

Abstract
We recently developed the first algorithms specifically for plants to predict proteins carrying peroxisome targeting signals type 1 (PTS1) from genome sequences.1 As validated experimentally, the prediction methods are able to correctly predict unknown peroxisomal Arabidopsis proteins and to infer novel PTS1 tripeptides. The high prediction performance is primarily determined by the large number and sequence diversity of the underlying positive example sequences, which mainly derived from EST databases. However, a few constructs remained cytosolic in experimental validation studies, indicating sequencing errors in some ESTs. To identify erroneous sequences, we validated subcellular targeting of additional positive example sequences in the present study. Moreover, we analyzed the distribution of prediction scores separately for each orthologous group of PTS1 proteins, which generally resembled normal distributions with group-specific mean values. The cytosolic sequences commonly represented outliers of low prediction scores and were located at the very tail of a fitted normal distribution. Three statistical methods for identifying outliers were compared in terms of sensitivity and specificity.” Their combined application allows elimination of erroneous ESTs from positive example data sets. This new post-validation method will further improve the prediction accuracy of both PTS1 and PTS2 protein prediction models for plants, fungi, and mammals.
Keywords: genome screens, machine learning methods, peroxisome, statistics, targeting
In the post-genomic era, accurate prediction tools are essential for identification of the proteomes of cell organelles.2-5 Such prediction methods have been developed for peroxisome-targeted proteins of animals and fungi,6-11 but had been missing specifically for plants. For development of a predictor for plant proteins carrying peroxisome targeting signals type 1 (PTS1), we assembled putatively orthologous plant sequences of Arabidopsis PTS1 proteins (so-called positive example sequences) and non-peroxisomal sequences (so-called negative example sequences) and applied a discriminative machine learning approach to derive two different prediction methods, both of which showed high prediction accuracy. Upon application of these methods to the Arabidopsis genome, a total of 392 gene models were predicted to be peroxisome-targeted. Extensive experimental validations revealed a high experimental verification rate of Arabidopsis proteins previously not known to be peroxisomal. Moreover, the prediction methods were able to correctly infer novel PTS1 tripeptides, which even included novel residues.1
The high performance of the new prediction methods was mainly based on the large size (> 2,500 sequences) and sequence diversity of the underlying data set of positive PTS1 protein example sequences, which mainly derived from EST databases. However, ESTs are known to contain a low but significant rate of sequencing errors. Indeed, among 32 positive example sequences validated experimentally, five reporter fusion constructs remained cytosolic.1 Two cytosolic sequences (terminating with LNL > and LCR >) could be identified bioinformatically as erroneous sequences because (1) they had been assigned extremely low position weight matrices (PWM) model prediction scores and posterior probabilities and (2) their C-terminal tripeptides deviated from the emerging general pattern of plant PTS1 tripeptides ([x(KR)(LMI)., (SA)y(LMI)., (SA)(KR)z., Table 1]. By contrast, the other three positive example sequences shown to be cytosolic1 could not be recognized as erroneous by bioinformatics methods. Their PWM model prediction scores and posterior probabilities were only slightly below threshold or in the prediction gray zone in which additional true positive sequences are found. Moreover, the sequences terminated with C-terminal tripeptides that matched the general pattern of plant PTS1 tripeptides (see above) and were closely related to recently identified plant PTS1 tripeptides (e.g., SEM > to SEL >; SGI > to SGL >, Table 1). Hence, it would be desirable to be able to apply bioinformatics including statistical methods to identify and eliminate putatively erroneous positive example sequences containing sequencing errors from starting data sets.
Table 1. Experimental validation of ambiguous positive example sequences. In addition to two obviously erroneous and four ambiguous positive example ESTs experimentally validated in Lingner et al.1 three additional SEL > ESTs were selected as putatively erroneous sequences in the present study. All three reporter protein constructs were shown to remain cytosolic (data not shown) and thereby validated as erroneous ESTs. Based on these data, three statistical methods were evaluated for their ability to identify such erroneous sequences as outliers in OG-specific histograms of PWM prediction scores. The following protein acronyms have been used: AGT, alanine (serine)-glyoxylate aminotransferase; DEG15, DEG15 endopeptidase; GOX, glycolate oxidase; GSTT1, glutathione S-transferase isoform theta 1; MLS, malate synthase; SDRb/DECR, short-chain dehydrogenase-reductase B/2,4-dienoyl-CoA reductase; SCP2, sterol carrier protein isoform 2; Uri, uricase. The following plant species acronyms have been used: Bd, Brachypodium distachyon; Ci, Cichorium intybus; Cpr, Chimonanthus praecox; Gr, Gossypium raimondii; Fv, Fragaria vesca subsp vesca; Pn, Populus nigra; Rs, Raphanus sativus; So, Saccharum officinarum; P, peroxisome; C, cytosol.
