

Abstract
Objective
Perihematomal edema results from disruption of the blood-brain barrier (BBB) by key mediators, such as thrombin, following intracerebral hemorrhage (ICH). Platelet derived growth factor receptor alpha (PDGFR-α), a tyrosine kinase receptor, was found in previous studies to play a role in orchestrating BBB impairment. In the present study, we investigated the role of PDGFR-α following ICH-induced brain injury in mice, specifically investigating its effect on BBB disruption.
Methods
Brain injury was induced by autologous arterial blood (30 μl) or thrombin (5 U)-injection into mice brains. A PDGFR antagonist (Gleevec) or agonist (PDGF-AA) was administered following ICH. PDGF-AA was injected with a thrombin inhibitor, hirudin in ICH mice. Thrombin-injected mice were given Gleevec or PDGF-AA neutralizing antibody. A p38 MAPK inhibitor, SB203580 was delivered with PDGF-AA in naïve animals. Post-assessment included neurological function tests, brain edema measurement, Evans blue extravasation, immunoprecipitation, western blot and immunohistology assay.
Results
PDGFR-α suppression prevented neurological deficits, brain edema and Evans blue extravasation at 24–72 hours following ICH. PDGFR-α activation led to BBB impairment and this was reversed by SB203580 in naïve mice. Thrombin inhibition suppressed PDGFR-α activation and exogenous PDGF-AA increased PDGFR-α activation, regardless of thrombin inhibition. Animals receiving a PDGF-AA neutralizing antibody or Gleevec showed minimized thrombin injection-induced BBB impairment.
Interpretation
PDGFR-α signaling may contribute to BBB impairment via p38 MAPK mediated MMP activation/expression following ICH and thrombin may be the key upstream orchestrator. The therapeutic interventions targeting the PDGFR-α signaling may be a novel strategy to prevent thrombin-induced BBB impairment following ICH.
Introduction
Spontaneous intracerebral hemorrhage (ICH) is the result of small vessel bleeds within the brain parenchyma and the subsequent formation and expansion of the hematoma. This process represents the deadliest and least treatable stroke subtype, accounting for close to 15–20% of all strokes 1. One of the main reasons for its devastating nature is the formation of perihematomal cerebral edema, a consequence that occurs from disruption of the blood-brain barrier (BBB). To this date, many factors have been implicated in orchestrating the disruption including thrombin, inflammatory mediators, hemoglobin degradation products 2, and matrix metalloproteinases (MMPs) 3. Yet the mechanism to explain how the process is carried out still remains to be elucidated.
Platelet derived growth factor receptors (PDGFRs) are a subfamily of tyrosine kinase receptors including two members, PDGFR-α and PDGFR-β, expressed throughout various cell-types in the brain, including astrocytes, neurons 4, and capillary endothelial cells 5. These receptors have extracellular domains which ligands, platelet derived growth factors (PDGFs) can bind to initiate downstream signaling pathways. Recently, several lines of evidence have suggested that PDGFRs, especially PDGFR-α may be involved in the stroke process, specifically orchestrating the disruption of the BBB 6–7. In one study the authors observed that PDGFR-α agonists injection into the CSF of naïve mice significantly increased Evans blue extravasation compared to just PBS injected animals 6.
As a result in the present study, we investigated the role of the PDGFR-α following an ICH-induced brain injury in mice, specifically investigating its position as a key orchestrator of BBB disruption. We hypothesize that PDGFR-α signal may contribute to BBB impairment via a p38 MAPK pathway mediated MMPs activation/expression following ICH injury and thrombin, an established mediator of BBB injury in ICH, may be the upstream regulator of PDGFR-α activation. In order to test this aim, first we investigated the expression of PDGFR-α and its ligand, PDGF-AA in brain following ICH. We next used both a PDGFR antagonist (Gleevec) and agonist (PDGF-AA) to manipulate PDGFR-α activation, and measured the phosphorylation level of the PDGFR-α while observing the pre-determined outcomes. We also gave a p38 MAPK inhibitor known as SB 203580 hydrochloride, to potentially reverse the BBB disruption induced by PDGFR-α activation. Because of our hypothesis that thrombin may act as the key upstream orchestrator, hirudin, a thrombin specific inhibitor was also administered into animals with or without PDGFR-α agonist injection following ICH. Furthermore, in an established thrombin injection model, PDGFR-α antagonist or PDGF-AA neutralizing antibody was introduced to determine the role of thrombin in activating and/or inhibiting the PDGFR-α pathway.
