

Abstract
The monoamine hypothesis of depression has dominated our understanding of both the pathophysiology of depression and the action of pharmacological treatments for the last decades, and it has led to the production of several generations of antidepressant agents. However, there are serious limitations to the current monoamine theory, and additional mechanisms, including hypothalamic–pituitary–adrenal (HPA) axis dysfunctions, as well as neurodegenerative and inflammatory alterations, are potentially associated with the pathogenesis of mood disorders. Moreover, new data have recently indicated that epigenetic mechanisms such as histone modifications and DNA methylation could affect diverse pathways leading to depression-like behaviours in animal models. In a transgenic mouse model of depression, in which a downregulation of glucocorticoid receptors (GR) causes a deficit in the HPA axis feedback control, besides alterations in monoamine neurotransmission and neuroplasticity, we found modifications in the expression of many proteins involved in epigenetic regulation, as well as clock genes, in the hippocampus and the frontal cortex, that might be central in the genesis of depressive-like behaviours.
Keywords: depression, glucocorticoid receptors, mice, epigenetics, stress, monoamines
1. Introduction
Although we are repeatedly exposed to adversity and stressful situations throughout life, inter-related hormonal, neurobiological and circadian system networks maintain physiological and behavioural homeostasis in most individuals. This functional equilibrium is determined by genetic- and environmentally driven epigenetic elements that interact in complex and still poorly understood ways. Disruptions in the interactions or imbalances between these systems could cause various neurobehavioural disorders such as depression. Depression is a chronic, recurring, life-threatening illness that affects up to 14 per cent of the population and causes a significant burden at both the individual and the society level [1]. Available treatments of depression are suboptimal and still unsatisfactory; only 50 per cent of treated depressed patients achieve full remission. Improvement of therapeutic strategies depends on the identification of underlying pathological processes and mechanisms of action of current treatments. Despite significant advances, the causes of depression and the molecular basis of treatments are still poorly understood. Various theories have been proposed to account for the overall pathophysiological state or particular symptoms of depression based on dysfunction of monoamine neurotransmission [2], the hypothalamic–pituitary–adrenal (HPA) axis [3], circadian rhythms [4] or neuroimmune processes [5]. Besides these major theories, there has recently been considerable interest in the ideas that epigenetic modifications could be biological markers of both depression and antidepressant (AD) efficacies, or that they could be directly involved in the pathophysiology of depression and in the action of AD compounds [6]. This review briefly summarizes the most prominent theories and focuses more specifically on a genetic model of the HPA axis dysregulation in depression, glucocorticoid receptor-impaired (GR-i) mice. We will see that these various hypotheses are far from being mutually exclusive.
2. Overview of the various hypotheses
(a). The monoamine hypothesis is central but remains insufficient
Our views on the pathophysiology of depression have been dominated by the monoamine hypothesis, based principally on the efficacy of both first- and second-generation ADs suggesting that an imbalance, mainly in serotonergic and noradrenergic neurotransmission, is at the core of the pathophysiology of depression. Several classes of ADs are currently available [7], and most of them affect monoaminergic transmissions in one way or another. Monoamine oxidase inhibitors (MAOIs), originally derived from drugs developed to treat tuberculosis (e.g. izoniazid) were observed to improve mood in patients. However, the first MAOIs had serious side effects, and their use was problematic because of the strict diet people needed to follow in order to prevent hypertensive reactions induced by food rich in tyramine. Tricyclic AD compounds (TCAs) such as imipramine, derived from structurally related molecules used to treat psychosis, also acted on the monoaminergic system and were very effective in relieving depressive symptoms. However, TCAs also induce aversive effects, presumably because of their action on other transmission systems (mainly histamine and acetylcholine). Drugs selectively blocking monoamine transporters, and thus leading to a specific increase in extracellular monoamine levels, were then developed. Compounds displaying selective serotonin (5-hydroxytryptamine, 5-HT) reuptake inhibition (SSRIs) were first produced in view of clinical findings, such as the strong relationship between suicide completion and low levels of the 5-HT metabolite (5-HIAA) in the cerebrospinal fluid of depressed patients [8]. The majority of ADs prescribed since the 1990s belong to this SSRI family, but more recently, drugs acting selectively on both the norepinephrine and the serotonine transporters (mixed 5-HT/norepinephrine reuptake inhibitors (SNRIs)) have also shown clinical benefits. In addition, atypical ADs having no direct effects on monoamine reuptake and primarily acting as antagonists at monoaminergic receptors are currently used. Overall, it remains reasonable to hypothesize that depression is caused by inadequate monoamine neurotransmission, as ADs act by increasing monoamine availability and by producing long-term adaptive changes in monoaminergic receptor sensitivity (5-HT1A autoreceptors, 5-HT2, β-adrenergic and α2-adrenergic heteroreceptors, etc.) [9]. However, although positive AD responses are transiently reversed in patients under low tryptophan diet leading to 5-HT depletion, this depletion does not worsen symptoms in unmedicated depressed patients [10]. In addition, 5-HT depletion by itself does not cause depression in healthy volunteers, undermining our view about the crucial role of a decrease in the serotonergic tone to trigger depressive episodes. Therefore, factors beyond monoamine deficiency or imbalance are most probably implicated in the development of major depression. Moreover, all available ADs exert their effects only after prolonged administration (several weeks to months), which suggests that their short-term effects on monoaminergic transmission are not directly responsible for the clinical efficacy of these drugs. Rather, long-term adaptations to AD treatment would appear to mediate their therapeutic action. Finally, it is necessary to search beyond the monoaminergic hypothesis because, albeit generally safe, monoamine-based ADs are far from being ideal drugs. The side effects of TCAs and SSRIs may be of sufficient severity to cause discontinuation of treatment in 19 per cent and 15 per cent of patients, respectively [11]. Moreover, in 30 per cent of the cases, there is little or no response to the medication, and almost 50 per cent of patients treated with a TCA do not show significant clinical improvements [12].
(b). The corticotrope hypothesis: an opening to the neuroimmune and the neuroplasticity theories of depression
Epidemiological data have provided strong support to the idea that stressful life events play a role in the aetiology of depression [13], and this would occur in interaction with genetic factors [14]. The involvement of stress in the development of depressive symptoms may involve several systems, including the monoamines and inflammation factors interacting with the HPA axis, which exerts a central role in the regulation of the stress response. By mediating the perceived stress response, the HPA axis is an essential component of an individual's ability to cope with stress. The activity of the HPA axis is governed by the secretion of corticotropin releasing hormone (CRH) and arginine–vasopressin (AVP) from the hypothalamus, which, in turn, activate the secretion of the adrenocorticotropic hormone (ACTH) from the pituitary. ACTH then stimulates the secretion of corticoids (cortisol in humans and corticosterone in rodents) from the adrenal cortex, and these hormones interact with their receptors (low-affinity glucocorticoid receptors (GRs) and high-affinity mineralocorticoid receptors (MRs)) in multiple target tissues throughout the body including the brain. Glucocorticoids exert a negative feedback control on CRH and AVP secretion through the activation of GRs expressed, among others, by hippocampal and paraventricular nucleus neurons (figure 1). Besides their involvement in the negative feedback regulation, these receptors also enhance hippocampal function and thereby promote certain cognitive abilities [15].
