gene products, a corresponding function of which is suppressed specifically in calcified versus non-calcified aortic valves. Among them, 40 proteins essential for energy metabolism are suppressed by aortic calcification. Furthermore, an expression of cytoskeleton-formation as well as ECM-building and tissue remodelling proteins (altogether 23 proteins) is completely suppressed in calcified valvular tissue. The above given protein core is switched off specifically in the case when the balance between cell growth and death in tissue homeostasis is shifted towards cellular death.
Table 1.
The data represent 63 gene products, the function of which is suppressed in calcified versus non-calcified degenerated aortic valves. There are following functional groups: energy metabolism, proteins responsible for cytoskeleton formation, matrix building, and tissue remodelling [7]
| GeneBank Accession / SwissProt Accession | Gene (protein) name / function | |
|---|---|---|
| I. Energy metabolism proteins (40 genes) | ||
| S70154 | Q16146 | acetyl-Coenzyme A acetyltransferase 2 (acetoacetyl Coenzyme A thiolase) |
| D90228 | P24752 | acetyl-Coenzyme A acetyltransferase 1 (acetoacetyl Coenzyme A thiolase) |
| L07033 | P35914 | 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase (hydroxymethylglutaricaciduria) |
| X83618 | P54868 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) |
| U62961 | P55809 | 3-oxoacid CoA transferase |
| M93107 | Q02338 | 3-hydroxybutyrate dehydrogenase (heart, mitochondrial) |
| X17025 | Q13907 | isopentenyl-diphosphate delta isomerase |
| X69141 | P37268 | farnesyl-diphosphate farnesyltransferase 1 |
| M88468 | Q03426 | mevalonate kinase (mevalonic aciduria) |
| U49260 | P53602 | mevalonate (diphospho) decarboxylase |
| D78130 | Q14534 | squalene epoxidase |
| D63807 | P48449 | lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase) |
| Q9UEZ1 | ||
| AF034544 | O60492 | 7-dehydrocholesterol reductase |
| U60205 | Q15800 | sterol-C4-methyl oxidase-like |
| M67466 | P14060 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 |
| Q14545 | ||
| P26439 | ||
| Y09501 | P00387 | diaphorase (NADH) (cytochrome b-5 reductase) |
| L21934 | P35610 | sterol O-acyltransferase (acyl-Coenzyme A: cholesterol acyltransferase) 1 |
| R07932 | diacylglycerol O-acyltransferase homolog 1 (mouse) | |
| M74047 | P31213 | steroid-5-alpha-reductase, alpha polypeptide 2 (3-oxo-5 alpha-steroid delta 4-dehydrogenase alpha 2) |
| L33179 | Q13713 | alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide |
| P40394 | ||
| M68895 | P28332 | alcohol dehydrogenase 6 (class V) |
| M63967 | P30837 | aldehyde dehydrogenase 1 family, member B1 |
| X05409 | P05091 | aldehyde dehydrogenase 2 family (mitochondrial) |
| Q03639 | ||
| M73704 | Q00169 | phosphotidylinositol transfer protein |
| L34081 | Q14032 | bile acid Coenzyme A: amino acid N-acyltransferase (glycine N-choloyltransferase) |
| U47105 | Q15738 | NAD(P) dependent steroid dehydrogenase-like; H105e3 |
| X05130 | P30037 | procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase), beta polypeptide (protein disulfide isomerase; thyroid hormone binding protein p55) |
| P32079 | ||
| Q15205 | ||
| P07237 | ||
| U12424 | P43304 | glycerol-3-phosphate dehydrogenase 2 (mitochondrial) |
| L34041 | P21695 | glycerol-3-phosphate dehydrogenase 1 (soluble) |
| D88308 | O14975 | fatty-acid-Coenzyme A ligase, very long-chain 1 |
| L09229 | P41215 | fatty-acid-Coenzyme A ligase, long-chain 1 |
| P33121 | ||
| X83368 | P48736 | phosphoinositide-3-kinase, catalytic, gamma polypeptide |
| S67334 | P42338 | phosphoinositide-3-kinase, catalytic, beta polypeptide |
| X66922 | P29218 | inositol(myo)-1(or 4)-monophosphatase 1 |
| M74161 | P32019 | inositol polyphosphate-5-phosphatase, 75kD |
| L08488 | P49441 | inositol polyphosphate-1-phosphatase |
| D16481 | P55084 | hydroxyacyl-Coenzyme A dehydrogenase/3-ketoacyl-Coenzyme A thiolase/enoyl-Coenzyme A hydratase (trifunctional protein), beta subunit |
| U40002 | Q05469 | lipase, hormone-sensitive |
| M72393 | P47712 | phospholipase A2, group IVA (cytosolic, calcium-dependent) |
| U20157 | Q15692 | phospholipase A2, group VII (platelet-activating factor acetylhydrolase, plasma) |
| Q13093 | ||
| II. Cytoskeleton formation, ECM-building & tissue-remodelling proteins (23 genes) | ||
| X58141 | P35611 | adducin 1 (alpha) |
| X58199 | P35612 | adducin 2 (beta) |
| M58018 | P12883 | myosin, heavy polypeptide 7, cardiac muscle, beta |
| Q14904 | ||
| Q16579 | ||
| M63603 | P26678 | Phospholamban |
| X92762 | Q16635 | tafazzin (cardiomyopathy, dilated 3A (X-linked); endocardial fibroelastosis 2; Barth syndrome) |
| X56134 | P08670 | vimentin |
| J03209 | P08254 P09238 | matrix metalloproteinase 3 (stromelysin 1, progelatinase) |
| D83646 | P51512 | matrix metalloproteinase 16 (membrane-inserted) |
| X75308 | P45452 | matrix metalloproteinase 13 (collagenase 3) |
| X07819 | P09237 | matrix metalloproteinase 7 (matrilysin, uterine) |
| J05070 | P14780 | matrix metalloproteinase 9 (gelatinase B, 92kD gelatinase, 92kD type IV collagenase) |
| X89576 | Q14850 | matrix metalloproteinase 17 (membrane-inserted) |
| L23808 | P39900 | matrix metalloproteinase 12 (macrophage elastase) |
| J05556 | P22894 | matrix metalloproteinase 8 (neutrophil collagenase) |
| J03210 | P08253 | matrix metalloproteinase 2 (gelatinase A, 72kD gelatinase, 72kD type IV collagenase) |
| X57766 | P24347 | matrix metalloproteinase 11 (stromelysin 3) |
| X03124 | P01033 | tissue inhibitor of metalloproteinase 1 (erythroid potentiating activity, collagenase inhibitor) |
| Q14252 | ||
| U14394 | P35625 | tissue inhibitor of metalloproteinase 3 (Sorsby fundus dystrophy, pseudoinflammatory) |
| U76456 | Q99727 | tissue inhibitor of metalloproteinase 4 |
| L00073 | P00797 | renin |
| J04144 | P12821 | angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 |
| L13977 | P42785 | prolylcarboxypeptidase (angiotensinase C) |
| K02566 | P01043 | kininogen |
Taking these data together, a well-coordinated programme of molecular events targeted in cellular death can be postulated considering the pathomechanisms of aortic valve calcification. However, before the end-point is reached when valve tissue is calcified, a long-time chronic process of degeneration occurs in the valve tissue.
Molecular attributes characteristic for the process of valvular degeneration
Altogether 99 genes have been reported earlier with the expression well detectable in calcified aortic valves (Table 2, [7]). Thereby, an expression level of 42 genes remains unaffected by the grade (calcified versus non-calcified) of degeneration severity such as albumin, specific receptors of oxidised low-density lipoprotein, advanced glycosylation end-products and natriuretic-peptide, potassium inwardly-rectifying channel-5, gap-junction proteins, particular integrins, tropins and cadherins [7]. However, the majority (57 proteins) detected was highly affected as a function of the degeneration grade: these are potassium voltage-gated channel-1, cardiotrophin, cardiac myosins, metalloproteinases, endothelins, neuropilins, caveolins, progesterone-, vasopressin-, tumour-necrosis-factor- and adrenergic-receptors. Moreover, whereas well-expressed hepatic lipase has been demonstrated in calcified valves, no traces of its expression could be detected in non-calcified tissue. Those gene products should be taken into account as the stage-specific targets in the cascade of cellular and molecular events that accompany chronic aortic degeneration for a predictive diagnosis and considering individualised therapeutic approaches.
