

Abstract
Molecular and cellular studies have demonstrated opposing actions of stress and antidepressant treatment on the expression of neurotrophic factors, particularly brain-derived neurotrophic factor, in limbic structures of the brain. These changes in neurotrophic factor expression and function result in structural alterations, including regulation of neurogenesis, dendrite length and spine density in hippocampus and prefrontal cortex (PFC). The deleterious effects of stress could contribute to the reduced volume of these brain regions in depressed patients. Conversely, the actions of antidepressant treatment could be mediated in part by blocking or reversing the atrophy caused by stress and depression. Recent studies have identified a novel, rapid-acting antidepressant, ketamine, in treatment-resistant depressed patients that addresses the limitations of currently available agents (i.e. delayed onset of action and low response rates). We have found that ketamine, an N-methyl-d-aspartate (NMDA) receptor antagonist, causes a rapid induction of synaptogenesis and spine formation in the PFC via stimulation of the mammalian target of the rapamycin signalling pathway and increased synthesis of synaptic proteins. These effects of ketamine rapidly reverse the atrophy of PFC neurons caused by chronic stress and correspond to rapid behavioural actions of ketamine in models of depression. Characterization of a novel signalling pathway also identifies new cellular targets that could result in rapid and efficacious antidepressant actions without the side effects of ketamine.
Keywords: ketamine, stress, glutamate, rapamycin, mammalian target of rapamycin, spine
1. Introduction
Depression is a widespread illness, affecting approximately 17 per cent of the population at some point in life, with tremendous personal and socioeconomic consequences [1]. The underlying causes of this heterogeneous illness as well as other mood disorders remain poorly understood. Moreover, the available pharmacological treatments for depression have significant limitations, including relatively low efficacy (i.e. approximately one-third of patients respond to the first agent prescribed), and time lag for treatment response (i.e. therapeutic effects are observed only after two to three weeks, and in many cases months of treatment) [2]. These limitations highlight a major unmet need for more efficacious and fast-acting antidepressant agents, particularly with the high rates of suicide in depressed subjects.
Despite these problems, recent studies have begun to elucidate the neurobiology of depression as well as treatment response, and have identified novel agents that have the potential to provide more efficacious and rapid response rates. In this review, we provide a brief update on the role of neurotrophic factors in the aetiology and treatment of depression- and stress-related illnesses. Then, we discuss the cellular and behavioural consequences of altered neurotrophic factor signalling in response to stress and antidepressant treatments. In particular, new evidence demonstrating that novel, rapid-acting N-methyl-d-aspartate (NMDA) receptor antagonists increase synaptogenesis, and the mechanisms underlying this effect are discussed.
2. Neurobiology of depression: atrophy and loss of neurons
Recent studies have begun to elucidate the pathophysiology of mood disorders, providing evidence for cell atrophy and loss in relevant limbic brain structures. Brain imaging studies demonstrate a reduction in the volume of limbic brain regions implicated in depression, notably the hippocampus and prefrontal cortex (PFC) [3,4]. Post-mortem studies report a reduction in the size of neurons and loss of glia [3,5], and preclinical studies show that exposure to repeated stress causes atrophy of neurons in the hippocampus and PFC, as well as loss of glia [6,7]. These studies provide strong evidence that atrophy and loss of neurons and glia are contributing factors to depression- and stress-related disorders.
A role for neurotrophic factors in cell atrophy and loss is supported by evidence that stress or depression decreases the expression of certain factors in limbic brain regions. One of the most highly studied factors is brain-derived neurotrophic factor (BDNF). Exposure to different types of physical or social stress decreases levels of BDNF in the hippocampus and PFC in rodent models [6–8]. Post-mortem studies also demonstrate a reduction of BDNF in these regions in post-mortem brains of depressed subjects [6]. This work has led to studies of growth factors in blood, which demonstrate decreased levels of BDNF in serum of depressed patients and reversal with antidepressant treatment, suggesting that BDNF is a biomarker of depression and treatment response [9,10]. In contrast to stress and depression, antidepressant treatment increases the expression of BDNF in the hippocampus and PFC [6,8]. Upregulation of BDNF is observed after chronic, but not acute, administration of different classes of antidepressants, including 5-hydroxytryptamine (5-HT) and norepinephrine-selective reuptake inhibitors. There is also evidence that antidepressant treatment increases BDNF in post-mortem brains of subjects on antidepressants at the time of death, as well as increasing blood levels of patients as discussed earlier [6,9,10].
In addition to BDNF, other neurotrophic/growth factors have been implicated in depression, including vascular endothelial growth factor (VEGF), fibroblast growth factor 2 and insulin-like growth factor 1 (IGF-1). Some of these factors have been best known for their effects on peripheral tissues (e.g. VEGF and IGF-1), but they are also expressed in neurons and glia and influence brain function [6,11,12]. Stress and antidepressant treatments have opposing effects on the expression of these factors. Moreover, functional studies demonstrate that altered levels of these neurotrophic/growth factors result in consequences in behavioural models of depression. However, this review will focus primarily on BDNF.
3. A neurotrophic hypothesis of depression and treatment response
Together, the preclinical and clinical gene expression and imaging studies support a neurotrophic hypothesis of depression and antidepressant response. This hypothesis proposes that depression results from decreased neurotrophic support, leading to neuronal atrophy, decreased hippocampal neurogenesis and loss of glia, and that antidepressant treatment blocks or reverses this neurotrophic factor deficit, and thereby reverses the atrophy and cell loss [6,13].
The neurotrophic hypothesis has been tested using various strategies for over-expression or knockdown of BDNF. These studies provide strong evidence that BDNF infusion is sufficient to produce an antidepressant response in behavioural models, and that BDNF is required for a response to antidepressant treatments [6,8]. However, there is much less evidence that BDNF depletion causes depressive behaviours. Most studies of BDNF-deletion mutant mice report normal behaviour in models of depression, with the exception that female conditional mutant mice show increased immobility in the forced swim test (FST) [14,15]. However, a recent study using RNA interference to knockdown BDNF expression in subregions of the hippocampus reports depressive behaviours in the forced swim and sucrose preference tests [16]. The discrepancy between these studies could be due to different knockdown approaches as well as behavioural methodology [16]. In addition, region-specific effects of BDNF (antidepressant effect in the hippocampus, but a pro-depressive effect in the nucleus accumbens) could influence behavioural outcomes particularly in mutant mouse models where knockout is global and not localized to a particular brain region [6,7].