Figure 1.
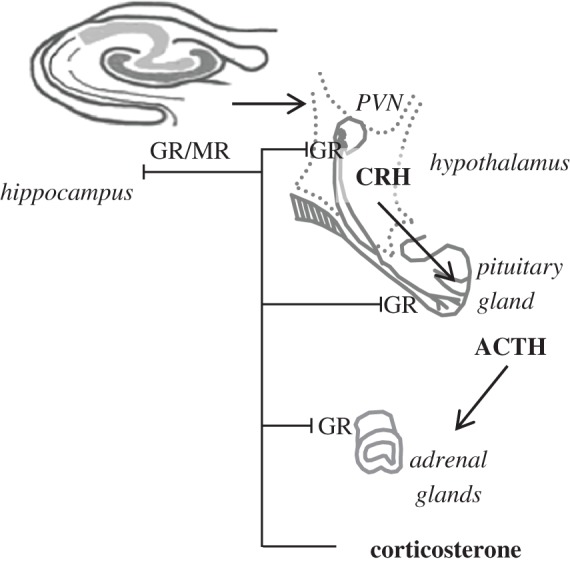
Regulation of the hypothalamic–pituitary–adrenal (HPA) axis. GRs, in particular in the hippocampus and the paraventricular nucleus of the hypothalamus (PVN) exert a negative feedback regulation on the secretion of ACTH and corticosterone. GR-i mice are deficient in GR expression. CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone; GR, low-affinity glucocorticoid receptor; MR, high affinity mineralocorticoid receptors.
The corticosteroid receptor hypothesis of depression is based on dysfunctions of the HPA axis observed in the majority of depressed patients [3,16]. HPA hyperactivity results from a deficit in the negative feedback regulation of the axis, as evidenced by the failure of GR activation to decrease plasma levels of cortisol in the ‘dexamethasone suppression test’ [17]. In this test, a low dose of dexamethasone, an exogenous steroid that activates GRs, normally suppresses cortisol secretion in healthy individuals. A high proportion of depressed patients are ‘non-suppressor’ in the dexamethasone test, in particular individuals with severe psychotic features and suicidal ideation [18]. In addition, dexamethasone administration suppresses mitogen-induced lymphocyte proliferation and the production of the pro-inflammatory cytokine interleukin-1β in healthy controls, but not in depressed patients [19]. Dexamethasone administration also induces an increase in the number of neutrophils and a decrease in the number of lymphocytes in healthy controls, but these effects are not observed in non-suppressors, suggesting that depression is also associated with changes in immune function which confer a resistance to the effects of glucocorticoids on immunity [20]. Other data indicate that cytokine-mediated inflammatory processes might play an important role in neurochemical changes associated with depression. In particular, both glucocorticoids and pro-inflammatory cytokines lead to a decrease in the synthesis of 5-HT and increase neurotoxic metabolites by enhancing the conversion of its precursor tryptophan into kynurenine [21] (figure 2). Depressive disorders are highly prevalent in infectious, autoimmune and neurodegenerative disorders [22] and, in turn, depressed patients show high levels of pro-inflammatory cytokines and other proteins involved in the immune response [23]. Interestingly, half of the patients treated with the cytokine interferon-α develop depressive symptoms that can be treated with classical ADs [24], further illustrating how inflammation factors may trigger depression. On the other hand, AD drugs were shown to reduce inflammation, possibly through inhibition of the release of pro-inflammatory cytokines from activated macrophages, via monoaminergic receptors located on immune cells [25].
Figure 2.
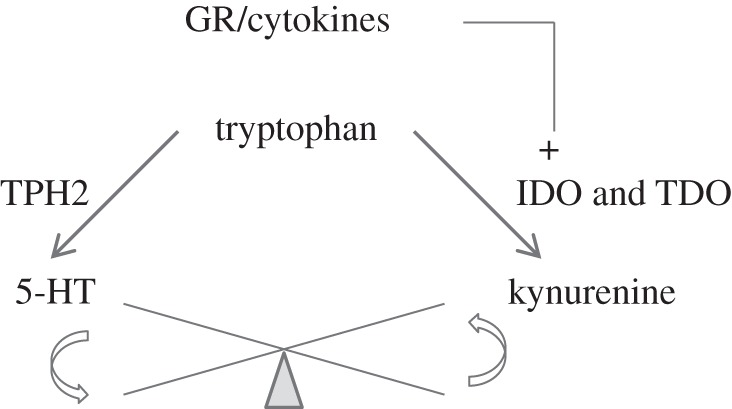
Imbalance in tryptophan metabolism caused by pro-inflammatory cytokines and glucocorticoids. The induction by pro-inflammatory cytokines of the enzyme indolamine 2,3-dioxygenase (IDO) and by glucocorticoids of the tryptophan 2,3-dioxygenase (TDO), which both convert tryptophan into kynurenine, leads to decreased 5-HT synthesis via tryptophan hydroxylase 2 (TPH2).
At the anatomo-functional level, the hippocampus seems to be especially sensitive to the deleterious effects of stress. Neuronal loss has been reported in the hippocampus of stressed or corticosterone-treated animals. Moreover, human brain imaging studies illustrate how stress and depression may cause reductions of the hippocampal volume, probably because of dendritic arborization atrophy and loss of neurons, as observed in post-mortem hippocampi of patients suffering from major depressive disorder [26]. The fact that these morphometric alterations are most often attenuated, or even reversed, by AD drugs further suggests that they are related to depression. They are probably under the regulation of both GRs and MRs since recent data also showed that prenatal stress in rodents impairs the morphological and functional maturation of hippocampal granule cells in adult offspring via MR downregulation [27]. Furthermore, convergent data obtained in several animal species showed that stress and glucocorticoids exert a drastic negative effect on the rate of cell proliferation, leading to a rapid and prolonged decrease in neurogenesis in the hippocampus of adult rodents [28]. Whether this effect is also observed in depressed patients is still unclear [29]. Regardless of the nature of hippocampal damages, these changes are likely to reduce the inhibitory control that the hippocampus exerts on the HPA axis, which would increase circulating glucocorticoid levels and lead to subsequent hippocampal dysfunction. Although ablation of neurogenesis has no effect on basal HPA axis activity, it prevents monoaminergic AD drugs from restoring the negative inhibitory function of the hippocampus when glucocorticoid levels become elevated in chronic stress conditions [30]. Hippocampal neurogenesis was also proposed to contribute to memory formation [31]. A reduction in neurogenesis can theoretically contribute to the cognitive symptoms of depression, even though by itself it is unlikely to produce mood disorders [32].
Neurogenesis is mainly under the control of factors controlling neuroplasticity. Extensive studies in both humans and validated animal models of depression have indicated that monoamines produce their effects through signalling pathways that regulate neuroplasticity and cell survival, such as the calcium/cyclic-AMP responsive-element-binding protein (CREB) and the brain-derived neurotrophic factor (BDNF) pathways. In rodents, both stress and glucocorticoids are known to reduce brain levels of neurotrophic factors, including BDNF mRNA and protein [33–35]. In contrast, different classes of AD drugs increase BDNF expression in the hippocampus [36]. In humans, post-mortem studies have shown that BDNF expression is reduced in the cerebral cortex of depressed patients compared with healthy controls, but increased in patients receiving an AD at the time of death [37]. All these data suggest that neurotrophic factors are involved in AD therapy. How much these neuroplasticity changes are important for mood regulation and adaptive behavioural responses to stress is still an open question. Interestingly, recent data support the hypothesis that hyperactivity of the HPA axis is not a simple consequence or an epiphenomenon of depression, but a risk factor predisposing the patient to the development of depression [38], brought about by genetic liability as well as by early life experiences which programme molecular changes including epigenetic modifications (see below) [14,39].