Table 2.
Among 99 gene reported to be expressed at the transcriptional level in human calcified degenerated aortic valves, there are 57 gene products listed below the expression level of which is specifically altered as compared to non-calcified valves [7]
| GeneBank Accession / SwissProt Accession | Gene (protein) name / function | |
|---|---|---|
| Increased | ||
| M65199 | P20800 | endothelin 2 |
| L25615 | P37288 | arginine vasopressin receptor 1A |
| Z11687 | P30518 | arginine vasopressin receptor 2 (nephrogenic diabetes insipidus) |
| D31833 | P47901 | arginine vasopressin receptor 1B |
| L02911 | Q04771 | activin A receptor, type I |
| AF015257 | Q99527 | G protein-coupled receptor 30 |
| Q99981 | ||
| O00143 | ||
| Q13631 | ||
| L35545 | Q14218 | protein C receptor, endothelial (EPCR) |
| AJ002962 | O15540 | fatty acid binding protein 7, brain |
| O14951 | ||
| M86917 | P22059 | oxysterol binding protein |
| L06133 | Q04656 | ATPase, Cu++ transporting, alpha polypeptide (Menkes syndrome) |
| U50743 | P54710 | FXYD domain-containing ion transport regulator 2 |
| U89364 | P51787 | potassium voltage-gated channel, KQT-like subfamily, member 1 |
| Q92960 | ||
| M93718 | P29474 | nitric oxide synthase 3 (endothelial cell) |
| U05291 | Q06828 | fibromodulin |
| Q15331 | ||
| S73813 | P49961 | ectonucleoside triphosphate diphosphohydrolase 1 |
| M90657 | P30408 | transmembrane 4 superfamily member 1 |
| D26512 | P50281 | matrix metalloproteinase 14 (membrane-inserted) |
| S39329 | P20151 | kallikrein 2, prostatic |
| M13143 | P03952 | kallikrein B, plasma (Fletcher factor) 1 |
| J05262 | P14324 | farnesyl diphosphate synthase (farnesyl pyrophosphate synthetase, dimethylallyltranstransferase, geranyltranstransferase) |
| X68505 | Q02078 | MADS box transcription enhancer factor 2, polypeptide A (myocyte enhancer factor 2A) |
| Q14223 | ||
| Q14224 | ||
| X07228 | P78529 | lipase, hepatic |
| P11150 | ||
| Decreased | ||
| M21121 | P13501 | small inducible cytokine A5 (RANTES) |
| O43646 | ||
| M31210 | P21453 | endothelial differentiation, sphingolipid G-protein-coupled receptor, 1 |
| U03865 | P35368 | adrenergic, alpha-1B-, receptor |
| AF016098 | O60462 | neuropilin 2 |
| AF016050 | O14786 | neuropilin 1 |
| O60461 | ||
| U41070 | Q15722 | leukotriene b4 receptor (chemokine receptor-like 1) |
| Q13305 | ||
| Q92641 | ||
| U01839 | Q16570 | Duffy blood group |
| Q16300 | ||
| Y12711 | O00264 | progesterone receptor membrane component 1 |
| L49399 | Q13772 | nuclear receptor coactivator 4 |
| J04739 | P17213 | bactericidal/permeability-increasing protein |
| L27213 | P48751 | solute carrier family 4, anion exchanger, member 3 |
| M20747 | P14672 | solute carrier family 2 (facilitated glucose transporter), member 4 |
| X52882 | P17987 | t-complex 1 |
| Q15556 | ||
| Z18951 | Q03135 | caveolin 1, caveolae protein, 22kD |
| AF035752 | P51636 | caveolin 2 |
| AF043101 | P56539 | caveolin 3 |
| X60592 | P25942 | tumor necrosis factor receptor superfamily, member 5 |
| AB000895 | O15098 | protocadherin 16 dachsous-like (Drosophila) |
| AF047826 | O60574 | cadherin 19, type 2 |
| AF016272 | P75309 | cadherin 16, KSP-cadherin |
| AB006757 | O60247 | BH-protocadherin (brain-heart) |
| L34954 | P36382 | gap junction protein, alpha 5, 40kD (connexin 40) |
| X87241 | Q14517 | FAT tumor suppressor homolog 1 (Drosophila) |
| M14993 | P11171 | erythrocyte membrane protein band 4.1 (elliptocytosis 1, RH-linked) |
| U49837 | P50461 | cysteine and glycine-rich protein 3 (cardiac LIM protein) |
| U43030 | Q16619 | cardiotrophin 1 |
| M94547 | Q01449 | myosin light chain 2a |
| X84075 | Q14896 | myosin binding protein C, cardiac |
| D00943 | P13533 | myosin, heavy polypeptide 6, cardiac muscle, alpha (cardiomyopathy, hypertrophic 1) |
| Q13943 | ||
| Q14906 | ||
| M86406 | P35609 | actinin, alpha 2 |
| U02031 | Q12772 | sterol regulatory element binding transcription factor 2 |
| L10413 | P49354 | farnesyltransferase, CAAX box, alpha |
| Y08200 | Q92696 | Rab geranylgeranyltransferase, alpha subunit |
| Y12856 | O00286 | protein kinase, AMP-activated, alpha 1 catalytic subunit |
| U16660 | Q13011 | enoyl Coenzyme A hydratase 1, peroxisomal |
A functional link between cell cycle-control and calcification of aortic valves: potential diagnostic and prognostic targets
A proper control over cell-cycle progression seems to be a crucial step in the maintenance of a physiological cell population. Although cardiac cells undergo terminal differentiation soon after birth, irreversibly withdrawing from the cell-cycle, growth stimulation induces cell hypertrophy, the first visible step of a developing imbalance in the maintenance of the cardiac cell population. The hypertrophic growth has been shown to be associated with the re-activation of the fetal gene programme in cardiac cells – the key event is the positive regulation of a cell-cycle progression [13–15]. This switch in the programme seems to be crucial for myocardial cell regulation. Such growth stimulation is responsible for the up-regulated activity of cyclin-dependent kinases, CDKs, that consist of a kinase-core and an associated cyclin-subunit acting as the positive regulator [16]. In the matter, different CDK inhibitors keep a negative control over CDK activities. CDK inhibitors are classified on the basis of their sequence homology and substrate specificity. A cardiac helicase CHAMP was described as inhibiting cell proliferation and cardiac hypertrophy [13]. The CHAMP-dependent inhibition of cardiac hypertrophy is accompanied by the strictly programmed up-regulation of the cyclin-dependent protein-kinase inhibitor P21WAF1/CIP1, a 21-kDa protein and member of the CIP/KIP family [16]. Furthermore, the targeted over-expression of P21WAF1/CIP1 prevents cell enlargement and suppresses a specific gene expression of cardiac hypertrophy markers in the cell population in vitro [17] indicating the key role of p21WAF1/CIP1 in the regulation of the hypertrophic response.
