

Abstract
The effect of placebo observed in schizophrenia clinical trials represents a growing problem that interferes with signal detection for treatments, increases costs of development, discourages investment in schizophrenia research and delays the introduction of new treatments. This paper seeks to clarify key issues related to this problem and identify potential solutions to them. Differences between placebo effect and response are characterized. Recent insights into the central nervous system mechanisms of placebo effect are described. This is followed by a description of protocol/study design and study conduct issues that are contributing to a growing placebo effect in clinical trials. Potential solutions to these problems are provided.
Keywords: Clinical trials, placebo effect, placebo response, schizophrenia
Introduction
In recent years there has been a trend towards increasing placebo effects in clinical trials whose data have been submitted for new drug applications (NDAs) (Kemp et al. 2008). This has been associated with diminishing drug–placebo differences in clinical trials, which, in turn, has interfered with signal detection for new therapies (Loebel et al. 2010). Consequences of this increasing placebo effect are increased costs for drug development, more inconclusive and failed trials, delays in the development of new antipsychotics or even the abandonment of the search for new therapies because the risks and costs are seen as too great. There may also be a reduction in the perceived value of newer therapies as poor signal detection is sometimes inappropriately interpreted as newer therapies being less potent relative to older therapies or that treatments are losing their effects over time (Lehrer, 2010). In addition, meta-analytic work based on trials conducted over an extended time frame may be biased or difficult to do well, as trials taken from different periods may not be directly comparable without addressing the progressive changes in placebo effect over time.
The following review offers a definition of terminology to provide the reader with an understanding of the distinction between placebo effect and placebo response and to clarify the mechanisms involved in placebo responses. A discussion of the value and limitations of placebo in clinical trials is also provided. This includes an explication of placebo-related effects and problems that have been identified in clinical trials involving patients with schizophrenia. Potential solutions to these problems are then discussed.
Defining placebo effect, placebo response and nocebo
‘Placebo effect’ and ‘placebo response’ are distinct entities, with a number of reviews discussing these often synonymously used terms in great detail (Benedetti, 2008a, b; Benedetti et al. 2007; Enck et al. 2008; Price et al. 2008; Zubieta & Stohler, 2009). Technically, ‘placebo effect’ is the response observed in the placebo arm of a clinical trial, which is produced by the totality of the placebo biological phenomenon combined with other potential factors contributing to symptom amelioration, such as natural history, regression to the mean, biases, judgement errors, etc. On the other hand, ‘placebo response’ designates the biological phenomenon in isolation, as can best be studied in specifically designed experimental protocols. This leads to a paradox, whereby the field seeks to enhance the beneficial effects of placebo response in clinical practice, while looking to reduce placebo effect in clinical trials.
From a neuroscientific perspective, to suggest that placebo (Latin ‘I shall please’), as associated with placebo response, is inert is not accurate. ‘Inert’ suggests that the substance or treatment is devoid of specific effects for the condition being treated. However, a placebo cannot be inert if it produces a response. A placebo response does not reflect a direct pharmacological effect, but rather the response of the brain to the perception of treatment. It is the symbolic meaning of the treatment, rather than the treatment itself, that triggers the placebo response. The placebo need not be a ‘treatment’ either. Its archetype is, of course, the sugar pill, but more general factors work equally well. For example, the stimulus eliciting the effect may be ascribed to one or all aspects of the context surrounding the therapeutic act and the simulation of a therapeutic situation may replace the sugar pill.
Mechanisms for placebo effect or response
Different explanatory mechanisms have been proposed for placebo effects or responses. Classical conditioning theory posits the placebo effects or responses as a result of Pavlovian conditioning. In this process, the repeated co-occurrence of an unconditioned response to an unconditioned stimulus (e.g. salivation after the sight of food) with a conditioned stimulus (e.g. a bell ringing) induces a conditioned response (i.e. salivation that is induced by bell ringing alone). Likewise, aspects of the clinical setting (e.g. taste, colour, shape of a tablet, as well as white coats or the peculiar hospital smell) can act as conditioned stimuli, eliciting a therapeutic response in the absence of an active principle, because they have been paired with it in the past. In the same way, the conditioned response can be a negative outcome, as in the case of nausea elicited by the sight of the environment where chemotherapy has been administered in the past. Classical conditioning seems to work best where unconscious processes are at play, as in placebo responses involving endocrine or immune systems.
Expectation theory conceives the placebo effect or response as the product of cognitive engagement, with the patient consciously foreseeing a positive/negative outcome, based on factors as diverse as verbal instructions, environmental clues, previous experience, emotional arousal and/or the interaction with care providers. This anticipation triggers internal changes resulting in specific experiences (e.g. analgesia/hyperalgesia). Desire, self-efficacy and self-reinforcing feedback all interact with expectation, potentiating its effects. Desire is the experiential dimension of wanting something to happen or wanting to avoid something happening (Price et al. 2008), while self-efficacy is the belief that one is able to personally manage the disease with one's own internal resources. Self-reinforcing feedback is a positive loop, whereby the subject attends selectively to signs of improvement, taking them as evidence that the placebo treatment has worked.