(c). Circadian system dysregulations: old observations and new concepts
Circadian rhythm abnormalities have been considered for years to play a role in mood disorders, but the exact nature and the mechanisms of these abnormalities are still poorly understood [40]. Biological clocks and the so-called clock genes regulate many behaviour and physiological functions, such as sleep or feeding. These functions are greatly altered in depression and the severity of depressive symptoms varies with a daily pattern. Circadian rhythms of many biological variables such as the daily profiles of body temperature, cortisol, thyrotropin, prolactin, growth hormone, melatonin and excretion of various metabolites are disrupted in depressed patients [41]. Although no clock gene polymorphism is apparently associated with major depression per se, a polymorphism has been linked to the recurrence of bipolar depression [42]. Most AD drugs and mood stabilizers influence endogenous rhythms. For instance, the AD agomelatine, a melatonin receptor agonist and a 5-HT2C receptor antagonist, improves disturbed sleep–wake rhythms in depressed patients and adjusts the circadian system by resynchronizing the body temperature and cortisol levels [43,44]. Moreover, the SSRI fluoxetine appears to modulate clock gene expression [45]. Finally, ‘chronotherapeutics’ such as sleep deprivation have been found to be efficient for clinical treatment of depressed patients [46]. Their rapid and transient effects can be stabilized with common AD treatments [47].
(d). Current issues about the role of epigenetics in depression
Aetiological studies of depression have traditionally focused primarily on genetic factors. However, epigenetic factors, independent of DNA sequence variations, appear to be of considerable interest in understanding the effect of early life stress in depression [48–50]. The epigenome that programmes the genome consists of covalent modifications of DNA by methylation of cytosines and a panel of different histone modifications and chromatin remodelling factors. Non-coding RNAs, including siRNA, antisense RNA and microRNAs are emerging as important regulators of stable gene expression and are integrated with both chromatin and DNA methylation epigenetic functions. Although the epigenetic programme is ruled by a highly organized developmental process that is tightly controlled to maintain tissue-specific patterns of gene expression and is generally similar in different individuals, recent data suggest that it might still be sensitive to input from the environment, especially in the early life period. Environmental conditions can modulate the pattern of DNA methylation, while maintaining the overall cell-type specificity. It has been hypothesized [51] that cues from social and physical environments early in life can cause variations in epigenetic programming that serve as an ‘adaptive’ response of the genome to the anticipated lifelong environment. A misfit between the ‘adaptive’ response and the actual environment later in life would result in maladaptation and an increased risk of developing diseases. In this context, certain environmental exposures early in life might result in persistent epigenetic reprogramming that contributes to the risk of later developing psychiatric diseases. For instance, the role of DNA methylation variations in sustaining the effects of early environmental experience in mood-related disorders, notably the dysregulation of the HPA axis activity, has been first demonstrated by Meaney and Szyf's laboratories in the context of postnatal mother–infant interactions [52]. Interestingly, this involves 5-HT-dependent synaptic transmission and binding of the NGFIA transcription factor to the GR promoter site [53]. Since then, other epigenetic modifications, notably of BDNF or arginine/vasopressin, were found to be linked to depression [54,55]. Epigenetic variations, in contrast to genetic variations, are potentially reversible by subsequent modulation of epigenetic enzymes [56]. Interestingly, various classical AD compounds display epigenetic effects, while histone deacetylases or DNA methylation inhibitors exhibit AD-like effects [57]. Therefore, the study of epigenetics in mood disorders bears the potential optimism to reshape our comprehension of the molecular aetiology of depression and the possibility of pharmacological as well as therapeutic interventions through the environment.
3. The glucocorticoid receptor-impaired mouse model of mood disorders
Investigations on depression have mainly focused on animal models of ‘mood-related behavioural disorders’ generated by exposure to chronic stress or gene manipulation in mice that can drive strong dysfunctions in HPA axis feedback regulation. Nevertheless, basic understanding of both molecular- and system-level perturbations resulting in the HPA axis dysregulations in depression is still lacking. A better comprehension of these mechanisms might possibly lead to the identification of novel therapeutic targets.
To investigate dysfunctions brought about by dysregulations of HPA axis, GR-i mice were developed as a genetic model of depressive disorders [58]. As a result of the expression of GR antisense mRNA under the control of a human neurofilament promoter element, GR-i mice exhibited a decrease in both GR-specific binding (approx. 50% in the hippocampus and the cerebral cortex) and GR mRNA levels (approx. 25% in the hippocampus and in the dorsal raphe nucleus, DRN) compared with paired wild-type (WT) mice [59]. Neuroendocrine regulation in these mice is heavily disturbed, as exemplified by a reduction in both glucocorticoid feedback efficiency and stress-induced increase in plasma ACTH, but not corticosterone [59–62]. Furthermore, it has been reported that GR-i mice have altered behavioural responses in models of anxiety and depression [62–64]. Most of these alterations are reduced by various AD treatments, such as TCAs, SSRIs or new generation ADs, such as agomelatine. Normalization by AD treatments of the hyperactive HPA system in GR-i mice is relevant to what occurs during successful AD therapy of depressive illness [63]. These observations fit well with the hypothesis that GR-i mice model a subgroup of depressed patients who do show a similar hyperactivity of the HPA axis. In addition, impaired GR functioning affects normal adaptive responses to chronic mild stress. Under non-stressful conditions, GR-i mutants present no alterations in 5-HT1A autoreceptor function in the dorsal DRN. However, whereas in WT mice, a chronic mild stress session produces a functional desensitization of DRN 5-HT1A autoreceptors, this is not observed in GR-i mice [59]. In addition, chronic mild stress was found to have a pro-cognitive effect in WT but not GR-i mice in a decision-making task [59]. When subjected to a psychological stressor, GR-i mice displayed hyper-responsiveness of the serotonergic system as shown by a profound rise in hippocampal 5-HT during a rat exposure, but no increase of free corticosterone and behavioural impairments when compared with WT mice [65]. These results demonstrate that impaired GR functioning affects normal adaptive responses to stress at both the HPA and the serotonergic levels and alters stress-related consequences on cognition.
Presumably, owing to a decrease in BDNF mRNA expression in the hippocampus of GR-i compared with WT mice, a reduction in cell proliferation and survival together with reduced neurogenesis have also been observed within the dentate gyrus of GR-i compared with WT mice [64]. These alterations were reversed by chronic AD treatments with either the MT1/MT2 receptor agonist and 5-HT2C receptor antagonist agomelatine or the SSRI fluoxetine [64]. Interestingly, GR-i mice have been shown to display numerous abnormalities regarding spatial learning and short-term memory [66], anxiety [62] and responses to stress [59,65]. All these alterations could be linked to the deficits observed in hippocampal cell proliferation and survival.