The physiological expression of p21WAF1/CIP1 shows a gradual increase during development in both rat and man, becoming maximal in adulthood [18]. A direct link between the Bcl-2 dependent down-regulation of p21WAF1/CIP1 and an increased myocyte density in the left ventricle has been shown in experimental work with transgenic mice [19]. These findings are in agreement with those achieved by examination of human tissue: the coordinated down-regulation of both G1 and G2 checkpoint genes p21WAF1/CIP1 and 14-3-3-sigma, respectively, correlates well with increasing cardiac cell density and the calcification appearance of aortic valve tissue [20]. The coordinated suppression of checkpoint genes in calcified aortic valves at both transcription (A) and translation (B) levels is represented in Fig. 3 [21]. Both cellularity and number of macrophages are significantly increased in calcified tissue (see Fig. 3c, d, respectively) [21]. According to the monitored CD68 positive signals, macrophages are localised predominantly in the sub-endothelial layer of the valvular fibrosa, whereas 14-3-3-sigma and p21WAF1/CIP1 can be observed in both sub-endothelial layer and valvular interstitium of non-calcified tissue, being mainly co-localised with alpha-actin in the valvular spongiosa and pointing to the target expression in myofibroblasts. There is a growing body of evidence that in response to stimulus/injury the heart valves undergo tissue remodelling including phenotypic modulation and transformation of fibroblast-like into myofibroblast-like cells [22]. Therefore, the target protein expression of 14-3-3-sigma and p21WAF1/CIP1 observed in degenerated valvular tissue, can originate predominantly from myofibroblasts.
Fig. 3.
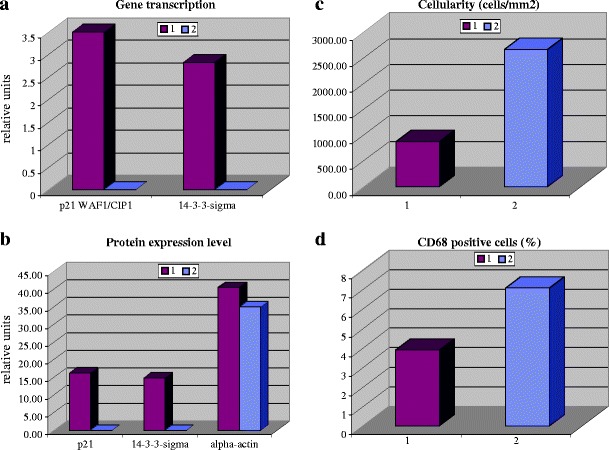
Comparative analysis in two groups of patients with non-calcified (1) versus calcified (2) degenerative aortic valves. All analyses have been performed as described earlier [20]. The corresponding mean values are presented with statistically significant differences between the groups of comparison. (a) Comparative gene expression analysis (mRNA level in relative units) of p21WAF1/CIP1 and 14-3-3-sigma. Quantitative Real-Time-PCR was applied. Beta-actin was used as the house-keeping gene for normalisation of corresponding values of the target gene expression rates. (b) Comparative analysis of protein expression levels (in relative units) of P21WAF1/CIP1, 14-3-3-sigma and alpha-actin. (c) Comparative analysis of cellular density (in relative units). (d) Comparative analysis of macrophages (in relative units)
Moreover, both the increased cell density and coordinated down-regulation of p21WAF1/CIP1 and 14-3-3-sigma gene expression were found to be characteristic for calcification, in contrast to non-calcified valvular tissue [23]. Therefore, the double-control via both check-point proteins over DNA quality and cell proliferation in valvular cells might be efficient only in non-calcified tissue, whereas in the calcifying one this function is getting suppressed at both G1 and G2 phases of cell-cycle. These findings give further evidence that the efficiency of cell-cycle control in human non-calcified valvular tissue depends not only on the positive/negative CDK regulation in the G1 phase but also on the coordinated regulation of both G1 and G2 dependent checkpoints. Further in vitro experiments on rat cardiac fibroblasts showed that a target up-regulation of inhibitors for G1 dependent CDKs effectively suppresses the DNA synthesis and may decrease a potential risk of cardiovascular diseases [23].
The dissociation of P21WAF1/CIP1 from the CDK complexes correlates well with the activation of CDK2, CDK4, CDK6, and the release from cell-cycle arrest, whereby the number of cardiac cells in S phase rises considerably [24]. Further, in contrast to P16 (a specific inhibitor of CDK4/6), the “universal” CDK inhibitor P21WAF1/CIP1 was shown to be able to block completely an E2F-1-induced G1 exit [25]. However, E1A binding activity to target protein complexes has effects on the cell-cycle progression beyond those produced by E2F-1 alone and can drive S-phase entry that is resistant to P21WAF1/CIP1 [24]. These facts explain the necessity of the coordinated regulation of both G1 and G2 dependent checkpoints, in order to keep the control over the cell population maintenance in cardiac tissue.
Pronounced up-regulation of both genes in non-calcified in contrast to their down-regulation in calcified degenerated valvular tissue indicates the central regulatory role of checkpoint genes in keeping functional the valvular cells. Blockade of cell-cycle progression results in a prolonged resistance to macrophage invasion and foam cell deposition [26]. Therefore, it is likely that reduced cell-cycle control in valvular tissue leads to the increased macrophage invasion that, in turn, can contribute to non-physiological calcification by both triggered unspecific inflammation and NO-toxicity [27–31]. Taken together, the coordinated activation of both G1 and G2 dependent checkpoint genes may be an attribute of the valvular tissue resistance against the calcification processes. These data should be taken into consideration to design novel therapeutic approaches targeted at pro-calcification mechanisms in the heart.
Risk assessment: factors involved in degenerative valve disease
Recent studies demonstrate an association between atherosclerosis and AS. Traditional cardiovascular risk factors such as lipid disorders, diabetes, arterial hypertension, smoking and male gender [32, 33] are reported to increase also the incidence of AS. At least one of these factors or, more frequently, even the combination of them is usually observed in this cohort of patients [20]. Although advanced age is the main risk factor, worldwide statistics indicate that degenerative aortic valve disease (DAVD) cannot be explained by ageing alone. No longer considered as a natural consequence of ageing, DAVD is the result of actively driven pathological processes including programmed (de)regulation of target genes, metabolic alterations, inflammatory cell infiltration, subcellular disruption, and consequent tissue degeneration, calcification and remodelling [20]. Due to extremely high morbidity and mortality caused by DAVD particularly in Western world, the central question has to be answered: Is an individual predisposition to the disease predictable? From this viewpoint a clear definition of disease specific risk factors is of particular interest.
Although the causal mechanisms are still largely unclear, all molecular as well as cellular processes attributed to DAVD are generally triggered secondarily to a central metabolic failure (diabetes, hypercholesterolemia, hypercalcaemia, leanness), hormonal deregulation (hyperparathyroidism), hypertension, and extreme stress conditions such as tobacco use and environmental stress factors [34–37]. Thus, an inverse relationship was demonstrated between body mass index and DAVD incidence: calcific changes were more frequently observed in lean people even independently of the risk factor of age, and, therefore, cannot be explained by leanness frequently observed in patients with highly advanced age. These facts indicate, further, an association of DAVD with metabolic disorders causing weight loss such as osteoporosis [36].
In diabetes, an increased production of highly aggressive reactive oxygen species (ROS) under hyperglycaemic conditions is considered as the main trigger for severe, chronic complications such as DAVD. Moreover, using advanced biomedical technologies such as clinical proteomics, individual stress reactions and resulting complications can be quite precisely predicted; disease specific molecular markers are already close to their clinical application specifically for the diabetic complication [38]. Similarly to diabetic patients, smokers also suffer from highly increased ROS production leading to enhanced incidence of DAVD, although specific pathomechanisms deserve further clarification. Deregulation of angiotensin-II metabolism and activity of angiotensin-specific receptors is considered to be the key molecule in the pathomechanisms that underlie DAVD in hypertension [37, 39, 40].
