

Abstract
Hyperglycemia is linked to increased heart failure among diabetic patients. However, the mechanisms that mediate hyperglycemia-induced cardiac damage remain poorly understood. Autophagy is a cellular degradation pathway that plays important roles in cellular homeostasis. Autophagic activity is altered in the diabetic heart, but its functional role has been unclear. In this study, we determined if mimicking hyperglycemia in cultured cardiomyocytes from neonatal rats and adult mice could affect autophagic activity and myocyte viability. High glucose (17 or 30 mM) reduced autophagic flux compared with normal glucose (5.5 mM) as indicated by the difference in protein levels of LC3-II (microtubule-associated protein 1 light chain 3 form II) or the changes of punctate fluorescence patterns of GFP-LC3 and mRFP-LC3 in the absence and presence of the lysosomal inhibitor bafilomycin A1. Unexpectedly, the inhibited autophagy turned out to be an adaptive response that functioned to limit high glucose cardiotoxicity. Indeed, suppression of autophagy by 3-methyladenine or short hairpin RNA-mediated silencing of the Becn1 or Atg7 gene attenuated high glucose-induced cardiomyocyte death. Conversely, upregulation of autophagy with rapamycin or overexpression of Becn1 or Atg7 predisposed cardiomyocytes to high glucose toxicity. Mechanistically, the high glucose-induced inhibition of autophagy was mediated at least partly by increased mTOR signaling that likely inactivated ULK1 through phosphorylation at serine 467. Together, these findings demonstrate that high glucose inhibits autophagy, which is a beneficial adaptive response that protects cardiomyocytes against high glucose toxicity. Future studies are warranted to determine if autophagy plays a similar role in diabetic heart in vivo.
Keywords: ULK1, autophagy, cardiomyocyte, diabetes, hyperglycemia, mTOR
Introduction
Hyperglycemia is an independent risk factor for diabetic cardiac injury.1-4 The development of diabetic cardiomyopathy and heart failure is closely related to the duration and severity of hyperglycemia.4-6 Indeed, the cardiotoxic effects of hyperglycemia have been corroborated in numerous in vitro and in vivo studies. High levels of glucose can directly induce cardiomyocyte death in culture,7-14 which has been attributed to excessive production of reactive oxygen species (ROS) triggered by high glucose. The critical role of oxidative stress in mediating diabetic cardiac injury has been strongly supported by the ability of various antioxidants to prevent or slow the development of diabetic cardiomyopathy in animal studies.8,15,16 However, antioxidants cannot effectively attenuate diabetic cardiomyopathy and heart failure in humans, suggesting that other mechanisms might also contribute to hyperglycemic cardiotoxicity in addition to the increased ROS generation.17,18
Autophagy is a highly conserved cellular pathway for lysosomal degradation and recycling of long-lived proteins and organelles, which plays an important role in cytoplasmic quality control and cardiac homeostasis under both normal and pathologic conditions.19-22 Autophagy has long been depicted as a survival mechanism that allows the cell to maintain energy homeostasis and viability under starvation and stress conditions.23 However, autophagy can also trigger cardiomyocyte death impairing cardiac function in other contexts, underscoring its nature as a double-edged sword that could be either protective or injurious depending on the cellular environment, the nature and intensity of the stimulus, and the levels of autophagy.24-27 Thus, the functional role of autophagy has to be determined individually within the specific context under study.
Insulin signaling activates the class I phosphatidylinositol 3-kinase (PtdIns3K), Akt/PKB and the mammalian target of rapamycin complex 1 (mTORC1) not only to stimulate glucose uptake and protein synthesis, but also to inhibit autophagy,23,28 presumably through phosphorylating and inactivating autophagy-related 1 (Atg1) protein kinase, whose mammalian homolog is known as ULK1.29 It is thus hypothesized that insulin deficiency (type 1 diabetes) or insulin resistance (type 2 diabetes) would upregulate autophagic activity, which in turn plays a protective role against diabetic injury.30 This hypothesis has been confirmed by the observation in pancreatic β-cells that autophagy attenuates diabetes-induced oxidative stress and mediates the clearance of protein aggregates.31 Also, β-cell-specific autophagy-deficient mice develop spontaneous diabetes, suggesting a necessary role for autophagy in maintaining normal β-cell function.32,33 However, the functional role of autophagy in other diabetic organs remains unknown. For example, in streptozotocin-diabetic rats, autophagy appears to be increased in the liver,34,35 but reduced in the kidney.36,37 In the OVE26 type 1 diabetic mouse heart, autophagy is inhibited, which is associated with cardiac dysfunction.38 In contrast, fructose-induced insulin resistance elevates autophagy, which is suggested to contribute to cardiac pathology.39 It is not clear why type 1 and type 2 diabetes can have completely different effects on cardiac autophagic activities.38-40 This dichotomy cannot be explained solely by hyperglycemia because both diabetic models display overt hyperglycemia. Thus, the observed changes of autophagy in the diabetic hearts likely reflect a net effect of insulin deficiency or resistance, varying degrees of hyperglycemia, and other diabetes-induced abnormalities such as dyslipidemia. Regardless, these observations demonstrate an association between altered autophagy and diabetic cardiac injury. However, whether this association represents a cause-and-effect relationship has not been determined.
In the present study, we sought to address three questions: (1) if high glucose can directly affect autophagic activity in cultured cardiomyocytes; (2) how altered autophagy has an impact upon cardiomyocyte death and survival in response to high glucose; and (3) what is the molecular mechanism underlying the observed change in autophagic activity? Our results demonstrated that high glucose inhibited autophagic flux in cardiomyocytes derived from both neonatal rats and adult mice. Surprisingly, however, the reduced autophagy turned out to be an adaptive response that acted to antagonize high glucose-induced cardiomyocyte injury. In addition, we showed that the enhanced mTORC1 signaling was likely responsible for autophagy inhibition under high glucose conditions.
Results
High glucose inhibits autophagy in neonatal rat cardiomyocytes
To determine whether and how high levels of glucose could affect autophagic activity in cardiomyocytes, we cultured neonatal rat ventricular myocytes in DMEM with normal (5.5 mM) and high doses (17 and 30 mM) of glucose for 72 h. Autophagy begins with the formation of an autophagosome. When autophagy is initiated, LC3 is processed from LC3-I (apparent molecular weight is 16 kDa) to LC3-II (14 kDa) and incorporated into autophagic vacuoles (AVs).41,42 Accompanied with this, ATG12 and ATG5 are conjugated by ATG7 and ATG10. The levels of LC3-II and ATG12–ATG5 conjugation are proportional to the number of accumulated AVs. SQSTM1/p62 is a polyubiquitin-binding protein that is degraded by autophagy. Thus, the protein levels of p62 are inversely related to autophagic activity.43-45 We used these three markers to assess the effect of high glucose on autophagy in cultured cardiomyocytes. As shown by western blot analysis in Figure 1A, compared with normal control, high glucose (17 or 30 mM) reduced the abundance of LC3-II and ATG12–ATG5 conjugates but increased p62 protein levels, suggesting that high glucose inhibited autophagy (Fig. 1A). This is not surprising given that glucose deprivation induces autophagy in cadiomyocytes.26 Because the steady-state levels of LC3-II or the number of AVs are the net effect of both AV formation and degradation, a more accurate measurement of autophagy activity is autophagic flux, which reflects the number of AVs that are delivered to and degraded in the lysosome and can be measured by the difference of LC3-II protein levels in the absence and presence of lysosomal inhibitors.42 To determine if the decrease in LC3-II was caused by a reduced AV formation rather than enhanced lysosome degradation, we cultured cells in the absence or presence of the lysosomal inhibitor bafilomycin A1 (BAF). Whereas BAF markedly increased LC3-II levels in cells cultured under normal glucose, it had only a small effect on LC3-II contents when cells were exposed to high glucose (Fig. 1A and B), suggesting that high glucose-mediated reduction in LC3-II levels was attributable mainly to an inhibited AV formation. The autophagic flux was plotted in Figure 1C as the difference of LC3-II protein levels in the absence and presence of BAF. The results clearly demonstrated that the autophagic flux in cardiomyocytes was inhibited by 48% and 56%, respectively, when cultured under 17 and 30 mM glucose (glucose 5.5 mM 1.04 ± 0.11 vs. 17 mM 0.54 ± 0.12 or 30 mM 0.45 ± 0.07, p < 0.01, n = 4). The glucose concentration-dependent differential effects of BAF were also recapitulated in the levels of p62 and ATG12–ATG5 (Fig. 1A). The inhibition of autophagy by high glucose was not due to increased osmolarity since it was normalized by mannitol.