(a). Genetic and epigenetic regulations in glucocorticoid receptor-impaired mice
We have recently investigated the genome-wide expression in GR-i mice [67]. Genes whose expression was modified in these mutants versus paired WT mice were deciphered using oligonucleotide-based microarrays in three brain areas relevant to mood disorders (anterior raphe area, hippocampus and frontal cortex). Important gene expression differences were observed in the hippocampus and in the frontal cortex, which are key sites for the negative feedback regulation of HPA axis activity. Among the genes differentially expressed in GR-i versus WT mice, those encoding Arc, NeuroD1, nocturnin, rev-erbB, Per 1–3 and NPAS2 are of particular interest because of their implications in synaptic plasticity, neurotransmission, neurogenesis and circadian rhythms (table 1). Some of the differences were confirmed using RT-PCR (figure 3 and see [68,69]). Interestingly, numerous mRNA-encoding proteins involved in epigenetic regulations such as the variant H2AZ of the histone 2A, the histone deacetylases HDAC2 and HDAC5, the MYST histone acetyltransferase 2 MYST2 and the nuclear receptor co-repressor NCor1 were downregulated in GR-i mutants (table 1 and figure 3). In contrast, methyl CpG-binding protein 2 (MeCP2) was upregulated. Finally, genes encoding various enzymes or complexes involved in cellular metabolism (cytochrome oxidase, NADH dehydrogenase, pyruvate dehydrogenase kinase, etc.) were also affected in the transgenic mice [67–69]. All these gene expression modulations might be involved in the depressive-like behaviours of the GR-i mice.
Table 1.
Changes in gene expression in the hippocampus of GR-i versus control WT paired mice. Microarray experiments were conducted using the Illumina MouseRef-8 Expression BeadChip [68].
| gene | full name | family/action | role | GR-i vs WT |
|---|---|---|---|---|
| Arc | activity-regulated cytoskeleton-associated protein | member of the immediate-early gene (IEG) family | plasticity | + |
| NeuroD1 | neurogenic differentiation 1 | member of the NeuroD family of basic helix-loop-helix (bHLH) transcription factors | plasticity | − |
| Nocturnin | CCR4 carbon catabolite repression 4-like | circadian deadenylase | post-transcriptional control of circadian gene expression | − |
| rev-erbB | nuclear receptor subfamily 1, group D, member 2 | member of the Rev-ErbB family of nuclear receptors | circadian transcriptional repression | − |
| Per 1 | period circadian protein homologue 1 | member of the Period family of genes | circadian transcriptional repression | + |
| Per 3 | period circadian protein homologue 3 | member of the Period family of genes | circadian transcriptional repression | + |
| NPAS2 | neuronal PAS domain-containing protein 2 i | member of the bHLH-PAS family of transcription factors | circadian transcriptional regulation | − |
| H2AZ | histone H2A variant histone family, member Z | variant of H2A localized to promoters | gene expression regulation | − |
| HDAC2 | histone deacetylase 2 HDAC Class I | removes acetyl groups from an N-acetyl lysine on a histone | transcriptional repression | − |
| HDAC5 | histone deacetylase 5 HDAC Class II A | removes acetyl groups from an N-acetyl lysine on a histone | transcriptional repression | − |
| MYST2 | MYST histone acetyltransferase 2 | acetylates lysine by transferring an acetyl group from acetyl CoA to form N-acetyl lysine | steroid receptor-mediated transcription regulation | − |
| NCor1 | nuclear receptor co-repressor 1 | recruits histone deacetylases to DNA promoter regions | transcriptional repression | − |
| MeCP2 | methyl CpG-binding protein 2 | binds methylated DNA | transcriptional regulation | + |
Figure 3.

Gene expression levels in GR-i mice compared with paired WT controls. (a) mRNA expression of various genes implicated in synaptic plasticity (a) and related to epigenetic regulations (b). Brains were removed between 9.00 and 11.00. Semiquantitative determinations were made with reference to reporter gene encoding hypoxanthine guanine phosphoribosyl transferase (HPRT). Data are mean ± s.e.m. (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001 GR-i (grey bars) versus WT (empty bars) mice, paired Student's t-test.
Emerging data underline that circadian rhythms, regulated at organ and cellular levels via the clock machinery, govern physiological and behavioural functions in most organisms [70]. Notably, the clock transcription–translation feedback loop system possesses intimate links with cellular metabolism. These connections are maintained by epigenetic mechanisms [71]. For instance, HDAC3 activation by Ncor1, the expression of which is decreased in GR-i mice, is critical for the epigenetic regulation of circadian and metabolic physiology [72]. Interestingly, data suggest that clock genes expression is regulated via direct interactions between NCor1 and rev-erbB [73], whose expression is also decreased in GR-i compared with WT mice. Moreover, the circadian CLOCK system and the HPA axis strongly interact. In particular, clock proteins directly regulate GR transcriptional activity [74] and glucocorticoids modulate energy homeostasis by entraining the clock machinery [75] via stimulation of rev-erbB and NPAS2 expressions that are also affected in GR-i mice. The dysregulation of NPAS2 in GR-i mice is of particular interest, since this homologue of the protein CLOCK operating in the mammalian forebrain [76] is involved in the acquisition of specific memory [77] and in the maintenance of circadian behaviours [78].
The observed downregulations of HDAC2 and HDAC5 expression in GR-i mice are consistent with results obtained by other groups. For example, Nestler's group reported that after AD treatment, HDAC5 expression is decreased in the hippocampus of stressed mice and might participate in the efficacy of the treatment [55]. The same group suggested that HDAC5 may regulate adaptation to chronic stress [79]. Therefore, the decrease in HDAC5 expression in GR-i mice may be a compensatory mechanism induced by the decrease in GR function. The downregulation in HDAC2 expression observed in the hippocampus of GR-i mice [69] may also be an adaptive mechanism, since it was found that, in the hippocampus, HDAC2 negatively regulates synaptic plasticity and morphological changes necessary for learning [80]. Furthermore, a decrease in HDAC2 protein expression in the nucleus accumbens was shown to be associated with both depression in humans and chronic social-stress-induced behavioural deficits in mice [81]. Increased expression of MeCP2 has also been observed in GR-i mice. MeCP2 was identified as a protein that binds specifically to methylated DNA and is primarily considered a transcriptional repressor through associated HDACs. However, MeCP2 appears to be a multi-functional protein. For example, an association with the transcriptional activator CREB1 was reported [82] and it is now believed that MeCP2 can function as both an activator and repressor of transcription. Moreover, MeCP2 has a widespread effect on gene expression [83] and could prevent aberrant transcriptional events [84]. A role of MeCP2 in regulating chromatin architecture has also been proposed [85]. It has recently been reported that a mouse line overexpressing MeCP2 in neurons only exhibits heightened anxiety, impairments in learning and memory, as well as hippocampal synaptic plasticity alterations that may be attributable to transcriptional repression via HDAC recruitment [86]. These results suggest that MeCP2 overexpression may directly cause some of the deficits observed in GR-i mice and strengthen the hypothesis that HDAC downregulation in GR-i mice could be a compensatory mechanism counteracting MeCP2 overexpression. However, detrimental or beneficial impacts of hippocampal MeCP2 overexpression in GR-i mice may be difficult to evaluate since repeated injections of fluoxetine in rats upregulate MeCP2 in the dentate gyrus of hippocampus [87].
