

OVERVIEW OF THE DISEASE
Hypoparathyroidism is an uncommon endocrine deficiency disease characterized by low serum calcium levels, elevated serum phosphorus levels, and absent or inappropriately low levels of parathyroid hormone (PTH) in the circulation.(1) Although much less common than its counterpart, primary hyperparathyroidism (PHPT), a disease characterized by overproduction of PTH,(1) hypoparathyroidism, like PHPT, also presents many therapeutic challenges. Over the past 10 years, we have gained greater understanding about the disorder with regard to epidemiology, genetics, skeletal disease, and therapies. A major therapeutic challenge is consistently effective management of the hypocalcemia while avoiding hypercalciuria and other complications. The challenges that patients with hypoparathyroidism face is recorded regularly by the chronicles of the Hypoparathyroidism Association.(2) Through its website, a newsletter, and an annual meeting, this group shares common experiences and coping strategies for patients and their doctors. Excerpting some of these experiences, patients repeatedly complain of fatigue, “brain fog,” tingling fingers and feet, cramping in the hands (“the claw”) and feet (“perching”), numbness around the mouth, twitching in the facial muscles, bone pain, general muscle cramps, chronic headaches, insomnia, and the misery of having to carry around so much calcium (“It’s heavy stuff!”). Besides these highly variable and subjective changes, many of which are specifically associated with hypocalcemia, affected individuals frequently have increased urinary calcium excretion, which can lead to nephrocalcinosis and kidney stones or even chronic kidney disease. Hyperphosphatemia can be associated with deposition of calcium-phosphate complexes in other soft tissues with further negative sequelae in target organs.
On the horizon is the possibility that PTH, the missing hormone, might be developed successfully to treat this disease. In fact, hypoparathyroidism is the last classical endocrine deficiency disease for which treatment with the missing hormone is not standard therapy. The purpose of this article is to review, in a comprehensive manner, major aspects of the disease. This review represents a summary of the First International Workshop on Hypoparathyroidism, which was held in November 2009. This review focuses primarily upon hypoparathyroidism in the adult, because the workshop did not focus upon hypoparathyroidism in infancy or in childhood. Also, issues surrounding the diagnosis and management of the condition in pregnancy and lactation are not addressed
CLINICAL PRESENTATION
Patients with hypoparathyroidism most often present with paresthesia, cramps, or tetany, but the disorder may also manifest acutely with seizures, bronchospasm, laryngospasm, or cardiac rhythm disturbances. In the postsurgical setting, the presentation can be acute with tetany, cramping, tachycardia, and altered mental status dominating the picture. The disorder occurs in both acquired and inherited forms.
CLASSIFICATION OF HYPOPARATHYROID DISORDERS
Classification of parathyroid disorders is helpful in diagnosis and management. The disease may appear as an isolated disorder or in association with other organ defects. Usually the disease is identifiable as hereditary. These inherited disorders of hypoparathyroidism are often classifiable according to the defined genetic defects, including abnormalities of PTH biosynthesis, PTH secretion, parathyroid gland development, or parathyroid tissue destruction. Genetic defects may also be associated with complex syndromes involving other organ defects. There are also rarely acquired reversible causes. Other classifications relate to hypoparathyroidism associated with complex syndromes involving resistance to PTH or other endocrine gland abnormalities (Table 1).
TABLE 1.
Classification of Congenital Hypoparathyroid Disorders With Genetic Characterization
| Disorder | Gene Defect/Chromosome Locus |
|---|---|
| Isolated Hypoparathyroidism | |
| Autosomal recessive | PTH/11p15 |
| GCMB/6p24.2 | |
| Autosomal dominant | PTH/11p15 |
| CaSR/3q21.1 | |
| GCMB/6p24.2 | |
| X-linked | SOX3/Xq26-27 |
| Hypoparathyroidism With Additional Features | |
| Polyglandular autoimmune syndrome | AIRE/21q22.3 |
| DiGeorge syndrome | TBX1/22q11 |
| Hypoparathyroidism-retardation-dysmorphism syndrome | TBCE/1q42-43 |
| Hypoparathyroidism-deafness-renal dysplasia syndrome | GATA3/10p13-14 |
| Mitochondrial Disorders Associated With Hypoparathyroidism | |
| Kearns-Sayre syndrome | Mitochondrial genome |
| Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes | Mitochondrial genome |
| Mitochondrial trifunctional protein deficiency syndrome | Unknown |
| Several Other Forms | |
| Defective PTH Action | |
| Pseudohypoparathyroidism | |
| Type 1a | GNAS/20q13.3 |
| Type 1b | GNAS/20q13.3 |
| Blomstrand chondrodysplasia | PTHR1/3p22-p21.1 |
Postsurgical Hypoparathyroidism
The most common acquired cause of hypoparathyroidism in adults is postsurgical hypoparathyroidism.(3) Surgery on the thyroid or parathyroid glands or adjacent neck structures or neck dissection surgery for malignancy may lead to acute or chronic hypoparathyroidism. Postsurgical hypoparathyroidism is usually due to inadvertent or unavoidable removal of or damage to the parathyroid glands and/or their blood supply. While transient hypoparathyroidism after neck surgery is relatively common, often called “stunning” of the glands, chronic partial hypoparathyroidism is less common, and chronic complete hypoparathyroidism relatively rare. The diagnosis of chronic hypoparathyroidism requires that features of hypoparathyroidism persist for at least 6 months after surgery. Most patients with postsurgical hypoparathyroidism recover parathyroid gland function within several weeks to months after surgery and, thus, do not develop permanent disease. Some patients with chronic hypoparathyroidism have a period of being relatively asymptomatic, and their biochemical abnormalities are found in a routine checkup or during the routine investigation of related but nonspecific symptoms (eg, fatigue, muscle aching). The development of postsurgical hypoparathyroidism, years after neck surgery, suggests that age-related compromise of the remaining parathyroid tissue eventually leads to gland hypofunction. The mechanism of this time-related process is not clear but eventual deficiency of the parathyroid blood supply is an attractive possibility.
The rates of postsurgical hypoparathyroidism vary across centers and with different procedures and surgical expertise. Surgical centers with experienced endocrine surgeons and a high case volume report rates of post-thyroid surgical permanent hypoparathyroidism of 0.9-1.6%.(4-6) Earlier reports had suggested that after thyroid surgery, permanent hypoparathyroidism can occur with a frequency as high as 6.6%.(7) These studies emphasize the importance of expertise and experience.
Transient hypoparathyroidism after thyroid surgery occurs with much higher frequency, ranging from 6.9% to 46%.(8-10) Parathyroid dysfunction after surgical manipulation of neck structures commonly occurs several days to weeks and even years after the procedure. Postoperative hypoparathyroidism is more likely to occur in patients who have undergone more than one neck operation and/or if extensive thyroid resection is required. Surgery for substernal goiter, head or neck malignancies involving the anterior neck structures, or Graves disease have all been shown to increase the risk of postoperative hypoparathyroidism.
Asari et al prospectively analyzed 170 patients undergoing total thyroidectomy for a variety of diagnoses.(11) Postoperative hypoparathyroidism was defined as a documented postsurgical serum calcium level of <1.9 mmol/L (<7.6 mg/dL), with or without symptoms, or postoperative serum calcium level of 1.0-2.1 mmol/L (4.0-8.4 mg/dL) with neuromuscular symptoms, 2 days after surgery. The study showed that a PTH level of ≤15 pg/mL, or postoperative serum calcium level of ≤1.9 mmol/L (≤7.6 mg/dL), on postoperative day 2, increased the risk of postoperative hypoparathyroidism. Richards et al compared the risk of postoperative hypoparathyroidism before and after the introduction of intraoperative PTH monitoring and showed that the risk of postoperative hypoparathyroidism is markedly reduced when intraoperative PTH monitoring is used.(12)
There is not at present an accepted classification of hypoparathyroidism that arises without prior neck surgery. The older terminology referred to all nonsurgical hypoparathyroidism as “idiopathic.” When a specific cause is identified, such as an autoimmune or genetic etiology, that etiology replaces the term “idiopathic”. However, idiopathic hypoparathyroidism is used when the underlying cause is not known or has not been investigated.
Autoimmune Hypoparathyroidism
After postsurgical hypoparathyroidism, autoimmune hypoparathyroidism is the next most common form of hypoparathyroidism in adults. Autoimmune hypoparathyroidism may be isolated or part of an autoimmune polyglandular syndrome (APS).(13) Blizzard et al first reported anti-parathyroid gland antibodies in 38% of 75 patients with idiopathic hypoparathyroidism, 26% of 92 patients with idiopathic Addison disease, 12% of 49 patients with Hashimoto thyroiditis, and 6% of 245 normal controls (14). These antibodies appeared to be specific for parathyroid tissue, because they were blocked by pre-absorption with parathyroid tissue extract but not with other tissue extracts. However, subsequent studies showed that some anti-parathyroid antibodies reacted with mitochondrial or endomysial antigens. Li et al reported that sera from 5 of 25 patients (25%) with autoimmune hypoparathyroidism, idiopathic hypoparathyroidism, or APS type 1 (APS-1) had immunoreactivity with the extracellular calcium-sensing receptor (CaSR).(15) Patients with autoimmune hypoparathyroidism for less than 5 years were more likely to have anti-CaSR antibodies. No anti-CaSR antibodies were seen in 22 controls or 50 patients with autoimmune disorders without hypoparathyroidism. Other studies subsequently showed varying rates of anti-CaSR antibody positivity, likely because of differences in technique. It is not yet clear whether the anti-CaSR antibodies play a causal role in the disease or serve as markers of tissue injury.(16)
Kifor et al reported two patients with activating anti-CaSR antibodies with direct functional actions on CaSR.(17) One patient had transient spontaneous mild hypoparathyroidism and Addison disease, and the other had severe hypoparathyroidism and Graves disease requiring thyroidectomy. Both patients had anti-CaSR antibodies detected by multiple techniques, that stimulated CaSR-transfected HEK293 (human embryonic kidney cell line 293) cells and inhibited PTH release by dispersed parathyroid adenoma cells. This study suggested that hypoparathyroidism resulted from a functional effect of the antibodies on the CaSR, and not irreversible parathyroid gland damage.
GENETIC DEFECTS
Hypocalcemia is observed in a variety of genetic disorders, including forms of hypoparathyroidism and pseudohypoparathyroidism (PHP). Hypoparathyroidism can be caused by mutations in one of several genes and may occur as an isolated disorder or associated with developmental defects. The two best-studied forms of PHP, PHP-Ia and PHP-Ib, are caused by mutations within or upstream of the GNAS locus on chromosome 20q13.3 that encodes the stimulatory G protein (Gsα) and several splice variants thereof. The various forms of hypoparathyroidism and PHP all share some common features, namely hypocalcemia and hyperphosphatemia, which are caused by low circulating levels of PTH or insensitivity to its action in the proximal renal tubules, respectively. In both disorders, serum 1,25(OH)2D is usually low, contributing to impaired intestinal calcium absorption. Fractional excretion of calcium is increased in hypoparathyroidism, but because of hypocalcemia, the filtered load of calcium and the 24-hour urinary calcium excretion may be reduced or inappropriately normal. In PHP, urinary excretion of calcium is lower than in hypoparathyroidism because renal resistance to hormone action is restricted to the proximal tubule; the elevated PTH is active in the distal renal tubules, where it promotes calcium reabsorption. Bone alkaline phosphatase activity is normal and bone resorption is diminished in hypoparathyroidism, while patients affected by PHP often have increased bone turnover and thus increased alkaline phosphate activity. In hypoparathyroidism and PHP, nephrogenous cyclic adenosine monophosphate (cAMP) excretion is low and renal tubular reabsorption of phosphate is high. However, while patients with hypoparathyroidism show a vigorous increase in urinary cAMP excretion when given PTH parenterally (Ellsworth-Howard test), patients with PHP show a blunted or absent response to exogenous PTH, which is consistent with resistance to PTH in the proximal tubule.
Hypoparathyroidism can be associated with a variety of syndromes or complex disorders that may be familial or due to a de novo mutation.(18-22) The genetic bases for some of these forms of hypoparathyroidism have been shown to be the disruption of one or more of the steps involved in the development of the parathyroid glands or in the production or secretion of PTH. These genetic studies have shed light on the pathogenesis of the hypoparathyroid disorders, leading to a classification based on whether they arise either from an abnormality in PTH synthesis or secretion or from insensitivity to PTH in the proximal renal tubules observed in PHP. These studies have made it possible to recognize previously unknown mechanisms regulating parathyroid gland development, PTH secretion, and PTH-mediated actions in target tissues (Table 1).
Isolated Hypoparathyroidism
Hypoparathyroidism can occur as a solitary endocrinopathy, referred to as isolated hypoparathyroidism. In most patients, no clear genetic basis is known. Familial occurrences of isolated hypoparathyroidism with autosomal dominant, autosomal recessive, or X-linked recessive modes of inheritance have been established. Autosomal forms of hypoparathyroidism are caused by mutations in the genes encoding PTH, GCMB ([glial cells missing homologue B] discussed below), or the CaSR,(18-22) but for most idiopathic cases of hypoparathyroidism, the genetic defect remains unknown (Table 1).
Other less common acquired causes of hypoparathyroidism include excessive accumulation of iron in the parathyroid glands due to thalassemia or hemochromatosis.(23) Excessive accumulation of copper in Wilson disease is estimated to have a prevalence of 1:50,000 to 1:100,000.(24) Acquired hypoparathyroidism has been reported to occur very rarely after iodine-131 therapy or metastatic infiltration of the parathyroid glands.(25)
Reversible hypoparathyroidism can occur with magnesium deficiency(26) due to malabsorption syndromes, alcoholism, and other states of poor nutrition. Proton pump inhibitor therapy can be associated with hypocalcemia in hypoparathyroidism(26a). Magnesium excess due to tocolytic therapy for preterm labor(27) may cause hypoparathyroidism because of magnesium-associated inhibition of PTH secretion. In the special situation of maternal hypercalcemia in pregnancy (i.e., primary hyperparathyroidism), the newborn can be hypocalcemic, and although usually a transient problem, prolonged suppression has occurred.