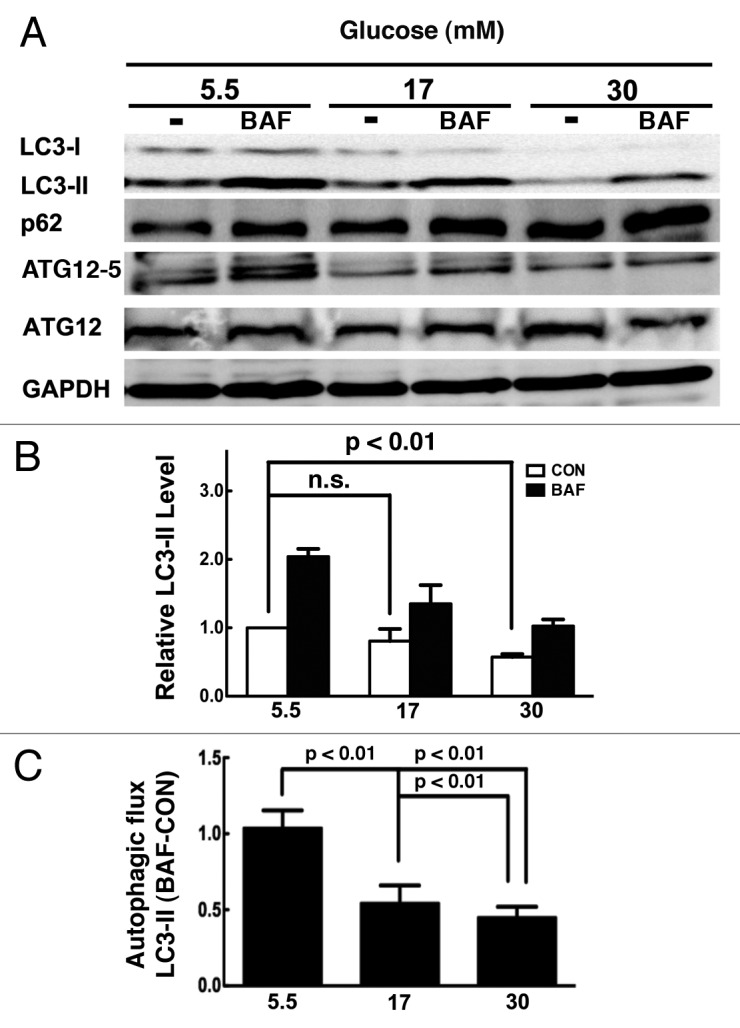
Figure 1. High glucose inhibited autophagy in cardiomyocytes. Neonatal rat cardiomyocytes were cultured in DMEM with indicated doses of glucose for 72 h. (A) Western blots show protein levels of LC3-II, p62 and ATG12–ATG5 conjugated form in the absence and presence of the lysosome inhibitor bafilomycin A1 (BAF 100 nM). (B) Quantification of the protein levels of LC3-II. The relative LC3-II levels were first normalized with GAPDH protein levels, and then compared with control (5.5 mM glucose with no BAF) which was defined as 1. (C) Quantification of autophagy flux, which is defined as the difference of the LC3-II levels in the absence and presence of BAF (BAF-CON). Data were expressed as the mean ± SD, and analyzed by one-way ANOVA (n = 4).
We also examined the effect of high glucose on autophagy using an alternative approach in which cardiomyocytes were infected with an adenovirus encoding green fluorescent protein (GFP)-LC3, a fusion protein that can incorporate into autophagic vacuoles forming punctate structures or dots. AVs include autophagosomes and autolysosomes. The latter is formed after an autophagosome is fused with a lysosome. As expected, an active basal autophagic flux was detected in the presence of 5.5 mM glucose as shown by increased GFP–LC3 dots with BAF (Fig. 2A, Control 17.7 ± 0.8 vs. BAF 29.8 ± 0.4, n = 3, p < 0.01). However, high glucose (17 or 30 mM) dramatically reduced the number of GFP-LC3 dots either with or without BAF, indicating that high glucose blocked autophagic flux, consistent with the results from western blot analysis. To simultaneously examine the effect of high glucose on the formation of both autophagosomes and autolysosomes, we took advantage of an adenovirus expressing mRFP-GFP tandem fluorescent-tagged LC3 (tfLC3).46 Monomeric RFP (mRFP), but not GFP, can produce fluorescence in the acidic environment of lysosomes. Therefore, the colocalization of GFP and mRFP, which appears yellow in the merged image, indicates autophagosomes, which can also be calculated by subtracting the number of total GFP dots from the number of total mRFP dots. The free mRFP signal that does not overlay with the GFP in the merged image is indicative of autolysosomes.47 As shown in Figure 2B, in cells cultured with 5.5 mM glucose, there were numerous GFP and RFP dots, and many of them are overlaid in the merged images, illustrating a baseline autophagy in which both autophagosomes and autolysosomes were present. However, 17 mM glucose markedly reduced the number of yellow dots but not the number of red dots in the merged image, suggesting that the formation of autophagosomes but not autolysosomes was inhibited by 17 mM glucose. Strikingly, further increasing glucose to 30 mM not only reduced the number of yellow dots but also that of red dots in the merged image, indicating an inhibited formation of both autophagosomes and autolysosomes. The quantification of GFP and mRFP dots in cardiomyocytes under these conditions was presented in the bar graph of Figure 2B. Together, these results provided strong evidence to support the conclusion that high glucose dose dependently inhibits autophagic flux in cardiomyocytes.

Figure 2. High glucose reduced the formation of autophagosome and autolysosome. (A) Cardiomyocytes were infected with AdGFP-LC3 for 24 h and cultured in DMEM with the indicated amounts of glucose in the absence or presence of BAF. GFP-LC3 formed punctate structures or dots indicative of AVs. (B) Cardiomyocytes were infected with an adenovirus expressing mRFP-GFP tandem fluorescent-tagged LC3 (tfLC3), and cultured with different levels of glucose. mRFP, but not GFP, can retain the ability to fluoresce in the acidic environment of lysosomes. Therefore, the colocalization of GFP and mRFP, which appears yellow in merged image, indicates autophagosome, whereas the free mRFP signal that does not overlay with the GFP in the merged image is indicative of autolysosomes. The scale bar represents 20 µm. The numbers of fluorescent puncta (GFP or mRFP dots) were counted manually from at least three independent experiments. At least 20 cells were scored in each experiment. Data in the bar graphs are means ± SD, and analyzed by one-way ANOVA (n = 3~4).
The inhibited autophagy is an adaptive response that promotes cardiomyocyte survival in response to high glucose
Autophagy has been thought to be a survival mechanism in response to various starvation and stress conditions,23 and high glucose is well known to induce cardiomyocyte death,7-14 raising the possibility that the inhibited autophagy may contribute to hyperglycemic toxicity. If this is true, restoring or enhancing autophagy would protect against high glucose-induced cardiomyocyte death, and conversely, inhibiting autophagy would predispose cardiomyocytes to high glucose injury. To test this hypothesis, we manipulated autophagic activity using pharmacological approaches, and then determined if altered autophagic activity would affect high glucose cytotoxicity. Rapamycin (Rap) is an mTOR inhibitor that has been commonly used to induce autophagy, whereas 3-methyladenine (3-MA) inhibits class III PtdIns3K, a positive regulator that forms a complex with several proteins including the autophagy-related protein BECN1 to promote autophagy. Cardiomyocytes were cultured with different levels of glucose for 72 h. Rap or 3-MA was added to the culture dishes in the last 24 h. Cardiomyocyte death was evaluated by propidium iodide (PI) staining, which estimates the number of dead cells regardless of the cause of death. Apoptosis was determined by DNA laddering and the cleavage of caspase 3 and poly(ADP-ribose) polymerase (PARP). As expected, 30 mM glucose led to increased PI-positive cells (18.3 ± 2.94% in 30 mM glucose vs. 2.1 ± 1.05% in 5.5 mM glucose; p < 0.01, n = 4, Fig. 3A and B), enhanced DNA laddering and elevated cleavage of caspase 3 and PARP (Fig. 3C and D). The ability of Rap or 3-MA to activate or inhibit autophagy was confirmed by western blot analysis of p62 protein levels, which were inversely related to autophagic activity (Fig. 3C and D). Of note, neither Rap nor 3-MA had a marked effect on the viability of cardiomyocytes cultured in 5.5 mM glucose. Surprisingly, however, high glucose-induced cell death was exacerbated by Rap and dramatically attenuated by 3-MA as shown by PI-positive cells (18.3 ± 2.94% in 30 mM glucose vs. 31.1 ± 2.90% in 30 mM glucose plus 50nM Rap and 8.22 ± 3.55% in 30 mM glucose plus 2 mM 3-MA, p < 0.01, n = 4), DNA laddering and cleaved caspase 3 and PARP. Although 17 mM glucose per se did not trigger significant cell death, adding Rap to the culture substantially increased PI-positive cells (16.1 ± 5.86% in 17 mM glucose plus Rap, vs. 5.13 ± 2.02% in 17 mM glucose alone, p < 0.01, n = 4). In summary, our results demonstrated that the inhibition of autophagy does not contribute to hyperglycemic cardiotoxicity. Instead, the downregulated autophagy appears to be an adaptive response that promotes cardiomyocyte survival, contrary to our initial hypothesis.