(a). Brain-derived neurotrophic factor gene × environment interactions
It is also important to consider the possibility that although BDNF depletion may not be sufficient to cause depressive behaviour it may result in a state of increased susceptibility. Recent basic research and clinical studies provide evidence for a BDNF gene × environment interaction. Heterozygous deletion mutant mice, which express approximately half the normal levels of BDNF, display normal behaviour under baseline conditions, but exhibit a depressive phenotype upon exposure to stress ([17] but see [18]).
Advances in human genetics also provide a means to examine the influence of BDNF on susceptibility and resilience. A BDNF single nucleotide polymorphism, Val66Met, which decreases the processing and activity-dependent release of BDNF has been identified [19]. The BDNF Met allele is associated with reduced episodic memory and executive function, and decreased hippocampal volume in normal and depressed patients [19]. Although there is no direct association with depression, the BDNF Met allele increases vulnerability to develop depression in subjects exposed to early life stress or trauma [20–22]. Mutant mice with a knockin of the BDNF Met allele display increased anxiety in behavioural models and are unresponsive to antidepressant treatment [23].
4. Regulation of neurogenesis by stress and antidepressant treatment
Alterations of BDNF, as well as other neurotrophic factors, indicate that stress and antidepressant treatment result in cellular changes, notably regulation of neurogenesis and complexity of neuronal processes (see below). A brief review of neurogenesis and a more extensive overview of recent studies of synaptogenic responses are provided.
Birth of new neurons or neurogenesis continues to occur in selected neurogenic zones in the adult brain. This includes the subventricular zone that gives rise to olfactory bulb neurons, and the subgranular zone that generates granule cells of the hippocampal dentate gyrus. Similar to regulation of BDNF in the dentate gyrus, stress and antidepressant treatments exert opposing effects on neurogenesis in the adult hippocampus (figure 1). Different types of acute or chronic physical and social stress decrease neurogenesis, while chronic antidepressant treatments, including serotonin-selective reuptake inhibitors (SSRIs) and norepinephrine-selective reuptake inhibitors (NSRIs), increase neurogenesis [6,24]. The relevance of neurogenesis to depression and antidepressant responses in humans has been examined, although the evidence is limited to a few post-mortem studies. A recent report shows that the rate of new cell birth is significantly increased in depressed subjects receiving antidepressant treatment prior to and at the time of death [25]. However, cell birth was not decreased in untreated depressed subjects.
Figure 1.

Opposing actions of stress and antidepressants on brain-derived neurotrophic factor (BDNF) and neurogenesis. Stress decreases and antidepressant treatment increases the expression of BDNF, as well as vascular endothelial growth factor (VEGF) in the dentate gyrus granule cell layer of the hippocampus. These changes in growth factor expression contribute to the regulation of neurogenesis by stress and antidepressants. The negative effects of stress are also mediated in part by interleukin-1 (IL-1). This model shows the proliferation of neural progenitor cells giving rise to new neurons in the adult hippocampus. Antidepressants influence both the proliferation and survival of new neurons via effects on BDNF and VEGF. See text for details.
The role of BDNF in the regulation of neurogenesis has been examined using a number of different approaches. Studies in BDNF deletion mice have been mixed, whereas deletion of TrkB in neural progenitor cells is reported to block the proliferation of newborn neurons [26,27]. Localized BDNF knockdown using RNA interference is reported to block the differentiation but not proliferation of newborn neurons [16]. Blockade of TrkB by the expression of dominant negative TrkB or deletion of TrkB in progenitor cells also blocks antidepressant-induction of neurogenesis [27,28]. Together, these studies provide evidence that alterations of BDNF contribute to the regulation of neurogenesis by stress and antidepressant treatments.
5. Regulation of neuronal processes and synaptogenesis by stress
In addition to regulation of neurogenesis, the complexity of the dendritic arbour of neurons is altered by stress and antidepressant treatments. The formation of spine synapses or synaptogenesis is a key form of neuroplasticity, and represents a fundamental characteristic of neurons. Synaptogenesis is a structural change at a subcellular level that takes place in response to synaptic activity, and provides a mechanism for processing and incorporating new information that can be used to make the appropriate, future adaptive response (figure 2). Cellular models of learning and memory, such as long-term potentiation (LTP), have been used to study the mechanisms underlying synaptogenesis. Increased neuronal activity leads to insertion of glutamate receptors and maturation of spine synapses [29].
Figure 2.

Model for activity-dependent stimulation of synaptogenesis and spine formation. Synaptic activity and increased glutamate transmission can lead to increased synapse formation and spine density. This occurs through insertion of glutamate-AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors into the postsynaptic membrane. The mechanisms underlying the regulation of synaptogenesis and spine formation have been studied using a cellular model of learning and memory, known as long-term potentiation (LTP). See text for details.
The dendrite branches of neurons can be visualized by using a number of approaches, including Golgi impregnation or by filling cells with a dye that diffuses throughout the processes. These approaches allow for analysis of the number and length of dendrite branch points, and even the number of spines, the points of synaptic contact between neuronal processes.
The complexity of neuronal dendrites and number of spine synapses is markedly decreased by chronic stress exposure. This includes decreased number and length of apical dendrites in the CA3 pyramidal cell layer of the hippocampus and layer V pyramidal neurons of the PFC [30,31]. Reductions of dendrites and spines are observed in the PFC after as little as 7 days of restraint stress [32], and have been associated with depressive behaviours after exposure to chronic unpredictable stress (CUS) [33]. These studies support the possibility that decreased dendrite complexity contributes to the reduced volume of hippocampus and PFC reported in depressed patients.