PTH Gene
The very rare cases of hypoparathyroidism caused by PTH gene mutations that lead to altered processing of the pre-pro-PTH molecule and/or to mRNA translation can follow either autosomal recessive or dominant inheritance.(28,29) Homozygous mutations in the pre-pro-PTH gene cause very low or undetectable levels of PTH leading to symptomatic hypocalcemia and hyperphosphatemia. DNA sequence analysis of the PTH gene from patients with autosomal dominant isolated hypoparathyroidism has revealed a single base substitution (thymine/cytosine) in codon 18 of exon 2, and the resulting mutant PTH molecule has a dominant negative effect that leads to no or very inefficient translocation of the nascent wild-type and mutant PTH molecule across the endoplasmic reticulum and to apoptosis.(30,31)
GCMB Gene
The human homologue of the Drosophila gene gcm (glial cells missing) and of the mouse gcm2 gene, is named GCMB in humans and is expressed almost exclusively in the parathyroid glands, suggesting an important role in parathyroid gland development.(32) Mice homozygous for deletion of gcm2 lack parathyroid glands and develop hypocalcemia and hyperphosphatemia.(32) Hypocalcemia in these animals can be corrected by PTH infusion, indicating that gcm2 does not affect the response to PTH. These observations prompted investigations regarding the potential role of the GCMB gene in the pathogenesis of congenital hypoparathyroidism. Ding et al identified a homozygous intragenic deletion of exon 5 in the GCMB gene in a family affected by severe hypocalcemia at an early age with no measurable circulating PTH.(33) Other cases of autosomal recessive isolated hypoparathyroidism were subsequently shown to be caused by homozygous point mutations.(34-36)
More recently, Mannstadt et al described two families in which hypocalcemia and low circulating levels of PTH were inherited as an autosomal dominant trait. In one family, the index case was discovered to have hypocalcemia as part of a routine checkup during pregnancy, but the woman did recall having had symptoms suggestive of mild hypocalcemia for several years.(37) The second kindred encompassed 10 affected family members in 4 generations. In both families, affected members carried a heterozygote, single base-pair deletion within the C-terminal portion of GCMB gene that resulted in a shift in the open reading frame, thus extending the encoded protein; the mutant GCMB molecules revealed a dominant negative effect when tested in vitro. However, most patients with isolated hypoparathyroidism do not appear to have GCMB mutations or mutations in the other genes known to cause hypoparathyroidism,(38) making it likely that additional genetic mutations that can cause isolated hypoparathyroidism will be identified.
CaSR Gene
The CaSR gene encoding the CaSR protein was another candidate gene whose sequence was examined in the search for the genetic bases of congenital hypoparathyroidism. Indeed, CaSR mutations that result in a gain of function lead to hypocalcemia with hypercalciuria, with the majority of activating CaSR mutations (more than 40 described) located within the extracellular domain of this G-protein coupled receptor.(39) In a survey carried out by Pidasheva et al, activating CaSR gene mutations were proposed as the most frequent cause of congenital hypoparathyroidism.(40) It is now recognized that certain patients with activating mutations of the CaSR gene can present with the phenotype of Bartter’s syndrome (classified as subtype 5), including wasting of calcium, magnesium, sodium, and chloride in the urine.(41)
Activating CaSR mutations cause a left-shifted set point for PTH secretion, defined as the extracellular calcium level required for half-maximal suppression of secretion, causing inappropriately normal or low PTH levels even at low serum calcium levels. Hypoparathyroidism due to activating CaSR mutations follows an autosomal dominant mode of inheritance with high penetrance. Consequently, about half of first-degree relatives present with mild hypocalcemia and inappropriately low PTH levels. Affected individuals generally exhibit normal PTH serum concentrations, and treatment with active vitamin D metabolites often results in marked hypercalciuria and nephrocalcinosis, potentially leading to impaired renal function. Treatment of patients with autosomal dominant hypoparathyroidism due to CaSR mutations should be performed with great care to increase the serum calcium level only into the low normal range to avoid episodes of severe hypercalciuria. Treatment with injections of synthetic PTH(1-34) has shown efficacy, especially if the peptide is given 2 or 3 times daily.(42)
X-linked Hypoparathyroidism
X-linked recessive hypoparathyroidism was originally reported in two multigenerational kindreds from Missouri, US,(43) who were later shown to be related.(44) In this disorder, only males are affected, with infantile onset of epilepsy and hypocalcemia; the responsible gene was localized to chromosome Xq26-27.(45) The insertion of genetic material from chromosomes 2p25.3 into the Xq27.1 region is thought to cause a position effect on possible regulatory elements controlling SOX3 gene transcription, thus impairing parathyroid gland development.(46)
Unknown
Clearly, more genes will be discovered in the characterization of the genetic bases of isolated hypoparathyroidism, as discussed in the analysis of large case series.(47)
Hypoparathyroidism With Additional Features
Polyglandular Autoimmune Hypoparathyroidism
Hypoparathyroidism is a prominent component of APS1, also known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED).(48) Polyglandular autoimmune syndrome type 1is also abbreviated as PGA1 and PAS1. The syndrome consists of hypoparathyroidism, Addison disease, candidiasis, and at least two of the following: insulin-dependent diabetes, primary hypogonadism, autoimmune thyroid disease, pernicious anemia, chronic active hepatitis, steatorrhea, alopecia, and vitiligo. More than 80% of APS1 patients exhibit hypoparathyroidism, which may be their sole endocrinopathy. Presentation in childhood and adolescence is typical, but patients with only one disease manifestation should be followed long-term for the appearance of other components of the syndrome. The incidence worldwide is 1 per million, but it is enriched in 3 genetically isolated populations: Finns (incidence 1:25,000), Sardinians (incidence 1:14,500), and Iranian Jews (incidence 1:9000).(49)
The syndrome is predominantly inherited as an autosomal recessive trait, although occasional cases with autosomal dominant pattern of inheritance have been reported. APS1 is caused by a mutation in an autoimmune regulator (AIRE) gene, a zinc-finger transcription factor present in thymus and lymph nodes and critical for mediating central tolerance by the thymus.(50,51) APS1, in contrast to other immune diseases, is monogenic and not associated with the major histocompatibility complex. To date, more than 58 APS1-causing mutations have been identified in the AIRE gene; in 9% of the affected patients, a mutation was identified on only one allele. There does not appear to be a genotype/phenotype correlation.(51)
NACHT leucine-rich-repeat protein 5 (NALP5), an intracellular signalling molecule strongly expressed in the parathyroid, may be a parathyroid-specific autoantigen present in APS1 patients with hypoparathyroidism. Antibodies to NALP5 are absent in patients who do not present with this disorder.(52) Interestingly, the extracellular domain of the CaSR has also been identified as an autoantigen in patients with autoimmune hypoparathyroidism. Activating antibodies to this portion of the receptor have been reported in both APS1 and in acquired hypoparathyroidism.(53-55) An important implication of these results is that, although the majority of APS1 patients do not have CaSR antibodies, there may be a small minority of patients in whom the hypoparathyroid state is the result of functional suppression of the parathyroid glands rather than their irreversible destruction.(56)
As B cells are required for AIRE-deficient mice to develop multiorgan inflammation, rituximab, a monoclonal antibody directed against B cells, was administered to these animals, with remission of the autoimmune disease.(57) This offers hope of applying this pharmacologic approach to human patients with APS1.
DiGeorge Syndrome
DiGeorge syndrome affects 1 in 4000-5000 live births.(58) The complete phenotype of DiGeorge syndrome includes usually asymptomatic hypocalcemia due to hypoparathyroidism (60% of cases), thymic aplasia or hypoplasia with immunodeficiency, congenital heart defects, cleft palate, dysmorphic facies, and renal abnormalities with impaired kidney function. The heterogeneity of defects observed in DiGeorge syndrome suggests a defect early during embryologic development. The syndrome most commonly arises from de novo mutations, but autosomal dominant inheritance has been reported. Molecular studies have shown that most cases (70-80%) of DiGeorge syndrome carry a hemizygous microdeletion within the 22q11.21-q11.23 chromosomal region.(58) Within this region, only the TBX1 gene has been shown to carry inactivating point mutations in some DiGeorge patients. This gene encodes a T-box transcription factor that is widely expressed in those embryonal tissues that give rise to many of the organs that can be clinically affected in this syndrome. Findings similar to or indistinguishable from those present in DiGeorge syndrome were reported in some patients with deletions of 10p13, 17p13, and 18q21.
In addition to the deletions leading to the complete DiGeorge syndrome, deletions within 22q11 can cause the conotruncal anomaly facies and velocardiofacial syndromes. In the latter condition, hypocalcemia has been found in up to 20% of cases. Because of the phenotypic variability of the different syndromes, these conditions are all included under the acronym CATCH-22, for complex of abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia with deletion of chromosome 22q11.
Hypoparathyroidism-Retardation-Dysmorphism Syndrome
Hypoparathyroidism-retardation-dysmorphism (HRD) syndrome is a rare form of autosomal recessive hypoparathyroidism encompassing two syndromes, the Sanjad-Sakati and the Kenny-Caffey syndromes.(59,60) Sanjad-Sakati syndrome is associated with parathyroid dysgenesis, short stature, mental retardation, microphthalmia, microcephaly, small hands and feet, and abnormal teeth. This disorder is seen almost exclusively in individuals of Arab descent. Kenny-Caffey syndrome is characterized by hypoparathyroidism, dwarfism, medullary stenosis of the long bones, and eye abnormalities. Both disorders are due to mutations in the TBCE gene on chromosome 1q42-43 that encodes a protein required for microtubule assembly in affected tissues, although a second gene locus for a closely related variant of the syndromes seems likely.
Hypoparathyroidism-Deafness-Renal Dysplasia Syndrome
Another syndrome that has been elucidated by molecular methods and clinical phenotyping is HDR syndrome, which is characterized by hypoparathyroidism, deafness, and renal dysplasia. It was first reported as an autosomal dominant syndrome in one family in 1992.(61) Usually patients had asymptomatic hypocalcemia with undetectable or inappropriately normal serum concentrations of PTH and normal response to PTH. After obtaining linkage to the chromosomal region 10p14-10pter, deletion mapping defined mutations or deletions in GATA3 that resulted in haploinsufficiency of the transcription factor GATA3, a protein that is critical for normal parathyroid, kidney, and otic vesicle development.(62)
Mitochondrial Disorders Associated With Hypoparathyroidism
Hypoparathyroidism has been described in 3 disorders characterized by mitochondrial dysfunction: the Kearns-Sayre syndrome; the mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome, and the mitochondrial trifunctional protein deficiency syndrome. Point mutations, deletions, rearrangements, and duplications of mitochondrial DNA maternally inherited have been described in these disorders.(63) The role of these mitochondrial DNA defects in the etiology of hypoparathyroidism remains to be further elucidated.
Defective PTH Action
Pseudohypoparathyroidism
Several clinical disorders characterized by end-organ resistance to PTH have been described that are collectively referred to as PHP, ie, hypocalcemia and hyperphosphatemia, in the presence of high plasma PTH level indicative of resistance in target tissues (chronic renal failure and magnesium deficiency or vitamin D deficiency states need to be excluded).(19) Consistent with PTH resistance, rather than deficiency, infusion of biologically active PTH fails to increase urinary cAMP and phosphate excretion(64-66) (Table 1). The blunted response to PTH in subjects with PHP type I is caused by maternally inherited, heterozygous GNAS mutations that lead to Gsα deficiency in the proximal renal tubules (and a few other cells or tissues). Thus, this is a deficiency of the most prominent signalling protein that couples the PTH/PTH-related protein (PTHrP) receptor (and other G protein-coupled receptors, such as the thyroid-stimulating hormone receptor and the growth hormone-releasing hormone [GHRH] receptor) to the adenylate cyclase enzyme.
1. Pseudohypoparathyroidism Type Ia
Patients show about a 50% reduction in Gsα activity, which is caused by maternally inherited, heterozygous GNAS mutations. The nucleotide changes lead to inactivation of the Gsα protein due to a shift in the open reading frame with a premature termination codon, to missense mutations, to abnormal splicing, or to large interstitial deletions/inversions that completely or partially eliminate Gsα expression. Because Gsα is derived in the proximal renal tubules only from the maternal allele (the paternal copy is imprinted and, therefore, silenced), these maternal GNAS mutations are expected to lead to a complete or nearly complete lack of this signalling protein in the proximal, but not the distal, renal tubules. In addition to the laboratory findings, patients affected by PHP-Ia can present with clinical features that are referred to as Albright hereditary osteodystrophy (round face, mental retardation, frontal bossing, short stature, obesity, brachydactyly, and/or ectopic ossification), presumably related to the deficiency of Gsα alleles in the relevant tissues during development. Hypothyroidism develops in the majority of patients because of resistance to thyrotropin; less frequently, hypogonadism, which occurs as a result of gonadotropin resistance; and frequently, GHRH resistance, explaining the short stature and thus the favorable response to recombinant human growth hormone.(67)
Subjects with paternally inherited GNAS mutations have some phenotypic features of Albright osteodystrophy but do not display hormonal resistance because the genetic imprinting (of the paternal allele) leaves the normal allele expressed. This condition is termed pseudo-PHP. It is not unusual to find extended families in which some members have pseudo-PHP (paternally inherited GNAS mutations), while others present with PHP-Ia (maternally inherited GNAS mutations). Because of this distinct mode of inheritance, both disorders never occur in the same sibship.
2. Pseudohypoparathyroidism Type Ib
Similar to individuals affected by PHP-Ia, individuals with PHP-Ib develop Gsα deficiency in the proximal renal tubules. For the autosomal dominant form of PHP-Ib, this deficiency is caused by maternally inherited microdeletions within or upstream of the GNAS locus that are associated with loss of one or several of the 4 maternal methylation imprints within GNAS. With the exception of a few cases with paternal uniparental isodisomy, most sporadic cases of PHP-1b, which also present with abnormal GNAS methylation and associated PTH resistance, have not been defined thus far at the molecular level. Of note, recent studies have shown that these sporadic cases of PHP-Ib can present with some of the clinical features of Albright osteodystrophy, including brachydactyly.(68) Typically, though, they do not present with the skeletal abnormalities characteristic of PHP-Ia.
3. Pseudohypoparathyroidism Type Ic
Pseudohypoparathyroidism type Ic (PHP-Ic) is a variant of PHP-Ia that follows the same mode of inheritance as the latter disorder, and affected individuals exhibit the same features of Albright osteodystrophy, in addition to resistance toward multiple hormones. However, because of the assay that is usually used to document Gsα deficiency, patients affected by PHP-Ic show no demonstrable defect in Gsα activity; the GNAS gene mutations leading to PHP-Ic usually reside in the last exon encoding Gsα.