The H2AZ variant of the histone 2A is involved in diverse biological functions, such as gene transcription [88,89]. H2AZ may prepare chromatin structure for recruitment of the transcriptional machinery and regulates promoter capacity to trigger transcription. Interestingly, this variant is enriched at GR promoter sites and this enrichment is modulated by GR receptor activity [90]. The decrease in H2AZ expression in GR-i mice could be a direct consequence of HPA axis activity dysregulation. However, it remains to be determined whether these H2AZ variations contribute to the regulation of the global expression pattern modulated by glucocorticoids, and whether it is of any relevance to depression or represents an adaptation mechanism similar to the changes observed in HDAC5, HDAC2 or MeCP2 expression levels.
Finally, GR-i mice also manifest immune–endocrine abnormalities. Indeed, in addition to HPA axis dysfunctions, GR-i mice show alterations in the trafficking and the responsiveness of T lymphocytes [91]. Hence, further work on these transgenic mice should be particularly informative regarding immune regulationrelated to depressive disorders. Moreover, the study of these mice in the context of gene × environment interactions appears particularly promising. It is indeed well demonstrated that adverse conditions early in life have a major influence on the normal development of GR– HPA interactions, and that events during development have long-term impact on the capacity of the glucocorticoid system to cope with stress. We should investigate whether early manipulations, such as handling and maternal separation in G-R-i mice, might alter the relationships between immune, neuroendocrine and epigenetic changes in the context of mood-related disorders.
4. Conclusion
Adaptation to environmental stresses is one of the most fundamental biological regulatory processes. Chronic stress entails long-term dysregulations at neurotransmitter, neurohormone and cellular levels leading to behavioural changes. Homeostasis is maintained by complex physiological and behavioural systems coordinated by the brain. The effectors are diverse and comprise endocrine and immune regulations, neural plasticity, circadian rhythms and chromatin modulations. Recent studies suggested that all these systems are inter-related and may underlie brain diseases, especially mood-related disorders. Perturbations of neurotransmitter metabolism, circadian clock genes and chromatin modifications interact and certainly determine, at least in part, behavioural disorders. Chromatin regulation, clock genes expression and neurotransmitter metabolism processes thought to be involved in mood disorders and HPA axis regulation are also found to be altered in GR-i mice. It is therefore of importance to determine the exact nature of such perturbations, as well as their relationships, in order to design new strategies for the treatment of depression.
Acknowledgements
This research was supported by Inserm (France), UPMC, the European Commission (FP6-LSHM-CT-2003-503474 and FP7-health-2007-A-201714) and IRIS (Courbevoie, France).
References
- 1.Sobocki P., Jonsson B., Angst J., Rehnberg C. 2006. Cost of depression in Europe. J. Ment. Health Policy Econ. 9, 87–98 [PubMed] [Google Scholar]
- 2.Prins J., Olivier B., Korte S. M. 2011. Triple reuptake inhibitors for treating subtypes of major depressive disorder: the monoamine hypothesis revisited. Expert. Opin. Investig. Drugs. 20, 1107–1130 10.1517/13543784.2011.594039 (doi:10.1517/13543784.2011.594039) [DOI] [PubMed] [Google Scholar]
- 3.Anacker C., Zunszain P. A., Carvalho L. A., Pariante C. M. 2011. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology 36, 415–425 10.1016/j.psyneuen.2010.03.007 (doi:10.1016/j.psyneuen.2010.03.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McClung C. A. 2007. Circadian genes, rhythms and the biology of mood disorders. Pharmacol. Ther. 114, 222–232 10.1016/j.pharmthera.2007.02.003 (doi:10.1016/j.pharmthera.2007.02.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller A. H. 2010. Depression and immunity: a role for T cells? Brain Behav. Immun. 24, 1–8 10.1016/j.bbi.2009.09.009 (doi:10.1016/j.bbi.2009.09.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnan V., Nestler E. J. 2008. The molecular neurobiology of depression. Nature 455, 894–902 10.1038/nature07455 (doi:10.1038/nature07455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slattery D. A., Hudson A. L., Nutt D. J. 2004. Invited review: the evolution of antidepressant mechanisms. Fundam. Clin. Pharmacol. 18, 1–21 10.1111/j.1472-8206.2004.00195.x (doi:10.1111/j.1472-8206.2004.00195.x) [DOI] [PubMed] [Google Scholar]
- 8.Asberg M., Traskman L., Thoren P. 1976. 5-HIAA in the cerebrospinal fluid: a biochemical suicide predictor? Arch. Gen. Psychiatry 33, 1193–1197 10.1001/archpsyc.1976.01770100055005 (doi:10.1001/archpsyc.1976.01770100055005) [DOI] [PubMed] [Google Scholar]
- 9.Mongeau R., Blier P., de Montigny C. 1997. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res. Brain Res. Rev. 23, 145–195 10.1016/S0165-0173(96)00017-3 (doi:10.1016/S0165-0173(96)00017-3) [DOI] [PubMed] [Google Scholar]
- 10.Delgado P. L. 2006. Monoamine depletion studies: implications for antidepressant discontinuation syndrome. J. Clin. Psychiatry 67(Suppl. 4), 22–26 [PubMed] [Google Scholar]
- 11.Montgomery S. A., Henry J., McDonald G., Dinan T., Lader M., Hindmarch I., Clare A., Nutt D. 1994. Selective serotonin reuptake inhibitors: meta-analysis of discontinuation rates. Int. Clin. Psychopharmacol. 9, 47–53 10.1097/00004850-199400910-00008 (doi:10.1097/00004850-199400910-00008) [DOI] [PubMed] [Google Scholar]
- 12.Nemeroff C. B. 1998. Psychopharmacology of affective disorders in the 21st century. Biol. Psychiatry 44, 517–525 10.1016/S0006-3223(98)00068-7 (doi:10.1016/S0006-3223(98)00068-7) [DOI] [PubMed] [Google Scholar]
- 13.Kendler K. S., Kessler R. C., Walters E. E., MacLean C., Neale M. C., Heath A. C., Eaves L. J. 1995. Stressful life events, genetic liability, and onset of an episode of major depression in women. Am. J. Psychiatry 152, 833–842 [DOI] [PubMed] [Google Scholar]
- 14.Caspi A., Moffitt T. E. 2006. Gene–environment interactions in psychiatry: joining forces with neuroscience. Nat. Rev. Neurosci. 7, 583–590 10.1038/nrn1925 (doi:10.1038/nrn1925) [DOI] [PubMed] [Google Scholar]