The role of BDNF in the regulation of dendrite complexity and spine formation has also been examined in mutant mice. A knockin of the BDNF Met polymorphism has been developed, and studies show that expression of even a single copy of this human variant decreases the number and length of apical dendrites of CA3 pyramidal neurons, similar to the effects of chronic stress on dendrites [23]. BDNF heterozygous deletion mutant mice also have reduced CA3 apical dendrites [23]. Further studies will be required to determine whether reduced BDNF is responsible for the dendritic atrophy caused by chronic stress, but the current findings are consistent with this hypothesis.
Although antidepressant medications increase the expression of BDNF, there is little evidence that these agents reverse the dendrite atrophy caused by chronic stress. There is one study demonstrating that an atypical antidepressant, tianeptine, reverses the effects of chronic stress on atrophy of CA3 pyramidal neurons [34]. The lack of evidence for antidepressant reversal could reflect the technical challenges and time commitment required to conduct these difficult studies. In addition, it is possible that although BDNF expression is increased, typical antidepressant treatments do not increase BDNF release, which is required for increased synaptogenesis [23,35,36].
6. Cellular actions of the rapid-acting antidepressant, ketamine
Recent studies demonstrate that a non-selective NMDA receptor antagonist, ketamine, could address the limited efficacy and time lag for therapeutic response to typical antidepressants. Ketamine is a psychotomimetic at low doses and a dissociative anaesthetic at high doses. The rapid antidepressant actions of ketamine were first reported by Krystal and co-workers [37], who found that a low dose of ketamine (0.5 mg kg−1, i.v.) produced a rapid antidepressant response, observed after only 4 h and that lasted for 3 days. Subsequent studies have confirmed and extended this finding, reporting rapid antidepressant responses to ketamine within 2 h with sustained effects for up to 7 days [37–41]. Moreover, these rapid effects of ketamine are observed in patients resistant to two or more typical antidepressants and considered treatment resistant.
These findings represent one of the most significant advances in the field of depression over the past 50 years: a novel, rapid-acting, efficacious antidepressant agent with a mechanism that is completely different from currently available medications.
(a). Ketamine rapidly increases synaptogenesis
The cellular and molecular mechanisms underlying the rapid antidepressant effects of ketamine are more complex than simple blockade of NMDA receptors. The fast actions of ketamine also indicate that the effects occur via regulation of synaptic transmission and/or neuronal plasticity. This could include increased synaptogenesis and spine density that could oppose the neuronal atrophy caused by chronic stress in PFC and hippocampus.
To address this possibility, we examined the influence of ketamine on the number and function of spine synapses on PFC neurons. We found that a single dose of ketamine increased the number of spines on the apical dendrites of layer V pyramidal neurons [42]. Ketamine administration also increased spine function, demonstrated by an increase in neurotransmitter-induced excitatory post-synaptic currents (EPSCs) of neurons. This included an increase in the frequency and amplitude of 5-HT- and hypocretin-induced EPSCs of layer V pyramidal neurons. Further analysis of spine morphology shows that ketamine treatment increases the number of ‘mushroom’-shaped spines, which are the mature and most functionally active spines [42] (figure 2). The increase in number of mushroom spines is consistent with the increase of synaptic function resulting from ketamine administration, as the glutamate receptors are incorporated into mature spines and underlie the increase in EPSC amplitude.
The initial studies of spine density and function were conducted 24 h after ketamine administration, but preliminary studies indicate that synaptogenesis may occur even faster, within hours after treatment. This possibility is supported by analysis of synaptic proteins that are required for synaptogenesis and new spine formation (figures 2 and 3). Levels of the synaptic proteins PSD95, GluR1 and synapsin I were measured in synaptoneurosome preparations of the PFC. Ketamine administration rapidly increased levels of these synaptic proteins, with significant increases observed after 2 h and sustained induction for up to 7 days [42]. This rapid time course for induction of synaptic proteins is consistent with the time course for the therapeutic actions of ketamine.
Figure 3.
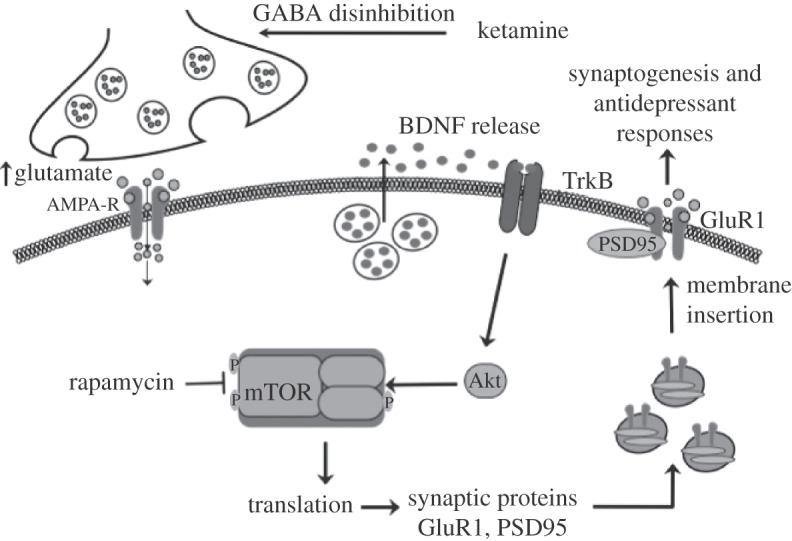
Regulation of mammalian target of rapamycin (mTOR) signalling by NMDA receptor antagonists. Ketamine increases extracellular glutamate, possibly via NMDA receptors on GABAergic interneurons resulting in disinhibition of glutamate transmission. This leads to activity-dependent release of BDNF and stimulation of signalling cascades, including Akt, that activate the mTOR translational system in dendrites of neurons. Induction of translation results in increased levels of GluR1 and other synaptic proteins, providing the machinery required for increased synaptogensis and spine formation. These effects contribute to the rapid and sustained antidepressant actions of ketamine. See text for further details.
(b). Ketamine rapidly reverses neuronal atrophy caused by chronic stress
The ability of ketamine to increase spine number and function observed in normal animals suggests that the atrophy of dendrites caused by chronic stress might be reversed by ketamine administration. To test this hypothesis, we used a CUS model of depression. This is considered one of the better animal models of depression because CUS exposure results in anhedonia or the inability to experience pleasure, a hallmark symptom of depression. Moreover, chronic stress exposure causes atrophy of apical dendrites and decreases spine density of PFC neurons [32].