| PTS1 protein |
Species |
Sequence type |
C-terminal decapeptide fused to EYFP |
PWM prediction model |
Experimental subcellular targeting |
Figure |
||
|---|---|---|---|---|---|---|---|---|
| Prediction score |
Posterior probability | Targeting prediction |
||||||
| Ambiguous positive examples (Lingner et al.)1: | ||||||||
|---|---|---|---|---|---|---|---|---|
| SDRb/DECR |
Gr |
EST |
TPVGVPSRKL > |
0.367 |
0.233 |
C |
Cytosol |
Lingner et al.,1Figure 2 Ae |
| SCP2 |
Fv |
EST |
SDIFPKPSEM > |
0.270 |
0.020 |
C |
Cytosol |
Lingner et al.,1Figure 2 Ad |
| AGT |
Gr |
EST |
NNIPMSPSGI > |
0.175 |
0.001 |
C |
Cytosol |
Lingner et al.,1Figure 2 Ac |
| DEG15 | Rs | EST | LSRDVIPSEL > | 0.410 | 0.485 | C | Peroxisome | Lingner et al.,1Figure 2 R |
| Obviously erroneous positive examples (Lingner et al., 2011): | ||||||||
|---|---|---|---|---|---|---|---|---|
| Uri |
So |
EST |
TSLDPPMLNL > |
−0.161 |
0.0 |
C |
Cytosol |
Lingner et al.,1Figure 2 Af |
| GOX_b | Pn | EST | TCSRWDHLCR > | −1.433 | 0.0 | C | Cytosol | Lingner et al.,1Figure 2 Ag |
| Ambiguous positive examples (this study): | ||||||||
|---|---|---|---|---|---|---|---|---|
| MLS |
Ci |
EST |
VHHPKGPSEL > |
0.682 |
0.997 |
P |
Cytosol |
This study (data not shown) |
| GSTT1 |
Cpr |
EST |
VRKQSTLSEL > |
0.386 |
0.339 |
C |
Cytosol |
This study (data not shown) |
| SCP2 | Bd | EST | PDIFTKPSEL > | 0.380 | 0.304 | C | Cytosol | This study (data not shown) |
Experimental Validation of Additional Positive Example Sequences
To identify additional erroneous ESTs among positive example sequences of plant PTS1 protein homologs, we validated subcellular targeting of further sequences in the present study. One positive example sequence terminating with the C-terminal tripeptide, SEL >, had previously been shown to target peroxisomes, characterizing the tripeptide as a novel functional plant PTS1.1 The presence of an acidic residue at position -2 of the PTS1 tripeptide, however, remained atypical, because typically positively charged residues, such as Arg and Lys, occur at the same position in the large majority of plant PTS1 sequences (92.03% of all positive example sequences).
In the present study we focused on experimental analysis of the three other SEL > sequences for two major reasons. First, SEL > can be created by single nucleotide sequencing errors from SKL > sequences by exchange of the first nucleotide (A-to-G) of the two lysine triplets (AAA and AAG) into glutamate triplets (GAA and GAG). Second, because SKL > is the prototypical PTS1 tripeptide and frequently found in high-abundance PTS1 proteins, the number of SKL > sequences was exceptionally high among positive example sequences (655 sequences, 26.65%). Codon similarity between SKL > and SEL > and the extremely high abundance of SKL > sequences were predicted to strongly increase the probability that some SEL > sequences were erroneous and cytosolic in experimental validation studies. The rational is that peroxisome targeting is generally expected to be abolished by SKL > -to-SEL > mutations because SEL > is considered a weak PTS1 tripeptide that requires specific targeting enhancing upstream elements for peroxisome targeting, which are generally not required and therefore missing in SKL > sequences.