Figure 3. Rapamycin (Rap) enhanced high-glucose-induced cardiomyocyte death, whereas 3-methyladenine (3-MA) attenuated it. Cardiomyocytes were treated with Rap (50 nM) or 3-MA (2 mM) for 24 h under the indicated glucose conditions. Cell death was determined by PI staining (A and B), cleavage of PARP (cPARP) and caspase 3 (cCasp3, C and D), and DNA laddering (C and D). Data in B were expressed as mean ± SD and analyzed by two-way ANOVA (n = 4). The scale bar in A represents 200 µm. The numbers under p62 western blots are fold changes of densities relative to the effect of 5.5 mM glucose (C and D). Numbers are means with no SD presented (n = 4, *p < 0.01 vs. 5.5 mM glucose; #p < 0.01 vs. 30 mM glucose).
To eliminate the potential nonspecific or autophagy-independent effects of Rap, we used adenoviral vectors to deliver a FLAG-tagged human BECN1 cDNA (AdBCN1) or ATG7 cDNA (AdATG7) to genetically achieve gain-of-function of autophagy in cardiomyocytes. Adenovirus expressing β-galactosidase (β-gal) was used as a control. Infection of cardiomyocytes with AdBCN1 led to increased BECN1 protein levels, which resulted in enhanced autophagic flux at baseline, as evidenced by the difference in protein levels of LC3-II in the absence and presence of BAF (β-gal 0.28 ± 0.04 vs. BECN1 0.55 ± 0.10, n = 3, p < 0.01, Fig. 4A). ATG7 overexpression tended to increase autophagic flux at 5.5 mM glucose (n = 3, p = 0.06, Fig. 4B). Regardless, overexpression of BECN1 and ATG7 each was able to overcome high glucose-mediated inhibition of autophagy, bringing autophagic flux back to the control levels in cells cultured in 5.5 mM glucose (lower panels of Fig. 4A and B). Although overexpression of either BECN1 or ATG7 under high glucose conditions did not further stimulate autophagic flux beyond basal levels, it reversed autophagy inhibition and predisposed cardiomyocytes to high glucose-induced cell death as indicated by PI staining (Fig. 4C and D), DNA laddering (Fig. 4E and F) and the cleavage of PARP and caspase 3 (Fig. 4G and H). These results indicated that upregulation of autophagy exaggerates hyperglycemia-induced cardiomyocyte injury, lending further support to the conclusion that inhibition of autophagy is an adaptive response conducive to cardiomyocyte survival under high glucose culture conditions.

Figure 4. Overexpression of BECN1 or ATG7 enhanced high glucose-induced cardiomyocyte death. Cardiomyocytes were infected with AdBCN1 or AdATG7 for 18 h and then cultured under the indicated glucose conditions for 72 h. Adenovirus-mediated overexpression of BECN1 (A) or ATG7 (B) accelerated autophagic flux indicated by the difference of LC3-II levels in the absence and presence of BAF, which predisposed cardiomyocytes to high glucose-induced cell death, as determined by PI staining (C and D), DNA laddering (E and F), and cleavage of PARP and caspase 3 (G and H). Data were expressed as the mean ± SD and were analyzed by two-way ANOVA (n = 4).
By the same token, although results obtained with 3-MA suggested a protective role of inhibited autophagy against high glucose toxicity, it remained possible that the protective effect of 3-MA was mediated through autophagy-independent mechanisms since the specificity of 3-MA is questionable.48 Therefore, we used adenoviral vector to deliver a short hairpin RNA (shRNA) against rat Becn1 mRNA (AdshBCN1) or rat Atg7 mRNA (AdshATG7) to genetically downregulate autophagy in cardiomyocytes. Adenovirus expressing a scrambled short hairpin RNA (shCon) was used as a control. When cells cultured under 5.5 mM glucose, shBCN1 and shATG7 reduced autophagic flux by 70% and 75% (n = 3, p < 0.01, the lower panels of Fig. 5A and B), respectively. Although there was no synergistic inhibition of autophagy between 30 mM glucose and shBCN1 or shATG7 (n = 3, p = n.s.), expression of shBCN1 or shATG7 attenuated high glucose-induced cardiomyocyte death as demonstrated by reduced PI-positive cells (Fig. 5C and D) and DNA laddering (Fig. 5E and F) as well as attenuated cleavage of PARP and caspase 3 (Fig. 5G and H). These results provided more unequivocal evidence to support a beneficial role of inhibited autophagy in limiting high glucose-induced cardiomyocyte injury.

Figure 5. Knockdown of Becn1 or Atg7 alleviated high glucose-induced cardiomyocyte death. Cardiomyocytes were infected with AdshBCN1 or AdshATG7 for 18 h, and then exposed to different amounts of glucose for 72 h. Knocking down Becn1 (A) or Atg7 (B) blocked autophagic flux indicated by the difference of LC3-II levels in the absence and presence of BAF, which prevented high-glucose-induced cell death, as determined by PI staining (C and D), DNA laddering (E and F), and cleavage of PARP and caspase 3 (G and H). Data were expressed as the mean ± SD and analyzed by two-way ANOVA (n = 4).
High glucose-induced activation of mTORC1 mediates autophagy inhibition and cardioprotection
Autophagy activity is tightly controlled by many positive and negative regulators.23,49 The intracellular energy sensor AMP-activated protein kinase (AMPK) is a positive regulator of autophagy. However, high glucose did not have a marked effect on AMPK activity as shown by western blot analysis of the phosphorylation of AMPKα at Thr172 and its downstream effector acetyl-CoA carboxylase (ACC) at Ser79 (Fig. 6), suggesting that AMPK might not be involved in high glucose-mediated autophagy inhibition. Akt and the mTOR complex 1 (mTORC1) are activated by insulin and many metabolic signals. We examined if and how these two pathways respond to high glucose treatment. Whereas Akt signaling was not affected by high glucose, the activity of the mTORC1 pathway was dramatically enhanced by high glucose, as indicated by the increased phosphorylation of mTORC1 downstream effectors including p70 ribosomal S6 subunit kinase (p70S6K), ribosomal S6 protein (S6) and elongation factor 4E binding protein (4EBP) (Fig. 6). Since mTORC1 senses nutrient availability and is well known to negatively regulate autophagy,50 the result suggested that the mTORC1 signaling pathway may be responsible for the suppressed autophagy under hyperglycemic conditions. Indeed, the mTORC1 inhibitor rapamycin antagonized high glucose-induced inhibition of autophagy as indicated by p62 levels (Fig. 3C), which was associated with enhanced cardiomyocyte death in response to high glucose (Fig. 3A–C). To further establish the role of mTOR in the regulation of autophagy and cardiomyocyte survival under hyperglycemic condition, we infected cardiomyocytes with an adenovirus expressing mTOR and then exposed the cells to high levels of glucose. As predicted, adenovirus-mediated overexpression of mTOR was sufficient to block autophagic flux regardless of glucose concentrations, as indicated by the LC3-II levels either with or without BAF (Fig. 7A). In addition, overexpression of mTOR substantially reduced 30 mM glucose-induced cardiomyocyte death, as demonstrated by decreased PI-positive cells (Fig. 7B), DNA laddering (Fig. 7C), and cleavage of caspase 3 and PARP (Fig. 7D). To determine if suppression of autophagy plays a major role in the protective effect of mTOR, we simultaneously overexpressed mTOR and BECN1 in cardiomyocytes. As indicated by the LC3-II levels in the absence and presence of BAF, BECN1 overexpression not only antagonized the inhibitory effect of mTOR on autophagic flux (Fig. 8A) but also the ability of mTOR to reduce 30 mM glucose-triggered cell death (PI-positive cells: β-gal 23.7 ± 1.2, mTOR 16.1 ± 2.2 vs. mTOR+BECN1 27.9 ± 3.2, n = 4, p < 0.01 or 0.05, Fig. 8B). Collectively, these results strongly suggest that the enhanced mTOR signaling is an adaptive response that protects cardiomyocytes against high glucose toxicity through autophagy inhibition.