Individualised treatment of aortic stenosis and prognosis
A large body of evidence indicates that aortic stenosis is an active process with a distinctive histological appearance, associated clinical factors, and, variable disease progression proposing that this disease may be amenable in terms of the variety of risk factors but also successful treatments by individualised therapeutic approaches to prevent or at least slow down the disease progression [41, 42]. Indeed, several retrospective studies have consistently demonstrated that statin-based treatments are associated with notably lower haemodynamic progression of aortic stenosis [43–46]; however, statins failed in the prospective SALTIRE trial. It was suggested that the beneficial effects by statin are independent of lowering cholesterol impacts [43, 44]. Interestingly, both CRP expression at the valvular tissue level and serum CRP levels were found to be significantly lower under statin-based treatments [47] suggesting its pleiotropic and/or anti-inflammatory properties. As demonstrated by several independent studies (SALTIRE, SEAS, ASTRONOMER) lowering LDL-cholesterol levels do not halt the progression of aortic stenosis in patients with mild to moderate aortic-valve disease [48, 49]. The fact that angiotensin converting enzyme (ACE) and angiotensin II can be found in sclerotic but not in normal aortic valves indicates an important role of the renin-angiotensin system (RAS) in the pathogenesis of AS [50]. Further, the RAS has already been shown to play an important role in atherosclerosis. Consequently, ACE inhibitors slow down the calcium accumulation in aortic valves [43]. However, studies evaluating the effects of ACE inhibitors [46] and angiotensin II type 1 receptor blockers [51] did not find any difference in haemodynamic progression of AS in untreated patients versus patients who were taking these drugs.
In conclusion, it is too early for recommendations in terms of prevention of AS progression by currently applied treatments: further studies are highly desired. The recommended approach to treat the symptomatic, advanced AS remains the prosthetic valve replacement. Moreover, there is a clear consensus that urgent valve replacement is required for symptomatic AS, while the management of asymptomatic patients with severe AS is still controversially discussed. In the matter, inhibitors of angiotensin-converting enzyme are currently under extensive consideration for their therapeutic application to effectively prevent both hypertension and DAVD [37, 39, 43, 52]. Independently from individual risk factors, the crucial role of metalloproteinases in the central pathomechanisms of the progressive tissue remodelling during the chronic development of DAVD is well recognised [20, 53]. Novel therapeutic interventions consider, therefore, metalloproteinases as the preferred target to delay or even prevent the progression of DAVD [37].
Aortic valve replacement: risk factors, geometry remodelling, complications
Dysfunction and bioactivity of implanted bioprostheses
Twenty percent to thirty percent of implanted bioprostheses show dysfunction after about 10 years post-implantation. Recent reports predict that a greater than 50% incidence of failure will be seen in bioprostheses at 12–15 years [54]. In addition, risk factors of atherosclerosis as well as chronic renal disease and parathyroid tumours might play a substantial role in the degeneration of bioprostheses. In order to improve the quality of life after cardiac valvular surgery, innovative procedures and new generations of prostheses have been developed in the past decade. The most frequently used porcine bioprostheses have been demonstrated to be bioactive in the human organism. DNA and RNA analysis of non-implanted bioprostheses before aortic valve replacement (AVR) has revealed sequences able to hybridise to as many as 112 human genes/transcripts relevant to cardiovascular pathologies [7]. Among those genes there are several overlapping sequences, the expression of which strictly depends on the grade of degeneration: endothelins, sodium / calcium exchangers, potassium voltage-gated channel-1, metalloproteinases, vasopressin- and adrenergic-receptors. Altogether, there are 74 genes found to be specifically altered by expression in human calcified degenerated aortic valves as summarised in Table 3.
Table 3.
DNA and RNA analysis of porcine bioprosthetic material before the aortic valve replacement revealed sequences able to hybridise to 74 human genes/transcripts, the expression of which is altered in human calcified degenerative aortic valves [7]
| GeneBank Accession / SwissProt Accession | Gene (protein) name / function | |
|---|---|---|
| M65199 | P20800 | endothelin 2 |
| M18185 | P09681 | gastric inhibitory polypeptide |
| AB010710 | P78380 | oxidised low density lipoprotein (lectin-like) receptor 1 |
| L25615 | P37288 | arginine vasopressin receptor 1A |
| Z11687 | P30518 | arginine vasopressin receptor 2 (nephrogenic diabetes insipidus) |
| D31833 | P47901 | arginine vasopressin receptor 1B |
| M31210 | P21453 | endothelial differentiation, sphingolipid G-protein-coupled receptor, 1 |
| U03865 | P35368 | adrenergic, alpha-1B-, receptor |
| L13436 | P20594 | natriuretic peptide receptor B/guanylate cyclase B (atrionatriuretic peptide receptor B) |
| X52282 | P17342 | natriuretic peptide receptor C/guanylate cyclase C (atrionatriuretic peptide receptor C) |
| L02911 | Q04771 | activin A receptor, type I |
| AF015257 | Q99527 | G protein-coupled receptor 30 |
| Q99981 | ||
| O00143 | ||
| Q13631 | ||
| Y10659 | P78552 | interleukin 13 receptor, alpha 1 |
| Q99656 | ||
| O95646 | ||
| M91211 | Q15109 | advanced glycosylation end product-specific receptor |
| Q15279 | ||
| L35545 | Q14218 | protein C receptor, endothelial (EPCR) |
| AF016050 | O14786 | neuropilin 1 |
| O60461 | ||
| U41070 | Q15722 | leukotriene b4 receptor (chemokine receptor-like 1) |
| Q13305 | ||
| Q92641 | ||
| AJ002962 | O15540 | fatty acid binding protein 7, brain |
| O14951 | ||
| M86917 | P22059 | oxysterol binding protein |
| S73197 | P41181 | aquaporin 2 (collecting duct) |
| L27213 | P48751 | solute carrier family 4, anion exchanger, member 3 |
| U89364 | P51787 | potassium voltage-gated channel, KQT-like subfamily, member 1 |
| Q92960 | ||
| M20747 | P14672 | solute carrier family 2 (facilitated glucose transporter), member 4 |
| U39195 | P48544 | potassium inwardly-rectifying channel, subfamily J, member 5 |
| Q92807 | ||
| M91368 | P32418 | solute carrier family 8 (sodium/calcium exchanger), member 1 |
| M23234 | P21439 | ATP-binding cassette, sub-family B (MDR/TAP), member 4 |
| J04456 | P09382 | lectin, galactoside-binding, soluble, 1 (galectin 1) |
| M93718 | P29474 | nitric oxide synthase 3 (endothelial cell) |
| X52882 | P17987 | t-complex 1 |
| Q15556 | ||
| X65784 | Q04762 | cell matrix adhesion regulator |
| U05291 | Q06828 | fibromodulin |
| Q15331 | ||
| M58664 | P25063 | CD24 antigen (small cell lung carcinoma cluster 4 antigen) |
| S57235 | P34810 | CD68 antigen |
| U85611 | Q99828 | calcium and integrin binding 1 (calmyrin) |
| Z34974 | Q15152 | plakophilin 1 (ectodermal dysplasia/skin fragility syndrome) |
| O00645 | ||
| U49240 | Q92797 | symplekin; Huntingtin interacting protein I |
| O00733 | ||
| O00689 | ||
| AB000897 | O15100 | protocadherin gamma subfamily A, 12 |
| AF047826 | O60574 | cadherin 19, type 2 |
| U07969 | Q12864 | cadherin 17, LI cadherin (liver-intestine) |
| U59325 | Q13634 | cadherin 18, type 2 |
| X52947 | P17302 | gap junction protein, alpha 1, 43kD (connexin 43) |
| M96789 | P35212 | gap junction protein, alpha 4, 37kD (connexin 37) |
| L34954 | P36382 | gap junction protein, alpha 5, 40kD (connexin 40) |
| U03493 | P36383 | gap junction protein, alpha 7, 45kD (connexin 45) |
| U34802 | P48165 | gap junction protein, alpha 8, 50kD (connexin 50) |
| X04325 | P08034 | gap junction protein, beta 1, 32kD (connexin 32, Charcot-Marie-Tooth neuropathy, X-linked) |
| M86849 | P29033 | gap junction protein, beta 2, 26kD (connexin 26) |
| X53416 | P21333 | filamin A, alpha (actin binding protein 280) |
| S73813 | P49961 | ectonucleoside triphosphate diphosphohydrolase 1 |
| M90657 | P30408 | transmembrane 4 superfamily member 1 |
| X82157 | Q14515 | SPARC-like 1 (mast9, hevin) |
| X87241 | Q14517 | FAT tumor suppressor homolog 1 (Drosophila) |
| Y00796 | P20701 | integrin, alpha L (antigen CD11A (p180), lymphocyte function-associated antigen 1; alpha polypeptide) |