Neurochemical and pharmacological effects
The last decade has witnessed the beginning of clarification of neurochemical and pharmacological details of placebo analgesia. Many studies have shown that the opiate antagonist naloxone is able to reduce or completely block the placebo effect/response (Amanzio & Benedetti, 1999; Eippert et al. 2009; Levine et al. 1978). Notably, placebo responders have levels of β-endorphin in the cerebrospinal fluid that are more than double those of non-responders; opioids released by a placebo procedure displayed the same side-effects as exogenous opiates; naloxone-sensitive cardiac effects could be observed during placebo-induced expectation of analgesia. Indirect support also comes from the possible placebo-potentiating role of the cholecystokinin (CCK) antagonist proglumide (Benedetti et al. 2007). Research suggests that the CCK system counteracts the effects of opioids, suggesting that the placebo effect may be under the opposing influences of facilitating opioids and inhibiting CCK. In some situations, a placebo effect/response can still occur despite blockade of the opioid mechanisms by naloxone. This suggests that systems other than opioids are implicated in the regulation of placebo effect/response. Little is currently known on these non-opioid systems and further research is needed to elucidate them. A detailed review may be found in Benedetti (2008b) .
The advent of neuroimaging techniques and their use for experimental purposes has added anatomical and temporal details to the neurochemical information regarding placebo effect/response. A study using positron emission tomography (PET) suggests that placebo effect/response in Parkinson's disease is mediated by dopamine (de la Fuente-Fernández et al. 2001). Placebo-induced changes in patients with Parkinson's disease were subsequently found to be associated with the reduction of bursting activity in subthalamic nucleus neurons (Fig. 1) (Benedetti et al. 2004, 2009). Subsequently, Petrovic et al. (2002) showed overlap in the brain activation pattern generated by opioid-induced analgesia and by placebo-induced analgesia. Both approaches activated areas in the rostral anterior cingulate cortex and the orbitofrontal cortex. Subsequently, in spite of some discrepancies likely explained by methodological and procedural differences, PET, functional magnetic resonance imaging and magnetoelectroencephalography studies have suggested that placebo effect/response is mediated through activation of the descending pain control system, with modulation of activity in areas such as periaqueductal grey, the ventromedial medulla, the parabrachial nuclei, the anterior cingulate cortex, the orbitofrontal cortex, the hypothalamus and the central nucleus of the amygdala (Zubieta & Stohler, 2009).
Fig. 1.

Differences in neural activity in placebo responders and placebo non-responders. These panels depict the relationship between clinical placebo response, as assessed through muscle rigidity at the wrist (a) and electrophysiological placebo responses, as measured by means of a single neuron recording (b), in Parkinson's disease. Note that in placebo responders (left), both muscle rigidity decreases and electrophysiological changes occur, whereas in placebo non-responders neither clinical nor electrophysiological changes take place (Benedetti et al. 2004, 2009).
Utility and feasibility of conducting placebo studies in schizophrenia
Discussion around placebo-controlled clinical trials in schizophrenia patients has mainly focused on ethical issues. The World Medical Association's Declaration of Helsinki (World Medical Association, 2008) is often cited in this context. This declaration stipulates that:
The use of placebo is acceptable in studies where no current proven intervention exists; or where for compelling and scientifically sound methodological reasons the use of placebo is necessary to determine efficacy or safety of an intervention and the patients who receive placebo or no treatment will not be subject to any risk of serious or irreversible harm. Extreme care must be taken to avoid abuse of this option.
Many investigators and, very importantly, regulatory agencies such as the Food and Drug Administration in the US and the European Medicines Agency have taken the position that a true appreciation of an intervention against schizophrenia, as well as the evaluation of such an intervention's safety, is not possible outside the methodology of a placebo-controlled design, with the sole exception that the experimental intervention shows superiority over existing treatments. This perspective has had a tremendous impact on drug development. Consequently, every antipsychotic that has been approved for the treatment of schizophrenia in either the US or Europe in the past 20 yr has been assessed in placebo-controlled clinical trials. This practice has been challenged by the increasing reluctance of clinician researchers (Fleischhacker & Burns, 2002) and patients (Hummer et al. 2003; Roberts, 1998) to participate in such studies. Ethical committees in many countries of the world are setting stricter standards, making it increasingly difficult to conduct placebo-controlled central nervous system (CNS) clinical trials. Where such studies are allowed, care must be taken to minimize any risks to subjects participating in these trials. Consequently, feasibility problems complicate the conduct of this research. All of these concerns are augmented by studies that have found large drop-out rates in clinical trials utilizing placebo controls (Kemmler et al. 2005) as well as a decrease of the placebo/drug difference (Kemp et al. 2008; Loebel et al. 2010) in clinical trials comparing both experimental molecules and approved antipsychotics with well-established efficacy to placebo.