4. Pseudohypoparathyroidism Type II
In subjects affected by pseudohypoparathyroidism type II (PHP-II), PTH resistance is characterized by a reduced phosphaturic response to administration of PTH, despite a normal increase in urinary cAMP excretion. This variant lacks a clear genetic or familial basis, and the possibility that it could be an acquired defect has been proposed. Indeed, a similar clinical and biochemical picture occurs in patients with severe deficiency of vitamin D that always needs to be excluded in PHP-II patients. However, it remains uncertain that the increase in PTH secretion associated with vitamin D deficiency leads to PTH-resistant hyperphosphatemia.
Blomstrand Lethal Chondrodysplasia
Blomstrand chondrodysplasia is a lethal, autosomal recessive disorder characterized by abnormal endochondral bone formation with prematurely occurring mineralization of the cartilaginous bone templates. This disorder is caused by homozygous or compound heterozygous mutations in the PTH/PTHrP receptor that impair its function.(69-71) Secondary hyperplasia of the parathyroid glands occurs as a result of presumed hypocalcemia.
DIFFERENTIAL DIAGNOSIS OF HYPOPARATHYROIDISM
The hypocalcemic patient may harbor one of a number of disorders. The different forms of hypoparathyroidism are the subject of this article but it is important to consider other etiologies that can also present with hypocalcemia. The major general distinction to be made is whether the hypocalcemia is associated with an absent or inappropriately low serum parathyroid hormone concentration (hypoparathyroidism) or whether the hypocalcemia is associated with an appropriate compensatory increase in parathyroid hormone. Figure 1 presents a practical schema whereby one can distinguish between these possibilities.
FIG. 1.
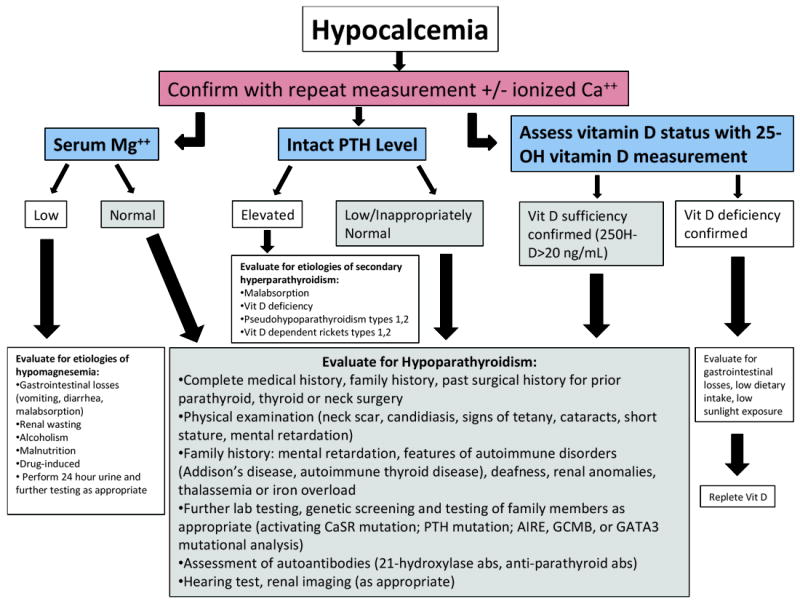
A clinical algorithm for the workup of the patient who presents with hypocalcemia, which should be confirmed with a repeat measurement, along with the specific steps in the workup to confirm the diagnosis of hypoparathyroidism. Additional etiologies for hypoparathyroidism are discussed in greater detail in the text, to which the reader is referred for further reference, and definitive genetic testing resources for explicit diagnoses and mutational analysis should be researched by the treating clinician or a geneticist.
Hypocalcemia along with a frankly low or inappropriately low-normal intact PTH level is the initial biochemical profile that leads the clinician to suspect hypoparathyroidism. Once suspected, it is important to confirm the presence of a low ionized calcium level and a normal serum level of magnesium. Hypomagnesemia can lower the PTH and total or ionized calcium levels and must be ruled out, because the etiologies and treatments are quite different for the two disorders. Although PHP also presents with hypocalcemia and hyperphosphatemia, like true hypoparathyroidism, the intact PTH levels are always elevated and sometimes dramatically so—confirming that the parathyroid glands are functional and appropriately responding to the hypocalcemia. The phenotypical features of PHP-1a, which include Albright hereditary osteodystrophy, would be a strong signal that PTH resistance and not hypoparathyroidism is present.
Because the most common etiology of hypoparathyroidism is inadvertent removal of or damage to the parathyroid glands during thyroid or parathyroid surgery, ascertaining any relevant surgical history is the critical first step in determining the etiology for the disorder. If there has been no prior surgery or external beam radiation or iodine-131 treatment (as much less common etiologies for gland destruction), one must consider other causes for low intact PTH levels. Autosomal dominant hypocalcemia due to mutations in the CaSR gene that cause constitutive activity is one of the first considerations as it may be one of the most common nonsurgical causes of hypoparathyroidism. These individuals manifest biochemical profiles similar to patients with idiopathic or postsurgical hypoparathyroidism. Their hypocalcemia is often milder and, in adults, rarely symptomatic. Biochemically, they tend to have hypercalciuria, and it has been shown that their levels of urinary calcium are higher than those of patients with hypoparathyroidism or other etiologies with comparable degrees of hypocalcemia, because of the constitutive activity of the CaSRs in the kidney. However, urinary calcium excretion may be difficult to use as a discriminating feature between the two disorders, especially if the patient is taking large amounts of calcium supplements and calcitriol at the time of evaluation. Family screening is helpful, because this condition follows an autosomal dominant pattern of inheritance and is highly penetrant. Mild hypocalcemia with inappropriately low normal or frankly low PTH levels may be evident in approximately half of first-degree relatives. However, there can be wide variability in the biochemical phenotype within a family. A definitive diagnosis can be made by sequencing the proband’s CaSR gene along with that of an unaffected family member(s). Numerous point mutations have been reported that render the CaSR hyperactive in the presence of low or low normal extracellular calcium concentrations. Definitive diagnosis can be difficult, however, if one encounters a new mutation (one that has not been previously reported or studied in vitro) in the case in question. The absence of the mutation in the unaffected family member would be reassuring that the mutation tracks with the disease, but further consultation should be sought with a genetics specialist and possibly research laboratories involved in the study of CaSR biology to determine the function of the new mutation in vitro. There are many presumably benign polymorphisms in the CaSR gene that have been encountered and others that may turn up in the course of sequencing the CaSR in patients. This must be borne in mind when interpreting results in a patient with hypocalcemia.
The next most likely etiology is autoimmune destruction of the parathyroid glands. This can occur sporadically as an isolated condition or as part of APS-1. The skin can also be involved with vitiligo and, more seriously, mucocutaneous candidiasis. The latter often (although not invariably) presents in childhood, as frequently as adrenal insufficiency and hypoparathyroidism. Securing a complete family history is essential in the next steps of differential diagnosis. APS-1 due to AIRE gene mutations is usually autosomal recessive, so the parental generation is generally skipped.(13,72,73) If hypoparathyroidism occurs in isolation without other autoimmune phenomena, the clinician must consider other autosomal dominant or recessive or even an X-linked recessive form of hypoparathyroidism.(7) These are exceedingly rare entities. Mutations in transcription factors that play a role in parathyroid gland development or in the pre-pro-PTH gene itself must be considered. The pattern of inheritance does not always determine the disorder because hypoparathyroidism due to AIRE mutations and GCMB mutations can rarely be autosomal dominant in their pattern of inheritance. GATA3 mutations usually produce a phenotype that follows an autosomal dominant pattern or presentation. Renal anomalies and hearing loss often accompany the hypoparathyroidism in patients with GATA3 mutations, so they should be looked for clinically.
In patients who present with hypoparathyroidism and multiple anomalies beyond the parathyroid gland, the following conditions should be considered: (1) DiGeorge syndrome(74,75); (2) mitochondrial gene mutations(76,77); and (3) HRD syndrome.(78) DiGeorge syndrome may present with cardiac, palatal, ocular, pharyngeal, immunologic, and cognitive defects due to developmental abnormalities in multiple tissues. Mitochondrial gene mutations may be associated not only with hypoparathyroidism but also diabetes, cardiomyopathy, ophthalmoplegia, and other complications.(76,77) HRD syndrome is also exceedingly rare. A careful clinical assessment of the patient, screening of the family, and judicious use of laboratory and genetic testing are used in concert to establish the diagnosis in a given patient with hypoparathyroidism.
MEASUREMENT OF PARATHYROID HORMONE
PTH Assays and the Diagnosis of Hypoparathyroidism
Successive advances in immunoassays of PTH have led to improvements in both the sensitivity and specificity the measurement of the clinically significant and secreted active form of PTH, namely PTH(1-84) (Figure 2). The first generation of PTH radioimmunoassays (RIAs) prevailed between 1963(79) and 1987. These initial RIAs used multivalent antibodies developed against partially or fully purified bovine PTH(1-84). Standards consisted of partially purified bovine PTH(1-84) or human serum containing high concentrations of PTH.(79) Most of these assays reacted with the C-region of PTH(1-84) and primarily identified biologically inactive C-fragments in the circulation. In addition to their lack of specificity for PTH(1-84), their varied characteristics resulted in the inability to compare results between RIAs for PTH.
FIG. 2.

Relationship between parathyroid hormone (PTH) structure, PTH assay generations, known PTH assay epitopes, and known circulating PTH molecular forms. With each generation, PTH assays have become more selective toward full-length PTH(1-84).
The second generation of assays for PTH was ushered in after 1987(80) by the advent of the immunoradiometric assay (IRMA). These much more useful second generation assays provided enhanced sensitivity and specificity. The principle of the PTH IRMA is to capture PTH molecules with one antibody directed against the carboxyl terminal region and detect PTH with a second radiolabeled or enzyme-labeled antibody directed toward the amino-terminus of PTH. This technique allowed for greater specificity and seemed to detect only the full-length molecule without interference by circulating carboxyl fragments. The enhanced specificity, moreover, led to more accurate discrimination of hyperparathyroidism from normal subjects–-a major diagnostic advance—and also allowed, for the first time, a direct comparison of results between laboratories.
Although second-generation IRMA assays were denoted as “intact” PTH assays, it was subsequently shown that they also detected nearly full-length but biologically inactive amino-terminally truncated PTH molecules, such as PTH(7-84), the existence of which had not previously been appreciated.(81-83) These large N-terminally truncated fragments represent approximately 20% of second-generation PTH immunoreactivity in normal subjects and up to 45% in renal failure, because the clearance of these larger fragments depends upon renal excretion.(81,84)
In order to eliminate from detection these large, inactive PTH fragments, third-generation PTH assays were developed. They accomplished the goal, to detect only biologically active PTH(1-84), by recognizing a detection epitope of PTH that was in the amino-terminal 1-7 region of the molecule.(83) Similar to the second-generation assays, they use solid phase C-terminal capture antibodies, but unlike the second-generation assays, they do not detect PTH(7-84) fragments. However, it subsequently became apparent that the third-generation assays detect another PTH form that represents a post-translational modification of hPTH(1-84). This circulating species, termed N-PTH, represents less than 10% of the immunoreactivity detected by these third-generation assays in normal subjects and up to 15% in advanced renal failure.(85) In parathyroid cancer, this species becomes even more predominant.(86) Although they provide a clear advantage over the second-generation assays, the third-generation PTH assays have not been shown to be more useful than second-generation assays in the diagnosis of PHPT.(87) It is likely that the third-generation assays are not more useful in the diagnosis of hypoparathyroidism,(88) although very few studies are available. Diagnosing patients with hypoparathyroidism can be improved by using stimulation tests rather than basal levels,(89) but they are generally not used.
SKELETAL DISEASE IN HYPOPARATHYROIDISM
Regardless of its etiology, the effects of chronic PTH deficiency on the human skeleton are profound. In normal adults, bone mass is regulated by a delicate balance between bone resorption and formation in a tightly regulated process termed remodeling. PTH is one of the key regulators of the rate of bone remodeling. A reduction or absence of circulating PTH leads initially to a decrease in bone resorption and then to a coupled reduction in bone formation. However, the balance between resorption and formation favors the latter as, over time, bone mass increases. This effect is manifested in both cancellous and cortical bone compartments.(90-94) Greater insight into the architectural basis of the increase in bone mass can be obtained by peripheral quantitative computed tomography (pQCT). Using this technique, Chen et al compared volumetric bone mineral density (vBMD) and geometry of the distal radius and midradius among postmenopausal women with postsurgical or idiopathic hypoparathyroidism, PHPT, and normal controls.(93) At the 4% distal radius site, which is enriched in cancellous bone, trabecular vBMD was higher in the rank order hypoparathyroidism>control>PHPT. At the 20% midradius site, cortical vBMD was also greater in the same rank order. The BMD differences among these 3 groups could be explained by differences in bone geometry. At both radial sites, total bone area and both periosteal and endosteal surfaces were greater in PHPT than in hypoparathyroidism, and controls and cortical thickness and area were higher in the rank order hypoparathyroidism>control>PHPT. Increased cancellous bone volume has been shown by high-resolution pQCT and increased mechanical strength has been suggested by microfinite element analysis (Figure 3).(95)
FIG. 3.

Images showing tissue stress levels under axial loading generated by microfinite element analysis based on microcomputed tomography of iliac crest bone biopsies (A, C, E) and high-resolution peripheral quantitative computed tomography (B, D, F) of the radius in a premenopausal woman with idiopathic osteoporosis (A and B), a normal premenopausal control (C and D), and a patient with hypoparathyroidism (E and F). Dark blue indicates the regions with lowest stress, and dark red indicates the regions with highest stress. The lowest stress levels are seen in the subject with hypoparathyroidism. Reproduced with permission.(95)
Although there are only two studies available, the most comprehensive information on the effects of hypoparathyroidism on the skeleton has come from histomorphometric analysis of the iliac crest bone biopsy. In the first of these studies, biopsies were obtained from 4 men and 8 women with vitamin D-treated hypoparathyroidism and 13 age- and gender-matched controls.(96) Nine of the subjects suffered from postoperative hypoparathyroidism of 2-53 years’ duration, and 3 had idiopathic disease. Ten of the patients were treated with 1-α-hydroxylated vitamin D (0.5-3.9 μg/d), and 2 received calciferol oil. There was a nonsignificant trend toward an increase in cancellous bone volume in the hypoparathyroid subjects, but the structural indexes, marrow star volume, trabecular star volume, and trabecular thickness were not different from controls. Bone forming surface and bone formation rate (BFR) were significantly reduced by 58% and 80%, respectively, in the hypoparathyroid subjects, and remodeling activation frequency was 0.13 per year, compared to 0.6 per year in controls. Initial mineral apposition rate was also lower, by a factor of 5, in the hypoparathyroid subjects, but this difference was not statistically significant. The total resorption period was prolonged from 26 to 80 days in the hypoparathyroid subjects and the resorption depth was reduced. The reconstructed remodeling cycles derived from these data are shown in Figure 4. The balance between the resorption depth and wall thickness of cancellous bone packets was slightly positive, by approximately 5 μm, in the hypoparathyroid subjects compared to the controls.
