

Abstract
The discovery of circulating cell-free fetal DNA (cffDNA) in maternal plasma allowed for the development of alternative methodologies that may facilitate safe non-invasive prenatal diagnosis (NIPD). The low concentration of cffDNA in maternal plasma, however, and the coexistence of maternal DNA limit its clinical application to the detection or exclusion of fetal targets that are not present in the mother, such as Y chromosome sequences, the RHD gene in a RhD-negative woman and genetic conditions inherited from the father. Strategies for NIPD of monogenic disorders and fetal chromosomal aneuploidies have also been achieved using next-generation sequencing and could be introduced to the clinics as soon as cost-effective and high throughput protocols are developed.
Keywords: Non-invasive prenatal diagnosis, Cell-free fetal nucleic acids, Monogenic disorders, Chromosomal aneuploidies, Fetal gender, Fetal RhD status
Introduction
Prenatal diagnosis is now part of established obstetric practice in many countries. To perform it, fetal genetic material is conventionally obtained by invasive techniques such as amniocentesis and chorionic villus sampling. Since procedural related miscarriage rate of about 1% has been reported, invasive prenatal diagnosis is reserved for pregnancies at risk for certain fetal genetic conditions. These include fetal chromosomal aneuploidies and monogenic disorders with relatively high prevalence in the relevant populations. For chromosomal aneuploidies, mainly Down Syndrome (DS), risk calculation is based on maternal age, ultrasonographic findings and maternal serum biochemical markers. For certain monogenic disorders, such as thalassaemia and cystic fibrosis, identification of a positive family history and confirmation of the carrier status of the parents are among the strategies used in order to identify high risk pregnancies that may be referred for invasive prenatal diagnosis.
Non-invasive testing, using maternal peripheral blood as a source of fetal genetic material, has long been the goal for early prenatal diagnosis avoiding the risk of miscarriage. In contrast to popular belief that placenta forms an impermeable barrier between mother and fetus, there is proof for bidirectional traffic between the fetus and the mother during pregnancy [1]. Multiple studies indicate that both intact fetal cells and cell-free fetal nucleic acids (cffNA) cross the placenta and can be found in maternal circulation. Intact-fetal cells present an attractive target for non-invasive prenatal diagnosis (NIPD) of fetal chromosomal abnormalities. Isolation and analysis of fetal cells from maternal circulation have been extensively investigated and several methods for fetal cell enrichment have been developed [2–4]. For the time being, results have been disappointing due to the scarcity of intact fetal cells in maternal circulation (1 cell per 1 ml of maternal blood) and the low efficiency of enrichment methods. In addition, chromosome analysis by Fluorescent In Situ Hybridization (FISH) is difficult due to the abnormally dense apoptotic nuclei of fetal cells [5, 6].
In 1997, Lo et al. reported the presence of cell-free fetal DNA (cffDNA) in maternal plasma and serum in amounts significantly increased, as compared to fetal DNA extracted from the cellular fraction of maternal blood [7, 8]. Following this discovery, a new area of research was developed.
This review summarizes the current status of NIPD using cffNA in maternal plasma and emphasizes recent developments that may allow in the future routine application of NIPD for assessment of chromosomal abnormalities and monogenic disorders.
Cell-free fetal nucleic acids in maternal plasma
Cell-free fetal DNA represents extracellular DNA which can be detected in maternal peripheral blood as early as 18 days following embryo transfer in in vitro fertilization pregnancies [9]. In contrast to fetal cells, it is cleared from maternal circulation shortly after delivery, with a mean half-life of 16 min [10]. The proportion of cffDNA, however, is only 3–6% of the total amount of cell-free DNA (cfDNA), the remaining portion of DNA being contributed by the mother, mainly from maternal blood cells. Later studies using digital PCR showed that the actual amount of cffDNA is somewhat higher (~19%) but still represents a minor fraction of the total amount of cfDNA in maternal plasma [11]. The concentration increases with gestational age, from the equivalent of 16 fetal genomes per millilitre of maternal blood in the first trimester to 80 in the third trimester, with a sharp peak during the last 8 weeks of pregnancy [8, 12].
It is generally accepted that cffDNA in maternal plasma is derived from syncytiotrophoblasts undergoing apoptosis and is, therefore, fragmented [13, 14]. Studies regarding the size distribution of cffDNA have confirmed that it is on the average 300 bp or smaller, in contrast to the maternal cfDNA fragments, which are considerably larger [15, 16].
In addition to cffDNA, cell-free fetal mRNA (cffmRNA) sequences of placental origin also exist in maternal plasma. The proof of principle of cffmRNA in maternal plasma was first demonstrated in pregnant women carrying a male child, with the detection of Y chromosome-specific zinc finger protein mRNA [17]. This finding was further confirmed a few years later, through the analysis of placental-specific mRNA, human placental lactogen (hPL) and the b subunit of human placental chorionic gonadotrophin [18].
Recent data have also demonstrated that placental miRNAs are also released from the placenta and are present in maternal plasma in detectable quantities. Quantification of placental miRNAs in maternal plasma may offer a non-invasive tool for monitoring gene regulation in the placenta. Placental miRNAs exhibit exceptional stability which is probably due to their association with subcellular particles such as syncytiotrophoblast microparticles [18–21].
Technical aspects
The scarity of cffDNA in maternal blood and its co-existence with maternal DNA represent the two major limitations for the use of cffDNA for diagnosis. Both maternal plasma and serum contain cffDNA, however, plasma is the material of choice for prenatal diagnosis since it contains less maternal background DNA. Usually, isolation of plasma cfDNA is performed manually with commercially available kits, although automation of the process has also been reported [22, 23]. It is important to note that special care should be taken to avoid external contamination during isolation and amplification.
Various methods have been used to overcome the presence of maternal background cfDNA, including methods based on the size difference of maternal fragments [15, 24]. Efforts to increase the relative proportion of fetal DNA compared to the larger maternal fraction have also included the use of formaldehyde, as a fixative, to prevent lysis of maternal cells during the isolation of the maternal plasma [25]. The formaldehyde enrichment technique, however, has not been reproducible by other laboratories.