Placebo effects in clinical schizophrenia treatment trials
Placebo effects, more broadly defined as any contributor to apparent symptom amelioration in clinical trials, appear to be increasing for the acute treatment studies in schizophrenia (Fig. 2) (Kemp et al. 2008). Supportive evidence for this was observed in a comparison of placebo effect observed in studies from two different phase III clinical development programmes that were used to support registration of two antipsychotic medications. These programmes were completed about 10 yr apart and had similar designs; therefore, giving us an opportunity to examine placebo effect over time and across different regions of the world. Placebo effect was determined based on the placebo group's least square mean change of the Positive and Negative Syndrome Scale (PANSS) total scores obtained from the analysis of covariance model with factors of treatment and region (when the trial was multi-regional) and the covariate of baseline PANSS total scores. As illustrated in Fig. 3, in these trials, placebo effects measured by amount of reduction in the PANSS total score increased over time (Kemp et al. 2008).
Fig. 2.

Placebo effect in acute schizophrenia trials over time. The mean change from baseline in total Positive and Negative Syndrome Scale (PANSS) scores for subjects receiving placebo across randomized, double-blind, placebo-controlled, clinical trials has increased in the direction of greater improvements that is correlated to the year that the studies were conducted. (Adapted from Kemp et al. 2008.)
Fig. 3.

Changes from baseline in total Positive and Negative Syndrome Scale (PANSS) scores in placebo-treated patients (baseline to endpoint) from two phase III antipsychotic development programmes. Analysis of the changes from baseline in total PANSS scores in placebo-treated patients enrolled in two phase III antipsychotic development trials, with earlier trials conducted nearly a decade prior to the later trials, revealed an increased effect of placebo over time.
Examination of potential drivers of placebo effects, including age, race, gender and baseline symptomatology, showed significant associations only for gender and age (Fig. 4 a, b). However, the more recent trials had an approximate 1.6-fold greater risk for placebo effects. Detailed evaluation of all participants in acute trials compared to those who completed these trials suggests that the differences in placebo effect may be driven by those subjects who completed the study and not by those who dropped out early. It was also observed that this placebo effect was most obvious in subjects originating from the USA. In this study, placebo effect was also present in other regions of the world and appeared to grow there over time.
Fig. 4.

Potential drivers of placebo effect in schizophrenia trials. Based upon multiple regression models and selection criteria of p⩽0.2, there were few variables associated with placebo effect in either the week 6 completer population (a) or the last observation carried forward (LOCF) analysis (b). With the exceptions of gender and age in the week 6 completer analysis, no other variables were associated with a placebo effect. There was, however, in more recent conducted trials a nearly 1.6-fold greater risk for placebo effect in both analyses.
In a recent publication, Chen et al. (2010) identified 10 schizophrenia drug programmes in support of NDAs that were submitted to the US Food and Drug Administration between December 1993 and December 2005. The investigators considered study data from all randomized, multi-region, multi-centre, double-blind, placebo-controlled trials. Through this, they identified 31 trials [22 positive and nine negative (i.e. none of the study drug groups showed significant results)] that included 12 585 patients from 37 countries (64% North America). In the US trials, placebo effects as measured by reduction in the PANSS total score increased over time, with no apparent trend over time observed in the non-US or ‘mixed’ trials. Placebo effect was associated with an estimated increase of 0.97 points in the reduction of the PANSS total score per year during the 12-yr span (nominal p=0.0015; Chen et al. 2010). These results (Chen et al. 2010) confirm the earlier finding (Kemp et al. 2008) that placebo effect has increased over time in schizophrenia trials and has been greater in trials performed in the US.
Factors potentially impacting upon placebo effect in clinical trials
Several potential protocol/study design and conduct-related factors may account for the placebo effect observed in schizophrenia trials. In a recently presented analysis of signal detection, Loebel et al. (2010) similarly noted that protocol/study design factors, patient's prior research involvement and duration of illness, recruitment methods and study site characteristics affect the likelihood of detecting treatment efficacy signals in schizophrenia trials. These potential contributors to placebo effect are summarized in Table 1 and discussed in more detail in the following sections.
Table 1.
Placebo effect in clinical trials of schizophrenia: potential factors and specifics
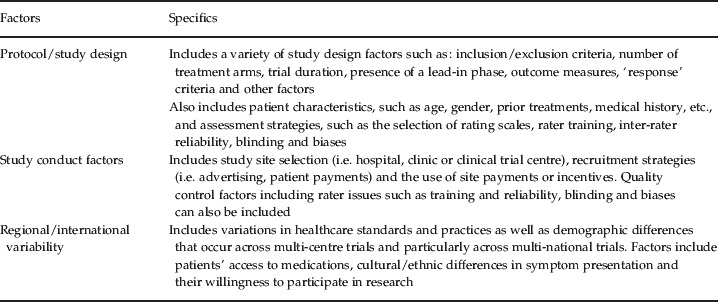
Protocol/Study design factors
Good protocol design is a critical component for mitigating placebo effect. Ironically, the desirability of placebo controls has become even more apparent as the placebo effect has become greater, more inconsistent and less predictable. These inconsistencies and the unpredictability of both placebo and drug effects over time have fuelled resistance to the use of non-inferiority trials against marketed agents as a substitute for placebo-controlled trials for assessing both the efficacy and safety of novel compounds. Although non-inferiority trials are reasonable and feasible alternatives for addressing important clinical questions (Fleischhacker et al. 2003), their design and conduct include features that are quite different from superiority trials, which must be clearly addressed during conception and interpretation.