Materials and Methods
Animals
All procedures for this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University. Please see details in Supplementary Text.
Intracerebral Hemorrhage Mouse Model
ICH was induced using the autologous arterial blood injection model (bICH) which was modified as previously described 8. Please see details in Supplementary Text.
Injection of Thrombin into Basal Ganglia
Animals were fixed in the same manner as the autologous blood injection model described above with the same coordinates used. Thrombin (Sigma) was dissolved in sterilized PBS and delivered into the right basal ganglia (5 U/5 μl per mouse). Control animals were given 5 μl of PBS.
Experimental Design
Four separate experiments were conducted (Fig 1, experiment 1–4) in two models. Experiment 1: Gleevec was administered (intraperitoneal injection) at three doses 1 hour following bICH. Post-assessment included western blot, zymography (6 hours), neurological deficits, brain edema and Evans blue extravasation (24 and 72 hours).
Figure 1.
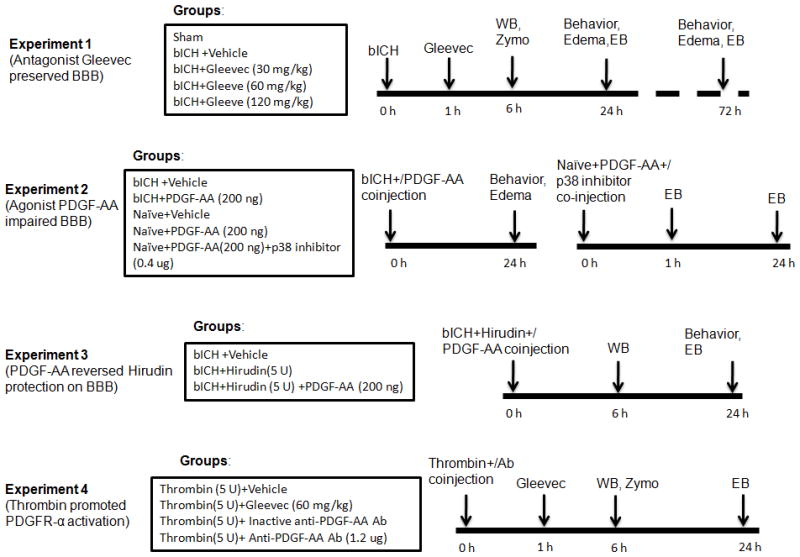
Experimental design and animal groups classification. bICH = autologous arterial blood-induced intracerebral hemorrhage; Zymo = zymography assay; WB = western blotting; EB = Evans blue assay; Anti-PDGF-AA Ab = Anti-PDGF-AA antibody.
Experiment 2: PDGF-AA was co-injected with blood into right basal ganglia. Neurological deficits and brain edema were determined at 24 hours; PDGF-AA was injected with or without a p38 MAPK inhibitor into right basal ganglia in naïve animals. Evans blue extravasation was detected at 1 and 24 hours.
Experiment 3: The thrombin specific inhibitor, hirudin with or without PDGF-AA was injected with blood into right basal ganglia. Post-assessment included western blot (6 hours), neurological deficits and Evans blue extravasation (24 hours).
Experiment 4: Gleevec was administered (intraperitoneal injection) 1 hour following thrombin injection. PDGF-AA antibody was co-injected with thrombin into right basal ganglia. Post-assessment included western blot, zymography (6 hours) and Evans blue extravasation (24 hours). Please see details in Supplementary Text.
Neurobehavioral Function Test
Neurobehavioral functions were evaluated by modified Garcia test 9–10 and corner turn test 11. Please see details in Supplementary Text.
Brain Water Content Measurement
Please see details in Supplementary Text.
BBB Permeability
BBB permeability was evaluated with Evans blue staining (250 μl of 4% solution in saline) as previously described 12. Please see details in Supplementary Text.
Immunoprecipitation
Please see details in Supplementary Text.
Western Blotting
Please see details in Supplementary Text.
Gelatin Zymography
MMP-2/9 activity was measured by gelatin zymography modified from previous study 13. Please see details in Supplementary Text.
Immunofluorescence
Please see details in Supplementary Text.