| U81984 | Q99814 | endothelial PAS domain protein 1 |
| Q99630 | ||
| X07897 | P02590 P04463 | troponin C, slow |
| S64668 | P45379 | troponin T2, cardiac |
| Q99596 | ||
| M14993 | P11171 | erythrocyte membrane protein band 4.1 (elliptocytosis 1, RH-linked) |
| M95627 | Q13685 | angio-associated, migratory cell protein |
| U49837 | P50461 | cysteine and glycine-rich protein 3 (cardiac LIM protein) |
| U43030 | Q16619 | cardiotrophin 1 |
| M86406 | P35609 | actinin, alpha 2 |
| D26512 | P50281 | matrix metalloproteinase 14 (membrane-inserted) |
| S39329 | P20151 | kallikrein 2, prostatic |
| M13143 | P03952 | kallikrein B, plasma (Fletcher factor) 1 |
| L19684 | P29622 | serine (or cysteine) proteinase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 4 |
| X14329 | P15169 | carboxypeptidase N, polypeptide 1, 50kD |
| M32313 | P18405 | steroid-5-alpha-reductase, alpha polypeptide 1 (3-oxo-5 alpha-steroid delta 4-dehydrogenase alpha 1) |
| U16660 | Q13011 | enoyl Coenzyme A hydratase 1, peroxisomal |
| X07228 | P78529 | lipase, hepatic |
| P11150 | ||
| U22662 | Q13133 | nuclear receptor subfamily 1, group H, member 3 |
| X02750 | Q16001 | protein C (inactivator of coagulation factors Va and VIIIa) |
| Q15190 | ||
| Q15189 | ||
| P04070 | ||
| M11723 | P00748 | coagulation factor XII (Hageman factor) |
| X68505 | Q02078 | MADS box transcription enhancer factor 2, polypeptide A (myocyte enhancer factor 2A) |
| Q14223 | ||
| Q14224 | ||
Currently, poor information is available concerning the bioactivity of prosthetic material when they are implanted in human valves. In vivo-hybridisation to human nucleic acids might be one feasible reason for several well-known complications triggered by implantation. Thus, worldwide statistics indicate that each kind of AVR is not rarely followed by different metabolic impairments and physiological complications such as progressively abnormal lipid profiles, a non-specific inflammation, blood trauma, haemorheologic changes or severe congestive heart failure and even death during individually long postoperative time [55–61]. After AVR, the wall thickness becomes significantly greater than normal for patients with aortic stenosis, and after 5 years of follow-up the remodelling of the left ventricular geometry is usually observed after AVR [62].
Tissue remodelling of replaced valves: matrix metalloproteinses as biomarkers and potential therapeutic targets
Matrix metalloproteinases (MMPs) play the key role in tissue remodelling under both physiological and pathological conditions. MMPs are produced as zymogens (pro-MMPs) that require proteolytic activation through the elimination of the N-terminal propeptide via membrane type-matrix metalloproteinase (MT-MMPs) activity. Tissue inhibitors of metalloproteinases (TIMPs) act to inhibit metalloproteinase activity by forming a non-covalent irreversible complex with MMPs. A shifted balance in resulting MMPs / TIMPs activity is well documented under stress conditions [58].
However, less is known about a regulation of ECM degrading enzymes in native degenerating aortic valves and in valvular tissue after replacement. Aortic valves tissue is characterised by considerable heterogeneity of the cellular population: endocardial, interstitial, smooth muscle cells as well as fibroblasts and myofibroblasts have been identified in highly sophisticated dynamic structures of cardiac valves [63]. The ECM is thought to be an integral component of this coordinated dynamism [64]. The cores of activated ECM degrading genes differ both qualitatively and quantitatively at each stage of valvular degeneration; after AVR it is regulated in a different manner [36]. The activation grade of the MMP cores is found to be specific for each stages of the valve degeneration: whereas MMP-9 activation differs quantitatively, an activation of MMP-2 was observed solely at the earliest stages of degenerative process [53, 65]. In contrast, the stage of progressive calcification is characterised by dropping of the ECM-degradation potential. Therefore, the highly activated ECM-degradation potential might be considered as an early marker for the triggered degeneration of valvular tissue. Consequently, ex vivo evaluation of the dynamic in the ECM-degradation potential, e.g. measured by comparative zymography in blood samples, seems to be of great prognostic value [66].
This is of note that the set-up of ECM-degrading enzymatic-core changes dramatically after AVR: in contrast to the expression rates well-detectable in native valvular tissue, neither MMP-2 expression nor this of MMP-9 was detected in the replaced tissue. In addition, TIMP-1 was shown to be activated in the valves after replacement. TIMP-1 represents the very last step in the negative regulation of collagenases, stromelysinases, and gelatinases [67, 68] and has been found to be highly expressed in actively resorbing tissue [69]. Also, the key-role is considered for MT1-MMP as a matrix degrading protease, specifically in geometry remodelling after AVR, and opens good perspectives for new targeted therapy approaches, in order to avoid the most common metabolic impairments and clinical complications well-known to be frequently developed by the patients after AVR [53].
Acute aortic insufficiency is a frequent complication after AVR: risk assessment
Besides cases with an acute injury, e.g. aortic dissection and thoracic injury, the main aetiologies of the progressive insufficiency are bioprosthesis degeneration and infectious endocarditis [70, 71]. In order to forestall a dysfunction of degenerating bioprostheses, patients without diagnosed risk factors undergo, on average, a re-operation 9–10 years after AVR. Against this, the period of time can be more than halved for patients demonstrating at least two of following risk factors: smoking, Diabetes mellitus, risk by gender (females), high cholesterol level [72]. Furthermore, these risk factors have a higher impact in bioprosthesis degeneration for younger patients than for the elderly. Therefore, targeted preventive measures such as proper (pre)diabetes care would be highly beneficial, in particular for subpopulations of young female diabetes-predisposed AVR-patients.
Diabetes mellitus as the risk factor for infectious endocarditis, accelerated valvular degeneration, dysfunction of bioprostheses valves and progressive aortic insufficiency
Diabetes mellitus is a well-acknowledged risk factor for progressive aortic insufficiency, accelerated degeneration of both native and prosthetic valves as well as infectious endocarditis [72–75]. Studies focused on the aetiology and prevalence of the latter demonstrated diabetic patients to be particularly predisposed (a relative increase of 40% compared to the general population) to infectious endocarditis mainly due to following reasons:
patients with DM are at highly increased risk of infections
most patients with infectious endocarditis have a history of pre-existing heart valve lesions, which DM patients are significantly predisposed to [73, 76].
Although, both causes are considered as independent risk factors for infectious endocarditis prevalence in DM [75], the synergistic effects can lead to a “vicious circle” in further progression of infectious endocarditis, heart valve lesions/degeneration and vulnerability of DM patients for infections (see Fig. 4) [21]. Due to a high symptomatic heterogeneity of the diabetic population, the better defined “metabolic syndrome” as a cluster of atherogenic, inflammatory, and atherothrombotic abnormalities linked to abdominal obesity and insulin resistance has been demonstrated to be a particularly strong independent predictor for poor prognosis in both degenerative valve disease and accelerated degeneration of bioprosthetic valves [73, 77]. The pro-atherogenic and pro-inflammatory pathomechanisms have been proposed to underlie the degenerative valvular processes, since statins-based treatment approaches are known to slow down the progression of valvular degeneration [73, 74, 78]. Identification of metabolic syndrome characteristic factors responsible for structural failure of a bioprosthesis is necessary for a development of individualised target-specific therapy approaches avoiding the need for re-operation after AVR. Improved (pre)Diabetes care is currently discussed as being one of the highest priorities of desirable healthcare worldwide [79–82].
Fig. 4.