- 15.Oitzl M. S., de Kloet E. R. 1992. Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav. Neurosci. 106, 62–71 10.1037/0735-7044.106.1.62 (doi:10.1037/0735-7044.106.1.62) [DOI] [PubMed] [Google Scholar]
- 16.Pariante C. M. 2003. Depression, stress and the adrenal axis. J. Neuroendocrinol. 15, 811–812 10.1046/j.1365-2826.2003.01058.x (doi:10.1046/j.1365-2826.2003.01058.x) [DOI] [PubMed] [Google Scholar]
- 17.Dam H. 1988. Dexamethasone suppression test. Acta Psychiatr. Scand. Suppl. 345, 38–44 [DOI] [PubMed] [Google Scholar]
- 18.Fountoulakis K. N., Gonda X., Rihmer Z., Fokas C., Iacovides A. 2008. Revisiting the dexamethasone suppression test in unipolar major depression: an exploratory study. Ann. Gen. Psychiatry 7, 22. 10.1186/1744-859X-7-22 (doi:10.1186/1744-859X-7-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maes M., Bosmans E., Suy E., Vandervorst C., DeJonckheere C., Raus J. 1991. Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1 beta and soluble interleukin-2 receptor production. Acta Psychiatr. Scand. 84, 379–386 10.1111/j.1600-0447.1991.tb03163.x (doi:10.1111/j.1600-0447.1991.tb03163.x) [DOI] [PubMed] [Google Scholar]
- 20.Maes M. 1994. Cytokines in major depression. Biol. Psychiatry 36, 498–499 10.1016/0006-3223(94)90652-1 (doi:10.1016/0006-3223(94)90652-1) [DOI] [PubMed] [Google Scholar]
- 21.Maes M., Leonard B. E., Myint A. M., Kubera M., Verkerk R. 2011. The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 702–721 10.1016/j.pnpbp.2010.12.017 (doi:10.1016/j.pnpbp.2010.12.017) [DOI] [PubMed] [Google Scholar]
- 22.Pollak Y., Yirmiya R. 2002. Cytokine-induced changes in mood and behaviour: implications for ‘depression due to a general medical condition’, immunotherapy and antidepressive treatment. Int. J. Neuropsychopharmacol. 5, 389–399 10.1017/S1461145702003152 (doi:10.1017/S1461145702003152) [DOI] [PubMed] [Google Scholar]
- 23.Raison C. L., Lin J. M., Reeves W. C. 2009. Association of peripheral inflammatory markers with chronic fatigue in a population-based sample. Brain Behav. Immun. 23, 327–337 10.1016/j.bbi.2008.11.005 (doi:10.1016/j.bbi.2008.11.005) [DOI] [PubMed] [Google Scholar]
- 24.Raison C. L., Capuron L., Miller A. H. 2006. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 27, 24–31 10.1016/j.it.2005.11.006 (doi:10.1016/j.it.2005.11.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard B. E. 2010. The concept of depression as a dysfunction of the immune system. Curr. Immunol. Rev. 6, 205–212 10.2174/157339510791823835 (doi:10.2174/157339510791823835) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stockmeier C. A., Mahajan G. J., Konick L. C., Overholser J. C., Jurjus G. J., Meltzer H. Y., Uylings H. B., Friedman L., Rajkowska G. 2004. Cellular changes in the postmortem hippocampus in major depression. Biol. Psychiatry 56, 640–650 10.1016/j.biopsych.2004.08.022 (doi:10.1016/j.biopsych.2004.08.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura M., Sajo M., Kakita A., Matsuki N., Koyama R. 2011. Prenatal stress inhibits neuronal maturation through downregulation of mineralocorticoid receptors. J. Neurosci. 31, 11 505–11 514 10.1523/JNEUROSCI.3447-10.2011 (doi:10.1523/JNEUROSCI.3447-10.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warner-Schmidt J. L., Duman R. S. 2006. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 16, 239–249 10.1002/hipo.20156 (doi:10.1002/hipo.20156) [DOI] [PubMed] [Google Scholar]
- 29.Reif A., Fritzen S., Finger M., Strobel A., Lauer M., Schmitt A., Lesch K. P. 2006. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol. Psychiatry 11, 514–522 10.1038/sj.mp.4001791 (doi:10.1038/sj.mp.4001791) [DOI] [PubMed] [Google Scholar]
- 30.Surget A., et al. 2011. Antidepressants recruit new neurons to improve stress response regulation. Mol. Psychiatry 16, 1177–1188 10.1038/mp.2011.48 (doi:10.1038/mp.2011.48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aasebo I. E., Blankvoort S., Tashiro A. 2011. Critical maturational period of new neurons in adult dentate gyrus for their involvement in memory formation. Eur. J. Neurosci. 33, 1094–1100 10.1111/j.1460-9568.2011.07608.x (doi:10.1111/j.1460-9568.2011.07608.x) [DOI] [PubMed] [Google Scholar]
- 32.Lucassen P. J., Meerlo P., Naylor A. S., van Dam A. M., Dayer A. G., Fuchs E., Oomen C. A., Czeh B. 2010. Regulation of adult neurogenesis by stress, sleep disruption, exercise and inflammation: implications for depression and antidepressant action. Eur. Neuropsychopharmacol. 20, 1–17 10.1016/j.euroneuro.2009.08.003 (doi:10.1016/j.euroneuro.2009.08.003) [DOI] [PubMed] [Google Scholar]
- 33.Duman R. S., Monteggia L. M. 2006. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127 10.1016/j.biopsych.2006.02.013 (doi:10.1016/j.biopsych.2006.02.013) [DOI] [PubMed] [Google Scholar]
- 34.Smith M. A., Makino S., Kvetnansky R., Post R. M. 1995. Effects of stress on neurotrophic factor expression in the rat brain. Ann. N. Y. Acad. Sci. 771, 234–239 10.1111/j.1749-6632.1995.tb44684.x (doi:10.1111/j.1749-6632.1995.tb44684.x) [DOI] [PubMed] [Google Scholar]
- 35.Gronli J., Bramham C., Murison R., Kanhema T., Fiske E., Bjorvatn B., Ursin R., Portas C. M. 2006. Chronic mild stress inhibits BDNF protein expression and CREB activation in the dentate gyrus but not in the hippocampus proper. Pharmacol. Biochem. Behav. 85, 842–849 10.1016/j.pbb.2006.11.021 (doi:10.1016/j.pbb.2006.11.021) [DOI] [PubMed] [Google Scholar]
- 36.Martinowich K., Lu B. 2008. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology 33, 73–83 10.1038/sj.npp.1301571 (doi:10.1038/sj.npp.1301571) [DOI] [PubMed] [Google Scholar]
- 37.Lee B. H., Kim Y. K. 2010. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Invest. 7, 231–235 10.4306/pi.2010.7.4.231 (doi:10.4306/pi.2010.7.4.231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pariante C. M., Lightman S. L. 2008. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 31, 464–468 10.1016/j.tins.2008.06.006 (doi:10.1016/j.tins.2008.06.006) [DOI] [PubMed] [Google Scholar]
- 39.Meaney M. J., Szyf M., Seckl J. R. 2007. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol. Med. 13, 269–277 10.1016/j.molmed.2007.05.003 (doi:10.1016/j.molmed.2007.05.003) [DOI] [PubMed] [Google Scholar]