Different methodologies applied for the detection of cffDNA include conventional PCR, restriction analysis, quantitative fluorescence real-time PCR (QF-PCR) and automated sequencing [11, 26–31].
Since QF-PCR is more sensitive compared to conventional PCR, enabling the detection of very low copy numbers of DNA, it represents the optimal method for reliable NIPD. The main advantage of QF-PCR is that it is quantitative and collects data in the exponential growth phase of the reaction, which is the most specific and precise one. The technique is less time consuming and offers an extra level of protection against contamination. A wide range of Ct values in each QF-PCR and poor repeatability of some replicates is reported, partly due to the variability of target copy number in maternal plasma. It is, therefore, recommended to perform several replicates from each maternal sample in order to increase the probability of fetal DNA detection and to avoid false-negative results [32].
Recently, new sophisticated molecular techniques such as mass spectrometry, massively parallel genomic sequencing (MPS) and digital PCR have emerged and are applied in the field of NIPD [33, 34]. They have higher sensitivity, but the expensive and complicated handling processes required by these techniques make them less useful for clinical practice.
Current clinical applications of non-invasive prenatal diagnosis using cffDNA
Fetal sex determination
Fetal sexing, the first clinical application of NIPD, is based on the detection of Y chromosome specific targets in maternal plasma [7]. Positive detection suggests that pregnancy involves a male fetus, while absence is interpreted as a female bearing pregnancy. The majority of the studies for non-invasive sex determination, uses QF-PCR to detect SRY and/or DYS14 sequences in maternal plasma [35–37]. Accuracy of fetal sex assessment has been confirmed by many research group to be better than 95%, so that its use has been adopted in a clinical setting [37, 38].
The most important clinical indication for fetal sex determination is an X-linked genetic disorder and it has been estimated that their cumulative incidence is around 5 in 10 000 live births [39]. Early determination of fetal sex non-invasively could limit the number of invasive diagnostic procedures required for each specific disease to male fetuses only, sparing most female bearing pregnancies from unnecessary invasive diagnostic testing.
Sex determination is also important in cases where development of external genitalia is ambiguous, in families with a history of conditions associated with ambiguous development of the external genitalia, some fetal ultrasound findings and occasionally, discrepancy between genetic sex and the appearance of the external genitalia on fetal ultrasound.
Non-invasive prenatal fetal sex detection has also been suggested for the management of some endocrine disorders, such as congenital adrenal hyperplasia, where masculinization of the female fetus can be suppressed by maternal administration of dexamethasone from the 6th week of gestation [35, 40]. This approach can allow either for slightly delayed administration of maternal steroids to women reluctant to expose fetuses unnecessarily to the potential side effects of dexamethasone, or for prompt cessation of treatment if the fetus is confirmed to be male.
Fetal RHD determination
Rhesus D (RhD) system incompatibility between a RhD-negative pregnant woman and her fetus can result in maternal alloimmunization and haemolytic disease of the fetus (HDFN) in subsequent pregnancies when the fetus is RhD-positive [41]. Routine postnatal injection of immunoglobin anti-D to all RhD-negative pregnant women, significantly prevents the occurrence of HDFN and has been successfully introduced in developed countries.
Prenatal determination of the fetal RhD status can be achieved by PCR amplification of RHD sequences in amniotic fluid or chorionic villus samples [42, 43]. These invasive procedures, however, carry not only the risk of miscarriage but most importantly, testing RhD-negative pregnant women may lead to immunization due to feto-maternal haemorrhage [44, 45].
The advent of NIDP using cffDNA to determine fetal RhD status offers significant potential for a change in the way RhD-negative pregnant women are managed. Since the RHD gene is usually completely absent from the genome of RhD-negative mothers, the detection of RHD sequences in maternal blood implies that the fetus must be RhD-positive.
There are multiple reports of high degrees of accuracy for the non-invasive prenatal determination of the fetal RhD status, but to date, clinical application has been confined to women known to be at high risk for HDFN [32, 46–48]. The benefits of mass testing antenatally for fetal RhD status by analysis of cffDNA in maternal plasma of RhD-negative mothers could reduce the use of anti-D and the number of anti-D donors exposed to blood products for hyperimmunization. Women carrying a RhD-negative fetus (approximately 40%) would be spared unnecessary exposure to anti-D with its associated discomfort and risk from viral (hepatitis C) or prion (variant Creutzfeld-Jacob disease) contamination.
QF-PCR technology is considered the optimal method for the reliable detection of RHD sequences using cffDNA [49, 50]. Efforts have also been made for non-invasive prenatal diagnosis of fetal RhD status by mass spectrometry (MS). Grill et al. used an automated system for the extraction of cell-free DNA from maternal plasma and detected the presence of fetal RHD exon 7 by MS technology provided by Sequenom to detect SNPs (Sequenom, Inc., San Diego, CA) [51]. Validation of the assay showed the presence of 2.5% RhD-positive genomic DNA in a background of RhD-negative genomic DNA. Five out of 178 samples examined were incorrectly diagnosed as RhD-negative. Grill’s report is the only one in the literature which applies MS for NIPD RhD testing. The main advantage of this approach over QF-PCR is that it has the potential of multiplex analysis of several different loci in a single assay but further studies are necessary in order to determine the clinical utility of the technique.
The most widespread approach used in prenatal RHD diagnosis is an assay that detects at least two different exons of the RHD gene. Many laboratories prefer to include amplification of exon 7, because it contains the most sequence differences to the highly homologous RHCE gene, thus improving specificity for RHD and allowing for the detection of fetal RHD even in the 7th week of gestation, without giving false-positive results [47, 52].
Fetal monogenic autosomal disorders
Many human diseases are caused by mutations in a single gene and it has been estimated that their combined occurrence is around 3.6 per 1000 live births [39]. Prenatal diagnosis of single gene disorders using invasive techniques is an accepted part of clinical practice and is performed when there is a positive family history for a particular disease.
NIPD for autosomal dominant disorders using maternal plasma has been reported, mainly based on the detection of paternally inherited DNA sequences in maternal sirculation. Such an approach can avoid conventional prenatal diagnosis in some cases. It is also important to note that detection of large-scale mutations, caused by expansion, insertion or duplication, is restricted to sequences less than 300 base-pairs in length, due to the fragmented nature of cffDNA [53]. NIPD of maternally transmitted autosomal dominant diseases is not as straightforward. The large maternal DNA background in maternal plasma renders it technically challenging to determine whether a fetus has inherited a mutation from its mother.