The results of these studies demonstrate that CUS exposure for three weeks significantly decreases the number and function of spines on layer V pyramidal neurons in the PFC [33]. This includes a decrease in the number of spines in the distal and proximal tufts of layer V neurons, and a decrease in 5-HT- and hypocretin-induced EPSCs. There was also a significant decrease in levels of synaptic proteins, including reduced levels of PSD95, GluR1 and synapsin I, consistent with the downregulation of synaptogenesis. Surprisingly, a single dose of ketamine caused a rapid and complete reversal of the deficit in spine number and function caused by three weeks of CUS exposure [33]. Ketamine administration also completely reversed the deficit in synaptic proteins, including PSD95, GluR1 and synapsin I, consistent with the reversal of synaptogenesis.
These findings demonstrate that ketamine rapidly reverses the deficits in spine number and function in the PFC resulting from chronic stress exposure. Ketamine may also cause a similar reversal of atrophy of hippocampal neurons resulting from stress, although further studies will be required to test this hypothesis. Nevertheless, the results suggest that the therapeutic actions of ketamine may result, at least in part, from reversal of neuronal atrophy caused by depression, and reinstatement of the limbic circuitry required for control of emotion and mood.
(c). Ketamine produces rapid antidepressant behavioural actions
To examine the possibility that the induction of synaptogenesis and synaptic proteins could contribute to the therapeutic actions of ketamine, studies of rodent behavioural models of depression were conducted. Previous studies have demonstrated that a single low dose of ketamine produces a rapid antidepressant response in behavioural models of depression, including the FST and learned helplessness (LH) paradigm [43]. In addition, a selective NMDA NR2B antagonist, Ro 25-6981, also produced an antidepressant effect in the FST and LH model similar to the effects of ketamine. The significance of these findings is supported by a clinical study demonstrating that another NR2B antagonist, CP101,606 also produces an antidepressant response in depressed patients [44].
We have confirmed these findings in the FST and LH paradigm, and also demonstrate that ketamine and Ro 25-6981 produce a rapid antidepressant response in a novelty suppressed feeding test (NSFT) [42]. The NSFT, which measures the latency to feed in an open field, is considered a model of anxiety but is responsive to chronic, but not acute administration of SSRI antidepressants. Because of the requirement for chronic, three-week, antidepressant treatment, NSFT provides a measure of the rapid actions of ketamine that cannot be determined with the FST and LH models, which are responsive to acute or subchronic (1 or 6 days, respectively) administration of a typical antidepressant.
We have also examined the influence of ketamine in the CUS model of depression and resulting anhedonic behaviour, measured by preference for a sweetened solution. Exposure to CUS for three weeks decreases sucrose preference and this effect is reversed by chronic administration (three weeks) of a typical antidepressant, another reason why the CUS paradigm is considered one of the better rodent models of depression. The CUS model thereby provides a rigorous test in rodents of the ability of ketamine to produce rapid antidepressant actions. The results demonstrate that a single dose of ketamine completely reverses the deficit in sucrose consumption caused by CUS exposure [33]. This rapid action of ketamine parallels the rapid reversal of the atrophy of PFC pyramidal neuron spine density caused by CUS exposure. Together, the results are consistent with the hypothesis that the rapid synaptogenic effects of ketamine underlie the therapeutic response to this agent.
7. Role of mammalian target of rapamycin signalling in the actions of ketamine
Studies of protein synthesis-dependent long-term memory have demonstrated that the induction of synaptogenesis requires protein synthesis and activation of the mammalian target of rapamycin (mTOR) [35]. The mTOR complex and translational machinery have been localized in dendrites and spines, as well as cell bodies, and are therefore available for regulation of new synaptic protein synthesis as needed [35,45]. Activation occurs via phosphorylation of specific residues in the kinase domain of mTOR. An adjacent domain, the FKBP12-rapamycin binding region, is also critical for rapamycin inhibition (figure 3). Activation of the mTOR complex 1, the rapamycin-sensitive complex, regulates two key components of translation initiation, p70 ribosomal S6 kinase (p70S6K) and eIF4E-binding proteins. Activation occurs via a number of pathways, most notably release of BDNF, stimulation of its receptor TrkB and downstream signalling cascades PI-3K-Akt and MEK-ERK [34,35].
Studies were conducted to determine whether ketamine increases the phosphorylated and activated forms of mTOR signalling proteins, including mTOR, p70S6K and 4E-BP1 (figure 3). The results demonstrate that a single dose of ketamine stimulates the mTOR cascade, increasing levels of phospho-mTOR, phospho-p70S6K and phospho-4E-BP1 [42]. Ketamine-induction of mTOR signalling is rapid, with induction of phosphorylated proteins observed at 30 and 60 min, but transient, as levels return to baseline by 2 h. A similar rapid and transient increase in mTOR was observed with the NR2 selective antagonist, Ro 25-6981. Although transient, the activation of mTOR signalling precedes the induction of synaptic proteins and could thereby underlie the increase in protein synthesis.
In contrast to ketamine, acute or chronic administration of fluoxetine or imipramine, two typical antidepressants that block the reuptake of 5-HT, did not influence levels of mTOR signalling in the PFC [42]. We also examined the influence of electroconvulsive seizures (ECSs), a model for one of the most effective therapies for treatment-resistant depressed patients that also has a slightly faster onset of action than typical antidepressant medications. However, ECS did not increase mTOR signalling in the PFC [42]. Together, these results indicate that the rapid induction of mTOR signalling and synaptic proteins is specific to ketamine.