The three SEL-terminating EST example sequences chosen for experimental validation derived from a homolog of malate synthase (MLS) from Cichorium intybus, a homolog of glutathione S- transferase isoform theta 1 (GSTT1) from Chimonanthus praecox, and a homolog of sterol carrier protein isoform 2 (SCP2) of Brachypodium distachyon. The proposed peroxisome targeting domains (PTDs), comprising the C-terminal decapeptide of the translated ESTs, were attached to the C-terminal end of the reporter protein, EYFP. The cDNAs were transiently expressed from the cauliflower mosaic virus (CaMV) 35S promoter in onion epidermal cells that had been biolistically transformed. Similar to EYFP alone, the reporter protein constructs extended by the three decapeptides terminating with SEL > remained in the cytosol and nucleus (data not shown). Extended expression times up to 1 week at reduced temperature (ca. 10°C) did not lead to peroxisome targeting (data not shown). Hence, the experimental data indicated that, consistent with the initial hypothesis, the three ESTs either contained sequencing errors that prevented peroxisome targeting or that they were not orthologous to the reference Arabidopsis PTS1 proteins.
In conclusion, the data by Lingner et al.1 supplemented by those presented in this publication confirmed that ESTs, even though valuable and indispensible resources to significantly enlarge data sets of positive example sequences (e.g., for plant PTS1 proteins approx. 8-fold, 87.2% ESTs1) contain a low rate of sequencing errors that might reduce the prediction accuracy of the PTS1 protein prediction methods.
Identification of Erroneous ESTs by Statistical Analysis of Prediction Score Distributions per Orthologous Group
To further improve the accuracy of PTS1 protein prediction methods, we aimed to identify erroneous ESTs by mathematical or bioinformatics methods. If the distribution of the PWM model prediction scores of all positive example sequences were analyzed (bin width 0.05), the histogram showed that the sequences clustered around a mean value, resembling a normal Gauss distribution with a relatively wide peak (Fig. 1A). Outliers, i.e., sequences with atypically low PWM prediction scores that are most likely caused by sequencing errors, were difficult to identify by this general analysis. However, we hypothesized that the PWM prediction scores of example sequences of a single orthologous group (OG) cluster around a mean value similar to a normal Gauss distribution with group-specific mean values. In this case, statistical methods might be applicable to identify outliers.

Figure 1A–F. Distribution of PWM model prediction scores of positive example sequences and outlier detection thresholds calculated by three different statistical methods for nine representative OGs. Each individual plot shows the histogram of prediction scores for positive examples associated with a particular orthologous group (OG), using the PWM prediction model (see also Table 1, data taken from ref. 1). The dashed red line represents the scaled density function of the normal distribution whose parameters have been estimated from the prediction scores. The solid red vertical corresponds to the mean of the distribution. The purple, green and black vertical lines represent the rejection thresholds for putative false positive examples corresponding to the three different statistical methods (see text). In the upper left corner of each plot the values of the thresholds are provided along with the respective number of rejected examples in parentheses. The following OGs are shown: (A) all sequences; (B) Acyl-CoA oxidase isoform 1 (ACX1); (C) Alanine (serine)-glyoxylate amiontransferase (AGT); (D) Acetyltransferase (ATF); (E) Quinone oxidoreductase (BSMDR); (F) Glutathione S-transferase isoform theta 1 (GSTT1); (G) Hydroxypyruvate reductase (HPR); (H) Malate synthase (MLS); (I) Short-chain dehydrogenase-reductase B/2,4-Dienoyl-CoA reductase (SDR-b/DECR); (J) sterol carrier protein isoform 2 (SCP2).
To this end, we analyzed the distribution of prediction scores separately for each OG containing more than ten sequences (i.e., 43 PTS1 protein groups). For several OGs such as acyl-CoA oxidase isoform 1 (ACX1) and quinone oxidoreductase (BSMDR, Fig. 1B and E) relatively sharp score distribution peaks were obtained, while for other groups such as alanine(serine)-glyoxylate aminotransferase (AGT) and acetyltransferase isoforms 1 and 2 (ATF1/2, Fig. 1C and D) the scores were distributed over a wider range of values. The histograms of a few OGs such as AGT and malate synthase (MLS) indicated atypical score distributions, partly with the presence of a second distribution peak clustering around a lower PWM value (Fig. 1C, H). A few obvious outliers of extremely low PWM prediction scores could be identified by visual histogram inspection. Notably, absolute PWM prediction scores appeared insufficient to identify outliers. For instance, one apparent BSMDR outlier had the same PWM score of 0.3 as several ATF sequences located within the main Gauss peak of ATF1/2 orthologs. Those six erroneous sequences that had been experimentally validated as cytosolic generally had been assigned exceptionally low prediction scores and were located as outliers outside of the fitted normal distribution (Tables 1 and 2; Fig. 1C, F, H and J).