Frequent, numerous or difficult assessments may impact placebo effects by exhausting the patient, such that they fail to complete assessments or provide invalid responses, leading to problems of both missing data and increased measurement variance. Poor choices in the selection of assessment instruments may also enhance placebo effect. Choice of a subjective, rather than an objective, endpoint may permit the introduction of increased inter- and intra-individual response variance as well as greater rater and/or patient bias. Similarly, the wording of questions used to gather information may result in biased responses. Poorly designed scales may not sensitively identify critical symptom differences or may be associated with high levels of rating variance/noise, which diminishes their effectiveness for discriminating among various treatments. The number of doses or treatment arms may enhance the placebo effect by increasing the perception of the likelihood for clinical success. Study duration may also have a role in the magnitude of a placebo effect. For instance, event-driven endpoints are likely to converge over long periods of time if the event is highly likely or inevitable over a long period (e.g. death or relapse in a chronic condition). On the other hand, for conditions such as schizophrenia, short studies may be more responsive to rater bias, Hawthorne effects and/or placebo response than longer studies, which allow the disease to fully manifest itself. If concomitant medications are permitted, they too may have small but measurable effects on clinical endpoints that obscure differences between treatment arms.
Clinical trials are often initiated with persons who are acutely ill. In such patients, the natural course of the disease, the initial acuity, the hospital milieu and the added attention provided by a research trial could lead to regression to the mean at subsequent evaluations and enhance an apparent placebo effect. Loebel et al. (2010) noted that a larger active treatment–placebo effect size was observed where subjects were more likely to be research naive and had longer illness durations. A number of the factors outlined above may compromise study completion by patients. Ensuing high drop-out rates jeopardize meaningful statistical analyses and generalizability of data (Kemmler et al. 2005).
Study conduct factors
The relationship of the subject to the study physician and increased attention from the research staff may result in improvement for all subjects in the trial. Incentives for patients and/or raters that may lead to culture-specific or compensation-specific increases in placebo effects include subjects enrolled from backgrounds where the clinician is particularly esteemed and subjects have a culturally driven incentive to please him or her as well as the availability of monetary rewards for participation. The conduct of trials in countries where patients have limited access to healthcare is likely to influence recruitment and, potentially, study outcomes. For treatments where clinical benefit is supported by prior results, both subjects and investigators/raters may be prone to look for and magnify clinical improvement after randomization into clinical trials, leading to expectation bias. There may also be instances in which it is either very desirable to demonstrate substantial improvement (e.g. as a consequence of the erroneous assumption that a sponsor is pleased by positive results) or no improvement (e.g. if patients fear to lose pensions or benefits if they get better).
Clinical trial recruitment strategies vary enormously from site to site and may be driven by financial factors or sponsors that emphasize rapid recruitment/enrolment methods – with these having an impact on placebo effects. The appropriateness of the recruitment/enrolment methods may vary depending upon the nature of the target population (e.g. acute exacerbation, persistent residual symptoms, refractory symptoms, etc.). Some recruiting agencies or sites pay fees to patients for their participation in the trial. While these fees are usually regulated by an Investigational Review Board at the site, there is considerable variation in what is considered appropriate or acceptable. Advertising to enrol patients has led to the phenomenon of ‘professional’ patients who will enrol in multiple trials sequentially, or even concurrently. These patients might exaggerate their symptoms in order to be eligible for a trial, which has a considerable impact on treatment outcomes.
Other study conduct issues may mask or obscure differences between treatments. For instance, if clinical raters are inadequately trained, they may not be able to discriminate clinically important differences between treatments. Many sponsors monitor sites to ensure the adequacy of study conduct; if this is not done with sufficient rigor, variance may be increased and the sensitivity of the study may be diminished. For some treatments, inadvertent unblinding of the treatment assignment may bias results. For example, side-effects or the taste of one test compound in a trial may be sufficiently different from other comparators to permit subjects to become aware of treatment assignment. Encapsulated medications present the possibility for unblinding by the inquisitive subject who opens the capsule. Finally, fraud, although infrequent, is a consideration for which study sponsors must be vigilant.
Regional/International variability factors
In studies conducted across various regions or countries healthcare access and utilization can vary widely. In addition to the patient's access to clinicians and medications, which may strongly impact patients’ willingness to participate in clinical trials, cultural and ethnic differences may result in differences in symptom presentation, description and ratings of severity. Taken together all of these factors may contribute to increased variability within each treatment arm and so make it more difficult to differentiate treatment responses between arms. Further, these local population differences may result in patient selection biases. On the other hand, broadly based clinical trials increase the generalizability of findings for that study.