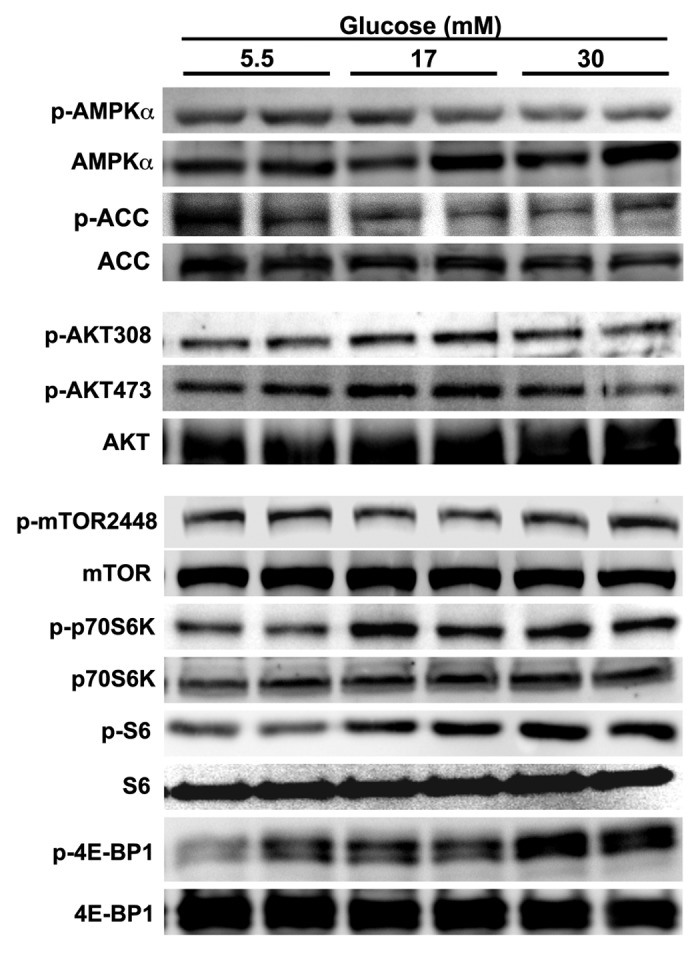
Figure 6. High glucose activated the mTORC1 signaling pathway. Cardiomyocytes were treated with different doses of glucose for 72 h. The protein expression levels and phosphorylation states of the components of several signaling pathways were determined by western blotting analysis.
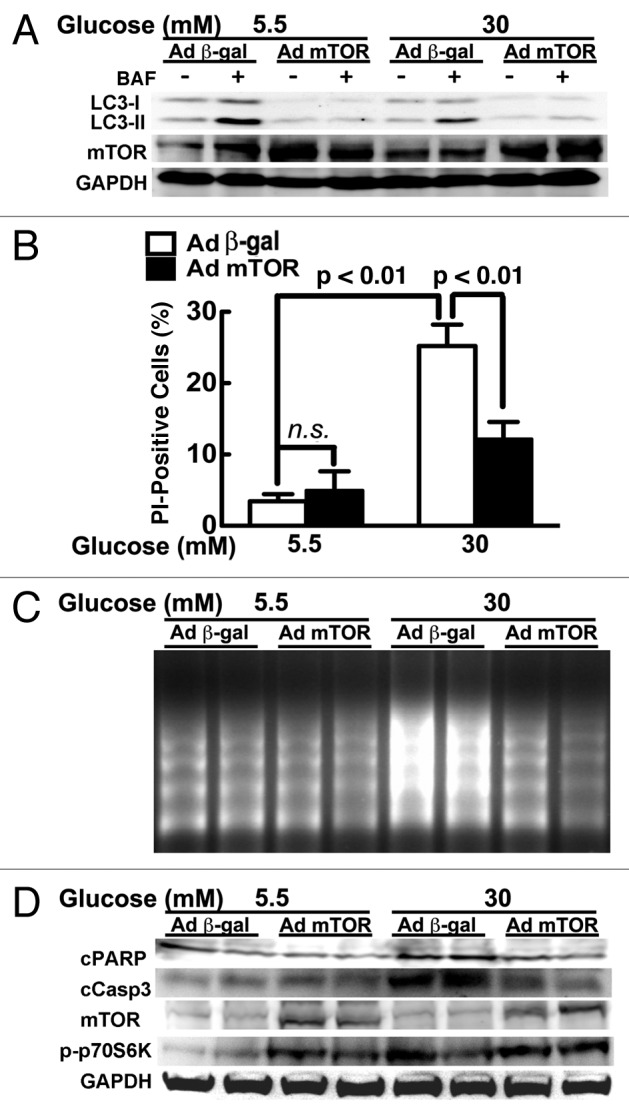
Figure 7. Overexpression of mTOR reduced high glucose cardiotoxicity. Cardiomyocytes were infected with AdmTOR or Ad β-gal and cultured under different glucose conditions. mTOR overexpression blocked autophagic flux as shown by the LC3-II levels (A), and reduced high glucose-induced cell death, as determined by PI staining (B), DNA laddering (C), and cleavage of PARP and caspase 3 (D). Data in (B) were expressed as the mean ± SD and analyzed by two-way ANOVA (n = 4).
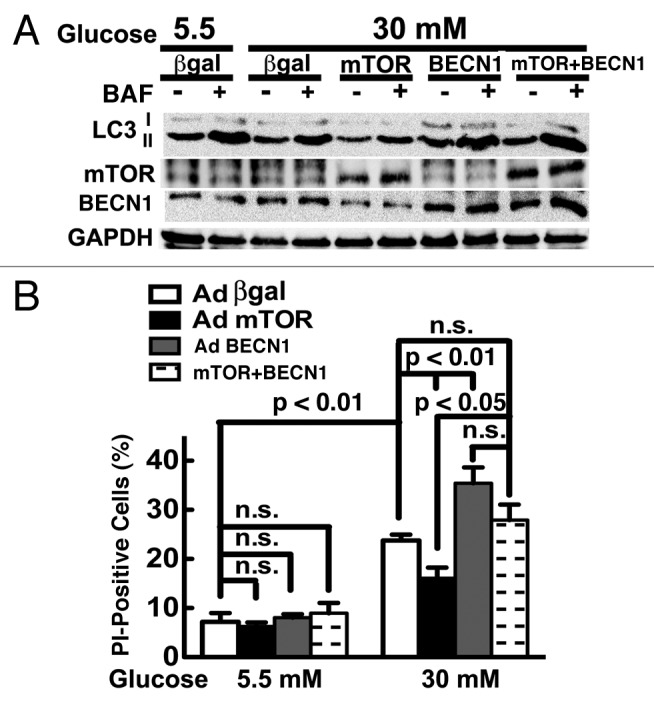
Figure 8. Overexpression of Becn1 diminished the ability of mTOR to reduce high glucose cardiotoxicity. Cardiomyocytes were infected with AdmTOR and/or AdBCN1 for 18 h and then cultured under the indicated glucose conditions for 72 h. (A) Overexpression of Becn1 reversed mTOR-mediated autophagy inhibition under 30 mM glucose as indicated by LC3-II levels in the absence and presence of BAF. (B) Increased BECN1 antagonized the protective effect of mTOR on cardiomyocyte death as determined by PI staining. Data were expressed as mean ± SD and analyzed by two-way ANOVA (n = 4).