FIG. 4.
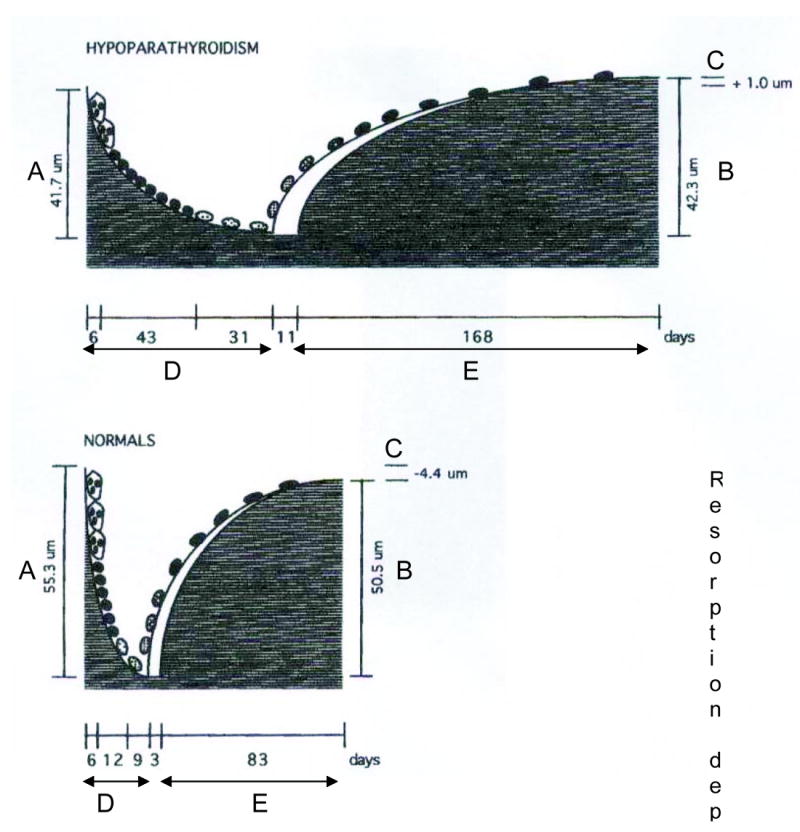
Reconstructed remodeling cycles in hypoparathyroid (upper) and normal (lower) subjects. Note the marked elongation of all phases of the remodeling cycle in hypoparathyroidism. Reproduced with permission.(95)
A more recent larger histomorphometric study involved 33 subjects (24 women and 9 men) with hypoparathyroidism treated with vitamin D and 33 age- and sex-matched control subjects.(94) The etiologies of the hypoparathyroid state were post-thyroid surgery (n=18), autoimmune (n=13), and DiGeorge syndrome (n=2), and the mean duration of the disease was 17±13 (SD) years. Vitamin D intake varied between 400 and 100,000 IU/d and calcium supplementation varied between 0 and 9 g/d. Ten of the 33 hypoparathyroid subjects were receiving thiazide diuretics. In contrast to the earlier smaller study,(96) cancellous bone volume was elevated in the hypoparathyroid subjects (Figures 5 and 6). The structural basis for the higher cancellous bone volume in hypoparathyroidism was an increase in trabecular width; trabecular number and trabecular spacing were both similar to controls. Cortical width was also significantly greater in the hypoparathyroid subjects, and cortical porosity was 17% lower than in controls, but this difference was not statistically significant. Remodeling activity was assessed separately on cancellous, endocortical, and intracortical skeletal envelopes. Osteoid surface and width were reduced in the hypoparathyroid subjects on all 3 envelopes. The tetracycline-based BFR was significantly lower on all 3 envelopes in the hypoparathyroid subjects with the most profound reduction (more than fivefold) seen on the cancellous envelope (Figure 7). The reduction in BFR was due to significant decreases in both mineralized surface and mineral apposition rate on all 3 envelopes. The eroded surface did not differ between the hypoparathyroid and normal subjects, but the bone resorption rate was significantly lower in the hypoparathyroid subjects on all 3 envelopes. As in the earlier study,(96) these findings are all indicative of a profound reduction in the bone turnover rate in hypoparathyroidism accompanied by an increase in bone mass in both cancellous and cortical compartments.
FIG. 5.

Low-power view of iliac crest bone biopsies from a control subject (left) and a subject with hypoparathyroidism (right). Goldner trichrome stain. Note the increase in cancellous bone volume and cortical thickness in the hypoparathyroid subject. Reproduced with permission from.(94)
FIG. 6.
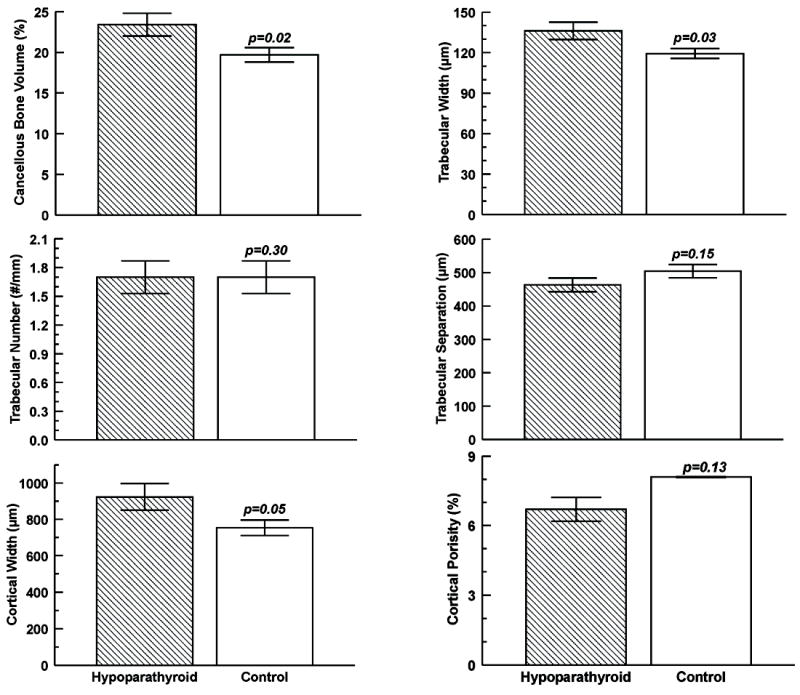
Cancellous and cortical bone parameters obtained by histomorphometry in subjects with hypoparathyroidism (hatched bars) and controls (open bars). Values are mean±SD. Drawn from data from Rubin et al.(94)
FIG. 7.
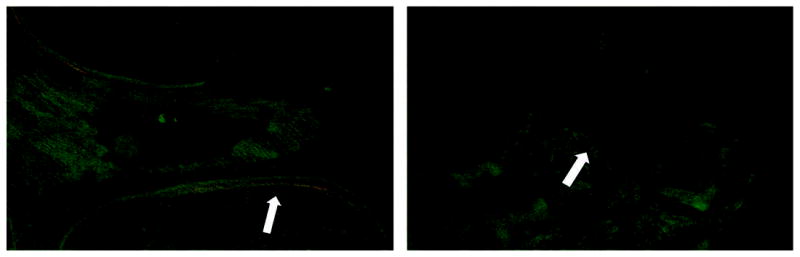
Tetracycline labels in a hypoparathyroid (left) and control subject (right). Tetracycline uptake was markedly reduced in the hypoparathyroid subject, reflecting the low turnover rate.
The effects of PTH deficiency on cancellous and cortical bone mass, which were initially observed by noninvasive imaging and by 2-dimensional histomorphometry, were recently confirmed by the 3-dimensional analytical capability afforded by microcomputed tomography.(97) Results from this study confirmed the increase in cancellous bone volume and trabecular thickness in hypoparathyroid subjects and demonstrated higher trabecular number and trabecular connectivity in comparison to matched control subjects. In addition, the structural model index was lower in hypoparathyroidism, indicating that the trabecular structure was more plate-like than rod-like (Figure 8).
FIG. 8.
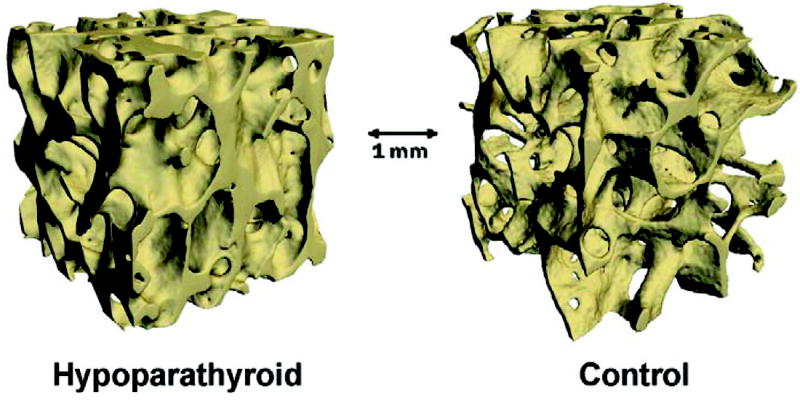
Reconstructed microcomputed tomographic images of cancellous bone from a hypoparathyroid (left) and a control subject (right). Note the dense trabecular structure in hypoparathyroidism. Reproduced with permission.(97)
Rubin and colleagues have also recently begun to explore the material composition of the bone matrix in hypoparathyroidism. Using backscatter electron imaging, they found that the mean mineralization density in iliac bone from subjects with hypoparathyroidism was similar to controls, although there was greater interindividual variation in mineralization parameters in the hypoparathyroidism subjects than in the controls.(98) This result is surprising as one might have expected that mineralization density would be enhanced in hypoparathyroidism because of the low turnover and the attendant increase in mineralization as the bone “ages.” It suggests that mineralization density is controlled by other factors, in addition to the degree of secondary mineralization, and indicates that the higher bone mineral density (BMD) by densitometry in hypoparathyroidism is due in large part to the increase in bone tissue volume rather than an increase in the amount of mineral within the tissue.
As evidenced by the work cited here, application of new research techniques to the study of hypoparathyroidism is leading to new insights regarding the skeletal abnormalities in this disease. We expect that this progress will continue at an even greater pace over the next 5 years and will lead to a much better understanding of the pathophysiology, as well as the biomechanical and metabolic effects, of a disease that has, until recently, received little attention.
CURRENT MANAGEMENT OF HYPOPARATHYROIDISM: APPROACHES AND ASSOCIATED COMPLICATIONS
Acute Management
In hypoparathyroidism, symptomatic hypocalcemia (carpal or pedal spasm, seizures, broncho- or laryngospasm) can be a medical emergency requiring acute intravenous administration of calcium. Although the actual value of the corrected serum calcium level is often regarded as a threshold for acute management (ie, 1.9 mmol/L [7.5 mg/dL]),(99-101) symptoms generally dictate the decision to administer acute therapy. Intravenous calcium gluconate should be used. Calcium chloride should be avoided because it is irritating and potentially sclerosing to veins.(99,102) Ten milliliters of 10% calcium gluconate diluted in 100 mL 5% dextrose (D5W) is infused intravenously over 5-10 minutes. This infusion provides 90 mg of elemental calcium and can be followed by a continuous infusion of a larger amount based on body weight: 15 mg/kg of elemental calcium generally translates to approximately 10 ampules (900 mg of elemental calcium) diluted in 1 L of D5W at 50 mL/h.(101-105) Over 8 hours, this infusion protocol will raise the serum calcium by approximately 2 mg/dL.(99) If the situation warrants (eg, digoxin therapy), the electrocardiogram can be monitored.(99,103,106) In the special situation of magnesium deficiency, calcium is given as described, but magnesium should also be administered.(99)
Chronic Management
In view of the fact that there are currently no formal guidelines, management of hypoparathyroidism is based upon experience and clinical judgment.(99)
The primary goals of chronic management include maintaining within an acceptable range the following indexes: (a) serum total calcium (usually in the low-normal range); (b) serum phosphorus (usually in the high-normal range), (c) 24-h urine calcium excretion (<7.5 mmol/d); (d) calcium-phosphate product under 55 mg2/dL2 (4.4 mmol2/L2).(99,106,107)
For chronic management, current treatment options include oral calcium, vitamin D (including its metabolites and analogs), and thiazide diuretics. In special situations, phosphate binders, low salt-diet, or a low-phosphate diet might be helpful adjuncts.(99)
Calcium
The recommended calcium supplements are calcium carbonate and calcium citrate, the latter being more consistently helpful in those with achlorhydria.(99,108,109) If calcium carbonate is to be used in subjects with achlorhydria, it should be taken with a protein-based meal to insure adequate absorption. (109a) The amount of calcium that is needed varies greatly among patients, from as little as 1 g/d to as much as 9 g/d.(99)
Vitamin D Metabolites
Along with calcium, therapy with native vitamin D and/or its active metabolites and analogs, is a cornerstone of management. 1,25(OH)2D3 (calcitriol), the active metabolite of vitamin D, maintains serum calcium, in part, by improving the efficiency of intestinal calcium absorption.(110) It also promotes bone remodeling through the RANKL signaling pathway.(108,110) Calcitriol is administered over a wide dosing range: 0.25-2.0 μg/d(99,103,106,111,118) and can increase serum calcium substantially within 3 days.(111-117) In modest doses (0.25-0.75 μg/d), a single daily dose is typically administered. When higher amounts are required, calcitriol is typically administered in divided doses. Vitamin D2 (ergocalciferol) or vitamin D3 (cholecalciferol) is often used along with the active metabolite of vitamin D. The longer half-life of the parent vitamin (2-3 weeks) helps to provide smoother control in view of the very short half-life of calcitriol, which is measured in hours.(114-117) The amount of parent vitamin D needed can be similar to amounts that euparathyroid individuals take (800-1500 IU daily) or can be in much higher doses (50,000 weekly or even more often). The analog alfacalcidol (1-alpha-hydroxyvitamin D3), which is rapidly converted to 1,25(OH)2D3 in vivo,(116) can be useful,(119). Dihydrotachysterol use, limited by availability and lower potency(108), is no longer available in the United States.