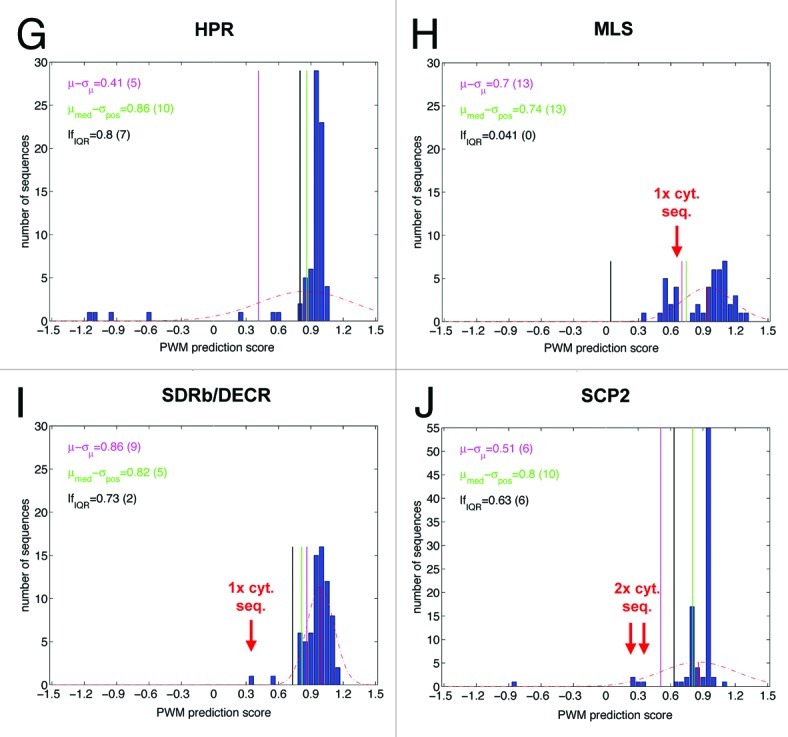
Figure 1G–J. For figure legend, see page 266.
Table 2. Comparative analysis of statistical methods for the identification of outliers representing erroneous ESTs of PTS1 proteins. Three statistical methods (see text) were evaluated to identify outliers in OG-specific PWM score histograms. Methods evaluated as too insensitive for an OG failed to identify additional apparent outliers and experimentally validated cytosolic sequences, while methods evaluated as too unspecific detected too many sequences as outliers. To achieve very good performance in outlier detection, it is recommended to apply all three methods and to eliminate outliers that are identified by at least two methods. ACX1, acyl-CoA oxidase; AGT, alanine (serine)-glyoxylate amiontransferase; ATF1/2, acetyltransferase; BSMDR, quinone oxidoreductase; GSTT1, glutathione S-transferase isoform theta 1; HPR, hydroxypyruvate reductase; MLS, malate synthase; SDRb/DECR, short-chain dehydrogenase-reductase B/2,4-dienoyl-CoA reductase; SCP2, sterol carrier protein isoform 2.