(a). Synaptogenic and behavioural actions of ketamine are blocked by rapamycin
The induction of mTOR signalling suggests that the ability of ketamine to increase synaptogenesis is mediated by stimulation of this protein synthesis regulatory pathway. To directly test this hypothesis, the influence of rapamycin, a selective inhibitor of mTOR (figure 3), on synaptogenesis was examined. Rapamycin pretreatment completely blocked ketamine-induction of spine number and function of layer V pyramidal neurons in the PFC [42]. In addition, rapamycin pretreatment completely blocked the induction of the synaptic proteins PSD95, GluR1 and synapsin I, resulting from ketamine administration. These findings provide direct evidence that ketamine-induction of synaptogenesis requires mTOR signalling and synaptic protein synthesis.
Next, studies were conducted to determine whether the behavioural actions of ketamine are also dependent on mTOR signalling. Pretreatment with rapamycin completely blocked the antidepressant effects of ketamine in the FST, LH and NSF test [42]. Moreover, the rapid antidepressant effects of ketamine on the deficit in sucrose preference caused by CUS were completely blocked by pretreatment with rapamycin [33]. The behavioural actions of the selective NR2B antagonist, Ro 25-6981, were also blocked by rapamycin pretreatment [33,42]. Together, these studies demonstrate that the rapid synaptogenic and antidepressant behavioural actions of ketamine are dependent on stimulation of mTOR signalling, induction of synaptic protein synthesis and increased synaptogenesis.
A recent study has reported that the behavioural actions of ketamine require BDNF protein synthesis, and that this effect is mediated by activation of eukaryotic elongation factor 2 (eEF2) [46]. The induction of eEF2, which plays an important role in the translocation of ribosomes during protein synthesis, is dependent on inhibition of eEF2 kinase. The activation of eEF2 could also synergize with the actions of ketamine on mTOR signalling. The requirement for BDNF and protein synthesis is consistent with the results of our study, although Monteggia and co-workers were unable to detect ketamine-induction of mTOR signalling or rapamycin blockade of the behavioural actions of ketamine [46]. There are several technical reasons that could explain these discrepancies. First, mTOR signalling was measured in crude homogenates of hippocampus, not synaptoneurosome-enriched fractions of PFC as previously reported by Li and co-workers [42]. This is critical as mTOR is expressed in neuronal and glial cell bodies, which could mask changes in the smaller dendritic compartment. Second, the behavioural analysis was conducted 30 min after ketamine administration, when ketamine levels are still high in brain and corresponds to the time when patients experience mild psychotomimetic and dissociative effects of ketamine [36,40]. This would also correspond to the time when ketamine increases levels of glutamate [47], which could underlie the increased activity observed in the FST. As pointed out by the authors [46], rapamycin would not be expected to block the effects of ketamine at this time point since the induction of synaptic proteins and synaptogenesis is delayed by approximately 2 h. This later time point corresponds more closely to the initial antidepressant response observed in depressed patients [36,40].
8. Potential synaptogenic targets
The rapid and efficacious actions of ketamine in treatment-resistant-depressed patients represent major advances for the treatment of depression. There are also reports that ketamine is effective for treatment of bipolar depression [48] and suicide [49,50], and it is possible that it will also be effective for other psychiatric illnesses (e.g. post-traumatic stress disorder). Despite this promise, the use of ketamine also has limitations. Ketamine is a street drug with abuse potential, and preclinical studies report that repeated daily administration of ketamine may have neurotoxic effects, particularly on the function of GABAergic interneurons [51,52]. However, characterization of the mechanisms underlying the actions of ketamine suggests potential targets that could lead to the development of medications with ketamine-like effects. It may also be possible that activation of certain targets could sustain the actions of ketamine so that repeated dosing is not needed.
One possibility that has already been discussed is a selective NMDA receptor antagonist. Basic and clinical studies have demonstrated that NR2B selective agents increase mTOR signalling and synaptic protein synthesis, and have antidepressant effects in rodent models and depressed subjects. Further studies in depressed patients will be needed to confirm the clinical results with CP101,606, in particular to determine whether this or other NR2B selective agents produce rapid and efficacious therapeutic responses in treatment-resistant depressed patients.
Other leads have come from basic research reports that the behavioural actions of ketamine are dependent on glutamate–AMPA receptor activation [42,43]. Ketamine is reported to increase levels of extracellular glutamate in the PFC, possibly via blockade of NMDA receptors on GABAergic interneurons resulting in disinhibition of glutamate transmission [47,53]. We have also found that ketamine induction of mTOR signalling and synaptic protein levels are dependent on AMPA receptor activation [42]. These studies suggest that pharmacological agents that increase glutamate–AMPA receptor transmission should produce a ketamine-like effect, or sustain the actions of ketamine. Two targets that influence glutamate–AMPA activity have already received attention: metabotropic glutamate receptor type II antagonists and AMPA receptor potentiating agents.
(a). Enhancing glutamate–AMPA receptor transmission
The mGlu II receptors, mGlu2/3, are located on presynaptic glutamate terminals and provide inhibitory control of glutamate release. Previous studies demonstrate that mGlu2/3 antagonists, notably MGS0039 and LY341,495, have antidepressant actions in standard behavioural models [54,55]. In addition, the antidepressant actions of mGlu2/3 antagonists are blocked by pretreatment with an AMPA receptor antagonist, similar to the blockade of ketamine [54,56]. These findings support the possibility that mGlu2/3 receptor antagonist would increase glutamate transmission and thereby stimulate mTOR signalling and synaptogenesis, effects which could underlie the antidepressant behavioural actions that have been reported. Studies are currently underway to determine whether mGlu2/3 antagonists stimulate mTOR and synaptic protein synthesis.
Positive AMPA receptor modulators increase AMPA receptor function by altering receptor kinetics (e.g. decrease receptor desensitization or deactivation) [57,58]. AMPA receptor potentiating drugs increase the size of EPSPs, enhance LTP, enhance learning and memory, and increase BDNF [59,60]. One AMPA receptor potentiating drug, CX614, is reported to stimulate mTOR signalling and dendritic protein synthesis in cultured neurons in a BDNF-dependent manner [36]. These findings are consistent with the possibility that AMPA receptor potentiating drugs could produce rapid ketamine-like effects. There are several AMPA receptor potentiating drugs available, including high- (CX614, LY451646) and low-impact (CX1739, Org 26576) agents, based on efficacy to increase current flow of AMPA receptors [57,58]. Moreover, these AMPA receptor potentiating drugs have shown promise in biochemical and behavioural studies, including induction of BDNF and antidepressant responses in the FST [61,62].