mTOR has been shown to inhibit autophagy under nutrient rich conditions by directly phosphorylating and inhibiting unc-51-like kinase 1 (ULK1), a mammalian homolog of yeast Atg1 that forms a complex with at least three other proteins (ATG13, FIP200 and ATG101) to initiate autophagosome formation.29,51,52 Phosphorylation of ULK1 at Ser757 by mTOR was essential for autophagy inhibition.29 However, we did not find that high glucose and mTOR signaling markedly affected ULK1 phosphorylation at Ser757 in cardiomyocytes. Instead, we found that ULK1 phosphorylation at another site, Ser467, was dramatically increased by high glucose (17 or 30 mM) compared with normal control (5.5 mM 1.00 ± 0.08 vs. 17 mM 1.58 ± 0.5 or 30 mM 2.26 ± 0.73, n = 6, p < 0.01, Fig. 9A). Strikingly, inhibition of mTORC1 with rapamycin reduced ULK1 phosphorylation at Ser467 under high glucose conditions (Fig. 9B), revealing a necessary role for mTORC1 in high glucose-induced ULK1 phosphorylation. Conversely, overexpression of mTOR not only was sufficient to induce ULK1 phosphorylation at normal glucose, but also produced synergistic effect with 30 mM glucose (β-gal 1.53 ± 0.23 vs. mTOR 2.02 ± 0.25, n = 6, p < 0.01, Fig. 9C). Together, these results suggested that mTOR may mediate high-glucose-induced autophagy inhibition through its effect on ULK1 phosphorylation at Ser467.

Figure 9. mTORC1 mediated high glucose-induced phosphorylation of ULK1. Cardiomyocytes were cultured under different doses of glucose for 72 h. (A) The phosphorylation of ULK1 at Ser467 was determined by western blot analysis. (B) Rapamycin (Rap) blocked high glucose-induced phosphorylation of ULK1. Rap (50 nM) was added to the culture media 24 h before cells were harvested for western blot analysis. (C) Cardiomyocytes were infected with AdmTOR or Ad-β-gal and then cultured under different glucose conditions for 72 h before western blot analysis. The numbers under phosphor-ULK1 western blots are fold changes of densities relative to the effect of 5.5 mM glucose. Data are means with no SD presented (n = 6, **p < 0.01 vs. 5.5 mM glucose; #p < 0.01 vs. 30 mM glucose).
Autophagy inhibition reduces high glucose toxicity in adult mouse cardiomyocytes
Since neonatal cardiomyocytes behave substantially different from adult cardiomyocytes,53 we repeated the key experiments above in adult cardiomyocytes isolated from 3-mo-old C57/6 mice and exposed to 40 mM glucose for 40 h. We used higher glucose and shorter exposure time because adult mouse cardiomyocytes can only be cultured for 2 d without losing their viability. As shown in Figure 10A, high glucose inhibited autophagic flux (5.5 mM 0.24 ± 0.01 vs. 40 mM 0.12 ± 0.02, n = 4, p < 0.01), which was reversed by rapamycin (Con 0.12 ± 0.02 vs. Rap 0.28 ± 0.07, n = 3, p < 0.01). In contrast, high glucose-mediated inhibition of autophagy was exaggerated by 3-MA (Con 0.17 ± 0.02 vs. 3-MA 0.08 ± 0.04, n = 4, p < 0.05, Fig. 10B). Importantly, high glucose-induced cell death as indicated by PI staining was markedly exacerbated by rapamycin but inhibited by 3-MA (Con 37.7 ± 4.5 vs. Rap 52.1 ± 5.1 and 3-MA 21.9 ± 2.5, n = 4, p < 0.05 or 0.01, Fig. 10C), suggesting that autophagy inhibition was protective in adult cardiomyocytes under high glucose. We also isolated cardiomyocytes from adult Becn1-overexpressing transgenic mice (Becn1 tg) and Becn1-deficient mice (Becn1+/−). Although overexpression of Becn1 did not markedly affect autophagic flux at baseline when cells were cultured under 5.5 mM glucose, it reversed 40 mM glucose-induced autophagy inhibition as shown by LC3-II levels with and without BAF (Fig. 11A), which was accompanied by enhanced cardiomyocyte death (PI-positive cells: wild-type 41.0 ± 3.3 vs. transgenic 58.0 ± 6.1, n = 4, p < 0.01, Fig. 11C). On the contrary, deletion of one copy of the Becn1 gene (Becn1+/−) reduced autophagic activity regardless of glucose concentrations (Fig. 11B). Notably, high glucose toxicity in wild-type (WT) cardiomyocytes was markedly reduced in Becn1+/− cells as determined by PI staining (WT 45.2 ± 5.9 vs. Becn1+/− 32.3 ± 2.5, n = 4, p < 0.01, Fig. 11D). Together, these data confirmed the results from neonatal rat cardiomyocytes, suggesting that autophagy inhibition is a beneficial adaptive response that limits high glucose cardiotoxicity.

Figure 10. Rapamycin (Rap) enhanced high glucose toxic effect on adult mouse cardiomyocytes, whereas 3-methyladenine (3-MA) attenuated it. Cardiomyocytes were isolated from adult C57B/6 mice, cultured under the indicated glucose conditions for 24 h, and then treated with Rap (50 nM) or 3-MA (1 mM) for another 16 h. Rap reversed 40 mM glucose-mediated autophagy inhibition (A), while 3-MA further reduced autophagic flux under 40 mM glucose (B), as quantified by the difference of LC3-II levels in the absence and presence of BAF. Correspondingly, high-glucose-induced cardiomyocyte death was exacerbated by Rap but attenuated by 3-MA as shown by PI staining (C). Data were expressed as mean ± SD and analyzed by two-way ANOVA (n = 4). The scale bar in (C) represents 200 µm.

Figure 11. Overexpression of Becn1 enhanced high glucose toxicity in adult mouse cardiomyocytes, whereas heterozygous knockout of Becn1 attenuated it. Cardiomyocytes were isolated from adult becn1 transgenic mice (BCN TG) or heterozygous Becn1 knockout mice (BCN+/−) and cultured under the indicated glucose conditions for 40 h. Autophagic flux was determined by western blot analysis of LC3-II levels with and without BAF in BCN TG cardiomyocytes (A) and BCN+/− (B) cardiomyocytes. Cardiomyocyte death was determined by PI staining (C and D). Data were expressed as mean ± SD and analyzed by two-way ANOVA (n = 4). The scale bar in (C and D) represents 200 µm.
Discussion
Uncontrolled hyperglycemia is intimately linked to increased heart failure among diabetic patients.4-6 Oxidative stress has been proposed as a major mechanism for hyperglycemia-induced diabetic cardiac damage.3 However, the failure of antioxidants to reduce diabetic cardiomyopathy in human patients17,18 has prompted the need to identify other molecular and cellular mechanisms that may contribute to hyperglycemic cardiotoxicity. In the present study, we demonstrated that high glucose was able to directly inhibit autophagic flux in neonatal rat cardiomyocytes, which appeared to be an adaptive response that functioned to limit high glucose-induce cardiomyocyte injury. This conclusion was supported by results obtained from experiments using both pharmacological and genetic gain- and loss-of-function approaches. Specifically, suppression of autophagy by either chemical inhibitor 3-MA or shRNA-mediated silencing of the Becn1 or Atg7 genes attenuated high glucose-induced cardiomyocyte death. Reversely, upregulation of autophagy with rapamycin or overexpression of Becn1 or Atg7 predisposed cardiomyocytes to high-glucose toxicity. The key results obtained from neonatal cardiomyocytes were confirmed in adult mouse cardiomyocytes. Overall, these observations suggest that inhibiting autophagy may be a potential therapeutic strategy for reducing hyperglycemic cardiotoxicity.
Autophagy is a conserved catabolic pathway in which cytoplasm, including damaged or aged organelles and long-lived protein or protein aggregates, is sequestered into double-membraned autophagosomes for degradation and recycling by lysosomes. Autophagy has received increasing attention due to its important role in various aspects of cell physiology, especially cell survival during nutrient or energy limitation. Given that glucose deprivation was able to induce autophagy,26 it is not surprising that high glucose, a condition of nutrient excess, would inhibit autophagy in cardiomyocytes. As autophagy protected against diabetes-induced oxidative injury in pancreatic β-cells,31 it was somewhat unexpected that the inhibited autophagy did not contribute to high glucose toxicity in the present study. Instead, this adaptive response constituted part of the mechanism that promotes cardiomyocyte survival during high glucose exposure. Supporting our results, glucose infusion reduced autophagy in the liver, which was accompanied by attenuated systemic inflammation induced by LPS.54 Together, these results underscore a well known fact that autophagy could be either protective or detrimental depending on the cell type and cellular environment.24-27 Thus, the functional significance of autophagy under different pathological conditions has to be individually determined.