Statistics
Data was expressed as mean ± standard error of the mean. Analysis was performed using GraphPad Prism software. For the rating scale data (modified Garcia test), data were expressed as median ± 25th–75th percentiles. Please see details in Supplementary Text.
Results
PDGFR-α and PDGF-AA were upregulated following bICH injury
Western blot was performed to determine the profile of PDGFR-α at 3, 6, 12, 24 and 72 hours and endogenous PDGF-AA level at 6 hours following bICH. Western blot results revealed that PDGFR-α level (Fig 2A, B) was increased 3 hours post bICH and reached a peak around 6 hours in which the PDGFR-α level was almost six times more than sham animals (p < 0.05). Following this peak, the level of PDGFR-α declined at 12 hours (p < 0.05) and 24 hours, returning close to normal level by 72 hours. Endogenous PDGF-AA (Fig 2C, D), a specific PDGFR-α ligand/agonist was significantly increased in the ipsilateral hemisphere (Ipsi) 6 hours post bICH compared to both contralateral (Contra) hemisphere (p < 0.05) and sham animals (p < 0.05). The double immunofluorescence staining revealed that the PDGFR-α immunoreactivity was mainly found on the neurovascular structure, including perivascular related astrocytes and the endothelial cells (Fig 2E).
Figure 2.
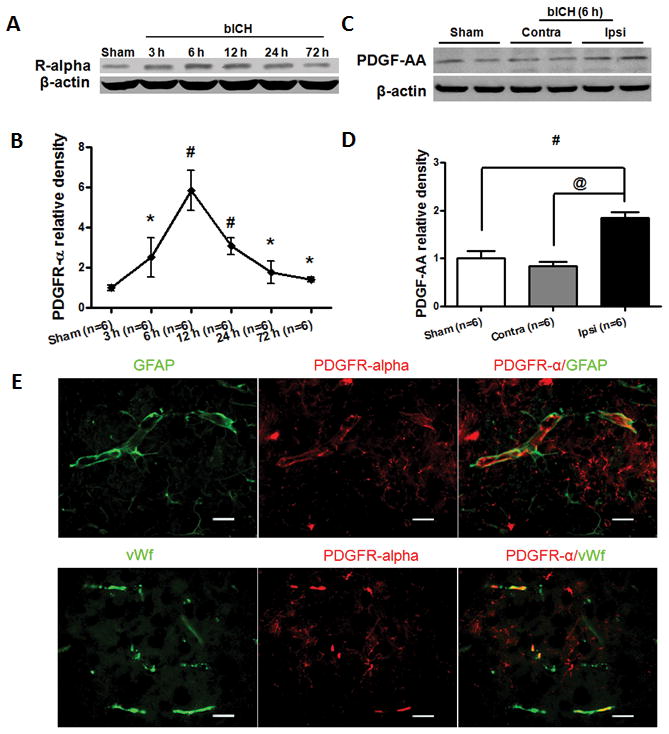
Expression of PDGFR-α and PDGF-AA after autologous arterial blood induced intracerebral hemorrhage (bICH). (A) Western blot assay for the profiles of PDGFR-α expression in the ipsilateral hemisphere in sham and bICH mice 3, 6, 12, 24 and 72 hours following operation. (C) Western blot assay for PDGF-AA expression in sham, ipsilateral (Ipsi) and contralateral (Contra) hemisphere in bICH mice 6 hours following operation; (E) Representative photographs of immunofluorescence staining for PDGFR-α (red) expression in astrocytes (GFAP, green) and endothelial cells (vWf, green) in the perihematomal area 6 hours following bICH. Scale bar: 50 μm. Quantification of A and C is shown in B and D, respectively. n = 6 mice per group and per time point. Error bars represent mean ± standard error of the mean. # p < 0.05 vs Sham; * p < 0.05 vs bICH (6 h); # p < 0.05 vs Sham; @ p < 0.05 vs Contra.