One potential caveat is that the function of these glutamate-modulating agents may be dependent on basal glutamate transmission. If synaptic glutamate levels are very low, then a presynaptic mGlu2/3 antagonist would not be expected to enhance glutamate transmission since there would be no negative tone to block. Similarly, the actions of an AMPA receptor potentiating agent would be dependent on the presence of sufficient synaptic glutamate to cause low levels of receptor activation that could be potentiated. Another potential problem is that these agents may produce global effects on glutamate transmission that could lead to toxicity or unwanted side effects. Direct tests will be required to determine whether these approaches rapidly stimulate mTOR and synaptogenesis and produce antidepressant actions without side effects. Alternatively, mGlu2/3 antagonists or AMPA receptor potentiating drugs could sustain the response to ketamine and still provide a critical unmet therapeutic need.
9. Summary and conclusions
Exposure to chronic stress and/or depression results in neuronal atrophy and decreased neurogenesis in limbic brain regions involved in regulation of mood and emotion. The mechanisms underlying the actions of stress have not been identified. Based on studies demonstrating that stress decreases BDNF, and that BDNF and other neurotrophic factors stimulate mTOR signalling, it will be interesting to determine whether downregulation of mTOR contributes to the reduction in synaptic proteins, spine number and dendrite branching in PFC. The ability of ketamine and NMDA receptor antagonists to rapidly increase synaptogenesis represents a fundamental shift in our understanding of the mechanisms underlying rapid acting antidepressants. Studies are underway to further characterize the mechanisms that lead to stimulation of mTOR signalling by ketamine, including experiments to determine the role of BDNF in activation of mTOR.
The advances made in our understanding of rapid-acting NMDA receptor antagonists could also lead to identification of novel drug targets for the treatment of depression. However, these novel agents do not come without risk, particularly when using approaches that enhance glutamate signalling that could lead to neurotoxic effects when over-activated. Investigating mechanisms and treatment strategies to optimize therapeutic response while limiting toxicity will be critical to the success of these approaches. Nevertheless, these exciting findings raise optimism for a new generation of rapid-acting agents with superior therapeutic efficacy.
Acknowledgements
This work is supported by US Public Health Service (grant nos MH93897 and MH45481), and the state of Connecticut, Department of Mental Health and Addiction Services.
References
- 1.Kessler R., Berlund P., Demler O., Jin R., Koretz D., Merikangas K. R. 2003. The epidemiology of major depressive disorder: results for the National Comorbidity Survey Replication (NCS-R). J. Am. Med. Assoc. 289, 3095–3105 10.1001/jama.289.23.3095 (doi:10.1001/jama.289.23.3095) [DOI] [PubMed] [Google Scholar]
- 2.Trivedi M., et al. , STAR*D Study Team 2006. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am. J. Psychiatry 163, 28–40 10.1176/appi.ajp.163.1.28 (doi:10.1176/appi.ajp.163.1.28) [DOI] [PubMed] [Google Scholar]
- 3.Drevets W., Price J. L., Furey M. L. 2008. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct. 213, 93–118 10.1007/s00429-008-0189-x (doi:10.1007/s00429-008-0189-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macqueen G., Yucel K., Taylor V. H., Macdonald K., Joffe R. 2008. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry 64, 880–883 10.1016/j.biopsych.2008.06.027 (doi:10.1016/j.biopsych.2008.06.027) [DOI] [PubMed] [Google Scholar]
- 5.Miguel-Hidalgo J., Rajkowska G. 2002. Morphological brain changes in depression: can antidepressants reverse them. CNS Drugs 16, 361–372 10.2165/00023210-200216060-00001 (doi:10.2165/00023210-200216060-00001) [DOI] [PubMed] [Google Scholar]
- 6.Duman R., Monteggia L. M. 2006. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127 10.1016/j.biopsych.2006.02.013 (doi:10.1016/j.biopsych.2006.02.013) [DOI] [PubMed] [Google Scholar]
- 7.Krishnan V., Nestler E. J. 2008. The molecular neurobiology of depression. Nature 455, 894–902 10.1038/nature07455 (doi:10.1038/nature07455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castren E., Rantamaki T. 2010. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev. Neurobiol. 70, 289–297 10.1002/dneu.20758 (doi:10.1002/dneu.20758) [DOI] [PubMed] [Google Scholar]
- 9.Bocchio-Chiavetto L., et al. 2010. Serum and plasma BDNF levels in major depression: a replication study and meta-analyses. World J. Biol. Psychiatry 11, 763–773 10.3109/15622971003611319 (doi:10.3109/15622971003611319) [DOI] [PubMed] [Google Scholar]
- 10.Sen S., Duman R. S., Sanacora G. 2008. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol. Psychiatry 64, 527–532 10.1016/j.biopsych.2008.05.005 (doi:10.1016/j.biopsych.2008.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akil H., Evans S. J., Turner C. A., Perez J., Myers R. M., Bunney W. E., Jones E. G., Watson S. J., Pritzker Consortium 2008. The fibroblast growth factor family and mood disorders. Novartis Foundation Symp. 289, 93–96 [DOI] [PubMed] [Google Scholar]
- 12.Fournier N. M., Duman R. S. 2011. Role of vascular endothelial growth factor in adult hippocampal neurogenesis: implications for the pathophysiology and treatment of depression. Behav. Brain Res. 227, 440–449 10.1016/j.bbr.2011.04.022 (doi:10.1016/j.bbr.2011.04.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duman R., Heninger G. R., Nestler E. J. 1997b. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597–606 10.1001/archpsyc.1997.01830190015002 (doi:10.1001/archpsyc.1997.01830190015002) [DOI] [PubMed] [Google Scholar]