The direct effect of high glucose on cultured cardiomyocytes may not be necessarily extrapolated to diabetic heart in vivo due to more complex metabolic changes and intricate regulatory mechanisms that operate at the whole animal level. A recent study demonstrated an inhibited autophagy in type 1 diabetic mouse heart, which is associated with cardiac dysfunction.38 Strikingly, metformin treatment not only improves cardiac function but also restores autophagy, suggesting a potentially beneficial role for autophagy in the diabetic heart,38 despite the fact that it remains to be determined whether or not the cardioprotective effect of metformin is mediated through enhanced autophagy. In contrast, another study found that cardiac autophagy is increased in mice fed a high-fructose diet that developed insulin resistance and hyperglycemia, two hallmark features of type 2 diabetes.39 The upregulated autophagy is associated with elevated cardiac superoxide production and fibrosis, raising the possibility that autophagy may contribute to diabetic cardiomyopathy in this type 2 diabetic model.39 Jointly, these two studies suggest that autophagy is differentially regulated in type 1 and type 2 diabetic hearts, and the functional roles of autophagy in diabetic cardiac injury may be fundamentally different between type 1 and type 2 diabetes. Interestingly, our present study showed that high glucose directly inhibited autophagy in cardiomyocytes, consistent with type 1,38 but not type 2, diabetes.39 This may suggest that the predominant metabolic defect in type 1 diabetes is hyperglycemia, which inhibits autophagy; whereas the enhanced autophagy in type 2 diabetic heart is a net effect of insulin resistance, hyperglycemia, and other metabolic abnormalities such as hyperlipidemia. Also, our results demonstrated a detrimental role for autophagy in high-glucose-induced cardiomyocyte death, which is in line with the suggestion from type 2 diabetes39 but not type 1 diabetes.38 Since these two studies only showed an association between altered autophagy and diabetic cardiac injury, the exact functional role of autophagy in either type 1 or type 2 diabetic hearts requires further investigation, desirably using both gain- and loss-of-function animal models. Nevertheless, based on our cell culture study, high glucose is able to directly inhibit autophagy, which attenuates cardiomyocyte injury triggered by high glucose itself.
As a nutrient-sensing kinase, mTORC1 is stimulated by high nutrient levels, which then coordinately activate anabolic processes such as protein synthesis, and inhibit the cellular catabolic pathways such as autophagy. It is thus not surprising that our data suggested that mTORC1 mediates the ability of high glucose to inhibit autophagy. The evidence for this is three-fold. First, high glucose activates mTORC1 as indicated by the increased phosphorylation of mTORC1 substrates p70S6K, S6 and 4EBP1 (Fig. 6), consistent with previous studies in both non-cardiomyocyte55,56 and adult rat cardiomyocyte.57 It is proposed that increased glucose content accelerates glycolytic flux and suppresses the interaction between GAPDH and RHEB, which allows RHEB to activate mTORC1.58 Second, the mTORC1 inhibitor rapamycin attenuated the inhibitory effect of high glucose on autophagy as indicated by p62 levels (Fig. 3C), suggesting that mTORC1 is required for high glucose to downregulate autophagy. Lastly, overexpression of mTOR was sufficient to block autophagic flux regardless of glucose concentrations, as indicated by the LC3-II levels either with or without BAF (Fig. 7A). Together, these results demonstrated that mTOR signaling is both sufficient and necessary for inhibiting autophagy in cardiomyocytes cultured under high-glucose conditions.
Recent studies have provided some insight into the mechanisms by which mTOR inhibits autophagy. mTOR has been proposed to negatively regulate p73, a member of the p53 family of transcription factors that induces autophagy and autophagy-related genes.59,60 However, the mode and the functional significance of this regulation remain unclear. More recently, another novel mechanism that mediates mTOR inhibition of autophagy has been characterized that hinges on ULK1, an autophagy initiating kinase that acts as a nodal point for regulation by multiple kinases. Specifically, under nutrient sufficient condition, mTORC1 phosphorylates ULK1 at Ser757, which disrupts the interaction between ULK1 and AMPK, preventing ULK1 phosphorylation by AMPK at Ser317 and Ser777, thereby inhibiting ULK1 and autophagosome formation.29 Not surprisingly, additional phosphorylation sites on ULK1 by either mTOR or AMPK have been reported, which may be equally important for regulating ULK1 and autophagic activity. For example, mTOR normally phosphorylates ULK1 at Ser638 and Ser758 in nutrient-rich media, whereas ULK1 undergoes dramatic dephosphorylation upon nutrient starvation.61 Also, AMPK is activated under nutrient starvation conditions to induce autophagy by directly phosphorylating ULK1 at Ser555.62,63 Of note, although phosphorylation of ULK1 at Ser467 is induced by the AMPK activator phenformin or expression of ULK1 with a constitutively active AMPKα1, it is not clear if the phosphorylation of endogenous ULK1 at Ser467 is mediated by AMPK.63 Thus, the kinase that phosphorylates Ser467 and the functional significance of this phosphorylation remain to be determined. Interestingly, in the present study, we found that endogenous ULK1 in cardiomyocytes was highly phosphorylated by high glucose treatment, and this effect was largely abolished by rapamycin, suggesting a necessary role for mTOR in high glucose-induced ULK1 phosphorylation at Ser467 (Fig. 8B). Since high glucose did not markedly affect the activity of AMPK (Fig. 6), it is unlikely that the increased ULK1 phosphorylation at Ser467 was mediated by AMPK. Moreover, overexpression of mTOR not only was sufficient to induce ULK1 phosphorylation on Ser467 in normal glucose, but also produced synergistic effect with high glucose (Fig. 9C). These results suggested that mTOR is both necessary and sufficient to phosphorylate ULK1 at Ser467, which may mediate high glucose-induced autophagy inhibition.
In conclusion, we showed that high glucose had a direct inhibitory effect on autophagy in cardiomyocytes, which was an adaptive response triggered by high glucose to attenuate cardiomyocyte injury. Mechanistically, the inhibition of autophagy was mediated, at least in part, by increased mTOR signaling that likely inactivated ULK1 through phosphorylation of Ser467. These findings suggested that a potential therapeutic strategy for reducing hyperglycemic cardiotoxicity is to enhance mTOR signaling thereby limiting autophagic activity and thus cardiomyocyte death. One limitation of this study is that the autophagic flux and other parameters were determined in the surviving cells, the number of which differs between different experiments. This may lead to misinterpretation or inappropriate inferences. Whether our conclusion holds true in either type 1 or type 2 diabetes requires further investigation using diabetic animal models with both gain- and loss-of-function of mTOR or autophagy in the heart.
Materials and Methods
Neonatal rat ventricular cardiomyocyte culture and high glucose treatment
Neonatal rat ventricular cardiomyocytes were isolated from 0- to 2-d old Harlan Sprague-Dawley rat neonates using a kit from Worthington Biochemical Corporation (LK003300) as described previously.12 After tissue digestion, cells were preplated for 1 h to remove non-myocytes and then plated on gelatinized dishes and cultured overnight in Dulbecco’s modified essential medium (DMEM; Cellgro, 10-017CV) with 15% bovine serum. The following day, the cells were washed in phosphate-buffered saline and cultured in serum-free DMEM containing 100 μM of 5-bromo 2′-deoxy-uridine (BrdU; Sigma, B5002). For high glucose studies, cardiomyocytes were cultured for 72 h in glucose-free DMEM (GIBCO, 11966) supplemented with 5.5, 17, or 30 mmol/liter of glucose (Sigma, G7021). The osmolarities of all media were made equal to 30 mM by adding different amounts of mannitol (Sigma, M9647). All media contained 100 units/ml penicillin and streptomycin (Sigma, P4333).