PDGFR-α suppression improved neurobehavioral functions, reduced brain edema, and preserved BBB integrity
A PDGFR-α antagonist, Gleevec was administered at three doses (30, 60, and 120 mg/kg) by intraperitoneal injection 1 hour following bICH. Neurobehavioral functions, brain edema and BBB permeability were evaluated at 24 and 72 hours following bICH. The results at 24 hours revealed that vehicle animals demonstrated severe deficits compared to sham animals in both modified Garcia test (p < 0.01; Fig 3A) and corner turn test (p < 0.01; Fig 3B). Following Gleevec administration at medium (60 mg/kg; p < 0.01) and high doses (p < 0.05; 120 mg/kg), there was a significant improvement in neurological score in modified Garcia test. With regards to corner turn test, the medium dose (60 mg/kg) significantly improved neurobehavioral function compared to vehicle animals (p < 0.05). We also evaluated neurobehavioral function at the delayed stage (72 hours) post bICH using the medium dose Gleevec treatment. The results demonstrated that the medium dose treatment could significantly improve neurobehavioral function following both modified Garcia test and corner turn test at 72 hours compared to vehicle animals (p < 0.05).
Figure 3.
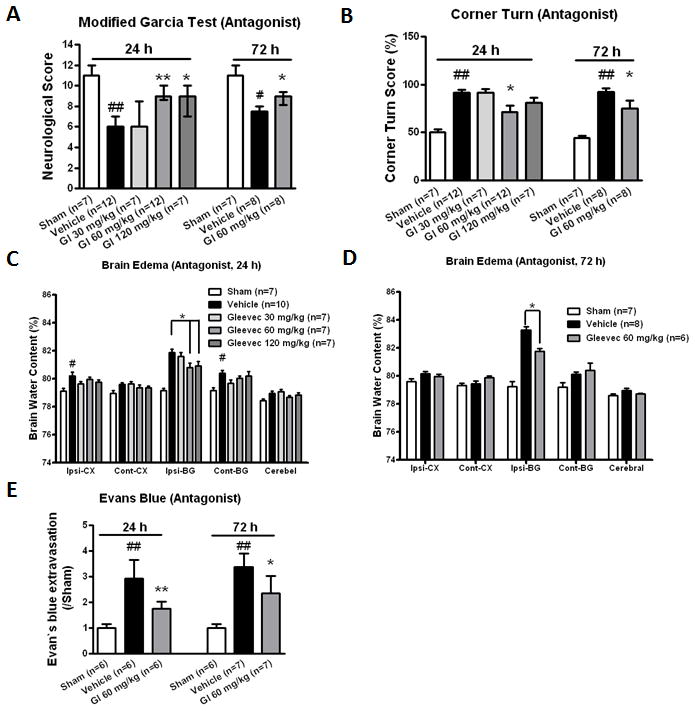
PDGFR-α suppression improved neurological functions, reduced brain edema and Evans blue extravasation at 24 and 72 hours following bICH. PDGFR-α antagonist, Gleevec was administered 1 hour following bICH. Modified Garcia test (A) and corner turn (B) at 24 and 72 hours following operation in sham, vehicle and Gl treatment groups (24 hours: 30, 60 and 120 mg/kg; 72 hours: 60 mg/kg). Brain edema at 24 hours (C) and 72 hours (D) following operation in sham, vehicle and Gl treatment groups (24 hours: 30, 60 and 120 mg/kg; 72 hours: 60 mg/kg). Brain sections (4 mm) were divided into four parts: ipsilateral basal ganglia (Ipsi-BG), ipsilateral cortex (Ipsi-CX), contralateral basal ganglia (Cont-BG), contralateral cortex (Cont-CX). Cerebellum (Cerebel) is the internal control. (E) Evans blue extravasation at 24 and 72 hours in the ipsilateral hemisphere following operations in sham, vehicle and Gl treatment groups (60 mg/kg). n = 6–12 mice per group. Error bars represent median ± 25th–75th percentiles (A) or mean ± standard error of the mean (B, C, D and E). # p < 0.05 vs Sham; ## p < 0.01 vs Sham; * p < 0.05 vs Vehicle; ** p < 0.01 vs Vehicle.
At 24 hours post bICH, the medium (60 mg/kg) and high-dose (120 mg/kg) treatment significantly decreased brain edema in the ipsilateral basal ganglia (ipsi-BG) compared to vehicle group (ipsi-BG: 60 mg/kg, 80.82 ± 0.30 vs vehicle, 81.88 ± 0.23, p < 0.05; 120 mg/kg, 80.92 ± 0.34 vs vehicle, 81.88 ± 0.23, p < 0.05; Fig 3C). In the ipsilateral cortex (ipsi-CX), brain edema was significantly increased in the vehicle group compared to sham group (ipsi-CX; vehicle, 80.22 ± 0.26 vs sham, 79.12 ± 0.21, p < 0.05). Although following Gleevec treatment the brain edema showed a trend towards reduction, there was no statistical significance reached. With regards to the 72 hours post bICH medium-dose (60 mg/kg) treatment, we found a significant reduction in brain edema in the ipsilateral basal ganglia compared to the vehicle group (ipsi-BG: 60 mg/kg, 81.75 ± 0.20 vs vehicle, 83.29 ± 0.23, p<0.05; Fig 3D). Evans blue extravasation (Fig 3E) was significantly increased at both 24 hours and 72 hours compared to sham groups (p < 0.01), and significantly reduced after medium-dose (60 mg/kg) Gleevec treatment (p < 0.05).