Potential solutions or remedies for placebo effects
Given the importance of finding safe and more effective treatments in efficiently designed CNS clinical trials, it is important to identify remedies that can be brought to bear on the problem of inconsistent, unpredictable and, at times, inordinately large placebo effects. Solutions to the problem of placebo effects in clinical trials depend heavily upon acknowledgement of the problem, followed by its accurate assessment and analysis.
Protocol/Study design
Suggestions to improve clinical trial design include selecting an optimal number of assessment scales with good psychometric properties and clear anchors. Frequency of scale administration should be limited so as to reduce both subject and rater fatigue. Study duration should provide optimal time to address the study question. Encouraging mutually shared accountability among persons who design the trials, persons who oversee the implementation and day-to-day conduct of clinical trials, e.g. contract research organizations, and investigators may improve both design and outcomes. Care should be taken to avoid unnatural trial environments that excessively minimize stress that will be seen in ‘real-world’ environments.
The value of lead-in phases to ascertain stability or non-response for eligibility to enter a prospective trial is complicated when investigators know what will determine eligibility. Investigators may be vulnerable to adjust ratings in order for patients to meet eligibility criteria, a phenomenon termed ‘baseline inflation’. A placebo lead-in phase has an inconsistent impact on reducing placebo effects and will vary depending upon the nature of the trial (e.g. acute treatment, augmentation, maintenance of effect, etc.). Some sponsors have tried to blind the sites (i.e. raters) in terms of the length of the placebo wash-out phase, the criteria for patient inclusion in analyses and other factors. Clearly, the ‘placebo’ effect also occurs among patients receiving active medications and, even in trials where there is no placebo arm, there can be a surprising degree of ‘placebo’ effect. Supporting this, a surprising degree of placebo improvement may occur when patients are switched to another agent or a second treatment is added, even in trials where patients have been selected on the basis of ‘stable persistent residual symptoms’ (Kane et al. 2009).
Patients’ eligibility criteria in terms of demographics, medical/treatment history and potential sites of recruitment are important elements in protocol design and the phenomenon of placebo effect. In acute schizophrenia trials, the intended patient is usually someone who has had a clear exacerbation/relapse with a marked, clinically significant worsening of symptoms. Such patients are most often found in acute care hospitals. However, patients frequently enrolled in clinical trials are those who have persistent residual symptoms that are severe enough to meet eligibility criteria, but have not had an acute exacerbation and have experienced continued symptoms despite adequate treatment. Such patients are identified in out-patient programmes, day/partial hospital programmes, adult homes and residential facilities or through advertising and are then admitted to hospital or professional clinical trial centres in order to participate in the clinical trial. It is not surprising to see either a high rate of placebo effect or a low rate of drug effect in such patients, although obviously each of those possibilities will be determined by different factors. Since it is generally left to the investigator's judgement as to whether there has been an acute exacerbation, and it is often difficult to find quantitative documentation of worsening symptoms retrospectively, this remains an important issue for sponsors. Therefore, methods to ensure that the desired target population is enrolled in the trial are essential. This is not to say that a signal for efficacy cannot be detected in a population of patients with persistent residual symptoms; however, this represents a distinct treatment-refractory population and that should be clearly identified as a desired characteristic of the study population. On the other hand, if such a population is not desired for the study, they should be excluded as their inclusion is likely to add considerable ‘noise’ to the data.
To manage placebo effects related to ascertainment bias, some have argued for designing trials with over-inclusive criteria, but having an ‘a priori criteria’ that define a specific subpopulation that will be utilized in the primary analysis. It is likely that the data collected on patients who were not technically ‘eligible’ can be put to good use and this might reduce the risk of various biases and misaligned incentives regarding patient recruitment. Clearly, this would involve greater resources from the sponsor and such an approach would only be appropriate to address questions where the additional resources use can be justified.
The use of an early response/non-response paradigm (Correll et al. 2003; Kinon et al. 2008, 2010) is another study design worth considering in order to enhance the selection of true drug responders, who can then participate in a double-blind, placebo-controlled discontinuation trial. This strategy involves treating all patients with the experimental drug (with or without an active control) and then identifying those subjects who have at least a minimal (i.e. 20%) improvement on a specified subset of items after 2 wk treatment. This offers the advantage of potentially eliminating a substantial proportion of the patients who are less likely to respond to any type of treatment or intervention. In recent retrospective and prospective studies employing this paradigm, approximately 70% of patients fall into the early ‘non-response’ group. Although these patients do go on to improve more over the next weeks or months, they never improve as much as the ‘early responders’. The assumption would be that the early responders would include a greater proportion of patients who are truly drug responsive, although it would also include some proportion of ‘placebo responders’. Because such patients represent a group enriched for drug responsiveness, they are an ideal subgroup to enter into a double-blind discontinuation study, where, following stabilization, patients are randomized to continued treatment or placebo and time to destabilization is the endpoint. Use of such an enriched population into these studies would likely increase the anticipated effect size for the study and reduce the overall number of individuals who are exposed to placebo.