To date, use of cffDNA for NIPD has been reported for the following dominant single gene disorders in at least one pregnancy:
▪ Huntington’s disease – detection of a paternally inherited expansion of 37 repeats [54]
▪ Achondroplasia – identification of a specific point mutation which accounts for more than 98% of cases [26, 27, 55]
▪ Myotonic dystrophy – a paternally inherited expansion of 70 repeats has been detected using cffDNA [56]
In autosomal recessive disorders, cffDNA can only be used to determine the carrier status of the fetus through detection of the paternally inherited disease allele, in cases where the maternal and paternal ones differ. This information could be used to reduce the number of invasive procedures required, either by increasing the risk for an affected fetus (from one in four to one in two), or by determining that the fetus has not inherited the paternal disease allele and therefore cannot be affected. To date, fetal carrier status has been performed using cffDNA in Cystic fibrosis and Congenital adrenal hyperplasia [57, 58]. For haemoglobinopathies, cffDNA has been used to detect paternally inherited mutations that cause β-thalassemia, in carriers of a different β-thalassemia or sickle cell mutation (leading to sickle β-thalassemia disease) and Hb Lepore. Sickle cell anaemia, however, the most common haemoglobinopathy, is not yet prenatally diagnosed using cffDNA, since it is caused by two identical copies of a single point mutation [29, 59, 60].
In order to extend NIPD to cases where the father and mother shared the same mutation Lum et al. developed a digital PCR based approach, called relative mutation dosage (RMD) [61]. RMD analysis can be used for diagnosis in cases of pregnant women heterozygous for a known mutation through determination of the dosages of the mutant and wild-type alleles of the disease-causing gene. For a woman carrying a fetus homozygous for the mutation there should be proportionally more mutant sequences than the non-mutant ones in maternal plasma, while in cases with an heterozygous fetus, equal amounts of mutant and non-mutant sequences should be detected in maternal plasma. Similarly, when the fetus is normal homozygous, there should be proportionally more non-mutant sequences than mutant sequences in maternal plasma.
Fetal markers
Significant effort has been made to detect fetal identifiers in maternal plasma samples. For fetal chromosome Y or RHD detection assays, failure to detect the targeted cffDNA sequences could be a result of failed or degrated fetal DNA. It is prudent therefore to confirm the presence of fetal material in maternal plasma before reporting a negative result. Detection of Y-chromosome specific sequences, such as SRY or DYS14, is used in order to confirm the presence of cffDNA in the sample tested [49]. This, however, can only be applied in pregnancies bearing a male fetus. An alternative approach involves detection of paternally inherited polymorphisms that are unique to the fetus, but insertion/deletion polymorphisms or single nucleotide polymorphisms (SNPs) are only useful as internal positive controls if they are absent in the maternal genome and the paternal-unique allele has been inherited by the fetus [62, 63]. Depending on the parental genotype and fetal inheritance, a particular polymorphism may not be applicable to all pregnancies. Consequently, a panel of polymorphisms is needed to ensure that at least one member of the panel is appropriate for any given pregnancy.
A major area of current research aims at finding universal fetal-specific markers, independent of sex or paternally inherited polymorphisms, that could be used either as diagnostic tests or to confirm the presence and quantify cffDNA. Recent approaches have targeted fetal DNA sequences in maternal plasma that are epigenetically different from maternal ones [64]. CpG methylation in the promoter regions of genes is involved in the regulation of gene expression. As tissues in the body have different gene expression profiles, the methylation status of certain genes also exhibits tissue-specific patterns. Chim et al. [64] studied the methylation profile of the promoter of serpin peptidase inhibitor, SERPINB5, and showed that it is hypomethylated in placental tissues but hypermethylated in maternal blood cells. Using methylation specific PCR, the placental-derived hypomethylated SERPINB5 could be detected and distinguished from maternally derived hypermethylated molecules in maternal plasma. The hypomethylated SERPINB5 sequences were shown to be pregnancy-associated, as they disappeared from maternal plasma within 24 h after delivery. SERPINB5 was the first universal circulating cffDNA marker that could be applied in all pregnancies regardless of fetal gender and genotype. However, methylation-specific PCR requires use of bisulphite conversion, which alters unmethylated cytosines to uracil nucleotides and thereby results in differences in the genetic sequence of methylated and unmethylated DNA molecules. Yet, bisulphite conversion degrades up to 95% of the DNA molecules in a sample [65]. This would substantially reduce the amount of fetal DNA in a maternal plasma sample and may result in false-negative results, particularly in early pregnancy when fetal DNA concentrations are very low.
For this reason, researchers looked for fetal epigenetic markers that could be detected in maternal plasma without bisulphite conversion. Hypomethylated RASSF1A sequences derived from maternal blood cells can be removed from maternal plasma using methylation-sensitive restriction enzyme digestion, revealing only the fetal hypomethylated target (Fig. 1) [66]. Several studies demonstrated the value of using digestion resistant RASSF1A DNA sequences as a positive control for NIPD of fetal RhD status [66–68]. False-negative diagnosis can be avoided in samples that are negative for both RHD and RASSF1A sequences, as failure to detect hypermethylated RASSF1A sequences signifies the lack of fetal DNA in maternal plasma sample. These developments improve the reliability of the applications of cffDNA analysis when used in clinical setting.
Fig. 1.