PDGFR-α suppression inhibited MMP activity and MMP-10/13 expression through orchestration of the p38 MAPK pathway post bICH
Phosphorylated PDGFR-α (Fig 4A, B) was significantly increased compared to sham animals (about seven times; p < 0.05) while Gleevec treatment (60 mg/kg) significantly reduced PDGFR-α phosphorylation level (p < 0.05) 6 hours post bICH. Gleevec treatment (60 mg/kg) also significantly reduced the active MMP-9 level (p < 0.05) but not MMP-2 compared to vehicle animals (Fig 4C–E), and reduced MMP-10 (p < 0.05; Fig 4F, G) and MMP-13 expression (p < 0.05; Fig 4H, I). The results also revealed that phosphorylated p38 MAPK was significantly reduced following Gleevec treatment (p < 0.05) yet, did not reduce the phosphorylation level of Erk1/2 and JNK1/2 (Fig 4J, K). Additionally, we also found that phosphorylated ATF-2, the substrate of p38 was also significantly reduced (p < 0.05; Fig 4L, M). Additionally, the cellular localization of PDGFR-α downstream mediators was determined by double immunofluorescence staining. Similar to the PDGFR-α, MMP-9, MMP-13 and phosphor-p38 immunoreactivity were mainly found in the neurovascular structure, including astrocytes and the endothelial cells, and MMP-10 was only found in the endothelial cells (Supplemental Fig 1).
Figure 4.
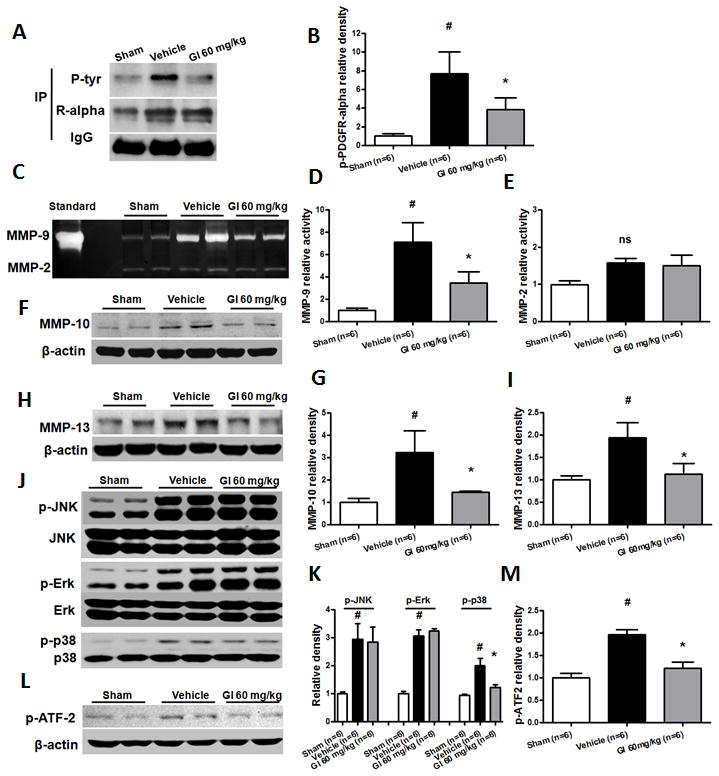
Characterization of PDGFR-α pathway at 6 hours following bICH in mice. PDGFR-α antagonist, Gleevec (60 mg/kg) was administered 1 hour following bICH. (A) Immunoprecipitation assay (IP) for phosphor-PDGFR-α level with phosphotyrosine-specific antibody (P-tyr) in the ipsilateral hemisphere in sham, vehicle and Gl treatment (60 mg/kg) mice. The precipitated protein was also visualized with PDGFR-α-specific antibodies (R-alpha). IgG was visualized as a loading control. (C) Gelatin zymography assay for MMP-9 and MMP-2 activity in the ipsilateral hemisphere in sham, vehicle and Gl treatment (60 mg/kg) mice; Western blot assay for MMP-10 (F), MMP-13 (H), JNK/p-JNK, Erk/p-Erk and p38/p-p38 (J), p-ATF-2 (L) in the ipsilateral hemisphere in sham, vehicle and G1 treatment (60 mg/kg) mice. Quantification of A, C, F, H, J, and L is shown in B, D, E, G, I, K, and M, respectively, n = 6 mice per group. Error bars represent mean ± standard error of the mean. # p < 0.05 vs Sham; * p < 0.05 vs Vehicle; ns indicates not significant.