Study conduct factors
Variability among sites with regard to patient recruitment techniques, as well as study participation/enrolment incentives, may drive a substantial portion of the placebo effect. For instance, individuals who have been hospitalized, but have already received treatment for several weeks without adequate response and are then referred or recruited for participation in a clinical trial can influence trial outcomes. This would likely increase the risk of enrolling poor or partially responsive (or even refractory) patients into the trial. Putting limits on the duration of the current ‘episode’ (although as previously indicated this would require careful documentation) or the duration of the current hospitalization (easier to document, but still does not confirm the presence of an acute exacerbation) or the duration of current treatment could help to reduce this risk. The variability in clinical trial recruitment strategies is a study conduct factor that may affect placebo effects and requires review in order to mitigate the impact. Recognition of the impact of these recruitment/enrolment variables and incentives and their minimization should be considered in order to reduce their effect on trial outcomes.
Other study conduct factors to be considered include clinical rater skill and training, validity and reliability of ratings in clinical trials, as well as the skill of monitors reviewing this work. True inter-rater reliability requires that different interviewers conduct interviews with the same patient and arrive at scores that (based on statistical tests) fall within a specified a priori range of agreement. In reliability studies based on the assessment of a videotape, or multiple raters assessing the same live patient, error variance is enormously reduced, because the questions are asked once by the same person and everyone uses the same interview to judge symptom severity. Such an approach assesses the rater's ability to agree on the severity and intensity of the reported symptoms. It does not establish the rater's ability to conduct a skilful interview that is thorough and unbiased. The type of rater training that is required to achieve true inter-rater reliability is rarely done in clinical trials because it is very time-consuming and expensive. Usually would-be raters are asked to rate a few videotapes (an insufficient number for meaningful statistical testing to be applied) and some predetermined level of agreement with a ‘gold standard’ rating score on each item is deemed sufficient to declare the rater eligible. The degree to which there is rater turnover and drift in rater performance is often not the focus of sufficient attention. Good inter-rater reliability is important because it has an enormous impact on statistical power and, therefore, sample size requirements. As summarized in Table 2, the use of raters with a high intraclass correlation coefficient [(ICC); a measure of how similar raters are to each other within a cohort of raters] is inversely related to the sample size needed to feel confident in the study results statistical power (i.e. higher ICC requires less participants per study arm) (Kobak et al. 2009).
Table 2.
Effect of interviewer reliability on study power

Adapted from Kobak et al. (2009) .
Study conduct should control for clinician as well as patient biases. Patient enrolment incentives play an important role when sites are paid on a per-patient basis. Many eligibility criteria are subjective and potentially influenced by bias. Examples include the inflation of baseline scores in order to meet patient eligibility requirements or deflation of scores if scores above a certain level are exclusionary. This is not only related to symptom severity but also to interpretation of selection criteria regarding whether patients are appropriate for inclusion in the trial. Expectation bias, demonstrated in clinical trial assessments (Davidson et al. 2009; Goldberg et al. 2007; Woods et al. 2005), may come from a variety of sources, including clinicians, patients, patient caregivers, etc. It has been shown that expectancy bias can be reduced when raters are well trained as well as when different raters with high inter-rater reliability evaluate the patient over time as compared to the same rater rating the patient at each visit. In order to effectively use different raters across time points to reduce bias, true inter-rater reliability must be established. Functional unblinding and resulting bias can also occur when certain side-effects develop that are more likely to occur in one treatment arm than another. This bias is reduced when different raters evaluate the patient over time.
Several approaches can be used to mitigate study conduct factors and enhance the quality and precision of the study assessments. One is to conduct extensive rater training, followed by careful follow-up supervision. Quality control measures can be instituted, such as videotaping or audiotaping some or all of the assessment sessions for review by an external expert, who can then provide feedback and further training to the rater if problems are identified. Other potential solutions include having an external expert randomly participate in the interview via telephone or two-way video or having the assessments conducted in their entirety by remote, centralized assessors using telephone or live two-way video. Other approaches include incentive payments for sites that are based on the quality of assessments rather than on merely the number of patients entered into the trial and keeping and publishing a registry of trial performance. The latter approach is similar to quality requirements that hospitals are increasingly required to meet regarding their outcome data. Such an approach would require that criteria for study conduct quality be established. Drug placebo differences would not be a reliable measure of quality as trials may include active controls that may have small or no clinical effects. Further, there is usually large variability in the study population, such that chance alone may lead to lower effect sizes at particular sites and, given the relatively small contribution of any site to the overall sample in large multi-centre studies, it is difficult to differentiate a response effect size that is below the mean from normal variation.
Regional/International variability
It is important to recognize regional differences in trial implementation and patient recruitment as drug development programmes are increasingly global. In some countries or regions, the opportunity to participate in a clinical trial might provide access to a more comprehensive standardized evaluation, as well as access to subsidized care and medication.
Cross-cultural differences in sites of multi-centre studies lead to the potential misinterpretation of the study selection criteria and goals. The spirit of the protocol may not be fully understood or adhered to, leading to technical adherence to the study design but loss of the spirit of the protocol. Personnel conducting the study may not have sufficient skills or time to ensure that the spirit of the protocol was always maintained. Different incentives or inadequate understanding may impact upon patient selection, patient ratings and trial outcomes.