Thiazide Diuretics
By enhancing distal renal tubular calcium reabsorption, thiazide diuretics reduce urinary calcium excretion and thus are sometimes of value in hypoparathyroidism.(99,102,120-124) The therapeutic class of benzothiadiazines, including chlorothiazide, hydrochlorothiazide, polythiazide, and chlorthalidone, all lower urinary calcium excretion by this mechanism.(99,125) The use of hydrochlorothiazide may help to limit the amount of vitamin D that is needed to maintain normal calcium levels in hypoparathyroidism.(125-128) The action of thiazide diuretic therapy to lower urinary calcium excretion can be seen as early as 3-4 days after starting treatment. The effects on serum calcium, which appear to be greater than what would be expected by reduced calcium excretion alone may occur also through an effect on gastrointestinal calcium absorption.(129-132)
Limitations of Currently Approved Treatment Options
Overall Sense of Well-being
A recent cross-sectional study compared well-being and mood using validated questionnaires in 25 women with postsurgical hypoparathyroidism who were on stable treatment with calcium and vitamin D (or analogs) and in 25 women with intact parathyroid function and a history of thyroid surgery.(133) Serum calcium remained in the accepted therapeutic range (2.00-2.35 mmol/L) in 18 of the 25 hypoparathyroid patients. Hypoparathyroid patients had significantly higher global complaint scores in the Giessen complaint list, von Zerssen symptom list, and Symptom Checklist-90, with increases in subscale scores for anxiety, phobic anxiety, and their physical equivalents. Current standard therapy for hypoparathyroidism failed to restore well-being in these patients.
Hypercalcemia and Hypercalciuria
With the need for high doses of calcium supplements and vitamin D and its analogues to maintain serum calcium levels, it is not surprising that hypercalcemia is an ever-present concern in some patients. Treatment with the vitamin D sterols carries the risk of vitamin D toxicity, which can manifest as hypercalcemia, hypercalciuria, and hyperphosphatemia.(108) Although calcitriol has an advantage over parent vitamin D therapy because of its short half-life and lack of appreciable storage in fat, it may be associated more often with hypercalcemia because of its greater potency.(112,126,134,135) If the hypercalcemia is due to calcitriol, rapid reversal can be expected because of its short half-life.(134) Other metabolites, such as alfacalcidol and dihydrotachysterol, as well as vitamin D itself, have longer half-lives. Hypercalcemia due to these forms of vitamin D may take longer to resolve.(108,116,134) Not surprisingly, hypercalciuria is a common complication of therapy.(99,102,120) Hypercalciuria will occur typically before the serum calcium increases. The complications of hypercalciuria include nephrolithiasis, nephrocalcinosis, and renal dysfunction.(99,136-139) Close monitoring of the laboratory profile is warranted in all patients being treated with large amounts of calcium and vitamin D preparations.(99,103)
Hyperphosphatemia
Vitamin D therapy can be associated with hyperphosphatemia, since active vitamin metabolites and analogs also increase intestinal phosphate absorption.(108) When it does occur, the hyperphosphatemia may be lowered by reducing dietary intake of phosphate. In extreme situations, phosphate binders can be used.(99,107) Presumably, if the serum calcium concentration can be controlled and the tendency for hyperphosphatemia is minimized, extraskeletal complications will be less likely to occur. Basal ganglia calcifications, however, are common but they are only rarely associated with movement disorders.
Hypokalemia and/or Hyponatremia
Limitations of thiazide diuretics are related to the risk of developing hypokalemia and/or hyponatremia.(128) A low-sodium diet is an effective and inexpensive adjunct.
USE OF PARATHYROID HORMONE IN THE TREATMENT OF HYPOPARATHYROIDISIM
Treatment of hypoparathyroidism with PTH is appealing because it provides the hormone that is missing. Moreover, reducing calcium and calcitriol requirements in hypoparathyroidism, an expected consequence of using PTH in this disorder, has important advantages with regard to safety and efficacy. Reduced calcium and vitamin D requirements potentially lessen the risk of hypercalcemia and hypercalciuria. An additional possible advantage is that, because of its phosphaturic properties, PTH use may reduce the risk of soft-tissue deposition of calcium in the kidneys (nephrocalcinosis, nephrolithiasis) and possibly in other soft tissues.
Use of Teriparatide [PTH(1-34)] in Hypoparathyroidism
Replacement therapy using synthetic human parathyroid hormone 1-34 [PTH(1-34)] in adults with hypoparathyroidism was initially investigated in a crossover pilot study of 10 subjects.(140) The results demonstrated that PTH(1-34) maintained both serum and urinary calcium in the normal range over a 24-hour period when given as a single daily subcutaneous injection for 10 weeks. PTH(1-34) resulted in a lower urinary calcium level than calcitriol for a given level of serum calcium. Subsequently, results of a randomized controlled-dose study showed that twice-daily PTH(1-34) given for 14 weeks provided effective short-term treatment for hypoparathyroidism with a reduced total daily dose, an apparent reduction in bone turnover, and a decreased incidence of bone pain compared to a once-daily regimen.(141) Twice-daily PTH(1-34) produced higher levels of serum calcium with fewer fluctuations into the hypocalcemic range. Markers of bone turnover were elevated above the normal range in response to both treatment regimens. However, twice-daily PTH(1-34) produced significantly lower serum marker levels. In a subsequent long-term study, 27 adults with hypoparathyroidism were randomized to either calcitriol or twice-daily PTH(1-34).(142) The findings demonstrated that twice-daily PTH(1-34) could maintain serum calcium in the low normal or just below the normal range over a 3-year period; there were no statistically significant differences in urine and serum calcium between the two groups. In the PTH(1-34) treated group, however, markers of bone turnover (osteocalcin, alkaline phosphatase) were significantly elevated during the 3-year study, at levels at least twofold greater than normal. Despite this increase in bone turnover, there was no change in the BMD, as measured with dual energy x-ray absorptiometry (DXA). This contrasts with the calcitriol-treated group, which experienced a rise in spinal BMD over time with no rise in bone markers.
PTH(1-34) replacement therapy has also been studied in children with hypoparathyroidism. A similar 14-week study in children showed better metabolic control with twice-daily PTH(1-34) versus a single daily injection, however, there were no significant differences in bone markers between the 2 groups.(143) Most recently, results of a 3-year study in 12 children randomized to twice-daily PTH(1-34) or calcitriol demonstrated that PTH(1-34) can effectively manage the hypocalcemia of hypoparathyroidism.(144) As in adults, 3 years of PTH(1-34) replacement therapy resulted in significant increases in bone turnover markers compared to calcitriol-treated controls. In addition, Z-scores of the 1/3 radius measured by DXA were significantly decreased in the PTH(1-34) group compared to the calcitriol group after 3 years. Both of these pediatric studies reported that urinary calcium excretion remained within the normal range; however, the investigators failed to normalize urinary calcium excretion to body weight.
A recent report describes a 20-year-old hypoparathyroid woman with a CaSR mutation who was treated with PTH(1-34) for 14 years.(145) PTH(1-34) treatment did not ameliorate her hypercalciuria nor did it prevent the development of nephrocalcinosis. However, bone biopsies revealed dramatic increases in cancellous bone volume.
Use of PTH(1-84) in Hypoparathyroidism
The effects of treatment with PTH(1-84) (100 ug in an every-other-day treatment regimen) were studied in 30 hypoparathyroid subjects for 24 months.(146) The majority (n=22) of the subjects were female, and the two major etiologies of hypoparathyroidism were postsurgical and autoimmune. Duration of hypoparathyroidism ranged from 3 to 45 years. Most subjects had normal serum calcium levels on replacement therapy with calcium and vitamin D (mean baseline serum calcium, 2.1±0.2 mmol/L; normal range, 2.1-2.6 mmol/L); baseline BMD T-scores were normal or above the normal range (lumbar spine T-score, 1.7±2; femoral neck T-score, 0.7±2; distal 1/3 radius T-score, -0.03±1.0).
Calcium and Vitamin D Supplementation With PTH(1-84)
The reductions in calcium and vitamin D supplementation with PTH(1-84) were notable. Requirements for supplemental calcium fell significantly from 3030±2325 to 1661±1267 mg/d (P<0.05; Figure 9). Even more telling, the number of subjects on calcium supplementation that was greater than 1500 mg/d decreased from 22 (73%) at study entry to only 12 (40%) at the study conclusion. Similarly, calcitriol supplementation declined from the baseline mean of 0.68±0.5 to 0.40±0.5 μg/d (P<0.05; Figure 10). Again, fewer patients needed high supplementation; the number of subjects on a dose of 1,25-dihydroxyvitamin D that was greater than 0.25 μg/d fell from 25 (83%) at study entry to 15 (50%) at the study conclusion.
FIG. 9.
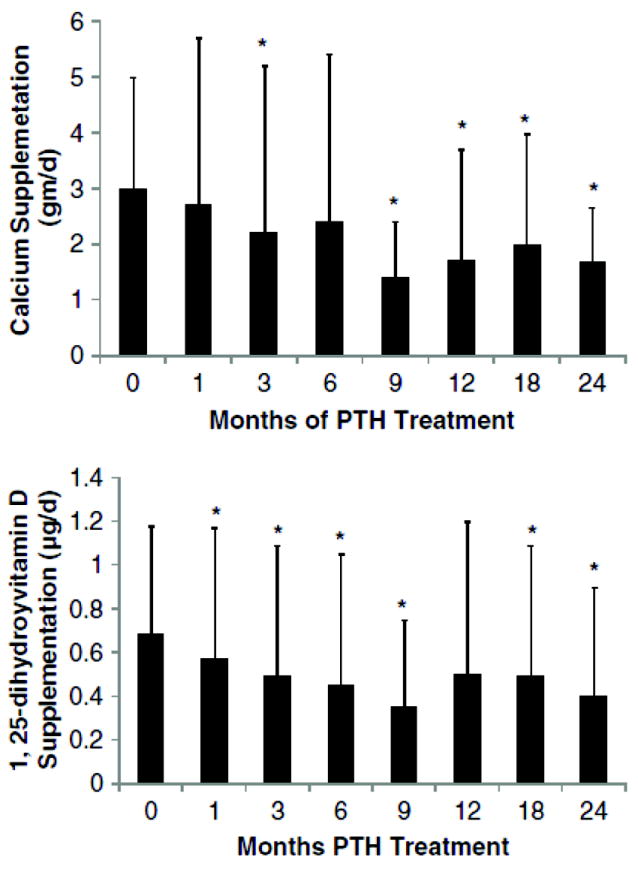
Changes in calcium and 1,25-dihydroxyvitamin D supplementation: Calcium requirements decreased at 3, 9, 12, 18, and 24 months from baseline while 1,25-dihydroxyvitamin D requirements decreased by 1 month. Data are mean±SD. *P<.05 as comparison to baseline. Reproduced with permission.(146)
FIG. 10.
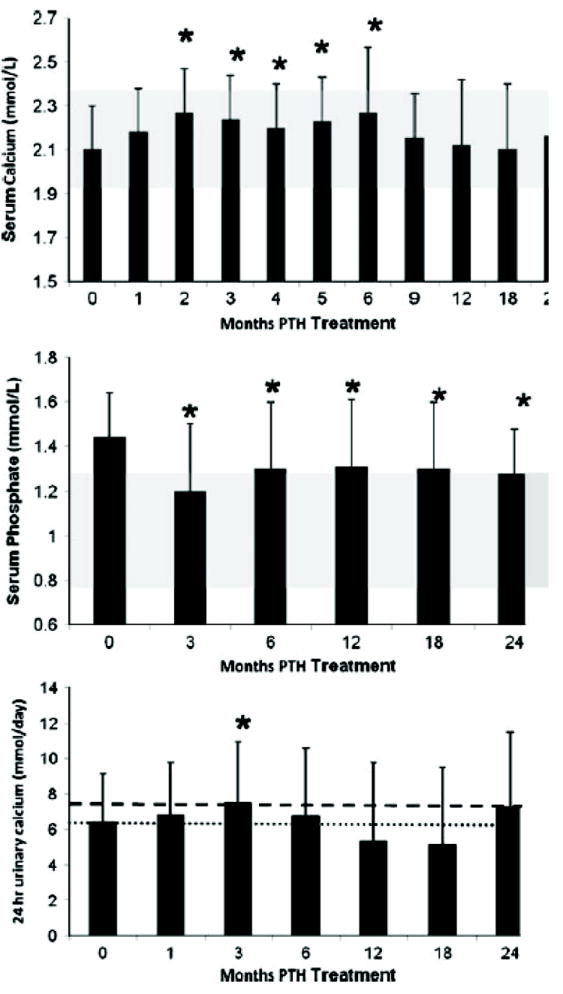
Changes in serum calcium, serum phosphate, and urinary calcium: Serum calcium increased at Months 2 through 6 but was no different from baseline at 1, 9, 12, 18, and 24 months. Serum phosphate decreased into the normal range at Month 3 and remained in the normal range through 24 months. The shaded area shows the normal ranges of serum calcium and phosphate. Urinary calcium increased at 3 months but otherwise did not change. The heavier and lighter dashed lines show the upper limit of normal urinary calcium levels in men and women, respectively. Data are mean±SD. *P<.05 as comparison to baseline. Reproduced with permission.(146)
Serum and Urinary Calcium Levels With PTH(1-84)
Hypercalcemia was uncommon with PTH(1-84) treatment. The serum calcium was maintained in the lower half of the normal range and during months 9 to 24 was not different from baseline values (Figure 10). During the first 6 months of the study, there were small but significant increases from baseline (eg, at 2 weeks: 2.1±0.2 to 2.2±0.3 mmol/L; P=0.03). Thereafter, serum calcium values were not different from baseline. Nine subjects (30%) developed a mild elevation in serum calcium at some point during the trial. In contrast to the studies of Winer et al with PTH(1-34),(142) 24-hour urinary calcium excretion changed significantly, but minimally, at only 1 time point with PTH(1-84) (3 months; Figure 10).