PDGFR-α activation increased brain edema post bICH
At 24 hours post PDGF-AA delivery, neurobehavioral deficits were evaluated using modified Garcia test (Supplemental Fig 2A) and corner turn test (Supplemental Fig 2B). Our results revealed no difference in deficit severity compared to vehicle treatment animals, although two out of nine animals with PDGF-AA injection died in 24 hours. We also found that the brain edema in the ipsilateral basal ganglia was significantly increased compared to vehicle animals (ipsi-BG: PDGF-AA, 82.59 ± 0.24 vs Vehicle, 81.87±0.23, p < 0.05; Supplemental Fig 2C) 24 hours after PDGF-AA delivery.
PDGFR-α activation impaired BBB integrity but was reversed using a p38 MAPK inhibitor in naïve mice
At 24 hours following PDGF-AA injection, Evans blue extravasation was significantly increased in the ipsilateral hemisphere compared to just PBS injection mice (p < 0.01). BBB permeability was also detected 1 hour following PDGF-AA injection. The results showed that the Evans blue extravasation was also increased compared to just PBS injection (p < 0.05; Fig 5A). A p38 MAPK inhibitor, SB 203580 hydrochloride was co-injected with PDGF-AA into the right basal ganglia of naïve mice. 24 hours later, we found that the Evans blue extravasation was significantly diminished compared to PDGF-AA injection animals (p < 0.05; Fig 5B).
Figure 5.
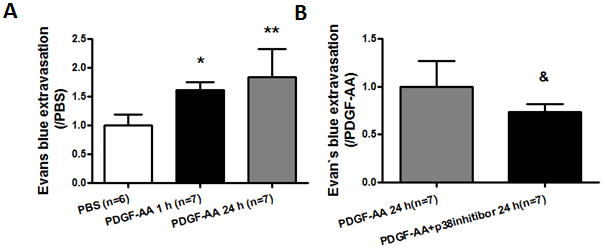
PDGFR-α activation by exogenous PDGF-AA increased Evans blue extravasation in naïve mice. (A) Evans blue extravasation in the ipsilateral hemisphere at 1 and 24 hours following PDGF-AA injection or 24 hours following PBS injection in naïve mice; (B) Evans blue extravasation in the ipsilateral hemisphere at 24 hours in PDGF-AA or PDGF-AA+p38 inhibitor co-injection naïve mice. n = 6–7 mice per group. Error bars represent mean ± standard error of the mean. * p < 0.05 vs PBS; ** p < 0.01 vs PBS; & p < 0.05 vs PDGF-AA (24 hours).
Thrombin inhibition preserved BBB integrity, while suppressing PDGFR-α activation and PDGF-AA expression post bICH
Thrombin inhibitor, hirudin was co-injected with autologous arterial blood into the right basal ganglia of mice. 24 hours following hirudin injection, Evans blue extravasation (Fig 6A) was significantly reduced in hirudin injected animals compared to vehicle animals (p < 0.05). Hirudin treatment also significantly improved neurological scores following modified Garcia test (p < 0.05; Supplemental Fig 3A), but failed to show improvement with corner turn test (Supplemental Fig 3B). Our results demonstrated that level of phosphorylated PDGFR-α (Fig 6B, C) and PDGF-AA (Fig 6D, E) were both significantly decreased in hirudin treated animals compared to vehicle animals (p < 0.05) 6 hours post bICH.
Figure 6.