Adult mouse cardiomyocyte culture
C57BL/6J mice, Becn1 transgenic mice, and Becn1 knockout mice were used for isolating adult cardiomyocytes. The human BECN1 cDNA was cloned behind a modified α-myosin heavy chain (α-MHC) promoter that contains nucleotide binding sites for tetracycline-controlled transactivator (tTA).64 To induce transgene expression, Becn1 transgenic mice were crossed with mice that expressed tTA in the heart.64 Mice harboring both BECN1 and tTA transgenes were referred to as BECN1 TG in this study, and mice having a single tTA transgene were used as the wild-type (WT) control. Becn1 heterozygous knockout (+/−) mice were provided by Dr. Zhenyu Yue.65 We used 10- to 15-week old mice. All protocols involving animal use were approved by the Institutional Animal Care and Use Committee at Sanford Research/University of South Dakota. This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US. National Institutes of Health (NIH Publication No. 85-23, revised 1996). Ventricular cardiomyocytes from adult mice were isolated as previously described.66,67 Briefly, hearts were removed, cannulated, and perfused with collagenase type II (Worthington Biochemical, LS004176) to dissociate ventricular myocytes. Cardiomyocytes were plated at a density of 50 rod-shaped myocytes per square millimeter on laminin-coated 6-well culture plates. Cardiomyocytes were cultured for 40 h in a 2% CO2 incubator at 37°C with 5.5 or 40 mM glucose supplemented Minimum Essential Medium (MEM: GIBCO, 11575).
Antibodies and reagents
The antibodies used in the study were purchased from the following vendors. Cell Signaling Technology: ATG7 (2631), ATG12 (2010), AKT (9272), AMPKα (2603), acetyl-CoA Carboxylase (ACC, 3662), Beclin1 (3738), cleaved caspase 3 (9661), LC3B (2775), mTOR (2972), p70 S6 Kinase (S6K, 9202), PARP (9542), ULK1 (R600, 4773), 4EBP (9644), phospho-4EBP (Ser65, 9451), phospho-AKT (Thr308, 9275), phospho-AKT (Ser473, 4058), phospho-AMPKα (Thr172, 2535), phospho-ACC (Ser79, 3661), phospho-mTOR (Ser2448, 2971), phospho-p70 S6K (Thr389, 9205), S6 Ribosomal Protein (S6, 2217) phospho-S6 (Ser240/244, 2215), and phospho-ULK1 (Ser467, 4634). Research Diagnostics: Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 10R-G109a). PROGEN: Anti-p62 c-terminus/SQSTM1 antibody (GP-62C). Rapamycin (Sigma, R0395) was dissolved in absolute ethanol (Fisher Scientific, AC61508), 3-methyladenine (Sigma, M9281) in DMEM (GIBCO, 11966), and Bafilomycin A1 (LC Laboratories, B-1080) in dimethyl sulfoxide (DMSO; Sigma, 472301). Each drug was added to the medium to final concentration as described in each experiment.
Western blot analysis
Protein extracts from cultured cardiomyocytes were prepared as described previously, except that 1% of protease inhibitor cocktail (Sigma, P8340) was added to the extraction buffer.68 Protein samples were subjected to PAGE, transferred to polyvinylidene difluoride membrane (Amersham Pharmacia, RPN3031 or Bio-Rad, 162-0177), and then blocked in 5% milk prepared with 0.1% Tween-20 Tris-buffered saline (TBST) solution for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C in 2.5% milk. The membrane was incubated with an appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotech, sc-2004, sc-2005, sc-2020, and sc-2438) for 1 h at room temperature in 2.5% milk and processed for chemiluminescent detection using ECL Advanced Western Blotting Kit (Amersham Biosciences UK Limited, RPN2135).
Replication-deficient adenoviruses
Adenovirus encoding GFP-LC3 and adenovirus expressing a short hairpin (sh) RNA directed against Atg7 were kindly provided by Dr. Tolkovsky69 and Dr. Kinya Otsu,43 respectively. Adenoviruses expressing β-galactosidase (Ad β-gal), human BECN1 (AdBCN1), the shRNA targeting rat BECN1 mRNA (AdshBCN1), and control shRNA (AdshCon) were created as previously described.68 Adenovirus expressing mTOR and adenovirus encoding mRFP-GFP tandem fluorescent-tagged LC3 (AdtfLC3) were the gifts of Dr. Chris Rhodes56 and Dr. Junichi Sadoshima,46 respectively. To generate adenoviral vector expressing ATG7, an insert containing Atg7 cDNA sequence was amplified by PCR from myc- and flag-tagged ORF clone of Mus musculus Atg7 (OriGene, MR210108) and integrated into the TA cloning vector (Invitrogen, KNM2000-01). The insert was then released from the TA vector and subcloned into the pShuttle-CMV vector via the BamHI-BglII and SalI restriction enzyme sites. Both BamHI and BglII sites were destroyed after compatible ligation. Recombinant adenoviruses expressing ATG7 were then generated and amplified using the AdEasy Adenoviral Vector System (Stratagene, 240009). Unless otherwise indicated, neonatal rat cardiomyocytes were infected with adenovirus at a multiplicity of infection (MOI) of 100 plaque forming unit (pfu) for 18 h before treatment with different doses of glucose.
Propidium iodide staining
Cardiomyocyte death was measured by staining with propidium iodide (PI, 1 μg/ml, Sigma 81845), which can penetrate cell membranes of dying or dead cells and intercalate into double-stranded nucleic acids. PI was added directly to the culture medium, and cardiomyocytes were examined and photographed with a fluorescence microscope (OLYMUS-IX71) under both phase contrast and fluorescent conditions.
DNA ladder assay
Cardiomyocyte apoptosis was measured by PCR-based DNA ladder Assay following the instruction manual of DNA Ladder Assay PCR kit (Maxim Biotech Inc., APO-DNA1).
Confocal microscopy for GFP-LC3 and mRFP-LC3
Cardiomyocytes in 2-well chamber slides were infected with AdGFP-LC3 or AdtfLC3, and then cultured in DMEM with different amounts of glucose for 72 h. Cells were fixed with 4% paraformaldehyde prepared in PBS for 10 min at room temperature. The slides were covered with coverslips and then observed with a laser scanning confocal microscope (OLYMPUS FV-1000). The confocal images were captured at 600× magnification to demonstrate the formation of GFP-LC3 and/or RFP-LC3 puncta or dots, indicative of autophagosomes and/or autolysosomes. The number of GFP and mRFP dots was determined as described.46 The numbers of fluorescent puncta were counted manually from at least three independent experiments. At least 20 cells were scored in each experiment.
Statistical analysis
Quantitative data were presented as the means ± SD. Differences between experimental groups were examined by one-way or two-way analysis of variance (ANOVA) followed by the Bonferroni post-test using Prism software (GraphPad). For all analysis, p < 0.05 were considered statistically significant.
Acknowledgments
This study was supported by a Career Development Grant (1-09-CD-09) from the American Diabetes Association. S.K. is supported by an Advanced Postdoctoral Fellowship from the Juvenile Diabetes Research Foundation (JDRF). The authors would like to thank Andy Cypher in Cell Culture Core Facility at the Sanford Research/USD for providing cultured neonatal rat and adult mouse cardiomyocytes, and Tricia Krueger and Yuan Huang in Virus Core Facility for preparing several adenoviruses used in this work. We also thank Imaging Core Facility for assistance in confocal microscopy. These core facilities are supported by NIH grant 2P20RR017662-06A1. We thank Dr. Zhenyu Yue for providing beclin 1 knockout mice and Dr. Jeff Robbins for providing the attenuated αMHC promoter and tTA expressing transgenic mice. We are grateful to Dr. Junichi Sadoshima, Dr. Aviva Tolkovsky, Dr. Kinya Otsu and Dr. Chris Rhodes for providing the adenoviruses expressing tfLC3, GFP-LC3, shATG7 and mTOR, respectively.