- 40.Schulz P. 2007. Biological clocks and the practice of psychiatry. Dialogues Clin. Neurosci. 9, 237–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marques A. H., Silverman M. N., Sternberg E. M. 2009. Glucocorticoid dysregulations and their clinical correlates. From receptors to therapeutics. Ann. N. Y. Acad. Sci. 1179, 1–18 10.1111/j.1749-6632.2009.04987.x (doi:10.1111/j.1749-6632.2009.04987.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benedetti F., Serretti A., Colombo C., Barbini B., Lorenzi C., Campori E., Smeraldi E. 2003. Influence of CLOCK gene polymorphism on circadian mood fluctuation and illness recurrence in bipolar depression. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 123B, 23–26 10.1002/ajmg.b.20038 (doi:10.1002/ajmg.b.20038) [DOI] [PubMed] [Google Scholar]
- 43.Gorwood P. 2010. Restoring circadian rhythms: a new way to successfully manage depression. J. Psychopharmacol. 24, 15–19 10.1177/1359786810372981 (doi:10.1177/1359786810372981) [DOI] [PubMed] [Google Scholar]
- 44.Popoli M. 2009. Agomelatine: innovative pharmacological approach in depression. CNS Drugs 23(Suppl 2), 27–34 10.2165/11318640-000000000-00000 (doi:10.2165/11318640-000000000-00000) [DOI] [PubMed] [Google Scholar]
- 45.Uz T., Ahmed R., Akhisaroglu M., Kurtuncu M., Imbesi M., Dirim Arslan A., Manev H. 2005. Effect of fluoxetine and cocaine on the expression of clock genes in the mouse hippocampus and striatum. Neuroscience 134, 1309–1316 10.1016/j.neuroscience.2005.05.003 (doi:10.1016/j.neuroscience.2005.05.003) [DOI] [PubMed] [Google Scholar]
- 46.Dallaspezia S., Benedetti F. 2011. Chronobiological therapy for mood disorders. Expert. Rev. Neurother. 11, 961–970 10.1586/ern.11.61 (doi:10.1586/ern.11.61) [DOI] [PubMed] [Google Scholar]
- 47.Benedetti F., Barbini B., Lucca A., Campori E., Colombo C., Smeraldi E. 1997. Sleep deprivation hastens the antidepressant action of fluoxetine. Eur. Arch. Psychiatry Clin. Neurosci. 247, 100–103 10.1007/BF02900200 (doi:10.1007/BF02900200) [DOI] [PubMed] [Google Scholar]
- 48.Mill J., Petronis A. 2007. Molecular studies of major depressive disorder: the epigenetic perspective. Mol. Psychiatry 12, 799–814 10.1038/sj.mp.4001992 (doi:10.1038/sj.mp.4001992) [DOI] [PubMed] [Google Scholar]
- 49.Schroeder M., Krebs M. O., Bleich S., Frieling H. 2010. Epigenetics and depression: current challenges and new therapeutic options. Curr. Opin. Psychiatry 23, 588–592 10.1097/YCO.0b013e32833d16c1 (doi:10.1097/YCO.0b013e32833d16c1) [DOI] [PubMed] [Google Scholar]
- 50.Tsankova N., Renthal W., Kumar A., Nestler E. J. 2007. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367 10.1038/nrn2132 (doi:10.1038/nrn2132) [DOI] [PubMed] [Google Scholar]
- 51.Szyf M. 2009. The early life environment and the epigenome. Biochim. Biophys. Acta 1790, 878–885 10.1016/j.bbagen.2009.01.009 (doi:10.1016/j.bbagen.2009.01.009) [DOI] [PubMed] [Google Scholar]
- 52.Weaver I. C., Cervoni N., Champagne F. A., D'Alessio A. C., Sharma S., Seckl J. R., Dymov S., Szyf M., Meaney M. J. 2004. Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854 10.1038/nn1276 (doi:10.1038/nn1276) [DOI] [PubMed] [Google Scholar]
- 53.Zhang T. Y., Meaney M. J. 2010. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 61, 439–466 10.1146/annurev.psych.60.110707.163625 (doi:10.1146/annurev.psych.60.110707.163625) [DOI] [PubMed] [Google Scholar]
- 54.Murgatroyd C., et al. 2009. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12, 1559–1566 10.1038/nn.2436 (doi:10.1038/nn.2436) [DOI] [PubMed] [Google Scholar]
- 55.Tsankova N. M., Berton O., Renthal W., Kumar A., Neve R. L., Nestler E. J. 2006. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525 10.1038/nn1659 (doi:10.1038/nn1659) [DOI] [PubMed] [Google Scholar]
- 56.Weaver I. C., Champagne F. A., Brown S. E., Dymov S., Sharma S., Meaney M. J., Szyf M. 2005. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 25, 11 045–11 054 10.1523/JNEUROSCI.3652-05.2005 (doi:10.1523/JNEUROSCI.3652-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boks M. P., de Jong N. M., Kas M. J., Vinkers C. H., Fernandes C., Kahn R. S., Mill J., Ophoff R. A. 2012. Current status and future prospects for epigenetic psychopharmacology. Epigenetics 7, 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pepin M. C., Pothier F., Barden N. 1992. Impaired type II glucocorticoid-receptor function in mice bearing antisense RNA transgene. Nature 355, 725–728 10.1038/355725a0 (doi:10.1038/355725a0) [DOI] [PubMed] [Google Scholar]
- 59.Froger N., et al. 2004. Neurochemical and behavioral alterations in glucocorticoid receptor-impaired transgenic mice after chronic mild stress. J. Neurosci. 24, 2787–2796 10.1523/JNEUROSCI.4132-03.2004 (doi:10.1523/JNEUROSCI.4132-03.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barden N., Stec I. S., Montkowski A., Holsboer F., Reul J. M. 1997. Endocrine profile and neuroendocrine challenge tests in transgenic mice expressing antisense RNA against the glucocorticoid receptor. Neuroendocrinology 66, 212–220 10.1159/000127240 (doi:10.1159/000127240) [DOI] [PubMed] [Google Scholar]
- 61.Karanth S., Linthorst A. C., Stalla G. K., Barden N., Holsboer F., Reul J. M. 1997. Hypothalamic–pituitary–adrenocortical axis changes in a transgenic mouse with impaired glucocorticoid receptor function. Endocrinology 138, 3476–3485 10.1210/en.138.8.3476 (doi:10.1210/en.138.8.3476) [DOI] [PubMed] [Google Scholar]
- 62.Montkowski A., et al. 1995. Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J. Neuroendocrinol. 7, 841–845 10.1111/j.1365-2826.1995.tb00724.x (doi:10.1111/j.1365-2826.1995.tb00724.x) [DOI] [PubMed] [Google Scholar]
- 63.Barden N., Shink E., Labbe M., Vacher R., Rochford J., Mocaer E. 2005. Antidepressant action of agomelatine (S 20098) in a transgenic mouse model. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 908–916 10.1016/j.pnpbp.2005.04.032 (doi:10.1016/j.pnpbp.2005.04.032) [DOI] [PubMed] [Google Scholar]
- 64.Paizanis E., et al. 2010. Behavioural and neuroplastic effects of the new-generation antidepressant agomelatine compared to fluoxetine in glucocorticoid receptor-impaired mice. Int. J. Neuropsychopharmacol. 13, 759–774 10.1017/S1461145709990514 (doi:10.1017/S1461145709990514) [DOI] [PubMed] [Google Scholar]
- 65.Linthorst A. C., Flachskamm C., Barden N., Holsboer F., Reul J. M. 2000. Glucocorticoid receptor impairment alters CNS responses to a psychological stressor: an in vivo microdialysis study in transgenic mice. Eur. J. Neurosci. 12, 283–291 10.1046/j.1460-9568.2000.00878.x (doi:10.1046/j.1460-9568.2000.00878.x) [DOI] [PubMed] [Google Scholar]