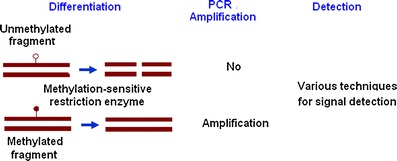
General schema of DNA methylation analysis using methylation-sensitive restriction enzymes that selectively digest unmethylated DNA, so that only methylated fragments remain available for detection
Pregnancy complications
Various problems associated with placental growth and development result in altered levels of cffDNA in maternal plasma. Elevated concentrations of cffDNA have been detected in pregnancy-related disorders associated with abnormal placentation such as preeclampsia, the leading cause of prematurity [69–71]. Preeclampsia complicates around 5–10% of pregnancies, and if it is not treated on time it can become life threatening for both mother and child. Although preeclampsia stems from a defective placenta, the underlying cause is unknown and the only available treatment is delivery of the fetus. Numerous studies have shown that the level of cffDNA (usually measuring Y chromosome DNA in pregnancies with a male fetus) is elevated by 2–3-fold before the onset of preeclampsia and 2–14-fold during preeclampsia [8, 71]. Elevations in cffDNA have also been reported in pregnancies with preterm contractions that did not respond to colytic treatment and resulted in premature deliveries, in contrast to those that responded to such treatment [72]. A significant number of additional pregnancy-related disorders have been linked to increased concentrations of cffDNA. These include hyperemesis gravidarum (severe morning sickness), invasive placentation (in which the placenta contacts the maternal bloodstream), intrauterine growth restriction, feto-maternal haemorrhage and polyhydramnios [73]. Hence, quantitative cffDNA analysis may assist in predicting pregnancy related complications. It should be noted, however, that the absolute level of circulating cffDNA fluctuates over short periods throughout pregnancy and varies with both ethnicity and maternal weight, raising important questions about the diagnostic utility of adding cffDNA concentration to the current panel of biomarkers [74–76].
Non-invasive prenatal diagnosis of fetal chromosomal aneuploidies
Since fetal and maternal alleles are by nature indistinguishable, detection of extra fetal chromosomes in maternal plasma poses a substantial challenge. Initially published work in the field revealed increased levels of circulating DNA in pregnancies known to carry DS or trisomy 13 fetuses as compared to chromosomally normal ones [77, 78]. It was therefore suggested that this increase of cffDNA levels in maternal plasma could be used as a marker for NIPD of fetal aneuploidies. Discordant results, however, among different research groups were reported given that the levels of cffDNA vary widely and, as mentioned previously, are also elevated in a number of pregnancy related complications [75]. More successful and specific diagnostic strategies are required, therefore, in order to identify non-invasively fetal aneuploidies.
An alternative more specific diagnostic strategy for NIPD of fetal aneuploidies focuses on the analysis of classes of nucleic acids in maternal plasma that are fetal-specific, because maternal blood cells do not express the target mRNA or have a different methylation profile for an epigenetic fetal marker. These fetal nucleic acids include as already mentioned, SERPINB5 and placenta-specific 4 (PLAC4) mRNA [79].
SERPINB5, one of the proposed universal fetal markers, has been studied in order to diagnose trisomy 18 as it is located on the specific chromosome [79]. Bisulphite modification was followed by methylation-specific PCR and primer extension to assess the allelic ratios. This generated a sensitivity of 100% and a false-positive rate of 9.7% Multiple differentially methylated genes have also been found on chromosome 21 [20]. This approach however requires several manipulations, thus making it less feasible for clinical practice.
Selective targeting by differentially methylated markers in placenta and maternal blood cells has recently been combined with microfluidics digital PCR for non-invasive detection of fetal trisomy 21 [80]. Chromosome dosage analysis was performed by comparing the dosage of an epigenetic chromosome 21 marker (HLCS, a hypermethylated fetal-DNA marker) with that of reference chromosomes, RASSF1A on chromosome 3 and ZFY on the Y chromosome. The ratio of HLCS to RASSF1A showed great overlap between euploid and trisomy 21 samples. The comparison between HLCS and ZFY can discriminate aneuploid fetuse, but its use is limited to women carrying male fetuses.
Lo et al. reported quantification of PLAC4 mRNA deriving from chromosome 21, for the NIPD of fetal DS [81]. Euploid cases have equal ratios of each allele (1 : 1 ratio), whereas if an aneuploidy is present, the ratio is 2 : 1 (Fig. 2). This approach appears to be quite promising, as the authors were able to detect fetal DS with a sensitivity of 90% and a specificity of 96%. The technique, however, is not applicable to all pregnancies, as it requires that the fetus has inherited two different SNP alleles in the region analyzed.
Fig. 2.
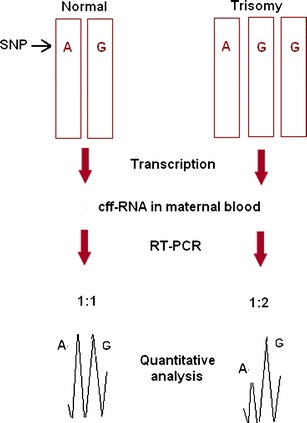
Calculation of the fetal chromosome dosage using the allelic ratio of a specific heterozygous SNP on cffmRNA
Recently developed single molecule counting techniques can be used for fetal aneuploidy detection without the restrictions of fetal-specific nucleic acids in maternal plasma:
Determination of chromosome dosage by digital PCR, a highly sensitive technique that uses limiting dilution to isolate single template DNA molecules to be amplified [81, 82]. Digital PCR was tested in a model system for molecular detection of fetal trisomy 21. A nonpolymorphic chromosome 21 locus was compared to one located on a reference chromosome. A change in the ratio of both chromosomes from 2:2 in an euploid fetus to 3:2 in a trisomic fetus was reported. It is noteworthy that the technique does not specifically distinguish fetal-derived from maternal DNA, thus the degree of increment depends on the concentration of cffDNA. The analytical platform would need to be quantitatively more precise to reliably determine the small expected increment.
Use of massively parallel, or next-generation, sequencing (MPS). MPS can analyse the nucleotide sequences of millions of DNA molecules in each run. The capacity of MPS to differentiate small quantitative alterations in genomic distributions of chromosomes, has allowed detection of higher amounts of chromosome 21 sequences in trisomy 21 pregnancies as compared to euploid pregnancies. Fan et al. tested this technique on cfDNA from plasma of pregnant women with a gestational age of 10–35 weeks [33]. Chiu et al. applied the same technique, but followed a different strategy for data analysis [83]. They used this strategy in order to detect trisomy 21 and the Y and X chromosomes difference between male and female fetuses. They tested an algorithm to calculate the percentage unique sequences for the chromosome of interest in the test sample and compared it with the reference population of that same chromosome. They were able to discriminate trisomy 21 from disomy 21 samples. Both studies demonstrated the feasibility of deep sequencing for NIPD. Measurements of the genomic representations for chromosomes 13 and 18 were less precise [84]. The important advantage of the MPS technique is that it is gender and polymorphism-independent, applicable in all pregnancies and likely to allow analysis of all frequent forms of aneuploidies in the same test. Currently the technique is technically demanding, the cost per tested sample is high and the throughput per instrument is low (16 samples per week). This prevents its use as a regular test for all pregnant women. Thus, although MPS is certainly one of the most promising approaches, population-based studies, involving prospective studies in low-risk populations are needed to indicate if the technique is robust and can be used for clinical diagnosis.