| OG acronym |
Total seq. number |
Method 1: „Standard deviation from mean value” |
Method 2: „Positive deviation from median score” |
Method 3: „Interquartile range” |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of seq. excluded | Number of seq. excluded (%) | Number of erroneous (cyt.) seq. excluded | Conclusion | Number of seq. excluded | Number of seq. excluded (%) | Number of erroneous (cyt.) seq. excluded | Conclusion | Number of seq. excluded | Number of seq. excluded (%) | Number of erroneous (cyt.) seq. excluded | Conclusion | ||
| ACX1 |
88 |
3 |
3.4 |
0/0 |
good |
3 |
3.4 |
0/0 |
good |
3 |
3.4 |
0/0 |
good |
| AGT |
94 |
15 |
16.0 |
1/1 |
good |
15 |
16.0 |
1/1 |
good |
14 |
14.9 |
1/1 |
good |
| ATF1/2 |
61 |
4 |
6.6 |
0/0 |
good |
5 |
8.2 |
0/0 |
good |
6 |
9.8 |
0/0 |
good |
| BSMDR |
52 |
3 |
5.8 |
0/0 |
good |
2 |
3.8 |
0/0 |
good |
2 |
3.8 |
0/0 |
good |
| GSTT1 |
54 |
4 |
7.4 |
1/1 |
good |
5 |
7.4 |
1/1 |
good |
4 |
7.4 |
1/1 |
good |
| HPR |
76 |
4 |
5.3 |
0/0 |
too insens. |
10 |
13.2 |
0/0 |
good |
7 |
9.2 |
0/0 |
good |
| MLS |
47 |
13 |
27.7 |
1/1 |
good |
13 |
27.7 |
1/1 |
good |
0 |
0 |
0/1 |
too insens. |
| SDRb/DECR |
72 |
9 |
12.5 |
1/1 |
too unspec. |
5 |
12.5 |
1/1 |
too unspec. |
2 |
2.8 |
1/1 |
good |
| SCP2 |
91 |
6 |
6.6 |
2/2 |
good |
10 |
11.0 |
2/2 |
too unspec. |
6 |
6.6 |
2/2 |
good |
|
total |
635 |
61 |
9.6 |
6/6 |
good |
68 |
10.7% |
6/6 |
good |
44 |
6.9% |
5/6 |
good |
| |
|
|
|
|
|
Combined method application |
|
|
|
|
|||
| 60 | 9.4% | 6/6 | very good | ||||||||||
Next, we applied three statistical methods to the histograms of the nine chosen example OGs and compared their ability in identifying apparent outliers including the six cytosolic sequences in terms of sensitivity and specificity. The first statistical method applies the most simple statistical rejection criterion, using the standard deviation from the mean value of a score distribution assuming a normal distribution of the scores. Here, the mean score value µ is estimated from all scores s1, …, sN associated with an OG according to 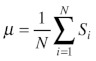 . For many OGs the mean PWM scores were high (i.e., between 0.7 to 0.9, e.g., ACX1, AGT, BSMDR, GSTT1, HPR, MLS, SDRb/DECR, SCP2), while for other OGs such as ATF1/2 the mean value (0.4) was significantly lower, confirming the hypothesis that the mean values of PWM prediction scores are often OG-specific, for instance, because the proteins of some OGs preferentially contain weak PTS1s. The (sample) standard deviation σ is then estimated according to
. For many OGs the mean PWM scores were high (i.e., between 0.7 to 0.9, e.g., ACX1, AGT, BSMDR, GSTT1, HPR, MLS, SDRb/DECR, SCP2), while for other OGs such as ATF1/2 the mean value (0.4) was significantly lower, confirming the hypothesis that the mean values of PWM prediction scores are often OG-specific, for instance, because the proteins of some OGs preferentially contain weak PTS1s. The (sample) standard deviation σ is then estimated according to 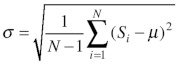 . In this case, examples with a score of s < µ-σ are rejected (Fig. 1). Upon method application to the nine representative OGs, between three (3.4%) to 13 sequences (27.7%) were excluded with 61 out of 635 (9.6%) in total (Table 2). The excluded sequences comprised all six cytosolic sequences, showing that the method was sufficiently sensitive on experimentally validated sequences. In general the method appeared well suitable in identifying outliers in most OGs but too insensitive for hydroxypyruvate reductase (HPR) and too unspecific for SDRb/DECR (Table 2).
. In this case, examples with a score of s < µ-σ are rejected (Fig. 1). Upon method application to the nine representative OGs, between three (3.4%) to 13 sequences (27.7%) were excluded with 61 out of 635 (9.6%) in total (Table 2). The excluded sequences comprised all six cytosolic sequences, showing that the method was sufficiently sensitive on experimentally validated sequences. In general the method appeared well suitable in identifying outliers in most OGs but too insensitive for hydroxypyruvate reductase (HPR) and too unspecific for SDRb/DECR (Table 2).