- 66.Rousse I., Beaulieu S., Rowe W., Meaney M. J., Barden N., Rochford J. 1997. Spatial memory in transgenic mice with impaired glucocorticoid receptor function. Neuroreport 8, 841–845 10.1097/00001756-199703030-00007 (doi:10.1097/00001756-199703030-00007) [DOI] [PubMed] [Google Scholar]
- 67.Hoyle D., et al. 2011. Shared changes in gene expression in frontal cortex of four genetically modified mouse models of depression. Eur. Neuropsychopharmacol. 21, 3–10 10.1016/j.euroneuro.2010.09.011 (doi:10.1016/j.euroneuro.2010.09.011) [DOI] [PubMed] [Google Scholar]
- 68.Massart R., Fabre V., Païzanis E., Hamon M., Lanfumey L. 2010. Epigenetic and clock genes’ expression perturbations in genetic and environmental models of depressive-like disorders. In 7th World Congress on Stress, 26 August 2010, Leiden, The Netherlands [Google Scholar]
- 69.Massart R., Stragier E., Boulle F., Païzanis E., Gabriel C., Mocaer E., Hamon M., Lanfumey L. 2011. Epigenetic modifications in genetic and environmental models of depressive-like disorder: reversal by chronic treatment with Agomelatine. In Program no. 394.18/WW16 Neuroscience Meeting Planner, 14 November 2011, Washington, DC Washington, DC: Society for Neuroscience [Google Scholar]
- 70.Turek F. W. 2008. Circadian clocks: tips from the tip of the iceberg. Nature 456, 881–883 10.1038/456881a (doi:10.1038/456881a) [DOI] [PubMed] [Google Scholar]
- 71.Grimaldi B., Nakahata Y., Kaluzova M., Masubuchi S., Sassone-Corsi P. 2009. Chromatin remodeling, metabolism and circadian clocks: the interplay of CLOCK and SIRT1. Int. J. Biochem. Cell Biol. 41, 81–86 10.1016/j.biocel.2008.08.035 (doi:10.1016/j.biocel.2008.08.035) [DOI] [PubMed] [Google Scholar]
- 72.Alenghat T., et al. 2008. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 456, 997–1000 10.1038/nature07541 (doi:10.1038/nature07541) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raghuram S., et al. 2007. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat. Struct. Mol. Biol. 14, 1207–1213 10.1038/nsmb1344 (doi:10.1038/nsmb1344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nader N., Chrousos G. P., Kino T. 2010. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol. Metab. 21, 277–286 10.1016/j.tem.2009.12.011 (doi:10.1016/j.tem.2009.12.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Balsalobre A., Brown S. A., Marcacci L., Tronche F., Kellendonk C., Reichardt H. M., Schutz G., Schibler U. 2000. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289, 2344–2347 10.1126/science.289.5488.2344 (doi:10.1126/science.289.5488.2344) [DOI] [PubMed] [Google Scholar]
- 76.Reick M., Garcia J. A., Dudley C., McKnight S. L. 2001. NPAS2: an analog of clock operative in the mammalian forebrain. Science 293, 506–509 10.1126/science.1060699 (doi:10.1126/science.1060699) [DOI] [PubMed] [Google Scholar]
- 77.Garcia J. A., Zhang D., Estill S. J., Michnoff C., Rutter J., Reick M., Scott K., Diaz-Arrastia R., McKnight S. L. 2000. Impaired cued and contextual memory in NPAS2-deficient mice. Science 288, 2226–2230 10.1126/science.288.5474.2226 (doi:10.1126/science.288.5474.2226) [DOI] [PubMed] [Google Scholar]
- 78.Dudley C. A., Erbel-Sieler C., Estill S. J., Reick M., Franken P., Pitts S., McKnight S. L. 2003. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science 301, 379–383 10.1126/science.1082795 (doi:10.1126/science.1082795) [DOI] [PubMed] [Google Scholar]
- 79.Renthal W., et al. 2007. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56, 517–529 10.1016/j.neuron.2007.09.032 (doi:10.1016/j.neuron.2007.09.032) [DOI] [PubMed] [Google Scholar]
- 80.Guan J. S., et al. 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 10.1038/nature07925 (doi:10.1038/nature07925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Covington H. E., III, et al. 2009. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29, 11 451–11 460 10.1523/JNEUROSCI.1758-09.2009 (doi:10.1523/JNEUROSCI.1758-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chahrour M., Jung S. Y., Shaw C., Zhou X., Wong S. T., Qin J., Zoghbi H. Y. 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229 10.1126/science.1153252 (doi:10.1126/science.1153252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ben-Shachar S., Chahrour M., Thaller C., Shaw C. A., Zoghbi H. Y. 2009. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442 10.1093/hmg/ddp181 (doi:10.1093/hmg/ddp181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guy J., Cheval H., Selfridge J., Bird A. 2011. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652 10.1146/annurev-cellbio-092910-154121 (doi:10.1146/annurev-cellbio-092910-154121) [DOI] [PubMed] [Google Scholar]
- 85.Chadwick L. H., Wade P. A. 2007. MeCP2 in Rett syndrome: transcriptional repressor or chromatin architectural protein? Curr. Opin. Genet. Dev. 17, 121–125 10.1016/j.gde.2007.02.003 (doi:10.1016/j.gde.2007.02.003) [DOI] [PubMed] [Google Scholar]
- 86.Na E. S., Nelson E. D., Adachi M., Autry A. E., Mahgoub M. A., Kavalali E. T., Monteggia L. M. 2012. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 32, 3109–3117 10.1523/JNEUROSCI.6000-11.2012 (doi:10.1523/JNEUROSCI.6000-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cassel S., Carouge D., Gensburger C., Anglard P., Burgun C., Dietrich J. B., Aunis D., Zwiller J. 2006. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol. Pharmacol. 70, 487–492 10.1124/mol.106.022301 (doi:10.1124/mol.106.022301) [DOI] [PubMed] [Google Scholar]
- 88.Draker R., Cheung P. 2009. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochem. Cell Biol. 87, 19–25 10.1139/O08-117 (doi:10.1139/O08-117) [DOI] [PubMed] [Google Scholar]
- 89.Zlatanova J., Thakar A. 2008. H2A.Z: view from the top. Structure 16, 166–179 10.1016/j.str.2007.12.008 (doi:10.1016/j.str.2007.12.008) [DOI] [PubMed] [Google Scholar]
- 90.John S., et al. 2008. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol. Cell. 29, 611–624 10.1016/j.molcel.2008.02.010 (doi:10.1016/j.molcel.2008.02.010) [DOI] [PubMed] [Google Scholar]
- 91.Marchetti B., Morale M. C., Testa N., Tirolo C., Caniglia S., Amor S., Dijkstra C. D., Barden N. 2001. Stress, the immune system and vulnerability to degenerative disorders of the central nervous system in transgenic mice expressing glucocorticoid receptor antisense RNA. Brain Res. Rev. 37, 259–272 10.1016/S0165-0173(01)00130-8 (doi:10.1016/S0165-0173(01)00130-8) [DOI] [PubMed] [Google Scholar]