Conclusions
New advances have been reported regarding NIPD using cffDNA from maternal plasma. Currently fetal sex assessment for X-linked disorders, and RhD incompatibility have been implemented in many diagnostic laboratories as a routine technique. Approaches for the NIPD of monogenic disorders, including both autosomal dominant and recessive, have also been developed. NIPD of fetal chromosomal aneuploidies, the main referral reason for prenatal diagnosis, has also been achieved with the use of next-generation sequencing, but issues related to the cost and throughput of this methodology should be solved before cffDNA can replace fetal genetic material obtained using invasive techniques.
References
- 1.Lo YM, Lo ES, Watson N, Noakes L, Sargent IL, Thilaganathan B, et al. Two-way cell traffic between mother and fetus: biologic and clinical implications. Blood. 1996;88:4390–5. [PubMed] [Google Scholar]
- 2.Bianchi DW. Fetal cells in the maternal circulation: feasibility for prenatal diagnosis. Br J Haematol. 1999;105:574–83. doi: 10.1046/j.1365-2141.1999.01383.x. [DOI] [PubMed] [Google Scholar]
- 3.Sekizawa A, Purwosunu Y, Farina A, Okai T, Takabayashi H, Kita M, et al. Development of noninvasive fetal DNA diagnosis from nucleated erythrocytes circulating in maternal blood. Prenat Diagn. 2007;27:846–8. doi: 10.1002/pd.1792. [DOI] [PubMed] [Google Scholar]
- 4.Jackson L. Fetal cells and DNA in maternal blood. Prenat Diagn. 2003;23:837–46. doi: 10.1002/pd.705. [DOI] [PubMed] [Google Scholar]
- 5.Babochkina T, Mergenthaler S, Napoli G, Hristoskova S, Tercanli S, Holzgreve W, et al. Numerous erythroblasts in maternal blood are impervious to fluorescent in situ hybridization analysis, a feature related to a dense compact nucleus with apoptotic character. Haematologica. 2005;90:740–5. [PubMed] [Google Scholar]
- 6.Bianchi DW, Williams JM, Sullivan LM, Hanson FW, Klinger KW, Shuber AP. PCR quantitation of fetal cells in maternal blood in normal and aneuploid pregnancies. Am J Hum Genet. 1997;61:822–9. doi: 10.1086/514885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 8.Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet. 1998;62:768–75. doi: 10.1086/301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guibert J, Benachi A, Grebille AG, Ernault P, Zorn JR, Costa JM. Kinetics of SRY gene appearance in maternal serum: detection by real time PCR in early pregnancy after assisted reproductive technique. Hum Reprod. 2003;18:1733–6. doi: 10.1093/humrep/deg320. [DOI] [PubMed] [Google Scholar]
- 10.Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64:218–24. doi: 10.1086/302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lun FM, Chiu RW. Allen Chan KC, Yeung Leung T, Kin Lau T, Dennis Lo YM. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–72. doi: 10.1373/clinchem.2008.111385. [DOI] [PubMed] [Google Scholar]
- 12.Birch L, English CA, O’Donoghue K, Barigye O, Fisk NM, Keer JT. Accurate and robust quantification of circulating fetal and total DNA in maternal plasma from 5 to 41 weeks of gestation. Clin Chem. 2005;51:312–20. doi: 10.1373/clinchem.2004.042713. [DOI] [PubMed] [Google Scholar]
- 13.Masuzaki H, Miura K, Yoshiura KI, Yoshimura S, Niikawa N, Ishimaru T. Detection of cell free placental DNA in maternal plasma: direct evidence from three cases of confined placental mosaicism. J Med Genet. 2004;41:289–92. doi: 10.1136/jmg.2003.015784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberry M, Maddocks D, Jones M, Abdel Hadi M, Abdel-Fattah S, Avent N, et al. Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn. 2007;27:415–8. doi: 10.1002/pd.1700. [DOI] [PubMed] [Google Scholar]
- 15.Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem. 2004;50:88–92. doi: 10.1373/clinchem.2003.024893. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Zimmermann B, Rusterholz C, Kang A, Holzgreve W, Hahn S. Size separation of circulatory DNA in maternal plasma permits ready detection of fetal DNA polymorphisms. Clin Chem. 2004;50:1002–11. doi: 10.1373/clinchem.2003.029835. [DOI] [PubMed] [Google Scholar]
- 17.Poon LL, Leung TN, Lau TK, Lo YM. Presence of fetal RNA in maternal plasma. Clin Chem. 2000;46:1832–4. [PubMed] [Google Scholar]
- 18.Ng EK, Tsui NB, Lau TK, Leung TN, Chiu RW, Panesar NS, et al. mRNA of placental origin is readily detectable in maternal plasma. Proc Natl Acad Sci USA. 2003;100:4748–53. doi: 10.1073/pnas.0637450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta AK, Holzgreve W, Huppertz B, Malek A, Schneider H, Hahn S. Detection of fetal DNA and RNA in placenta-derived syncytiotrophoblast microparticles generated in vitro. Clin Chem. 2004;50:2187–90. doi: 10.1373/clinchem.2004.040196. [DOI] [PubMed] [Google Scholar]
- 20.Chim SS, Jin S, Lee TY, Lun FM, Lee WS, Chan LY, et al. Systematic search for placental DNA-methylation markers on chromosome 21: toward a maternal plasma-based epigenetic test for fetal trisomy 21. Clin Chem. 2008;54:500–11. doi: 10.1373/clinchem.2007.098731. [DOI] [PubMed] [Google Scholar]