As a second statistical method for outlier detection, we used the maximum positive deviation from the median score of the distribution. Here, the (so-called robust) median μmed corresponds to the midpoint value of the sorted list of example scores. In case of an even number of examples μmed is computed as the mean value of the two mid-point scores in this list. Compared with the mean value (see method 1), the median is robust toward outliers in the score distribution, in particular if the outliers are located asymmetrically, as is generally the case for positive PTS1 protein example sequences (e.g., HPR). The maximum positive deviation σpos is calculated as the difference of the highest example score smax and the median value (σpos = smax − μmed). Example sequences with scores s < μmed − σpos are considered potential outliers and are rejected. Upon application to the nine representative OGs, between two (3.8%) to 15 sequences (16.0%) were excluded with 68 (10.7%) out of 635 in total. In general, this method performed similar to the first method in terms of correct exclusion of all six cytosolic sequences and the total number of excluded sequences (68 compared with 61). This second method, however, appeared to perform better for HPR in being more sensitive in identifying outliers, while it seemed too unspecific for SDRb/DECR and SCP2 (Fig. 1 and Table 2).
The third statistical method uses quartiles of the score distribution to define an acceptable minimum score value. A quartile is defined as one of three points that divide the (sorted) list of prediction scores into four equally sized sets. As an example, the first quartile border q25 incorporates the prediction scores associated with the first 25% of data points in an (ascendingly) ordered list of values. Outliers on the left-hand size of the score distribution can then be identified by the so-called “lower fence” (lf). The lower fence value is defined as q25 − 1.5*IQR, whereby IQR represents the interquartile range between the third and the first quartile border (q75-q25). Between zero to 14 sequences (14.9%) were excluded with a relatively low total number of 44 rejected sequences (6.9%). This statistical method generally appeared well suitable in identifying outliers but too insensitive for MLS in terms of both total sequences and cytosolic sequences (Fig. 1 and Table 2).
In summary, histogram analysis of PWM score distributions is an important method to computationally validate the identification of both putatively orthologous sequences per se and of sequences containing sequencing errors. All three statistical methods performed well on most OGs but none on all OGs because they were too insensitive or too unspecific on a few OGs whose histogram shape morphology significantly differed from an approximate Gauss curve. However, for each representative OG at least one of the three statistical methods appeared to perform well. Hence, by testing all three statistical methods on all OGs and selecting the most appropriate for exclusion of outliers, very good performance in outlier identification can be achieved.
Conclusions
Highly accurate prediction tools have been developed for plant PTS1 proteins. The development of similar tools for plant PTS2 proteins and animal and fungi PTS1 and PTS2 proteins is in progress. The development of these prediction tools essentially relies of ESTs to increase the number and sequence diversity of positive example sequences. The drawback that ESTs contain a low sequencing error rate can be overcome to large extent by post-validation of positive example sequences in an OG-specific manner. According to the data presented in this study, the application of the three statistical methods allows identification and elimination of erroneous ESTs from the present1 and future data sets of positive example sequences (Fig. 2). This post-validation will be instrumental in further improving the prediction accuracy of both PTS1 and PTS2 proteins from genome sequences.
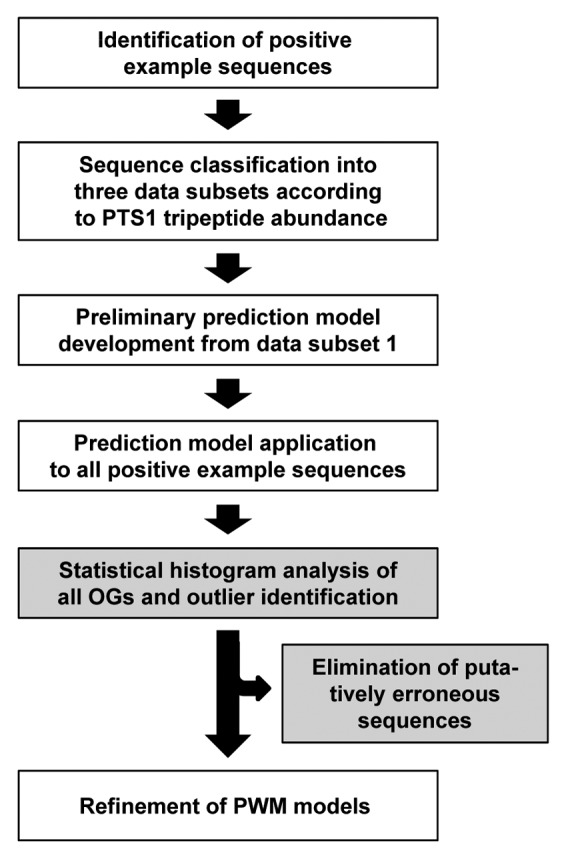
Figure 2. Improved prediction of plant PTS1 proteins by PWM prediction models made possible by post-validation of positive example ESTs applying OG-specific statistical methods. Outliers are identified and eliminated by application of a combination of three different statistical methods to each OG (gray boxes).