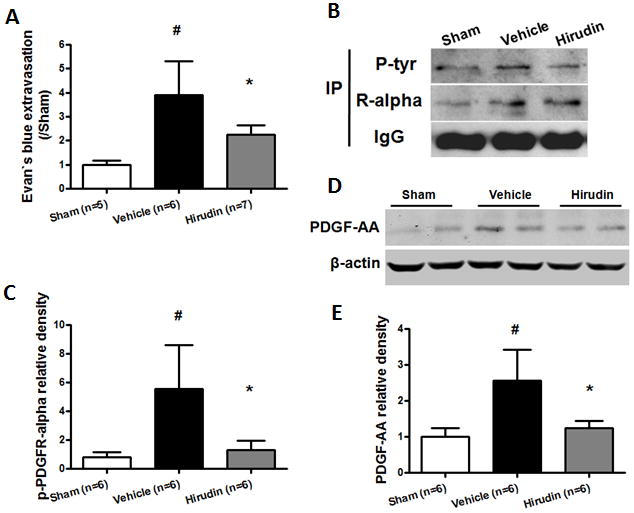
Thrombin inhibition reduced Evans blue extravasation, phosphor-PDGFR-α and PDGF-AA levels following bICH injury. Thrombin inhibitor, hirudin (5 U) was co-injected with autologous arterial blood. (A) Evans blue extravasation in the ipsilateral hemisphere 24 hours following operation in sham, vehicle and hirudin treatment (5 U) mice; (B) Immunoprecipitation assay (IP) for phosphor-PDGFR-α level with phosphotyrosine-specific antibody (P-tyr) in the ipsilateral hemisphere 6 hours following operation in sham, vehicle and hirudin treatment (5 U) mice. The precipitated protein was also visualized with PDGFR-α-specific antibodies (R-alpha). IgG was visualized as a loading control. (D) Western blot assay for PDGF-AA level in the ipsilateral hemisphere 6 hours following operation in sham, vehicle and hirudin treatment (5 U) mice. Quantification of B and D is shown in C and E, respectively, n = 5–7 mice per group. Error bars represent mean ± standard error of the mean. # p < 0.05 vs Sham; * p < 0.05 vs Vehicle.
PDGFR-α activation reversed the protective effects of thrombin inhibition on BBB integrity post bICH
Our results demonstrated that Evans blue extravasation was significantly increased compared to only hirudin treated mice (p < 0.05) 24 hours following hirudin and PDGF-AA co-injection (Fig 7A). The protection asserted by hirudin on neurobehavioral function was reversed following PDGF-AA administration in modified Garcia test (p < 0.05; Supplemental Fig 4A) but not in corner turn test (Supplemental Fig 4B) 24 hours after injection. Additionally, we also observed that the level of phosphorylation of PDGFR-α significantly increased by PDGF-AA compared to just hirudin treated mice (p < 0.05) 6 hours after injection (Fig 7B, C).
Figure 7.
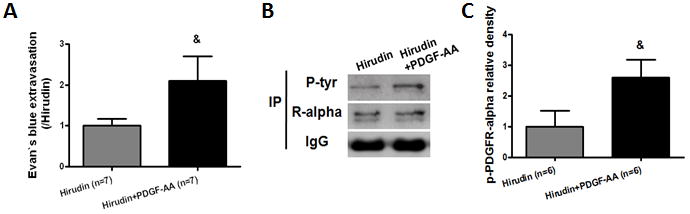
Activation of PDGFR-α by PDGF-AA reversed thrombin inhibition by hirudin following bICH. Thrombin inhibitor, hirudin (5 U) with or without PDGF-AA (200 ng) was co-injected with autologous arterial blood. (A) Evans blue extravasation in the ipsilateral hemisphere 24 hours following bICH in hirudin (5 U) and hirudin (5 U) + PDGF-AA (200 ng) mice; (B) Immunoprecipitation assay (IP) for phosphor-PDGFR-α level with phosphotyrosine-specific antibody (P-tyr) in the ipsilateral hemisphere 6 hours after bICH in hirudin (5 U) and hirudin (5 U) + PDGF-AA (200 ng) mice. The precipitated protein was also visualized with PDGFR-α-specific antibodies (R-alpha). IgG was visualized as a loading control. Quantification of B is shown in C. n = 6–7 mice per group. Error bars represent mean ± standard error of the mean. & p < 0.05 vs Hirudin.