- 21.Chim SS, Shing TK, Hung EC, Leung TY, Lau TK, Chiu RW, et al. Detection and characterization of placental microRNAs in maternal plasma. Clin Chem. 2008;54:482–90. doi: 10.1373/clinchem.2007.097972. [DOI] [PubMed] [Google Scholar]
- 22.Clausen FB, Krog GR, Rieneck K, Dziegiel MH. Improvement in fetal DNA extraction from maternal plasma. Evaluation of the NucliSens Magnetic Extraction system and the QIAamp DSP Virus Kit in comparison with the QIAamp DNA Blood Mini Kit. Prenat Diagn. 2007;27:6–10. doi: 10.1002/pd.1605. [DOI] [PubMed] [Google Scholar]
- 23.Legler TJ, Liu Z, Mavrou A, Finning K, Hromadnikova I, Galbiati S, et al. Workshop report on the extraction of foetal DNA from maternal plasma. Prenat Diagn. 2007;27:824–9. doi: 10.1002/pd.1783. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Naro E, Vitucci A, Zimmermann B, Holzgreve W, Hahn S. Detection of paternally inherited fetal point mutations for beta-thalassemia using size-fractionated cell-free DNA in maternal plasma. JAMA. 2005;293:843–9. doi: 10.1001/jama.293.7.843. [DOI] [PubMed] [Google Scholar]
- 25.Dhallan R, Au WC, Mattagajasingh S, Emche S, Bayliss P, Damewood M, et al. Methods to increase the percentage of free fetal DNA recovered from the maternal circulation. JAMA. 2004;291:1114–9. doi: 10.1001/jama.291.9.1114. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Page-Christiaens GC, Gille JJ, Holzgreve W, Hahn S. Non-invasive prenatal detection of achondroplasia in size-fractionated cell-free DNA by MALDI-TOF MS assay. Prenat Diagn. 2007;27:11–7. doi: 10.1002/pd.1608. [DOI] [PubMed] [Google Scholar]
- 27.Saito H, Sekizawa A, Morimoto T, Suzuki M, Yanaihara T. Prenatal DNA diagnosis of a single-gene disorder from maternal plasma. Lancet. 2000;356:1170. doi: 10.1016/S0140-6736(00)02767-7. [DOI] [PubMed] [Google Scholar]
- 28.Chiu RW, Lau TK, Cheung PT, Gong ZQ, Leung TN, Lo YM. Noninvasive prenatal exclusion of congenital adrenal hyperplasia by maternal plasma analysis: a feasibility study. Clin Chem. 2002;48:778–80. [PubMed] [Google Scholar]
- 29.Chiu RW, Lau TK, Leung TN, Chow KC, Chui DH, Lo YM. Prenatal exclusion of beta thalassaemia major by examination of maternal plasma. Lancet. 2002;360:998–1000. doi: 10.1016/S0140-6736(02)11086-5. [DOI] [PubMed] [Google Scholar]
- 30.Ding C, Chiu RW, Lau TK, Leung TN, Chan LC, Chan AY, et al. MS analysis of single-nucleotide differences in circulating nucleic acids: Application to noninvasive prenatal diagnosis. Proc Natl Acad Sci USA. 2004;101:10762–7. doi: 10.1073/pnas.0403962101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bustamante-Aragones A, Gallego-Merlo J, Trujillo-Tiebas MJ, Alba MR, Gonzalez-Gonzalez C, Glover G, et al. New strategy for the prenatal detection/exclusion of paternal cystic fibrosis mutations in maternal plasma. J Cyst Fibros. 2008;7:505–10. doi: 10.1016/j.jcf.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Minon JM, Gerard C, Senterre JM, Schaaps JP, Foidart JM. Routine fetal RHD genotyping with maternal plasma: a four-year experience in Belgium. Transfusion. 2008;48:373–81. doi: 10.1111/j.1537-2995.2007.01533.x. [DOI] [PubMed] [Google Scholar]
- 33.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA. 2008;105:16266–71. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan HC, Blumenfeld YJ, El-Sayed YY, Chueh J, Quake SR. Microfluidic digital PCR enables rapid prenatal diagnosis of fetal aneuploidy. Am J Obstet Gynecol. 2009;200:543 e1–7. doi: 10.1016/j.ajog.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyett JA, Gardener G, Stojilkovic-Mikic T, Finning KM, Martin PG, Rodeck CH, et al. Reduction in diagnostic and therapeutic interventions by non-invasive determination of fetal sex in early pregnancy. Prenat Diagn. 2005;25:1111–6. doi: 10.1002/pd.1284. [DOI] [PubMed] [Google Scholar]
- 36.Costa JM, Benachi A, Gautier E, Jouannic JM, Ernault P, Dumez Y. First-trimester fetal sex determination in maternal serum using real-time PCR. Prenat Diagn. 2001;21:1070–4. doi: 10.1002/pd.219. [DOI] [PubMed] [Google Scholar]
- 37.Scheffer PG, van der Schoot CE, Page-Christiaens GC, Bossers B, van Erp F, de Haas M. Reliability of fetal sex determination using maternal plasma. Obstet Gynecol. 115:117–26. [DOI] [PubMed]
- 38.Bustamante-Aragones A, Rodriguez de Alba M, Gonzalez-Gonzalez C, Trujillo-Tiebas MJ, Diego-Alvarez MD, Vallespin E, et al. Foetal sex determination in maternal blood from the seventh week of gestation and its role in diagnosing haemophilia in the foetuses of female carriers. Haemophilia. 2008;14:593–8. doi: 10.1111/j.1365-2516.2008.01670.x. [DOI] [PubMed] [Google Scholar]
- 39.Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet. 1988;42:677–93. [PMC free article] [PubMed] [Google Scholar]
- 40.Bartha JL, Finning K, Soothill PW. Fetal sex determination from maternal blood at 6 weeks of gestation when at risk for 21-hydroxylase deficiency. Obstet Gynecol. 2003;101:1135–6. doi: 10.1016/S0029-7844(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 41.Bowman J. Rh-immunoglobulin: Rh prophylaxis. Best Pract Res Clin Haematol. 2006;19:27–34. doi: 10.1016/j.beha.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Bennett PR. Le Van Kim C, Colin Y, Warwick RM, Cherif-Zahar B, Fisk NM, Cartron JP. Prenatal determination of fetal RhD type by DNA amplification. N Engl J Med. 1993;329:607–10. doi: 10.1056/NEJM199308263290903. [DOI] [PubMed] [Google Scholar]