In an attempt to mitigate regional and international variability factors, researchers may consider stratification of the trial findings by regions. Examination of salient differences by regions may also be beneficial.
Summary
The design and conduct of clinical trials presents a complex array of challenging problems, one of which is that of the placebo effect. The first step in addressing the issue of placebo effect is acknowledgement of its existence. We must then focus on its potential causes in order to adjust clinical trial design elements. Clearly, the sources of placebo response are diverse. Understanding placebo response as a neurobiological effect is different from the sources of ‘placebo response’ in a population that includes a much broader range of issues that relate to trial design, conduct and factors such as ascertainment bias and regression to the mean. The latter may be associated with strong regional differences. All of these factors should be taken into consideration when interpreting results from clinical trials.
Increasing placebo response is frequently associated with increased variance around study endpoint measurement, leading to poor signal detection. This, in turn, has led to increasing sample sizes, increasing numbers of failed studies and much higher treatment development costs. Therefore, failure to address these issues threatens the support for investments in and the success of CNS drug development.
Acknowledgements
The development of this manuscript was funded by Ortho-McNeil Janssen Scientific Affairs, Titusville, New Jersey, USA. The authors acknowledge the editorial and technical support that was provided by Susan Ruffalo, PharmD, MedWrite, Inc., Newport Coast, California in combining the individual author's sections into this presentation.
Each author is entirely responsible for the overall scientific content of the manuscript. All authors read and approved the final manuscript. All authors contributed equally to the manuscript.
Statement of Interest
Dr Alphs is an employee of Janssen Scientific Affairs, LLC. Dr Kane is a shareholder of MedAvante and has been a consultant to Alkermes, Amgen, Astra-Zeneca, Bristol-Myers-Squibb, Cephalon, Dainippon Sumitomo, Intracellular Therapeutics, Johnson & Johnson, Janssen, Lilly, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Pierre Fabre, Proteus, Sunovion, and Targacept.
References
- Amanzio M, Benedetti F. Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems vs. conditioning-activated subsystems. Journal of Neuroscience. 1999;19:484–494. doi: 10.1523/JNEUROSCI.19-01-00484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F 2008aMechanisms of placebo and placebo-related effects across diseases and treatments Annual Reviews Pharmacology and Toxicology 4833–60. [DOI] [PubMed] [Google Scholar]
- Benedetti F 2008bPlacebo Effects: Understanding the Mechanisms in Health and Disease Oxford, UK: Oxford University Press [Google Scholar]
- Benedetti F, Colloca L, Torre E, Lanotte M. et al. Placebo-responsive Parkinson patients show decreased activity in single neurons of subthalamic nucleus. Nature Neuroscience. 2004;7:587–588. doi: 10.1038/nn1250. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Lanotte M, Colloca L, Ducati A. et al. Electrophysiological properties of thalamic, subthalamic and nigral neurons during the anti-Parkinsonian placebo response. Journal of Physiology. 2009;587:3869–3883. doi: 10.1113/jphysiol.2009.169425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F, Lanotte M, Lopiano L, Colloca L. When words are painful: unraveling the mechanisms of the nocebo effect. Neuroscience. 2007;147:260–271. doi: 10.1016/j.neuroscience.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Chen Y-F, Wang S-J, Khin NA, Hung MJ. et al. Trial design issues and treatment effect modeling in multi-regional schizophrenia trials. Pharmaceutical Statistics. 2010;9:217–229. doi: 10.1002/pst.439. [DOI] [PubMed] [Google Scholar]
- Correll CU, Malhotra AK, Saurabh K, McMeniman M. et al. Early prediction of antipsychotic response in schizophrenia. American Journal of Psychiatry. 2003;160:2063–2065. doi: 10.1176/appi.ajp.160.11.2063. [DOI] [PubMed] [Google Scholar]
- Davidson M, Galderisi S, Weiser M, Werbeloff N. et al. Cognitive effects of antipsychotic drugs in first-episode schizophrenia and schizophrenia disorder: a randomized, open-label clinical trial (EUFEST) American Journal of Psychiatry. 2009;166:675–682. doi: 10.1176/appi.ajp.2008.08060806. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Fernández R, Ruth TJ, Sossi V, Schulzer M. et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson's disease. Science. 2001;293:1164–1166. doi: 10.1126/science.1060937. [DOI] [PubMed] [Google Scholar]