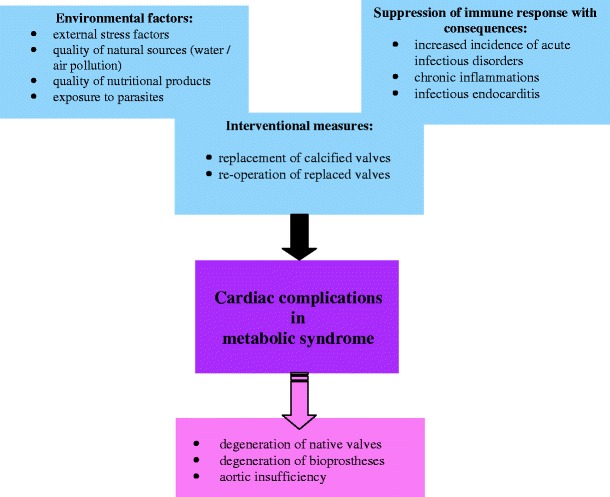
Various factors, burden and pathologic processes, contributing to cardiac complications in metabolic syndrome [20]. The crucial role of environmental factors as increasing the overall risk is discussed in our previous reviews [7, 15, 21 38]
Concluding remarks and Outlook
There is a long period of time during which patients predisposed to valvular degeneration remain asymptomatic. In this period a pathology progression can and must be detected followed by targeted therapeutic measures. Molecular attributes characteristic for early stages of valvular degeneration represent reliable predictive biomarkers and – at the same time – the targets for more effective individualised treatment approaches before the pathology is clinically manifested. Risk factors should be considered individually. The characteristic molecular signature is one of them.
Besides several kinds of acute injury (aortic dissection, thoracic injury) the main aetiology of the aortic insufficiency in patients after AVR is a bioprosthesis dysfunction and infectious endocarditis. On average, patients without diagnosed risk factors undergo a re-operation 9–10 years after AVR. Against this, the period of time can be more than halved for patients demonstrating at least two of following risk factors: smoking, Diabetes mellitus, risk by gender (females), high cholesterol levels. Therefore, individualised targeted measures would be highly effective in prevention of AVD and re-operation after AVR. Pathology- and stage-specific molecular patterns should be taken into consideration for the reliable prediction, individualised treatment algorithms and correct prognosis.
References
- 1.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111:3316–26. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 2.Baumgartner H. Aortic stenosis: medical and surgical management. Heart. 2005;91:1483–8. doi: 10.1136/hrt.2004.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faggiano P, Antonini-Canterin F, Erlicher A, Romeo C, Cervesato E, Pavan D, Piazza R, Huang G, Nicolosi GL. Progression of aortic valve sclerosis to aortic stenosis. Am J Cardiol. 2003;91:99–101. doi: 10.1016/S0002-9149(02)03011-4. [DOI] [PubMed] [Google Scholar]
- 4.Rosenhek R, Maurer G, Baumgartner H. Should early elective surgery be performed in patients with severe but asymptomatic aortic stenosis? Eur Heart J. 2002;23:1417–21. doi: 10.1053/euhj.2002.3163. [DOI] [PubMed] [Google Scholar]
- 5.Bonow RO, Carabello B, Leon AC, Edmunds LH, Fedderly BJ, Freed MD, Gaasch WH, McKay CR, Nishimura RA, O’Gara PT, O’Rourke RA, Rahimtoola SH, Ritchie JL, Cheitlin MD, Eagle KA, Gardner TJ, Garson A, Jr, Gibbons RJ, O’Rourke RA, Russell RO, Ryan TJ, Smith SC., Jr ACC/AHA guidelines for the management of patients with valvular heart disease. A report of the American College of Cardiology/American Heart Association task force on practice guidelines (committee on management of patients with valvular heart disease) J Am Coll Cardiol. 1998;32:1486–588. doi: 10.1016/S0735-1097(98)00454-9. [DOI] [PubMed] [Google Scholar]
- 6.Srivatsa SS, Harrity PJ, Maercklein PB, Kleppe L, Veinot J, Edwards WD, Johnson CM, Fitzpatrick LA. Increased cellular expression of matrix proteins that regulate mineralization is associated with calcification of native human and porcine xenograft bioprosthetic heart valves. J Clin Invest. 1997;99:996–1009. doi: 10.1172/JCI119265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeghiazaryan K, Skowasch D, Bauriedel G, Schild HH, Golubnitschaja O. Prediction of degeneration of native and bioprosthetic aortic valves. In: Golubnitschaja O, editor. Predictive Diagnostics & Personalized Treatment: Dream or Reality. New York: Nova Science Publishers; 2009. pp. 73–101. [Google Scholar]
- 8.Kim KM. Apoptosis and calcification. Scanning Microsc. 1995;9:1137–78. [PubMed] [Google Scholar]
- 9.Skowasch D, Schrempf S, Wernert N, Steinmetz M, Jabs A, Tuleta I, Welsch U, Preusse CJ, Likungu JA, Welz A, Luderitz B, Bauriedel G. Cells of primarily extra-valvular origin in degenerative aortic valves and bioprostheses. Eur Heart J. 2005;26:2576–80. doi: 10.1093/eurheartj/ehi458. [DOI] [PubMed] [Google Scholar]
- 10.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–9. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cipollone F, Prontera C, Pini B, Marini M, Fazia M, Cesare D, Iezzi A, Ucchino S, Boccoli G, Saba V, Chiarelli F, Cuccurullo F, Mezzetti A. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation. 2001;104:921–7. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- 12.Jian B, Jones PL, Li Q, Mohler ER, 3rd, Schoen FJ, Levy RJ. Matrix metalloproteinase-2 is associated with tenascin-C in calcific aortic stenosis. Am J Pathol. 2001;159:321–7. doi: 10.1016/S0002-9440(10)61698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu ZP, Olson EN. Suppression of proliferation and cardiomyocyte hypertrophy by CHAMP, a cardiac-specific RNA helicase. Proc Natl Acad Sci USA. 2002;99:2043–8. doi: 10.1073/pnas.261708699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bär MDH, Kreuzer J, Cojoc A, Jahn L. Upregulation of embryonic transcription factors in right ventricular hypertrophy. Basic Res Cardiol. 2003;98:285–94. doi: 10.1007/s00395-003-0410-2. [DOI] [PubMed] [Google Scholar]
- 15.Golubnitschaja O. Cell cycle checkpoints: the role and evaluation for early diagnosis of senescence, cardiovascular, cancer, and neurodegenerative diseases. Amino Acids. 2007;32:359–71. doi: 10.1007/s00726-006-0473-0. [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 17.Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda MA. Essential roles for G1 cyclin-dependent kinase activity in development of cardiomyocyte hypertrophy. Am J Physiol. 1998;275:H2036–40. doi: 10.1152/ajpheart.1998.275.6.H2036. [DOI] [PubMed] [Google Scholar]
- 18.Burton PB, Yacoub MH, Barton PJ. Cyclin-dependent kinase inhibitor expression in human heart failure. A comparison with fetal development. Eur Heart J. 1999;20:604–11. doi: 10.1053/euhj.1998.1231. [DOI] [PubMed] [Google Scholar]
- 19.Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, Nadal-Ginard B, Izumo S, Anversa P. bcl-2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci USA. 2002;99:6257–62. doi: 10.1073/pnas.092672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golubnitschaja O, Yeghiazaryan K, Skowasch D, Schild H, Bauriedel G. p21WAF1/CIP1 and 14-3-3 sigma gene expression in degenerated aortic valves: a link between cell cycle checkpoints and calcification. Amino Acids. 2006;31:309–16. doi: 10.1007/s00726-006-0365-3. [DOI] [PubMed] [Google Scholar]
- 21.Yeghiazaryan K, Bauriedel G, Schild HH, Golubnitschaja O. Prediction of degeneration of native and bioprosthetic aortic valves: issue-related particularities of diabetes mellitus. Infect Disord Drug Targets. 2008;8:88–99. doi: 10.2174/187152608784746547. [DOI] [PubMed] [Google Scholar]
- 22.Schoen FJ. Cardiac valves and valvular pathology: update on function, disease, repair, and replacement. Cardiovasc Pathol. 2005;14:189–94. doi: 10.1016/j.carpath.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Mercier I, Colombo F, Mader S, Calderone A. Ovarian hormones induce TGF-beta(3) and fibronectin mRNAs but exhibit a disparate action on cardiac fibroblast proliferation. Cardiovasc Res. 2002;53:728–39. doi: 10.1016/S0008-6363(01)00525-9. [DOI] [PubMed] [Google Scholar]