- 43.Colin Y, Cherif-Zahar B, Le Van Kim C, Raynal V, Van Huffel V, Cartron JP. Genetic basis of the RhD-positive and RhD-negative blood group polymorphism as determined by Southern analysis. Blood. 1991;78:2747–52. [PubMed] [Google Scholar]
- 44.Aubin JT, Le Van Kim C, Mouro I, Colin Y, Bignozzi C, Brossard Y, et al. Specificity and sensitivity of RHD genotyping methods by PCR-based DNA amplification. Br J Haematol. 1997;98:356–64. doi: 10.1046/j.1365-2141.1997.2193040.x. [DOI] [PubMed] [Google Scholar]
- 45.Daniels G, Finning K, Martin P, Soothill P. Fetal blood group genotyping from DNA from maternal plasma: an important advance in the management and prevention of haemolytic disease of the fetus and newborn. Vox Sang. 2004;87:225–32. doi: 10.1111/j.1423-0410.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- 46.Kolialexi A, Tounta G, Mavrou A. Noninvasive fetal RhD genotyping from maternal blood. Expert Rev Mol Diagn. 2010;10:285–96. doi: 10.1586/erm.10.5. [DOI] [PubMed] [Google Scholar]
- 47.Schoot CE, Hahn S, Chitty LS. Non-invasive prenatal diagnosis and determination of fetal Rh status. Semin Fetal Neonatal Med. 2008;13:63–8. doi: 10.1016/j.siny.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Finning K, Martin P, Daniels G. A clinical service in the UK to predict fetal Rh (Rhesus) D blood group using free fetal DNA in maternal plasma. Ann NY Acad Sci. 2004;1022:119–23. doi: 10.1196/annals.1318.019. [DOI] [PubMed] [Google Scholar]
- 49.Finning KM, Martin PG, Soothill PW, Avent ND. Prediction of fetal D status from maternal plasma: introduction of a new noninvasive fetal RHD genotyping service. Transfusion. 2002;42:1079–85. doi: 10.1046/j.1537-2995.2002.00165.x. [DOI] [PubMed] [Google Scholar]
- 50.Daniels G, Schoot CE, Olsson ML. Report of the Second International Workshop on molecular blood group genotyping. Vox Sang. 2007;93:83–8. doi: 10.1111/j.1423-0410.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 51.Grill S, Banzola I, Li Y, Rekhviashvili T, Legler TJ, Muller SP, et al. High throughput non-invasive determination of foetal Rhesus D status using automated extraction of cell-free foetal DNA in maternal plasma and mass spectrometry. Arch Gynecol Obstet. 2009;279:533–7. doi: 10.1007/s00404-008-0774-5. [DOI] [PubMed] [Google Scholar]
- 52.Finning K, Martin P, Summers J, Massey E, Poole G, Daniels G. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. BMJ. 2008;336:816–8. doi: 10.1136/bmj.39518.463206.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norbury G, Norbury CJ. Non-invasive prenatal diagnosis of single gene disorders: how close are we? Semin Fetal Neonatal Med. 2008;13:76–83. doi: 10.1016/j.siny.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Gonzalez MC, Trujillo MJ, Rodriguez de Alba M, Garcia-Hoyos M, Lorda-Sanchez I, Diaz-Recasens J, et al. Huntington disease-unaffected fetus diagnosed from maternal plasma using QF-PCR. Prenat Diagn. 2003;23:232–4. doi: 10.1002/pd.570. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Holzgreve W, Page-Christiaens GC, Gille JJ, Hahn S. Improved prenatal detection of a fetal point mutation for achondroplasia by the use of size-fractionated circulatory DNA in maternal plasma–case report. Prenat Diagn. 2004;24:896–8. doi: 10.1002/pd.1030. [DOI] [PubMed] [Google Scholar]
- 56.Amicucci P, Gennarelli M, Novelli G, Dallapiccola B. Prenatal diagnosis of myotonic dystrophy using fetal DNA obtained from maternal plasma. Clin Chem. 2000;46:301–2. [PubMed] [Google Scholar]
- 57.Gonzalez-Gonzalez MC, Garcia-Hoyos M, Trujillo MJ, Rodriguez de Alba M, Lorda-Sanchez I, Diaz-Recasens J, et al. Prenatal detection of a cystic fibrosis mutation in fetal DNA from maternal plasma. Prenat Diagn. 2002;22:946–8. doi: 10.1002/pd.439. [DOI] [PubMed] [Google Scholar]
- 58.Nasis O, Thompson S, Hong T, Sherwood M, Radcliffe S, Jackson L, et al. Improvement in sensitivity of allele-specific PCR facilitates reliable noninvasive prenatal detection of cystic fibrosis. Clin Chem. 2004;50:694–701. doi: 10.1373/clinchem.2003.025981. [DOI] [PubMed] [Google Scholar]
- 59.Lazaros L, Hatzi E, Bouba I, Paraskevaidis E, Georgiou I. Non-invasive prenatal detection of paternal origin hb lepore in a male fetus at the 7th week of gestation. Fetal Diagn Ther. 2006;21:506–9. doi: 10.1159/000095662. [DOI] [PubMed] [Google Scholar]
- 60.Fucharoen G, Tungwiwat W, Ratanasiri T, Sanchaisuriya K, Fucharoen S. Prenatal detection of fetal hemoglobin E gene from maternal plasma. Prenat Diagn. 2003;23:393–6. doi: 10.1002/pd.607. [DOI] [PubMed] [Google Scholar]
- 61.Lun FM, Tsui NB, Chan KC, Leung TY, Lau TK, Charoenkwan P, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:19920–5. doi: 10.1073/pnas.0810373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Page-Christiaens GC, Schoot CE, Haas DE. Use of bi-allelic insertion/deletion polymorphisms as a positive control for fetal genotyping in maternal blood: first clinical experience. Ann NY Acad Sci. 1075;2006:123–9. doi: 10.1196/annals.1368.016. [DOI] [PubMed] [Google Scholar]