- 14.Adachi M., Barrot M., Autry A. E., Theobald D., Monteggia L. M. 2008. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry 63, 642–649 10.1016/j.biopsych.2007.09.019 (doi:10.1016/j.biopsych.2007.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heldt S., Stanek L., Chhatwal J. P., Ressler K. J. 2007. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol. Psychiatry 12, 656–670 10.1038/sj.mp.4001957 (doi:10.1038/sj.mp.4001957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taliaz D., Stall N., Dar D. E., Zangen A. 2010. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol. Psychiatry 15, 80–92 10.1038/mp.2009.67 (doi:10.1038/mp.2009.67) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duman C., Russell D. A., Duman R. S. 2007. Blockade of ERK produces a pro-depressive effect and blocks the behavioral actions of antidepressants. Biol. Psychiatry 61, 661–670 10.1016/j.biopsych.2006.05.047 (doi:10.1016/j.biopsych.2006.05.047) [DOI] [PubMed] [Google Scholar]
- 18.Ibarguen-Vargasa Y., Surgeta A., Vourc'ha P., Lemana S., Andresa C. R., Gardierc A. M., Belzunga C. 2009. Deficit in BDNF does not increase vulnerability to stress but dampens antidepressant-like effects in the unpredictable chronic mild stress. Behav. Brain Res. 202, 245–251 10.1016/j.bbr.2009.03.040 (doi:10.1016/j.bbr.2009.03.040) [DOI] [PubMed] [Google Scholar]
- 19.Casey B., et al. 2009. Brain-derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neuroscience 164, 108–120 10.1016/j.neuroscience.2009.03.081 (doi:10.1016/j.neuroscience.2009.03.081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gatt J., Nemeroff C. B., Dobson-Stone C., Pauyl R. H., Bryant R. A., Schofield P. R., Gordon E., Kemp A. H., Williams L. M. 2009. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol. Psychiatry 14, 681–695 10.1038/mp.2008.143 (doi:10.1038/mp.2008.143) [DOI] [PubMed] [Google Scholar]
- 21.Kaufman J., Yang B. Z., Douglas-Palumberi H., Grasso D., Lipschitz D., Houshyar S., Krystal J. H., Gelernter J. 2006. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol. Psychiatry 59, 673–680 10.1016/j.biopsych.2005.10.026 (doi:10.1016/j.biopsych.2005.10.026) [DOI] [PubMed] [Google Scholar]
- 22.Kim J., Stewart R., Kim S. W., Yang S. J., Shin I. S., Kim Y. H., Yoon J. S. 2007. Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biol. Psychiatry 62, 423–428 10.1016/j.biopsych.2006.11.020 (doi:10.1016/j.biopsych.2006.11.020) [DOI] [PubMed] [Google Scholar]
- 23.Chen Z.-Y., et al. 2006b. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143 10.1126/science.1129663 (doi:10.1126/science.1129663) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahay A., Hen R. 2007. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 10, 1110–1115 10.1038/nn1969 (doi:10.1038/nn1969) [DOI] [PubMed] [Google Scholar]
- 25.Boldrini M., Underwood M. D., Hen R., Rosoklija G. B., Dwork A. J., Mann J., Arango V. 2009. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34, 2376–2389 10.1038/npp.2009.75 (doi:10.1038/npp.2009.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergami M., Rimondini R., Santi S., Blum R., Gotz M., Canossa M. 2008. Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc. Natl Acad. Sci. USA 105, 15 570–15 575 10.1073/pnas.0803702105 (doi:10.1073/pnas.0803702105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Luikart B. W., Birnbaum S., Chen J., Kwon C. H., Kernie S. G., Bassel-Duby R., Parada L. F. 2008. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59, 399–412 10.1016/j.neuron.2008.06.023 (doi:10.1016/j.neuron.2008.06.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sairanen M., Lucas G., Ernfors P., Castren M., Casren E. 2005. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 25, 1089–1094 10.1523/JNEUROSCI.3741-04.2005 (doi:10.1523/JNEUROSCI.3741-04.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessels H., Malinow R. 2009. Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350 10.1016/j.neuron.2009.01.015 (doi:10.1016/j.neuron.2009.01.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEwen B. 2007. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 87, 873–904 10.1152/physrev.00041.2006 (doi:10.1152/physrev.00041.2006) [DOI] [PubMed] [Google Scholar]
- 31.Shansky R., Morrison J. H. 2009. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 1293, 108–113 10.1016/j.brainres.2009.03.062 (doi:10.1016/j.brainres.2009.03.062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu R.-J., Aghajanian G. K. 2008. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc. Natl Acad. Sci. USA 105, 359–364 10.1073/pnas.0706679105 (doi:10.1073/pnas.0706679105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li N., Liu R.-J., Dwyer J., Banasr M., Lee B., Son J., Li X.-Y., Aghajanian G., Duman R. S. 2011. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 69, 754–761 10.1016/j.biopsych.2010.12.015 (doi:10.1016/j.biopsych.2010.12.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe Y., Gould E., Daniels D. C., Cameron H., McEwen B. S. 1992. Tianeptine attenuates stress-induced morphological changes in the hippocampus. Eur. J. Pharmacol. 222, 157–162 10.1016/0014-2999(92)90830-W (doi:10.1016/0014-2999(92)90830-W) [DOI] [PubMed] [Google Scholar]
- 35.Hoeffer C., Klann E. 2010. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 10.1016/j.tins.2009.11.003 (doi:10.1016/j.tins.2009.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jourdi H., Hsu Y. T., Zhou M., Qin Q., Bi X., Baudry M. 2009. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J. Neurosci. 29, 8688–8697 10.1523/JNEUROSCI.6078-08.2009 (doi:10.1523/JNEUROSCI.6078-08.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berman R., Cappiello A., Anand A., Oren D. A., Charney D. S., Krystal J. H. 2000. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354 10.1016/S0006-3223(99)00230-9 (doi:10.1016/S0006-3223(99)00230-9) [DOI] [PubMed] [Google Scholar]
- 38.aan het Rot M., Collins K. A., Murrough J. W., Perez A. M., Reich D. L., Charney D. S., Mathew S. J. 2010. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67, 139–145 10.1016/j.biopsych.2009.08.038 (doi:10.1016/j.biopsych.2009.08.038) [DOI] [PubMed] [Google Scholar]