- 24.Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin-dependent kinases in the presence of insulin-like growth factor I. Circ Res. 1999;85:128–32. doi: 10.1161/01.res.85.2.128. [DOI] [PubMed] [Google Scholar]
- 25.Akli S, Zhan S, Abdellatif M, Schneider MD. E1A can provoke G1 exit that is refractory to p21 and independent of activating cdk2. Circ Res. 1999;85:319–28. doi: 10.1161/01.res.85.4.319. [DOI] [PubMed] [Google Scholar]
- 26.Mann MJ, Gibbons GH, Tsao PS, Leyen HE, Cooke JP, Buitrago R, Kernoff R, Dzau VJ. Cell cycle inhibition preserves endothelial function in genetically engineered rabbit vein grafts. J Clin Invest. 1997;99:1295–301. doi: 10.1172/JCI119288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt HH, Walter U. NO at work. Cell. 1994;78:919–25. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang JC, Wogan GN. Growth and viability of macrophages continuously stimulated to produce nitric oxide. Proc Natl Acad Sci USA. 1997;94:11875–80. doi: 10.1073/pnas.94.22.11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutgens E, Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–9. doi: 10.1016/S0008-6363(98)00311-3. [DOI] [PubMed] [Google Scholar]
- 30.Sanders DB, Hunter K, Wu Y, Jablonowski C, Bahl JJ, Larson DF. Modulation of the inflammatory response in the cardiomyocyte and macrophage. J Extra-Corpor Technol. 2001;33:167–74. [PubMed] [Google Scholar]
- 31.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/S0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 32.Rajamannan NM, Gersh B, Bonow RO. Calcific aortic stenosis: from bench to the bedside-emerging clinical and cellular concepts. Heart. 2003;89:801–5. doi: 10.1136/heart.89.7.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohler ER., III Mechanisms of aortic valve calcification. Am J Cardiol. 2004;94:1396–402. doi: 10.1016/j.amjcard.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 34.Roberts WC, Waller BF. Effect of chronic hypercalcemia on the heart. An analysis of 18 necropsy patients. Am J Med. 1981;71:371–84. doi: 10.1016/0002-9343(81)90163-7. [DOI] [PubMed] [Google Scholar]
- 35.Niederle B, Stefenelli T, Glogar D, Woloszczuk W, Roka R, Mayr H. Cardiac calcific deposits in patients with primary hyperparathyroidism: preliminary results of a prospective echocardiographic study. Surgery. 1990;108:1052–7. [PubMed] [Google Scholar]
- 36.Lindroos M, Kupari M, Valvanne J, Strandberg T, Heikkilä J, Tilvis R. Factors associated with calcific aortic valve degeneration in the elderly. Eur Heart J. 1994;15:865–70. doi: 10.1093/oxfordjournals.eurheartj.a060602. [DOI] [PubMed] [Google Scholar]
- 37.Goldbarg SH, Elmariah S, Miller MA, Fuster V. Insights into degenerative aortic valve disease. J Am Coll Cardiol. 2007;50:1205–13. doi: 10.1016/j.jacc.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 38.Golubnitschaja O. Clinical proteomics in application to predictive diagnostics and personalized treatment of diabetic patients. Curr Proteomics. 2008;5:35–44. doi: 10.2174/157016408783955092. [DOI] [Google Scholar]
- 39.Couet J, Gaudreau M, Lachance D, Plante E, Roussel E, Drolet MC, Arsenault M. Treatment of combined aortic regurgitation and systemic hypertension: Insights from an animal model study. Am J Hypertens. 2006;19:843–50. doi: 10.1016/j.amjhyper.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 40.Orlowska-Baranowska E, Placha G, Baranowski R, Michalek P, Gora J, Gaciong Z, Stepinska J. Can angiotensin II +1675 G/A type 2 receptor gene polymorphism be a marker of left ventricular hypertrophy in patients with aortic stenosis? J Heart Valve Dis. 2007;16:495–503. [PubMed] [Google Scholar]
- 41.Rajamannan NM, Otto CM. Targeted therapy to prevent progression of calcific aortic stenosis. Circulation. 2004;110:1180–2. doi: 10.1161/01.CIR.0000140722.85490.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liebe V, Brueckmann M, Borggrefe M, Kaden JJ. Statin therapy of calcific aortic stenosis: hype or hope? Eur Heart J. 2006;27:773–8. doi: 10.1093/eurheartj/ehi697. [DOI] [PubMed] [Google Scholar]
- 43.O'Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, Wu AH, Zhao XQ, Budoff MJ. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–62. doi: 10.1001/archinte.165.8.858. [DOI] [PubMed] [Google Scholar]
- 44.Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001;104:2205–9. doi: 10.1161/hc4301.098249. [DOI] [PubMed] [Google Scholar]
- 45.Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol. 2002;40:1723–30. doi: 10.1016/S0735-1097(02)02496-8. [DOI] [PubMed] [Google Scholar]
- 46.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–5. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- 47.Skowasch D, Schrempf S, Preusse CJ, Likungu JA, Welz A, Lüderitz B, Bauriedel G. Tissue resident C reactive protein in degenerative aortic valves: correlation with serum C reactive protein concentrations and modification by statins. Heart. 2006;92:495–8. doi: 10.1136/hrt.2005.069815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan KL, Teo K, Dumesnil JG, Ni A. ASTRONOMER Investigators. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis. Circulation. 2010;121:306–14. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 49.Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Bärwolf C, Holme I, Kesäniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K. SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–56. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 50.O'Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, Probstfield JL. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2006;106:2224. doi: 10.1161/01.CIR.0000035655.45453.D2. [DOI] [PubMed] [Google Scholar]
- 51.Olsen MH, Wachtell K, Bella JN, Liu JE, Boman K, Gerdts E, Papademetriou V, Nieminen MS, Rokkedal J, Dahlöf B, Devereux RB. Effect of losartan versus atenolol on aortic valve sclerosis (a LIFE substudy) Am J Cardiol. 2004;94:1076–80. doi: 10.1016/j.amjcard.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 52.Lorell BH. Role of angiotensin AT1, and AT2 receptors in cardiac hypertrophy and disease. Am J Cardiol. 1999;83:48H–52. doi: 10.1016/S0002-9149(99)00258-1. [DOI] [PubMed] [Google Scholar]
- 53.Yeghiazaryan K, Skowasch D, Bauriedel G, Schild H, Golubnitschaja O. Could activated tissue remodeling be considered as early marker for progressive valve degeneration? Comparative analysis of checkpoint and ECM remodeling gene expression in native degenerating aortic valves and after bioprosthetic replacement. Amino Acids. 2007;32:109–14. doi: 10.1007/s00726-006-0376-0. [DOI] [PubMed] [Google Scholar]
- 54.Schoen FJ, Levy RJ. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg. 2005;79:1072–80. doi: 10.1016/j.athoracsur.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 55.Hattori T, Sakai A, Lhashi K, Kasahara S, Oosawa M. Mitral and aortic valve replacement associated with aortitis syndrome: a case report. Kyobu Geka. 1994;47:1016–9. [PubMed] [Google Scholar]