- Eippert F, Bingel U, Schoell ED, Yacubian J. et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron. 2009;63:533–543. doi: 10.1016/j.neuron.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Enck P, Benedetti F, Schedlowski M. New insights into the placebo and nocebo responses. Neuron. 2008;59:195–206. doi: 10.1016/j.neuron.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Fleischhacker WW, Burns T. Feasibility of placebo-controlled clinical trials of antipsychotic compounds in Europe. Psychopharmacology. 2002;162:82–84. doi: 10.1007/s00213-002-1060-z. and The European Group for Research in Schizophrenia. [DOI] [PubMed] [Google Scholar]
- Fleischhacker WW, Czobor P, Hummer M, Kemmler G. et al. Placebo or active control trials of antipsychotic drugs? Archives of General Psychiatry. 2003;60:458–464. doi: 10.1001/archpsyc.60.5.458. [DOI] [PubMed] [Google Scholar]
- Goldberg GE, Goldman RS, Burdick KE, Malhotra AK. et al. Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: is it a practice effect? Archives of General Psychiatry. 2007;64:1115–1122. doi: 10.1001/archpsyc.64.10.1115. [DOI] [PubMed] [Google Scholar]
- Hummer M, Holzmeister R, Kemmler G, Eder U. et al. Attitudes of patients with schizophrenia toward placebo-controlled clinical trials. Journal of Clinical Psychiatry. 2003;64:277–281. doi: 10.4088/jcp.v64n0308. [DOI] [PubMed] [Google Scholar]
- Kane JM, Correll CU, Goff DC, Kirkpatrick B. et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. Journal of Clinical Psychiatry. 2009;70:1348–1357. doi: 10.4088/JCP.09m05154yel. [DOI] [PubMed] [Google Scholar]
- Kemmler G, Hummer M, Widschwendter C, Fleischhacker WW. Dropout rates in placebo-controlled and active-control clinical trials of antipsychotic drugs. A meta-analysis. Archives of General Psychiatry. 2005;62:1305–1312. doi: 10.1001/archpsyc.62.12.1305. [DOI] [PubMed] [Google Scholar]
- Kemp AS, Schooler NR, Kalali AH, Alphs L. et al. What is causing the reduced drug-placebo difference in recent schizophrenia clinical trials and what can be done about it? Schizophrenia Bulletin. 2008;36:504–509. doi: 10.1093/schbul/sbn110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinon BJ, Chen L, Ascher-Svanum H, Stauffer VL. et al. Predicting response to atypical antipsychotics based on early response in the treatment of schizophrenia. Schizophrenia Research. 2008;102:230–240. doi: 10.1016/j.schres.2008.02.021. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Chen L, Ascher-Svanum H, Stauffer V. et al. Early response to antipsychotic drug therapy as a clinical marker of schizophrenia response in the treatment of schizophrenia. Neuropsychopharmacology. 2010;35:581–590. doi: 10.1038/npp.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobak KA, Brown B, Sharp I, Levy-Mack H. et al. Sources of unreliability in depression ratings. Journal of Clinical Psychopharmacology. 2009;29:82–85. doi: 10.1097/JCP.0b013e318192e4d7. [DOI] [PubMed] [Google Scholar]
- Lehrer J 2010). The truth wears off. Is there something wrong with the scientific method? The New Yorker 13 December 2010 (www.newyorker.com/reporting/2010/12/13/101213fa_fact_lehrer?currentPage=all) Accessed 29 November 2011.
- Levine JD, Gordon NC, Fields HL. The mechanisms of placebo analgesia. Lancet. 1978;2:654–657. doi: 10.1016/s0140-6736(78)92762-9. [DOI] [PubMed] [Google Scholar]
- Loebel A, Cucchairo J, Siu C, Daniel D, Baltimore, MD: 2010. ). Signal detection in clinical trials: a post-study survey of schizophrenia trial sites. In ISCTM 2010 Autumn Conference, 13–14 October, [Google Scholar]
- Petrovic P, Kalso E, Petersson KM, Ingvar M. Placebo and opioid analgesia – imaging a shared neuronal network. Science. 2002;295:1737–1740. doi: 10.1126/science.1067176. [DOI] [PubMed] [Google Scholar]
- Price DD, Finniss DG, Benedetti F. A comprehensive review of the placebo effects: recent advances and current thought. Annual Reviews of Psychology. 2008;59:565–590. doi: 10.1146/annurev.psych.59.113006.095941. [DOI] [PubMed] [Google Scholar]
- Roberts LW. The ethical basis of psychiatric research: conceptual issues and empirical findings. Comprehensive Psychiatry. 1998;39:99. doi: 10.1016/s0010-440x(98)90068-2. [DOI] [PubMed] [Google Scholar]
- Woods SW, Gueorguieva RV, Baker CB, Makuch RW. Control group bias in randomized atypical antipsychotic medication trials for schizophrenia. Archives of General Psychiatry. 2005;62:961–970. doi: 10.1001/archpsyc.62.9.961. [DOI] [PubMed] [Google Scholar]
- World Medical Association (WMA) 2008. http://www.wma.net/en/30publications/10policies/b3/index.html. http://www.wma.net/en/30publications/10policies/b3/index.html ) Declaration of Helsinki-Ethical principles for medical research involving human subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended by the 59th WMA General Assembly, Seoul, October 2008 ( ). Accessed 11 January 2011.
- Zubieta JK, Stohler CS. Neurobiological mechanisms of placebo responses. Annals of New York Academy of Science. 2009;1156:198–210. doi: 10.1111/j.1749-6632.2009.04424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]