Bone Mineral Density With PTH(1-84)
Compared to baseline, BMD increased at the lumbar spine—by 2.9±4% (P<0.05) from 1.24±0.3 to 1.27±0.3 g/cm2 (T-score, 1.7±2 to 1.9±2; Figure 11)—a site that is enriched in cancellous bone. Because PTH is known to be anabolic for cancellous bone, this observation could indicate that new, younger bone is being formed as a result of PTH treatment. A more detailed examination of skeletal features, using high-resolution imaging or bone biopsy, would be necessary to elucidate which changes in microarchitectural parameters contribute to the increase in trabecular BMD. Such results would also be of great interest in terms of a comparison between the effects of PTH(1-84) as a therapy for osteoporosis or as replacement therapy for hypoparathyroidism. Along with the increase in lumbar spine BMD, a decrease in the distal 1/3 radius, a site of cortical bone, was observed, decreasing by 2.4±4% (P<0.05) from 0.72±0.1 to 0.70±0.1 g/cm2 (T-score, 0.03±2 to 0.26±1; Figure 11). These results speak to the effects of PTH to cause endosteal resorption. These data do not imply that bone is weakened, because salutary effects on microarchitecture and bone size could well provide biomechanical advantages, despite the reduction in BMD. A more detailed skeletal assessment would be required to answer this question. Overall, these changes in trabecular and cortical skeletal compartments recall the pattern seen with PTH treatment of osteoporosis in individuals who do not have hypoparathyroidism.(147)
FIG. 11.
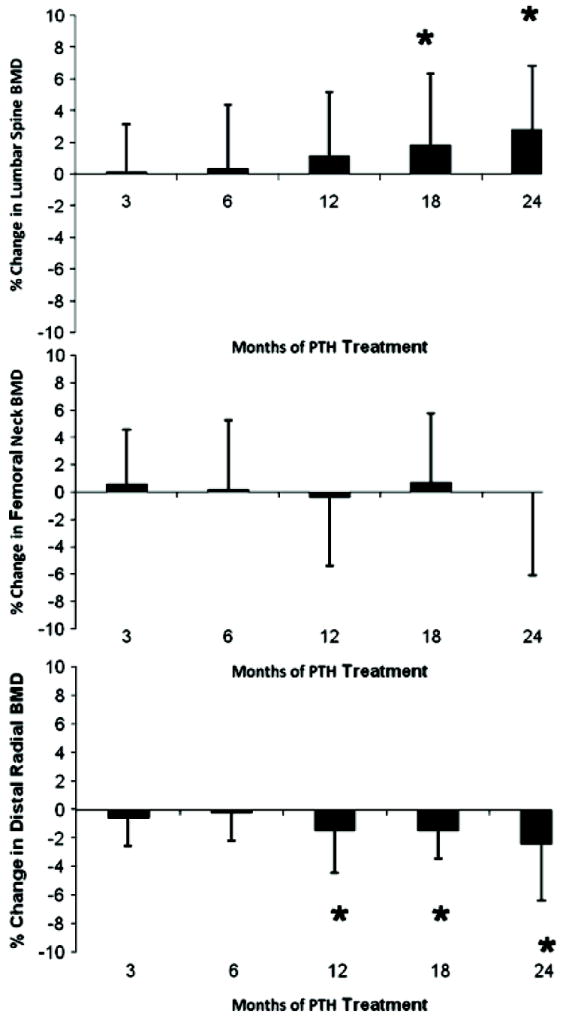
Changes in bone mineral density (BMD): Lumbar spine BMD increased, while the femoral neck did not change and the distal 1/3 radius BMD decreased. Data are mean ± SD. *P<.05 as comparison to baseline. Reproduced with permission.(146)
RESEARCH DIRECTIONS
1. Classification of Hypoparathyroidism
Hypoparathyroidism has been defined with respect to the specific causes, mutations, and complex genetic abnormalities that cause the disorder, in addition to the most common variety due to postsurgical complications. With greater knowledge of these other etiologies, a clearer differential diagnosis can be generated and more efficient workup can be formulated, when one encounters a new index case and family. Understanding the molecular etiology of the disease in a patient and family has the potential for tailored treatment, family screening, and genetic counseling. The term “idiopathic hypoparathyroidism” should be reserved for hypoparathyroidism in which no cause is known. An accurate classification system for the hypoparathyroid disorders, based on their hereditary patterns and genetic defects, when known, would provide a more scientifically rigorous framework in which to approach patients clinically. In addition, basic research in this area would establish a knowledge base that would inform clinicians and scientists about genes important in parathyroid/endocrine cell development and in the regulation of the PTH secretory pathway within the cell.
2. Epidemiology
More complete information is needed on the incidence of postsurgical hypoparathyroidism with attention to comparing centers where surgical expertise is extensive versus limited. More complete information about the incidence of autoimmune and other variants of hypoparathyroidism is also needed. Data on the risk of fractures are needed.
3. Diagnostic Criteria
Absent or inappropriately low PTH in the face of hypocalcemia is the diagnostic criterion of hypoparathyroidism. However, the nature of the immunoactive material detected by the PTH assay in some subjects with hypoparathyroidism has not been elucidated. Can tests be performed to clarify whether this material is truly PTH(1-84), ie, authentic hormone? Secondly, if it is PTH(1-84), can its production be stimulated with calcium lowering or the pharmacologic surrogate -- a calcium receptor antagonist (“calcilytic”)-- or is secretory control lost? When is genetic testing recommended?
4. Clinical Features of the Hypoparathyroid Disorders
Signs and symptoms may be associated not only with the extent, chronicity, and therapeutic endpoints in hypoparathyroidism but also with the different etiologies of the disease. Signs and symptoms in postsurgical hypoparathyroidism might be different from those with the autoimmune or genetic variants. Can clinical phenotypes be identified on the basis of whether or not there is or is not any circulating PTH?
5. Designing an Optimal Long-term Treatment Strategy
The effects of therapy with different forms of treatment (PTH peptides, vitamin D and its analogs, and thiazide diuretics) on BMD and bone quality need more investigation. A comparison between PTH(1-34) or the longer-lived PTH(1-84) would be of interest with regard to detailed structural and densitometric effects at specific skeletal sites. Does PTH improve skeletal microstructure and the properties of bone mineral in hypoparathyroidism? Do changes in circulating markers of bone turnover after PTH administration relate to changes in bone quality as determined by bone biopsy or noninvasive high resolution skeletal imaging?
More information is needed about the chronic effects of PTH treatment. Is the kidney protected by PTH action? Are there salutary effects on the cardiovascular system? What is the effect of PTH treatment on quality of life; are changes in quality of life only related to normalization of calcium and phosphate per se? Are there quality-of-life measures that can be specifically attributable to giving the missing hormone PTH, independent of the normalization of circulating calcium and phosphate levels? Will these effects vary with different formulations of PTH and/or modes of hormone delivery? What is the ideal dosing and delivery system to achieve sustained effects on serum calcium and phosphate, while protecting the kidney from the potential adverse events?
SUMMARY
This report summarizes our current state of knowledge of hypoparathyroidism, a major disorder of parathyroid function. It emphasizes not only what we know about the disease but also what additional knowledge is needed in order for us to understand more completely its epidemiology, etiologies and molecular pathogenesis, and clinical manifestations. In a disorder characterized by absent or inadequate parathyroid function, hypoparathyroidism presents an opportunity to appreciate the effects of absent or inadequate PTH on the skeleton and other target tissues. Finally, one looks forward to a time when the missing hormone, namely PTH, will become the standard option for therapy of this disease.
Acknowledgments
Dr. Shoback acknowledges funding from NIH RO1 and the Research Service of the Department of Veterans Affairs; Dr Brandi acknowledges support from F.I.R.M.O. Fondazione Raffaella Becagli; Dr. Jüppner acknowledges funding from NIH R37 DK46718; Drs. Bilezikian, Rubin, and Dempster acknowledge support from NIH (DK 069350) and the FDA (FD 002525); and Drs. Collins and Gafni acknowledge support by the Division of Intramural Research, NIDCR, NIH, and state: “This article represents the opinions of the authors and not of the NIH or the US federal government.”
References
- 1.Bilezikian JP, Khan A, Potts JT., Jr Guidelines for the management of asymptomatic primary hyperparathyroidism: Summary statement from the Third International Workshop. J Clin Endocrinol Metab. 2009;94:335–339. doi: 10.1210/jc.2008-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hypoparathyroidism Association Inc website. HPTH Association Inc; Idaho Falls, ID, USA: [2011]. Available at: http://www.hpth.org. [Google Scholar]
- 3.Marx SJ. Hyperparathyroid and hypoparathyroid disorders. New Engl J Med. 2000;343:1863–1875. doi: 10.1056/NEJM200012213432508. [DOI] [PubMed] [Google Scholar]
- 4.Thomusch O, Machens A, Sekulla C, et al. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: A multivariate analysis of 5846 consecutive patients. Surgery. 2003;133:180–195. doi: 10.1067/msy.2003.61. [DOI] [PubMed] [Google Scholar]
- 5.Zarnegar R, Brunaud L, Clark OH. Prevention, evaluation, and management of complications following thyroidectomy for thyroid carcinoma. Endocrinol Metab Clin North Am. 2003;32:483–502. doi: 10.1016/s0889-8529(03)00009-4. [DOI] [PubMed] [Google Scholar]
- 6.Page C, Strunski V. Parathyroid risk in total thyroidectomy for bilateral, benign, multinodular goiter: Report of 351 surgical cases. J Laryngol Otol. 2007;121:237–241. doi: 10.1017/S0022215106003501. [DOI] [PubMed] [Google Scholar]
- 7.Shoback D. Clinical practice: Hypoparathyroidism. New Engl J Med. 2008;359:391–403. doi: 10.1056/NEJMcp0803050. [DOI] [PubMed] [Google Scholar]
- 8.Falk SA, Birken EA, Baran DT. Temporary postthyroidectomy hypocalcemia. Arch Otolaryngol Head Neck Surg. 1998;114:168–174. doi: 10.1001/archotol.1988.01860140066023. [DOI] [PubMed] [Google Scholar]
- 9.Percival RC, Hargreaves AW, Kanis JA. The mechanism of hypocalcemia following thyroidectomy. Acta Endocrinol. 1985;109:220–226. doi: 10.1530/acta.0.1090220. [DOI] [PubMed] [Google Scholar]
- 10.See ACH, Soo KC. Hypocalcemia following thyroidectomy for thyrotoxicosis. Br J Surg. 1997;84:95–97. [PubMed] [Google Scholar]
- 11.Asari R, Koperek O, Kaczirek K, et al. Hypoparathyroidism after total thyroidectomy: A prospective study. Arch Surg. 2008;143:132–137. doi: 10.1001/archsurg.2007.55. [DOI] [PubMed] [Google Scholar]
- 12.Richards ML, Thompson GB, Farley DR, Grant CS. Reoperative parathyroidectomy in 228 patients during the era of minimal-access surgery and intraoperative parathyroid hormone monitoring. Am J Surg. 2008;196:937–943. doi: 10.1016/j.amjsurg.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 13.Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. New Engl J Med. 2004;350:2068–2079. doi: 10.1056/NEJMra030158. [DOI] [PubMed] [Google Scholar]
- 14.Blizzard RM, Chee D, Davis W. The incidence of parathyroid and other antibodies in the sera of patients with idiopathic hypoparathyroidism. Clin Exp Immunol. 1966;1:119–128. [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Song YH, Rais N, et al. Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest. 1996;97:910–914. doi: 10.1172/JCI118513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown EM. Anti-parathyroid and anti-calcium sensing receptor antibodies in autoimmune hypoparathyroidism. Endocrinol Metab Clin North Am. 2009;38:437–445. doi: 10.1016/j.ecl.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kifor O, McElduff A, LeBoff MS, et al. Activating antibodies to the calcium-sensing receptor in two patients with autoimmune hypoparathyroidism. J Clin Endocrinol Metab. 2004;89:548–556. doi: 10.1210/jc.2003-031054. [DOI] [PubMed] [Google Scholar]
- 18.Thakker RV. Principles of Bone Biology. Academic Press; New York, NY, USA: 1996. Molecular basis of PTH underexpression; pp. 837–851. [Google Scholar]
- 19.Bilezikian JP, Thakker RV. Hypoparathyroidism. Curr Opin Endocrinol Diabetes. 1998;4:427–432. [Google Scholar]
- 20.Marx SJ. Hyperparathyroid and hypoparathyroid disorders. N Engl J Med. 2000;343:1863–1875. doi: 10.1056/NEJM200012213432508. [DOI] [PubMed] [Google Scholar]
- 21.Thakker RV. Genetic developments in hypoparathyroidism. Lancet. 2001;357:974–976. doi: 10.1016/s0140-6736(00)04254-9. [DOI] [PubMed] [Google Scholar]
- 22.Thakker RV, Juppner H. Genetic disorders of calcium homeostasis caused by abnormal regulation of parathyroid hormone secretion or responsiveness. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 4. WB Saunders Company; Philadelphia, PA, USA: 2001. pp. 1062–1074. [Google Scholar]
- 23.Toumba M, Sergis A, Kanaris C, et al. Endocrine complications in patients with thalassemia major. Pediatr Endocrinol Rev. 2007;5:642–648. [PubMed] [Google Scholar]
- 24.Carpenter TO, Carnes DL, Jr, Anast CS. Hypoparathyroidism in Wilson’s disease. New Engl J Med. 1983;309:873–877. doi: 10.1056/NEJM198310133091501. [DOI] [PubMed] [Google Scholar]
- 25.Goddard CJ. Symptomatic hypocalcaemia associated with metastatic invasion of the parathyroid glands. Br J Hosp Med. 1990;43:72. [PubMed] [Google Scholar]
- 26.Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med. 2005;20:3–17. doi: 10.1177/0885066604271539. [DOI] [PubMed] [Google Scholar]
- 26a.Milman S, Epstein EJ. Proton pump inhibitor-induced hypocalcemic seizure in a patient with hypoparathyroidism. Endocr Pract. 2011;17:104–107. doi: 10.4158/EP10241.CR. [DOI] [PubMed] [Google Scholar]
- 27.Cholst IN, Steinberg SF, Tropper PJ, et al. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. New Engl J Med. 1984;310:1221–1225. doi: 10.1056/NEJM198405103101904. [DOI] [PubMed] [Google Scholar]
- 28.Sunthornthepvarakul T, Churesigaew S, Ngowngarmratana S. A novel mutation of the signal peptide of the pre-pro-parathyroid hormone gene associated with autosomal recessive familial isolated hypoparathyroidism. J Clin Endocrinol Metab. 1999;84:3792–3796. doi: 10.1210/jcem.84.10.6070. [DOI] [PubMed] [Google Scholar]
- 29.Parkinson DB, Thakker RV. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet. 1992;1:149–153. doi: 10.1038/ng0592-149. [DOI] [PubMed] [Google Scholar]