- 63.Chow KC, Chiu RW, Tsui NB, Ding C, Lau TK, Leung TN, et al. Mass spectrometric detection of an SNP panel as an internal positive control for fetal DNA analysis in maternal plasma. Clin Chem. 2007;53:141–2. doi: 10.1373/clinchem.2007.079335. [DOI] [PubMed] [Google Scholar]
- 64.Chim SS, Tong YK, Chiu RW, Lau TK, Leung TN, Chan LY, et al. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc Natl Acad Sci USA. 2005;102:14753–8. doi: 10.1073/pnas.0503335102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chan KC, Ding C, Gerovassili A, Yeung SW, Chiu RW, Leung TN, et al. Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin Chem. 2006;52:2211–8. doi: 10.1373/clinchem.2006.074997. [DOI] [PubMed] [Google Scholar]
- 67.Hyland CA, Gardener GJ, Davies H, Ahvenainen M, Flower RL, Irwin D, et al. Evaluation of non-invasive prenatal RHD genotyping of the fetus. Med J Aust. 2009;191:21–5. doi: 10.5694/j.1326-5377.2009.tb02668.x. [DOI] [PubMed] [Google Scholar]
- 68.Tounta G, Vrettou C, Kolialexi A, Papantoniou N, Destouni A, Tsangaris GT, et al. A multiplex PCR for non-invasive fetal RHD genotyping using cell-free fetal DNA. In Vivo. 2011; in press. [PubMed]
- 69.Bauer M, Hutterer G, Eder M, Majer S, Leshane E, Johnson KL, et al. A prospective analysis of cell-free fetal DNA concentration in maternal plasma as an indicator for adverse pregnancy outcome. Prenat Diagn. 2006;26:831–6. doi: 10.1002/pd.1513. [DOI] [PubMed] [Google Scholar]
- 70.Bischoff FZ, Lewis DE, Simpson JL. Cell-free fetal DNA in maternal blood: kinetics, source and structure. Hum Reprod Update. 2005;11:59–67. doi: 10.1093/humupd/dmh053. [DOI] [PubMed] [Google Scholar]
- 71.Hahn S, Huppertz B, Holzgreve W. Fetal cells and cell free fetal nucleic acids in maternal blood: new tools to study abnormal placentation? Placenta. 2005;26:515–26. doi: 10.1016/j.placenta.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 72.Leung TN, Zhang J, Lau TK, Hjelm NM, Lo YM. Maternal plasma fetal DNA as a marker for preterm labour. Lancet. 1998;352:1904–5. doi: 10.1016/S0140-6736(05)60395-9. [DOI] [PubMed] [Google Scholar]
- 73.Wright CF, Burton H. The use of cell-free fetal nucleic acids in maternal blood for non-invasive prenatal diagnosis. Hum Reprod Update. 2009;15:139–51. doi: 10.1093/humupd/dmn047. [DOI] [PubMed] [Google Scholar]
- 74.Gerovassili A, Nicolaides KH, Thein SL, Rees DC. Cell-free DNA levels in pregnancies at risk of sickle-cell disease and significant ethnic variation. Br J Haematol. 2006;135:738–41. doi: 10.1111/j.1365-2141.2006.06357.x. [DOI] [PubMed] [Google Scholar]
- 75.Hahn S, Zhong XY, Burk MR, Troeger C, Kang A, Holzgreve W. Both maternal and fetal cell-free DNA in plasma fluctuate. Ann NY Acad Sci. 2001;945:141–4. doi: 10.1111/j.1749-6632.2001.tb03875.x. [DOI] [PubMed] [Google Scholar]
- 76.Wataganara T, Peter I, Messerlian GM, Borgatta L, Bianchi DW. Inverse correlation between maternal weight and second trimester circulating cell-free fetal DNA levels. Obstet Gynecol. 2004;104:545–50. doi: 10.1097/01.AOG.0000137352.93110.15. [DOI] [PubMed] [Google Scholar]
- 77.Lo YM, Lau TK, Zhang J, Leung TN, Chang AM, Hjelm NM, et al. Increased fetal DNA concentrations in the plasma of pregnant women carrying fetuses with trisomy 21. Clin Chem. 1999;45:1747–51. [PubMed] [Google Scholar]
- 78.Zhong XY, Burk MR, Troeger C, Jackson LR, Holzgreve W, Hahn S. Fetal DNA in maternal plasma is elevated in pregnancies with aneuploid fetuses. Prenat Diagn. 2000;20:795–8. doi: 10.1002/1097-0223(200010)20:10<795::AID-PD897>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 79.Tong YK, Ding C, Chiu RW, Gerovassili A, Chim SS, Leung TY, et al. Noninvasive prenatal detection of fetal trisomy 18 by epigenetic allelic ratio analysis in maternal plasma: Theoretical and empirical considerations. Clin Chem. 2006;52:2194–202. doi: 10.1373/clinchem.2006.076851. [DOI] [PubMed] [Google Scholar]
- 80.Tong YK, Jin S, Chiu RW, Ding C, Chan KC, Leung TY, et al. Noninvasive prenatal detection of trisomy 21 by an epigenetic-genetic chromosome-dosage approach. Clin Chem. 2010;56:90–8. doi: 10.1373/clinchem.2009.134114. [DOI] [PubMed] [Google Scholar]
- 81.Lo YM, Tsui NB, Chiu RW, Lau TK, Leung TN, Heung MM, et al. Plasma placental RNA allelic ratio permits noninvasive prenatal chromosomal aneuploidy detection. Nat Med. 2007;13:218–23. doi: 10.1038/nm1530. [DOI] [PubMed] [Google Scholar]
- 82.Lo YM, Lun FM, Chan KC, Tsui NB, Chong KC, Lau TK, et al. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc Natl Acad Sci USA. 2007;104:13116–21. doi: 10.1073/pnas.0705765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:20458–63. doi: 10.1073/pnas.0810641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiu RW, Sun H, Akolekar R, Clouser C, Lee C, McKernan K, et al. Maternal plasma DNA analysis with massively parallel sequencing by ligation for noninvasive prenatal diagnosis of trisomy 21. Clin Chem. 2010;56:459–63. doi: 10.1373/clinchem.2009.136507. [DOI] [PubMed] [Google Scholar]