- 39.Price R., Nock M. K., Charney D. S., Mathew S. J. 2009. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol. Psychiatry 66, 522–526 10.1016/j.biopsych.2009.04.029 (doi:10.1016/j.biopsych.2009.04.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salvadore G., et al. 2010. Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology 35, 1415–1422 10.1038/npp.2010.24 (doi:10.1038/npp.2010.24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarate C. J., Singhm J. B., Carlson P. J., Brutsche N. E., Ameli R., Luckenbaugh D. A., Charney D. S., Manji H. K. 2006. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864 10.1001/archpsyc.63.8.856 (doi:10.1001/archpsyc.63.8.856) [DOI] [PubMed] [Google Scholar]
- 42.Li N., Lee B. Y., Liu R. J., Banasr M., Dwyer J., Iwata M., Li X. Y., Aghajanian G., Duman R. S. 2010. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 10.1126/science.1190287 (doi:10.1126/science.1190287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeng S., Zarate C. A., Du J., Schloesser R. J., McCammon J., Chen G., Manji H. K. 2007. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63, 349–352 10.1016/j.biopsych.2007.05.028 (doi:10.1016/j.biopsych.2007.05.028) [DOI] [PubMed] [Google Scholar]
- 44.Preskorn S., Baker B., Kolluri S., Menniti F. S., Krams M., Landen J. W. 2008. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-asparate antagonist, CP-101, 606, in patients with treatment-refractory major depressive dosorder. J. Clin. Psychopharmacol. 28, 631–637 10.1097/JCP.0b013e31818a6cea (doi:10.1097/JCP.0b013e31818a6cea) [DOI] [PubMed] [Google Scholar]
- 45.Livingston M., Atas E., Meller A., Sonenberg N. 2010. Mechanisms governing the control of mRNA translation. Phys. Biol. 7, 021001. 10.1088/1478-3975/7/2/021001 (doi:10.1088/1478-3975/7/2/021001) [DOI] [PubMed] [Google Scholar]
- 46.Autry A., Adachi M., Nosyreva E., Na E. S., Los M. F., Cheng P. F., Kavalali E. T., Monteggia L. M. 2011. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95 10.1038/nature10130 (doi:10.1038/nature10130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moghaddam B., Adams B., Verma A., Daly D. 1997. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2912–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diazgranados N., et al. 2010. A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67, 793–802 10.1001/archgenpsychiatry.2010.90 (doi:10.1001/archgenpsychiatry.2010.90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiazGranados N., Ibrahim L. A., Brutsche N. E., Ameli R., Henter I. D., Luckenbaugh D. A., Machado-Vieira R., Zarate C. A., Jr 2010. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-d-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 71, 1605–1611 10.4088/JCP.09m05327blu (doi:10.4088/JCP.09m05327blu) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larkin G., Beautrais A. L. 2011. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int. J. Neuropsychopharmacol. 14, 1127–1131 10.1017/S1461145711000629 (doi:10.1017/S1461145711000629) [DOI] [PubMed] [Google Scholar]
- 51.Behrens M., Ali S. S., Dao D. N., Lucero J., Shekhtman G., Quick K. L., Dugan L. L. 2007. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318, 1645–1647 10.1126/science.1148045 (doi:10.1126/science.1148045) [DOI] [PubMed] [Google Scholar]
- 52.Behrens M., Sejnowski T. J. 2009. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex. Neuropharmacology 57, 193–200 10.1016/j.neuropharm.2009.06.002 (doi:10.1016/j.neuropharm.2009.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Homayoun H., Moghaddam B. 2007. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 27, 11496–11500 10.1523/JNEUROSCI.2213-07.2007 (doi:10.1523/JNEUROSCI.2213-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pałucha-Poniewiera A., Wierońska J. M., Brański P., Stachowicz K., Chaki S., Pilc A. 2010. On the mechanism of the antidepressant-like action of group II mGlu receptor antagonist, MGS0039. Psychopharmcology 212, 523–535 10.1007/s00213-010-1978-5 (doi:10.1007/s00213-010-1978-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pilc A., Chaki S., Nowak G., Witkin J. M. 2007. Mood disorders: regulation by metabotropic glutamate receptors. Biochem. Pharmacol. 75, 997–1006 10.1016/j.bcp.2007.09.021 (doi:10.1016/j.bcp.2007.09.021) [DOI] [PubMed] [Google Scholar]
- 56.Karasawa J., Shimazaki T., Kawashima N., Chaki S. 2005. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 1042, 92–98 10.1016/j.brainres.2005.02.032 (doi:10.1016/j.brainres.2005.02.032) [DOI] [PubMed] [Google Scholar]
- 57.Arai A., Kessler M. 2007. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr. Drug Targets 8, 583–602 10.2174/138945007780618490 (doi:10.2174/138945007780618490) [DOI] [PubMed] [Google Scholar]
- 58.Lynch A., Walsh C., Delaney A., Nolan Y., Campbell V. A., Lynch M. A. 2004. Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10: a role for IL-1β? J. Neurochem. 88, 635–646 10.1046/j.1471-4159.2003.02157.x (doi:10.1046/j.1471-4159.2003.02157.x) [DOI] [PubMed] [Google Scholar]
- 59.Arai A., Kessler M., Rogers G., Lynch G. 2000. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol. Pharmacol. 58, 802–813 [DOI] [PubMed] [Google Scholar]
- 60.Lauterborn J., Lynch G., Vanderklish P., Arai A., Gall C. M. 2000. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J. Neurosci. 20, 8–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai F., Li X., Clay M., Lindsrom T., Skolnick P. 2001. Intra- and interstrain differences in models of ‘behavior despair’. Pharmacol. Biochem. Behav. 70, 187–192 10.1016/S0091-3057(01)00599-8 (doi:10.1016/S0091-3057(01)00599-8) [DOI] [PubMed] [Google Scholar]
- 62.Bai J., Ramos R. L., Ackman J. B., Thomas A. M., Lee R. V., LoTurco J. J. 2003. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 6, 1277–1283 10.1038/nn1153 (doi:10.1038/nn1153) [DOI] [PubMed] [Google Scholar]