- 30.Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM. Mutations in the signal peptide encoding region of preproparathyroid hormone gene in isolated hypoparathyroidism. J Clin Invest. 1990;86:1084–1087. doi: 10.1172/JCI114811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datta R, Waheed A, Shah GN, Sly WS. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci. 2007;104:19989–19994. doi: 10.1073/pnas.0708725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gunther T, Chen ZF, Kim J, et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. 2000;406:199–203. doi: 10.1038/35018111. [DOI] [PubMed] [Google Scholar]
- 33.Ding C, Buckingham B, Levine MA. Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J Clin Endocrinol. 2001;108:1215–1220. doi: 10.1172/JCI13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomée C, Schubert SW, Parma J, et al. GCMB mutation in familial isolated hypoparathyroidism with residual secretion of parathyroid hormone. J Clin Endocrinol Metab. 2005;90:2487–2492. doi: 10.1210/jc.2004-2450. [DOI] [PubMed] [Google Scholar]
- 35.Baumer L, Tufarelli C, Patel S, et al. Identification of a novel mutation disrupting the DNA binding activity of GCM2 in autosomal recessive familial isolated hypoparathyroidism. J Med Genet. 2005;42:443–448. doi: 10.1136/jmg.2004.026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowl MR, Mirczuk SM, Grigorieva IV, et al. Identification and characterization of novel parathyroid-specific transcription factor Glial cells missing homolog B (GCMB) mutations in eight families with autosomal recessive hypoparathyroidism. Hum Mol Genet. 2010;19:2028–2038. doi: 10.1093/hmg/ddq084. [DOI] [PubMed] [Google Scholar]
- 37.Mannstadt M, Bertrand G, Muresan M, et al. Dominant-negative GCMB mutations cause an autosomal dominant form of hypoparathyroidism. J Clin Endocrinol Metab. 2008;93:3568–3576. doi: 10.1210/jc.2007-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maret A, Ding C, Kornfield SL, Levine MA. Analysis of the GCM2 gene in isolated hypoparathyroidism: a molecular and biochemical study. J Clin Endocrinol Metab. 2008;93:1426–1432. doi: 10.1210/jc.2007-1783. [DOI] [PubMed] [Google Scholar]
- 39.Pollak MR, Brown EM, Estep HL, et al. Autosomal dominant hypocalcemia caused by a calcium-sensing receptor gene mutation. Nature Genet. 1994;8:303–307. doi: 10.1038/ng1194-303. [DOI] [PubMed] [Google Scholar]
- 40.Pidasheva S, D’Souza-Li L, Canaff L, Cole DE, Hendy GN. CASRdb: calcium-sensing receptor locus-specific database for mutations causing familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mut. 2004;24:107–111. doi: 10.1002/humu.20067. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe S, Fukumoto S, Chang H, et al. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet. 2002;360:692–694. doi: 10.1016/S0140-6736(02)09842-2. [DOI] [PubMed] [Google Scholar]
- 42.Winer KK, Sinaii N, Reynolds J, Peterson D, Dowdy K, Cutler GB., Jr Long-term treatment of 12 children with chronic hypoparathyroidism: a randomized trial comparing synthetic human parathyroid hormone 1-34 versus calcitriol and calcium. J Clin Endocrinol Metab. 2010;95:2680–2688. doi: 10.1210/jc.2009-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whyte MP, Weldon VV. Idiopathic hypoparathyroidism presenting with seizures during infancy: X-linked recessive inheritance in a large Missouri kindred. J Pediatr. 1981;99:608–611. doi: 10.1016/s0022-3476(81)80272-7. [DOI] [PubMed] [Google Scholar]
- 44.Mumm S, Whyte MP, Thakker RV, Buetow KH, Schlessinger D. mtDNA analysis shows common ancestry in two kindreds with X-linked recessive hypoparathyroidism and reveals a heteroplasmic silent mutation. Am J Hum Genet. 1997;60:153–159. [PMC free article] [PubMed] [Google Scholar]
- 45.Thakker RV, Davies KE, Whyte MP, Wooding C, Riordan JL. Mapping the gene causing X-linked recessive idiopathic hypoparathyroidism to Xq26-Xq27 by linkage studies. J Clin Invest. 1990;86:40–45. doi: 10.1172/JCI114712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowl MR, Nesbit MA, Harding B, et al. An interstitial deletion-insertion involving chromosome 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest. 2005;115:2822–2831. doi: 10.1172/JCI24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maret A, Ding C, Levine Kornfield S, Levine MA. Analysis of the GCM2 gene in isolated hypoparathyroidism: A molecular and biochemical study. Clin Endocrinol Metab. 2008;93:1426–1432. doi: 10.1210/jc.2007-1783. [DOI] [PubMed] [Google Scholar]
- 48.Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiadis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 49.Lankisch TO, Jaeckel E, Strassburg CP. The autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy or autoimmune polyglandular syndrome type 1. Sem Liver Dis. 2009;29:307–314. doi: 10.1055/s-0029-1233535. [DOI] [PubMed] [Google Scholar]
- 50.Bjorses P, Halonen M, Palvimo JJ, et al. Mutations in the AIRE gene: Effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J Hum Genet. 2000;66:378–392. doi: 10.1086/302765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su MA, Giang K, Zumer K, et al. Mechanisms of autoimmunity syndrome in mice caused by a dominant mutation in Aire. J Clin Invest. 2008;118:1712–1726. doi: 10.1172/JCI34523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alimohammadi M, Bjorklund P, Hallgren A, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. 2008;358:1018–1028. doi: 10.1056/NEJMoa0706487. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Song YH, Rais N, et al. Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest. 1996;97:910–914. doi: 10.1172/JCI118513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gavalas NG, Kemp EH, Krohn KJE, Brown EM, Watson PF, Weetman AP. The calcium-sensing receptor is a target of autoantibodies in patients with autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2007;92:2107–2114. doi: 10.1210/jc.2006-2466. [DOI] [PubMed] [Google Scholar]
- 55.Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Int Med. 2009;265:514–529. doi: 10.1111/j.1365-2796.2009.02090.x. [DOI] [PubMed] [Google Scholar]
- 56.Kemp EH, Gavalas NG, Krohn KJE, Brown EM, Watson PF, Weetman AP. Activating autoantibodies against the calcium-sensing receptor detected in two patients with autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2009;94:4749–4756. doi: 10.1210/jc.2009-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc Natl Acad Sci USA. 2008;105:13009–13014. doi: 10.1073/pnas.0806874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 59.Parvari R, Diaz GA, Hershkovitz E. Parathyroid development and the role of tubulin chaperone E. Horm Res. 2007;358:12–21. doi: 10.1159/000095944. [DOI] [PubMed] [Google Scholar]
- 60.Sanjad SA, Sakati NA, Abu-Osba YK, Kaddoura R, Nilner RDG. A new syndrome of congenital hypoparathyroidism, severe growth failure, and dysmorphic features. Arch Dis Child. 1991;66:193–196. doi: 10.1136/adc.66.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bilous RW, Murty G, Parkinson DB, et al. Brief report: Autosomal dominant familial hypoparathyroidism, sensineural deafness and renal dysplasia. N Engl J Med. 1992;327:1069–1074. doi: 10.1056/NEJM199210083271506. [DOI] [PubMed] [Google Scholar]
- 62.Ali A, Christie PT, Grigorieva IV, et al. Functional characterization of GATA3 mutations causing the hypoparathyroid-deafness-renal (HDR) dysplasia syndrome: Insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet. 2007;16:265–275. doi: 10.1093/hmg/ddl454. [DOI] [PubMed] [Google Scholar]
- 63.Thakker RV. Genetics of endocrine and metabolic disorders: parathyroid. Rev End Metab Dis. 2004;5:37–51. doi: 10.1023/B:REMD.0000016123.21743.fe. [DOI] [PubMed] [Google Scholar]
- 64.Chase RL, Melson GL, Aurbach GD. Pseudohypoparathyroidism: defective excretion of 3’,5’-AMP in response to parathyroid hormone. J Clin Invest. 1969;48:1832–1844. doi: 10.1172/JCI106149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinstein LS, ShuHua Y, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- 66.Bastepe M. The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol. 2008;626:27–40. doi: 10.1007/978-0-387-77576-0_3. [DOI] [PubMed] [Google Scholar]
- 67.Mantovani G, Ferrante E, Giavoli C, et al. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type 1a: First study on the effect on growth. J Clin Endocrinol Metab. 2010;95:5011–5017. doi: 10.1210/jc.2010-1649. [DOI] [PubMed] [Google Scholar]
- 68.Mantovani G, de Sanctis L, Barbieri AM, et al. Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010;95:651–658. doi: 10.1210/jc.2009-0176. [DOI] [PubMed] [Google Scholar]
- 69.Jobert AS, Zhang P, Couvineau A, et al. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34–40. doi: 10.1172/JCI2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang P, Jobert AS, Couvineau A, Silve C. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand’s chondrodysplasia. J Clin Endocrinal Metab. 1998;83:3373–3376. doi: 10.1210/jcem.83.9.5245. [DOI] [PubMed] [Google Scholar]
- 71.Karaplis AC, He B, Nguyen MT, et al. Inactivating mutation in the human parathyroid hormone receptor type I gene in Blomstrand’s chondrodysplasia. Endocrinology. 1998;139:5255–5258. doi: 10.1210/endo.139.12.6522. [DOI] [PubMed] [Google Scholar]
- 72.Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91:2843–2850. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 73.Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. New Engl J Med. 1990;322:1829–1836. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 74.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 75.Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clinic N Am. 2008;28:353–366. doi: 10.1016/j.iac.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 76.Cassandrini D, Savasta S, Bozzola M, et al. Mitochondrial DNA deletion in a child with mitochondrial encephalomyopathy, growth hormone deficiency, and hypoparathyroidism. J Child Neurol. 2006;21:983–985. doi: 10.1177/08830738060210111001. [DOI] [PubMed] [Google Scholar]
- 77.Labarthe E, Benoist JF, Brivet M, et al. Partial hypoparathyroidism associated with mitochondrial trifunctional protein deficiency. Eur J Pediatr. 2006;165:389–391. doi: 10.1007/s00431-005-0052-5. [DOI] [PubMed] [Google Scholar]
- 78.Parvari R, Hershkovitz E, Grossman N, et al. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet. 2002;32:448–452. doi: 10.1038/ng1012. [DOI] [PubMed] [Google Scholar]
- 79.Berson SA, Yalow RS, Aurlach GD, Potts JT., Jr Immunoassay of bovine and human parathyroid hormone. Proc Natl Acad Sci. 1963;49:613–617. doi: 10.1073/pnas.49.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nussbaum SR, Zahradnik RJ, Lavigne JR, et al. Highly sensitive two-site immunoradiometric assay of parathyrin, and its clinical utility in evaluating patients with hypercalcemia. Clin Chem. 1987;33:1364–1367. [PubMed] [Google Scholar]
- 81.D’Amour P, Räkel A, Brossard JH, Rousseau L, Albert C, Cantor T. Acute regulation of circulating parathyroid hormone (PTH) molecular forms by calcium: Utility of PTH fragments/PTH(1-84) ratios derived from three generations of PTH assays. J Clin Endocrinol Metab. 2006;91:283–289. doi: 10.1210/jc.2005-1628. [DOI] [PubMed] [Google Scholar]
- 82.Brossard JH, Cloutier M, Roy L, Lepage R, Gascon-Barré M, D’Amour P. Accumulation of a non-(1-84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: importance in the interpretation of PTH values. J Clin Endocrinol Metab. 1996;61:3923–3929. doi: 10.1210/jcem.81.11.8923839. [DOI] [PubMed] [Google Scholar]
- 83.Gao P, Scheibel S, D’Amour P, et al. Development of a novel immunoradiometric assay exclusively for biologically active whole parathyroid hormone 1-84: Implications for improvement of accurate assessment of parathyroid function. J Bone Miner Res. 2001;16:605–614. doi: 10.1359/jbmr.2001.16.4.605. [DOI] [PubMed] [Google Scholar]
- 84.Lepage R, Roy L, Brossard JH, et al. A non-(1-84) circulating parathyroid hormone (PTH) fragment interferes significantly with intact PTH commercial assay measurements in uremic samples. Clin Chem. 1998;44:805–809. [PubMed] [Google Scholar]
- 85.D’Amour P, Brossard JH, Rousseau L, Roy L, Gao P, Cantor T. Amino-terminal form of parathyroid hormone (PTH) with immunologic similarities to hPTH(1-84) is overproduced in primary and secondary hyperparathyroidism. Clin Chem. 2003;49:2037–2044. doi: 10.1373/clinchem.2003.021592. [DOI] [PubMed] [Google Scholar]
- 86.Rubin MR, Silverberg SJ, D’Amour P, Bilezikian JP. An aminoterminal molecular form of parathyroid hormone distinct from PTH (1-84) is overproduced in parathyroid carcinoma. Clin Chem. 2007;53:1470–1476. doi: 10.1373/clinchem.2007.085506. [DOI] [PubMed] [Google Scholar]
- 87.Eastell R, Arnold A, Brandi ML, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab. 2009;94:340–350. doi: 10.1210/jc.2008-1758. [DOI] [PubMed] [Google Scholar]
- 88.Inaba M, Nakatsuka K, Imanishi Y, et al. Technical and clinical characterization of the Bio-PTH (1-84) immunochemiluminometric assay and comparison with a second-generation assay for parathyroid hormone. Clin Chem. 2004;50:385–390. doi: 10.1373/clinchem.2003.026831. [DOI] [PubMed] [Google Scholar]