Acknowledgments
The research was supported by UiS funding and a grant by the Deutsche Forschungsgemeinschaft (DFG) to T.L. (Li-2050), and UiS and FUGE funding to S.R.
Glossary
Abbreviations:
- aa
amino acid
- ACX1
acyl-CoA oxidase isoform 1
- AGT
alanine(serine)-glyoxylate aminotransferase
- ATF1/2
acetyltransferase
- BSMDR
quinone oxidoreductase
- CaMV
cauliflower mosaic virus
- DEG15
DEG15 endopeptidase
- GOX
glycolate oxidase
- EST
expressed sequence tag
- GSTT1
glutathione S-transferase isoform theta 1
- HPR
hydroxypyruvate reductase
- MLS
malate synthase
- OG
orthologous group
- PTD
peroxisome targeting domain
- PTS1/2
peroxisome targeting signal type 1/2
- PWM
position weight matrix
- SCP2
sterol carrier protein isoform 2
- SDRb/DECR
short-chain dehydrogenase-reductase B/2,4-dienoyl-CoA reductase
- Uri
uricase
- YFP
yellow fluorescent protein
Footnotes
Previously published online: www.landesbioscience.com/journals/psb/article/18720
References
- 1.Lingner T, Kataya AR, Antonicelli GE, Benichou A, Nilssen K, Chen XY, et al. Identification of novel plant peroxisomal targeting signals by a combination of machine learning methods and in vivo subcellular targeting analyses. Plant Cell. 2011;23:1556–72. doi: 10.1105/tpc.111.084095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider G, Fechner U. Advances in the prediction of protein targeting signals. Proteomics. 2004;4:1571–80. doi: 10.1002/pmic.200300786. [DOI] [PubMed] [Google Scholar]
- 3.Mitschke J, Fuss J, Blum T, Höglund A, Reski R, Kohlbacher O, et al. Prediction of dual protein targeting to plant organelles. New Phytol. 2009;183:224–35. doi: 10.1111/j.1469-8137.2009.02832.x. [DOI] [PubMed] [Google Scholar]
- 4.Nair R, Rost B. Protein subcellular localization prediction using artificial intelligence technology. Methods Mol Biol. 2008;484:435–63. doi: 10.1007/978-1-59745-398-1_27. [DOI] [PubMed] [Google Scholar]
- 5.Mintz-Oron S, Aharoni A, Ruppin E, Shlomi T. Network-based prediction of metabolic enzymes’ subcellular localization. Bioinformatics. 2009;25:i247–52. doi: 10.1093/bioinformatics/btp209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emanuelsson O, Elofsson A, von Heijne G, Cristo´bal S. In silico prediction of the peroxisomal proteome in fungi, plants and animals. J Mol Biol. 2003;330:443–56. doi: 10.1016/S0022-2836(03)00553-9. [DOI] [PubMed] [Google Scholar]
- 7.Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J Mol Biol. 2003;328:581–92. doi: 10.1016/S0022-2836(03)00319-X. [DOI] [PubMed] [Google Scholar]
- 8.Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Motif refinement of the peroxisomal targeting signal 1 and evaluation of taxon-specific differences. J Mol Biol. 2003;328:567–79. doi: 10.1016/S0022-2836(03)00318-8. [DOI] [PubMed] [Google Scholar]
- 9.Reumann S. Specification of the peroxisome targeting signals type 1 and type 2 of plant peroxisomes by bioinformatics analyses. Plant Physiol. 2004;135:783–800. doi: 10.1104/pp.103.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bode´n M, Hawkins J. Prediction of subcellular localization using sequence-biased recurrent networks. Bioinformatics. 2005;21:2279–86. doi: 10.1093/bioinformatics/bti372. [DOI] [PubMed] [Google Scholar]
- 11.Hawkins J, Mahony D, Maetschke S, Wakabayashi M, Teasdale RD, Bode´n M. Identifying novel peroxisomal proteins. Proteins. 2007;69:606–16. doi: 10.1002/prot.21420. [DOI] [PubMed] [Google Scholar]