- 56.Sugiyama Y, Hirai H, Ohta A, Inoue T, Lee T, Harada M, Suzuki M, Tamura S, Shiroma K, Ebine K, Takahashi K, Yamaguchi T. Severe aortic regurgitation with marked thickening of the aortic annulus: a case report. J Cardiol. 1997;29:81–6. [PubMed] [Google Scholar]
- 57.Yun KL, Sintek CF, Fletcher AD, Pfeffer TA, Kochamba GS, Hyde MR, Torpoeo JO, Khonsari S. Aortic valve replacement with the freestyle stentless bioprosthesis: five-year experience. Circulation. 1999;100:II17–23. doi: 10.1161/01.cir.100.suppl_2.ii-17. [DOI] [PubMed] [Google Scholar]
- 58.Chen BPC, Li Y-S, Zhao Y, Chen K-D, Li S, Lao J, Yuan S, Shyy JY, Chien S. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol Genomics. 2001;7:55–63. doi: 10.1006/geno.2001.6511. [DOI] [PubMed] [Google Scholar]
- 59.Vrandecic M, Fantini FA, Filho BG, Oliveira OC, Costa Junior IM, Vrandecic E. Retrospective clinical analysis of stented vs. stentless porcine aortic bioprostheses. Eur J Cardiothorac Surg. 2000;18:46. doi: 10.1016/S1010-7940(00)00416-4. [DOI] [PubMed] [Google Scholar]
- 60.Novaro GM, Pearce GL, Sprecher DL, Griffin BP. Comparison of cardiovascular risk and lipid profiles in patients undergoing aortic valve surgery versus those undergoing coronary artery bypass grafting. J Heart Valve Dis. 2001;10:19–24. [PubMed] [Google Scholar]
- 61.Horiguchi K, Ohtake S, Matsumiya G, Sawa Y, Nishimura M, Satou H, Kawai N, Matsuda H. Aortic valve replacement combined with endoventricular circulatory patch plasty (Dor operation) in a patient with aortic valve stenosis and severe ischemic cardiomyopathy. Ann Thorac Cardiovasc Surg. 2001;7:170–4. [PubMed] [Google Scholar]
- 62.Murakami T, Kikugawa D, Endou K, Fukuhiro Y, Ishida A, Morita I, Masaki H, Inada H, Fujiwara T. Changes in patterns of left ventricular hypertrophy after aortic valve replacement for aortic stenosis and regurgitation with St. Jude Medical cardiac valves. Artif Organs. 2000;24:953–8. doi: 10.1046/j.1525-1594.2000.06641.x. [DOI] [PubMed] [Google Scholar]
- 63.Taylor PM, Allen SP, Yacoub MH. Phenotypic and functional characterization of interstitial cells from human heart valves, pericardium and skin. J Heart Valve Dis. 2000;9:150–8. [PubMed] [Google Scholar]
- 64.Yacoub MH, Kilner PJ, Birks EJ, Misfeld M. The aortic outflow and root: a tale of dynamism and crosstalk. Ann Thorac Surg. 1999;68:S37–43. doi: 10.1016/S0003-4975(99)00745-6. [DOI] [PubMed] [Google Scholar]
- 65.Kaden JJ, Vocke DC, Fischer CS, Grobholz R, Brueckmann M, Vahl CF, Hagl S, Haase KK, Dempfle CE, Borggrefe M. Expression and activity of matrix metalloproteinase-2 in calcific aortic stenosis. Z Kardiol. 2004;93:124–30. doi: 10.1007/s00392-004-1021-0. [DOI] [PubMed] [Google Scholar]
- 66.Blankenberg S, Rupprecht HJ, Poirier O, Bickel C, Smieja M, Hafner G, Meyer J, Cambien F, Tiret L. AtheroGene Investigators. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation. 2003;107:1579–85. doi: 10.1161/01.CIR.0000058700.41738.12. [DOI] [PubMed] [Google Scholar]
- 67.McGeehan GM, Becherer JD, Bast RC, Jr, Boyer CM, Champion B, Connolly KM, Conway JG, Furdon P, Karp S, Kidao S, McElroy AB, Nichols J, Pryzwansky KM, Schoenen F, Sekut L, Truesdale A, Verghese M, Warner J, Ways JP. Regulation of tumour necrosis factor-alpha processing by a metalloproteinase inhibitor. Nature. 1994;370:558–61. doi: 10.1038/370558a0. [DOI] [PubMed] [Google Scholar]
- 68.Gearing AJH, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, Drummond AH, Galloway WA, Gilbert R, Gordon JL, Leber TM, Mangan M, Miller K, Nayee P, Owen K, Patel S, Thomas W, Wells G, Wood LM, Woolley K. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–7. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 69.Leco KJ, Khokha R, Pavloff N, Hawkes SP, Edwards DR. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is an extracellular matrix-associated protein with a distinctive pattern of expression in mouse cells and tissues. J Biol Chem. 1994;269:9352–60. [PubMed] [Google Scholar]
- 70.Jamet B, Chabert JP, Metz D, Elaerts J. Acute aortic insufficiency. Ann Cardiol Angeiol (Paris) 2000;49:183–6. [PubMed] [Google Scholar]
- 71.Ichihara T, Fujii G, Sasaki M, Kawaguchi O, Ueda Y. Clinical characteristics of bicuspid aortic valves in surgical patients. Asian Cardiovasc Thorac Ann. 2006;14:210–2. doi: 10.1177/021849230601400308. [DOI] [PubMed] [Google Scholar]
- 72.Nollert G, Miksch J, Kreuzer E, Reichart B. Risk factors for atherosclerosis and the degeneration of pericardial valves after aortic valve replacement. J Thorac Cardiovasc Surg. 2003;126:965–8. doi: 10.1016/S0022-5223(02)73619-2. [DOI] [PubMed] [Google Scholar]
- 73.Briand M, Pibarot P, Després JP, Voisine P, Dumesnil JG, Dagenais F, Mathieu P. Metabolic syndrome is associated with faster degeneration of bioprosthetic valves. Circulation. 2006;114:I512–7. doi: 10.1161/CIRCULATIONAHA.105.000422. [DOI] [PubMed] [Google Scholar]
- 74.Colli A, Gherli T, Mestres CA, Pomar JL. Degeneration of native and tissue prosthetic valve in aortic position: do statins play an effective role in prevention? Int J Cardiol. 2007;116:144–52. doi: 10.1016/j.ijcard.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 75.Movahed MR, Hashemzadeh M, Jamal MM. Increased prevalence of infectious endocarditis in patients with type II diabetes mellitus. J Diabetes its Complicat. 2007;21:403–6. doi: 10.1016/j.jdiacomp.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 76.Gouriet F, Lepidi H, Habib G, Collart F, Raoult D. From cat scratch disease to endocarditis, the possible natural history of Bartonella henselae infection. BMC Infect Dis. 2007;7:30. doi: 10.1186/1471-2334-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Briand M, Lemieux I, Dumesnil JG, Mathieu P, Cartier A, Després JP, Arsenault M, Couet J, Pibarot P. Metabolic syndrome negatively influences disease progression and prognosis in aortic stenosis. J Am Coll Cardiol. 2006;47:2229–36. doi: 10.1016/j.jacc.2005.12.073. [DOI] [PubMed] [Google Scholar]
- 78.Baghdasarian SB, Jneid H, Hoogwerf BJ. Association of dyslipidemia and effects of statins on nonmacrovascular diseases. Clin Ther. 2004;26:337–51. doi: 10.1016/S0149-2918(04)90031-8. [DOI] [PubMed] [Google Scholar]
- 79.Golubnitschaja O. Time for new guidelines in advanced diabetes care: Paradigm change from delayed interventional approach to predictive, preventive & personalized medicine. EPMA J. 2010;1:3–12. doi: 10.1007/s13167-010-0014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.George B, Cebioglu M, Yeghiazaryan K. Inadequate diabetic care: global figures cry for preventive measures and personalized treatment. EPMA J. 2010;1:13–8. doi: 10.1007/s13167-010-0006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abebe W, Mozaffari M. Endothelial dysfunction in diabetes: potential application of circulating markers as advanced diagnostic and prognostic tools. EPMA J. 2010;1:32–45. doi: 10.1007/s13167-010-0012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sena CM, Bento CF, Pereira P, Seiça R. Diabetes mellitus: new challenges and innovative therapies. EPMA J. 2010;1:138–63. doi: 10.1007/s13167-010-0010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]