- 89.Lepage R, Whittom S, Bertrand S, Bahsali G, D’Amour P. Superiority of dynamic over static reference intervals for intact, midmolecule, and C-terminal parathyrin in evaluating calcemic disorders. Clin Chem. 1992;38:2129–2135. [PubMed] [Google Scholar]
- 90.Abugassa S, Nordenstrom J, Eriksson S, Sjoden G. Bone mineral density in patients with chronic hypoparathyroidism. J Clin Endocrinol Metab. 1993;76:1617–1621. doi: 10.1210/jcem.76.6.8501170. [DOI] [PubMed] [Google Scholar]
- 91.Fujiyama K, Kiriyama T, Ito M, et al. Attenuation of postmenopausal high turnover bone loss in patients with hypoparathyroidism. J Clin Endocrinol Metab. 1995;80:2135–2138. doi: 10.1210/jcem.80.7.7608266. [DOI] [PubMed] [Google Scholar]
- 92.Duan Y, De Luca V, Seeman E. Parathyroid hormone deficiency and excess: Similar effects on trabecular bone but differing effects on cortical bone. J Clin Endocrinol Metab. 1999;84:718–722. doi: 10.1210/jcem.84.2.5498. [DOI] [PubMed] [Google Scholar]
- 93.Chen Q, Kaji H, Iu MF, et al. Effects of an excess and a deficiency of endogenous parathyroid hormone on volumetric bone mineral density and bone geometry determined by peripheral quantitative computed tomography in female subjects. J Clin Endocrinol Metab. 2003;88:4655–4658. doi: 10.1210/jc.2003-030470. [DOI] [PubMed] [Google Scholar]
- 94.Rubin MR, Dempster DW, Zhou H, et al. Dynamic and structural properties of the skeleton in hypoparathyroidism. J Bone Miner Res. 2008;23:2018–2024. doi: 10.1359/JBMR.080803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen A, Dempster DW, Müller R, et al. Assessment of trabecular and cortical architecture and mechanical competence of bone by high-resolution peripheral computed tomography: comparison with transiliac bone biopsy. Osteoporos Int. 2010;21:263–273. doi: 10.1007/s00198-009-0945-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langdahl BL, Mortensen L, Vesterby A, Eriksen EF, Charles P. Bone histomorphometry in hypoparathyroid patients treated with vitamin D. Bone. 1996;18:103–108. doi: 10.1016/8756-3282(95)00443-2. [DOI] [PubMed] [Google Scholar]
- 97.Rubin MR, Dempster DW, Kohler T, et al. Three dimensional cancellous bone structure in hypoparathyroidism. Bone. 2010;46:190–195. doi: 10.1016/j.bone.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rubin, Roschger, Klaushofer, et al. J Bone Miner Res. abstract. [Google Scholar]
- 99.Shoback D. Hypoparathyroidism. N Engl J Med. 2008;359:391–403. doi: 10.1056/NEJMcp0803050. [DOI] [PubMed] [Google Scholar]
- 100.Reber PM, Heath H. Hypocalcemic emergencies. Med Clin North Am. 1995;79:93–106. doi: 10.1016/s0025-7125(16)30086-4. [DOI] [PubMed] [Google Scholar]
- 101.Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am. 1993;22:363–375. [PubMed] [Google Scholar]
- 102.Murphy E, Williams GR. Hypocalcaemia. Medicine. 2009;37:465–468. [Google Scholar]
- 103.Cooper MS, Gittoes NJL. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298–1302. doi: 10.1136/bmj.39582.589433.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rude RK. Hypocalcemia and hypoparathyroidism. Curr Ther Endocrinol Metab. 1997;6:546–551. [PubMed] [Google Scholar]
- 105.Thakker R. Parathyroid Disorders and Diseases Altering Calcium Metabolism. 4. Oxford University Press; Oxford, England, UK: 2003. [Google Scholar]
- 106.Maeda SS, Fortes EM, Oliveira UM, Borba VC, Lazaretti-Castro M. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bras Endocrinol Metab. 2006;50:664–673. doi: 10.1590/s0004-27302006000400012. [DOI] [PubMed] [Google Scholar]
- 107.Noordzij M, Voormolen NMC, Boeschoten EW, et al. Disordered mineral metabolism is not a risk factor for loss of residual renal function in dialysis patients. Nephrol Dial Transplant. 2009;24:1580–1587. doi: 10.1093/ndt/gfn768. [DOI] [PubMed] [Google Scholar]
- 108.Harris VW, Jan De Beur S. Postoperative hypoparathyroidism: Medical and surgical therapeutic options. Thyroid. 2009;19:967–973. doi: 10.1089/thy.2008.0306. [DOI] [PubMed] [Google Scholar]
- 109.Harvey JA, Zobitz MM, Pak CY. Dose dependency of calcium absorption: A comparison of calcium carbonate and calcium citrate. J Bone Miner Res. 1988;3:253–258. doi: 10.1002/jbmr.5650030303. [DOI] [PubMed] [Google Scholar]
- 109a.Recker RR. Calcium absorption and achlorhydria. N Eng J Med. 1985;313:70–73. doi: 10.1056/NEJM198507113130202. [DOI] [PubMed] [Google Scholar]
- 110.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 111.Russell RGG, Walton RJ, Smith R, et al. 1,25-Dihydroxycholecalciferol and 1[alpha]-hydroxycholecalciferol in hypoparathyroidism. Lancet. 1974;304:14–17. doi: 10.1016/s0140-6736(74)91348-8. [DOI] [PubMed] [Google Scholar]
- 112.Broadus A, Horst R, Lang R, Littledike E, Rasmussen H. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renal-stone formation in primary hyperparathyroidism. N Engl J Med. 1980;302:421–426. doi: 10.1056/NEJM198002213020801. [DOI] [PubMed] [Google Scholar]
- 113.Mortensen L, Hyldstrup L, Charles P. Effect of vitamin D treatment in hypoparathyroid patients: A study on calcium, phosphate and magnesium homeostasis. Eur J Endocrinol. 1997;136:52–60. doi: 10.1530/eje.0.1360052. [DOI] [PubMed] [Google Scholar]
- 114.Neer RM, Holick MF, DeLuca HF, Potts JT. Effects of 1alpha-hydroxy-vitamin D3 and 1,25-dihydroxy-vitamin D3 on calcium and phosphorus metabolism in hypoparathyroidism. Metab Clin Exp. 1975;24:1403–1413. doi: 10.1016/0026-0495(75)90055-4. [DOI] [PubMed] [Google Scholar]
- 115.Kooh S, Fraser D, DeLuca H, et al. Treatment of hypoparathyroidism and pseudohypoparathyroidism with metabolites of vitamin D: Evidence for impaired conversion of 25-hydroxyvitamin D to 1 alpha,25-dihydroxyvitamin D. N Engl J Med. 1975;293:840–844. doi: 10.1056/NEJM197510232931702. [DOI] [PubMed] [Google Scholar]
- 116.Quack I, Zwernemann C, Weiner SM, et al. Dihydrotachysterol therapy for hypoparathyroidism: consequences of inadequate monitoring: Five cases and a review. Exp Cli Endocrinol Diabetes. 2005;113:376–380. doi: 10.1055/s-2005-865724. [DOI] [PubMed] [Google Scholar]
- 117.Favus MJ, Coe FL, Kathpalia SC, Porat A, Sen PK, Sherwood LM. Effects of chlorothiazide on 1,25-dihydroxyvitamin D3, parathyroid hormone, and intestinal calcium absorption in the rat. Am J Physiol Gastrointest Liver Physiol. 1982;242:G575–G581. doi: 10.1152/ajpgi.1982.242.6.G575. [DOI] [PubMed] [Google Scholar]
- 118.Callies F, Arlt W, Scholz HJ, Reincke M, Allolio B. Management of hypoparathyroidism during pregnancy—Report of twelve cases. Eur J Endocrinol. 1998;139:284–289. doi: 10.1530/eje.0.1390284. [DOI] [PubMed] [Google Scholar]
- 119.Halabe A, Arie R, Mimran D, Samuel R, Liberman UA. Hypoparathyroidism—A long-term follow-up experience with 1 alpha-vitamin D3 therapy. Clin Endocrinol (Oxf) 1994;40:303–307. doi: 10.1111/j.1365-2265.1994.tb03923.x. [DOI] [PubMed] [Google Scholar]
- 120.Horwitz MJ, Stewart AF. Hypoparathyroidism: Is it time for replacement therapy? J Clin Endocrinol Metab. 2008;93:3307–3309. doi: 10.1210/jc.2008-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yendt ER, Gagné RJ, Cohanim M. The effects of thiazides in idiopathic hypercalciuria. Trans Am Clin Climatol Assoc. 1966;77:96–110. [PMC free article] [PubMed] [Google Scholar]
- 122.Yendt ER, Guay GF, Garcia DA. The use of thiazides in the prevention of renal calculi. Can Med Assoc J. 1970;102:614–620. [PMC free article] [PubMed] [Google Scholar]
- 123.Costanzo L, Weiner I. Relationship between clearances of Ca and Na: Effect of distal diuretics and PTH. Am J Physiol. 1976;230:67–73. doi: 10.1152/ajplegacy.1976.230.1.67. [DOI] [PubMed] [Google Scholar]
- 124.Lamberg B, Kuhlback B. Effect of chlorothiazide and hydrochlorothiazide on the excretion of calcium in urine. Scand J Clin Lab Invest. 1959;11:351–357. doi: 10.3109/00365515909060464. [DOI] [PubMed] [Google Scholar]
- 125.Alon U, Wellons MD, Chan JC. Reversal of vitamin-D2-induced hypercalciuria by chlorothiazide. Pediatr Res. 1983;17:117–119. doi: 10.1203/00006450-198302000-00007. [DOI] [PubMed] [Google Scholar]
- 126.Santos F, Smith MJV, Chan JCM. Hypercalciuria associated with long-term administration of calcitriol (1,25-dihydroxyvitamin D3) action of hydrochlorothiazide. Am J Dis Child. 1986;140:139–142. doi: 10.1001/archpedi.1986.02140160057032. [DOI] [PubMed] [Google Scholar]
- 127.Kurzel RB, Hagen GA. Use of thiazide diuretics to reduce the hypercalciuria of hypoparathyroidism during pregnancy. Am J Perinatol. 1990;7:333–336. doi: 10.1055/s-2007-999516. [DOI] [PubMed] [Google Scholar]
- 128.Porter R, Cox B, Heaney D, Hostetter T, Stinebaugh B, Suki W. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med. 1978;298:577–581. doi: 10.1056/NEJM197803162981101. [DOI] [PubMed] [Google Scholar]
- 129.Caldwell J, Adioli L, Boisseau C. Hydrochlorothiazide and calcium homeostasis in idiopathic hypercalciuria. Clin Res. 1971;19:676. [Google Scholar]
- 130.Ehrig U, Harrison JE, Wilson DR. Effect of long-term thiazide therapy on intestinal calcium absorption in patients with recurrent renal calculi. Metab Clin Exp. 1974;23:139–149. doi: 10.1016/0026-0495(74)90111-5. [DOI] [PubMed] [Google Scholar]
- 131.Jørgensen FS, Transbøl I. The effect of bendroflumethiazide on the intestinal absorption of calcium in normocalcaemic renal stone formers and in hyperparathyroidism. Acta Medica Scandinavica. 1974;195:33–36. [PubMed] [Google Scholar]
- 132.Donath A, Nordio S, Macagno F, Gatti R. The effect of hydrochlorothiazide on calcium and strontium transport in intestine and kidney. Helv Paediatr Acta. 1970;25:293–300. [PubMed] [Google Scholar]
- 133.Arlt W, Fremerey C, Callies F, et al. Well-being, mood and calcium homeostasis in patients with hypoparathyroidism receiving standard treatment with calcium and vitamin D. Eur J Endocrinol. 2002;146:215–222. doi: 10.1530/eje.0.1460215. [DOI] [PubMed] [Google Scholar]
- 134.Haussler MR, Cordy PE. Metabolites and analogues of vitamin D: Which for what? JAMA. 1982;247:841–844. [PubMed] [Google Scholar]
- 135.Chan JCM, Young RB, Alon U, Mamunes P. Hypercalcemia in children with disorders of calcium and phosphate metabolism during long-term treatment with 1,25-dihydroxyvitamin-D3. Pediatrics. 1983;72:225–233. [PubMed] [Google Scholar]
- 136.Winterborn MH, Mace PJ, Heath DA, White RH. Impairment of renal function in patients on 1alpha-hydroxycholecalciferol. Lancet. 1978;2:150–151. doi: 10.1016/s0140-6736(78)91530-1. [DOI] [PubMed] [Google Scholar]
- 137.Kanis JA, Russell RG. Rate of reversal of hypercalcaemia and hypercalciuria induced by vitamin D and its 1alpha-hydroxylated derivatives. Br Med J. 1977;1:78–81. doi: 10.1136/bmj.1.6053.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nordin BEC. Diagnostic procedures in disorders of calcium metabolism. Clin Endocrinol. 1978;8:55–67. doi: 10.1111/j.1365-2265.1978.tb01350.x. [DOI] [PubMed] [Google Scholar]
- 139.Farrington K, VArghese Z, Moorhead JF. Vitamin-D analogues and renal function. Lancet. 1978;2:941. doi: 10.1016/s0140-6736(78)91656-2. [DOI] [PubMed] [Google Scholar]
- 140.Winer KK, Yanovski JA, Cutler GB., Jr Synthetic human parathyroid hormone 1-34 vs calcitriol and calcium in the treatment of hypoparathyroidism. JAMA. 1996;276:631–636. [PubMed] [Google Scholar]
- 141.Winer KK, Yanovski JA, Sarani B, Cutler GB., Jr A randomized, cross-over trial of once-daily versus twice-daily parathyroid hormone 1-34 in treatment of hypoparathyroidism. J Clin Endocrinol Metab. 1998;83:3480–3486. doi: 10.1210/jcem.83.10.5185. [DOI] [PubMed] [Google Scholar]
- 142.Winer KK, Ko CW, Reynolds JC, et al. Long-term treatment of hypoparathyroidism: A randomized controlled study comparing parathyroid hormone-(1-34) versus calcitriol and calcium. J Clin Endocrinol Metab. 2003;88:4214–4220. doi: 10.1210/jc.2002-021736. [DOI] [PubMed] [Google Scholar]
- 143.Winer KK, Sinaii N, Peterson D, Sainz B, Jr, Cutler GB., Jr Effects of once versus twice-daily parathyroid hormone 1-34 therapy in children with hypoparathyroidism. J Clin Endocrinol Metab. 2008;93:3389–3395. doi: 10.1210/jc.2007-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Winer KK, Sinaii N, Reynolds J, Peterson D, Dowdy K, Cutler GB. Long-term treatment of 12 children with chronic hypoparathyroidism: A randomized trial comparing synthetic human parathyroid hormone 1-34 versus calcitriol and calcium. J Clin Endocrinol Metab. 2010;95:2680–2688. doi: 10.1210/jc.2009-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Theman TA, Collins MT, Dempster DW, et al. PTH(1-34) replacement therapy in a child with hypoparathyroidism caused by a sporadic calcium receptor mutation. J Bone Miner Res. 2009;24:964–973. doi: 10.1359/JBMR.081233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rubin MR, Sliney J, Jr, McMahon DJ, Silverberg SJ, Bilezikian JP. Therapy of hypoparathyroidism with intact parathyroid hormone. Osteoporos Int. 2010;21:1927–1934. doi: 10.1007/s00198-009-1149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]