Glossary
Abbreviations:
- DMEM
Dulbecco’s modified essential medium
- 3-MA
3-methyladenine
- BAF
bafilomycin A1
- GFP
green fluorescent protein
- AVs
autophagic vacuoles
- LC3
microtubule-associated protein 1 light chain 3
- mRFP
monomeric red fluorescent protein
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- BECN1
Beclin 1
- ULK1
unc-51-like kinase 1
- PI
propidium iodide
- Rap
rapamycin
- AMPK
AMP-activated protein kinase
- ACC
acetyl-CoA carboxylase
- S6K
p70 ribosomal protein S6 kinase
- 4EBP
elongation factor 4E binding protein
- PARP
Poly (ADP-ribose) polymerase
- shRNA
short hairpin RNA
- β-gal
β-galactosidase
Declaration of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/18980
References
- 1.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405–12. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 3.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- 4.Iribarren C, Karter AJ, Go AS, Ferrara A, Liu JY, Sidney S, et al. Glycemic control and heart failure among adult patients with diabetes. Circulation. 2001;103:2668–73. doi: 10.1161/01.cir.103.22.2668. [DOI] [PubMed] [Google Scholar]
- 5.Bhatia V, Wilding GE, Dhindsa G, Bhatia R, Garg RK, Bonner AJ, et al. Association of poor glycemic control with prolonged hospital stay in patients with diabetes admitted with exacerbation of congestive heart failure. Endocr Pract. 2004;10:467–71. doi: 10.4158/EP.10.6.467. [DOI] [PubMed] [Google Scholar]
- 6.van Melle JP, Bot M, de Jonge P, de Boer RA, van Veldhuisen DJ, Whooley MA. Diabetes, glycemic control, and new-onset heart failure in patients with stable coronary artery disease: data from the heart and soul study. Diabetes Care. 2010;33:2084–9. doi: 10.2337/dc10-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–48. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 8.Fiordaliso F, Bianchi R, Staszewsky L, Cuccovillo I, Doni M, Laragione T, et al. Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats. J Mol Cell Cardiol. 2004;37:959–68. doi: 10.1016/j.yjmcc.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Malhotra A, Kang BP, Hashmi S, Meggs LG. PKCepsilon inhibits the hyperglycemia-induced apoptosis signal in adult rat ventricular myocytes. Mol Cell Biochem. 2005;268:169–73. doi: 10.1007/s11010-005-3858-6. [DOI] [PubMed] [Google Scholar]
- 10.Shizukuda Y, Reyland ME, Buttrick PM. Protein kinase C-delta modulates apoptosis induced by hyperglycemia in adult ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H1625–34. doi: 10.1152/ajpheart.00783.2001. [DOI] [PubMed] [Google Scholar]
- 11.Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal-Ginard B, et al. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes. 2001;50:2363–75. doi: 10.2337/diabetes.50.10.2363. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi S, Mao K, Zheng H, Wang X, Patterson C, O'Connell TD, et al. Diminished GATA4 protein levels contribute to hyperglycemia-induced cardiomyocyte injury. J Biol Chem. 2007;282:21945–52. doi: 10.1074/jbc.M703048200. [DOI] [PubMed] [Google Scholar]
- 13.Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341–51. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. 2011;14:425–37. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–43. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 16.Liang Q, Carlson EC, Donthi RV, Kralik PM, Shen X, Epstein PN. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes. 2002;51:174–81. doi: 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- 17.Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care. 2002;25:1919–27. doi: 10.2337/diacare.25.11.1919. [DOI] [PubMed] [Google Scholar]
- 18.Sacco M, Pellegrini F, Roncaglioni MC, Avanzini F, Tognoni G, Nicolucci A. Primary prevention of cardiovascular events with low-dose aspirin and vitamin E in type 2 diabetic patients: results of the Primary Prevention Project (PPP) trial. Diabetes Care. 2003;26:3264–72. doi: 10.2337/diacare.26.12.3264. [DOI] [PubMed] [Google Scholar]
- 19.Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–65. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Takagi H, Matsui Y, Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal. 2007;9:1373–81. doi: 10.1089/ars.2007.1689. [DOI] [PubMed] [Google Scholar]
- 21.Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51:584–93. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20:445–62. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–41. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 24.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 25.Kang C, Avery L. To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation. Autophagy. 2008;4:82–4. doi: 10.4161/auto.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 27.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–75. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 28.Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med. 2006;27:411–25. doi: 10.1016/j.mam.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meijer AJ, Codogno P. Macroautophagy: protector in the diabetes drama? Autophagy. 2007;3:523–6. doi: 10.4161/auto.4449. [DOI] [PubMed] [Google Scholar]
- 31.Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-Protein Aggregates Form in Pancreatic {beta}-Cells During Diabetes-Induced Oxidative Stress and Are Regulated by Autophagy. Diabetes. 2007;56:930–9. doi: 10.2337/db06-1160. [DOI] [PubMed] [Google Scholar]
- 32.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–24. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Hur KY, Jung HS, Lee MS. Role of autophagy in beta-cell function and mass. Diabetes Obes Metab. 2010;12(Suppl 2):20–6. doi: 10.1111/j.1463-1326.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- 34.Amherdt M, Harris V, Renold AE, Orci L, Unger RH. Hepatic autography in uncontrolled experimental diabetes and its relationships to insulin and glucagon. J Clin Invest. 1974;54:188–93. doi: 10.1172/JCI107742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagiwara S, Iwasaka H, Koga H, Hasegawa A, Kudo K, Kusaka J, et al. Stimulation of autophagy in the liver by lipopolysaccharide-induced systemic inflammation in a rat model of diabetes mellitus. Biomed Res. 2010;31:263–71. doi: 10.2220/biomedres.31.263. [DOI] [PubMed] [Google Scholar]
- 36.Han K, Lehringer-Polzin M, Zhou H, Pfeifer U. Cellular autophagy in proximal tubules of early diabetic rats following insulin treatment and islet transplantation. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61:367–73. doi: 10.1007/BF02890440. [DOI] [PubMed] [Google Scholar]
- 37.Han K, Zhou H, Pfeifer U. Inhibition and restimulation by insulin of cellular autophagy in distal tubular cells of the kidney in early diabetic rats. Kidney Blood Press Res. 1997;20:258–63. doi: 10.1159/000174155. [DOI] [PubMed] [Google Scholar]
- 38.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–8. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50:1035–43. doi: 10.1016/j.yjmcc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Mellor KM, Reichelt ME, Delbridge LM. Autophagy anomalies in the diabetic myocardium. Autophagy. 2011;7:1263–7. doi: 10.4161/auto.7.10.17148. [DOI] [PubMed] [Google Scholar]
- 41.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–18. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 43.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 44.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 45.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 46.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–82. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 48.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 50.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 51.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 53.Rothen-Rutishauser BM, Ehler E, Perriard E, Messerli JM, Perriard JC. Different behaviour of the non-sarcomeric cytoskeleton in neonatal and adult rat cardiomyocytes. J Mol Cell Cardiol. 1998;30:19–31. doi: 10.1006/jmcc.1997.0596. [DOI] [PubMed] [Google Scholar]
- 54.Hagiwara S, Iwasaka H, Hasegawa A, Kudo K, Kusaka J, Oyama Y, et al. Infusion of a Glucose Solution Reduces Autophagy in the Liver after LPS-induced Systemic Inflammation. Inflammation. 2012;35:249–58. doi: 10.1007/s10753-011-9311-y. [DOI] [PubMed] [Google Scholar]
- 55.Mariappan MM, Feliers D, Mummidi S, Choudhury GG, Kasinath BS. High glucose, high insulin, and their combination rapidly induce laminin-beta1 synthesis by regulation of mRNA translation in renal epithelial cells. Diabetes. 2007;56:476–85. doi: 10.2337/db05-1334. [DOI] [PubMed] [Google Scholar]
- 56.Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem. 2005;280:2282–93. doi: 10.1074/jbc.M412179200. [DOI] [PubMed] [Google Scholar]
- 57.Li SY, Fang CX, Aberle NS, 2nd, Ren BH, Ceylan-Isik AF, Ren J. Inhibition of PI-3 kinase/Akt/mTOR, but not calcineurin signaling, reverses insulin-like growth factor I-induced protection against glucose toxicity in cardiomyocyte contractile function. J Endocrinol. 2005;186:491–503. doi: 10.1677/joe.1.06168. [DOI] [PubMed] [Google Scholar]
- 58.Tsai IC, Liang KW, Lee WL. Penumbra imaging as a prognostic indicator for acute myocardial infarction using MDCT. AJR Am J Roentgenol. 2009;192:W75–6. doi: 10.2214/AJR.08.1458. [DOI] [PubMed] [Google Scholar]
- 59.Rosenbluth JM, Pietenpol JA. mTOR regulates autophagy-associated genes downstream of p73. Autophagy. 2009;5:114–6. doi: 10.4161/auto.5.1.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenbluth JM, Mays DJ, Jiang A, Shyr Y, Pietenpol JA. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci USA. 2011;108:2076–81. doi: 10.1073/pnas.1011936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA. 2011;108:4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440:283–91. doi: 10.1042/BJ20101894. [DOI] [PubMed] [Google Scholar]
- 63.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–16. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 65.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, et al. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007;115:763–72. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- 67.O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–96. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. 2010;285:793–804. doi: 10.1074/jbc.M109.070037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]