PDGFR-α suppression reduced thrombin-induced BBB impairment through the PDGFR-α/ p38/MMPs pathway
Our results showed that Gleevec treatment significantly diminished Evans blue extravasation compared to thrombin injected animals (p < 0.05; Fig 8A). Phosphorylated PDGFR-α was significantly increased 6 hours following thrombin injection and significantly reduced in the Gleevec treated mice compared to just thrombin injected mice (p < 0.05; Fig 8B, C). Gleevec treatment significantly reduced MMP-9 level (p < 0.05) but not MMP-2 (Supplemental Fig 5A–C) 6 hours following thrombin injection. Similarly, MMP-10 (Supplemental Fig 5D, E) and MMP-13 (Supplemental Fig 5F, G) expression were also significantly reduced after treatment (p < 0.05). Additionally, Gleevec treatment significantly diminished the phosphorylation level of p38 MAPK (p < 0.05; Supplemental Fig 5H, I) as well as p38 MAPK substrate, ATF2 (p < 0.05; Supplemental Fig 5J, K) compared to just thrombin injected mice.
Figure 8.

Gleevec and PDGF-AA neutralizing antibody reduced Evans blue extravasation 24 hours following thrombin injection in mice. PDGFR-α antagonist, Gleevec (60 mg/kg) was administered 1 hour following thrombin (5 U) injection. Inactive PDGF-AA antibody (PDGF-AA Ab) or PDGF-AA antibody (PDGF-AA Ab, 1.2 μg) was co-injected with thrombin (5 U) into right basal ganglia. (A) Evans blue extravasation in the ipsilateral hemisphere 24 hours following operation in sham, thrombin (5 U) and Gl treatment (60 mg/kg) groups; (B) Immunoprecipitation assay (IP) for phosphor-PDGFR-α level with phosphotyrosine-specific antibody (P-tyr) in the ipsilateral hemisphere 6 hours following thrombin injection in sham, thrombin (5 U) and Gl treatment (60 mg/kg) mice. The precipitated protein was also visualized with PDGFR-α-specific antibodies (R-alpha). IgG was visualized as a loading control. (D) Western blot assay for PDGF-AA in Sham, ipsilateral (Ipsi) and contralateral (Contra) hemisphere in thrombin injection mice 6 hours following operation; (F) Evans blue extravasation in the ipsilateral hemisphere 24 hours following operation in thrombin (5 U), thrombin (5 U)+inactive PDGF-AA antibody (PDGF-AA Ab), and PDGF-AA antibody (PDGF-AA Ab, 1.2 μg) mice. Quantification of B and D is shown in C and E, respectively. n = 5–8 mice per group. Error bars represent mean ± standard error of the mean. # p < 0.05 vs Sham; * p < 0.05 vs Thrombin; @ p < 0.05 vs Contra; & p < 0.05 vs Thrombin+inactive Ab.
Neutralization of PDGF-AA with anti-PDGF-AA antibody reduced thrombin-induced BBB impairment
PDGF-AA level was significantly increased 6 hours in ipsilateral hemisphere following thrombin injection compared to contralater hemisphere and sham (p < 0.05; Fig 8D, E). 24 hours after PDGF-AA antibody injection, Evans blue extravasation level was significantly diminished compared to either control (Thrombin+inactive antibody) or just thrombin injected mice (p < 0.05; Fig 8F).
Discussion
Intracerebral hemorrhage is a fatal stroke subtype that currently has no effective treatment option. Even if patients survive the initial attack, the growing hematoma triggers a series of life threatening events leading to accumulation of cerebral edema, progression of neurobehavioral deficits, and possibly death 14. In the present study, we investigated the effects of the PDGFR-α and its ability to orchestrate BBB disruption following an ICH injury. Our findings suggest that therapeutic interventions targeting the PDGF-AA/PDGFR-α system may be a novel strategy to prevent BBB impairment and thus attenuate the subsequent accumulation of brain edema responsible for both structural and functional damage following ICH injury.
In order to determine the role of PDGFR-α on BBB disruption in ICH, a PDGFR antagonist, Gleevec was used to suppress PDGFR-α activity, which has showed protective effect on BBB integrity in ischemic stroke model 6. Gleevec represented a new class of anticancer drugs and has been approved by US Food and Drug Administration for the therapy on chronic myelogenous leukemia and other cancers by inhibition of several tyrosine kinase, including PDGFR-α. It was regarded as a new gold standard for treatment of chronic myeloid leukemia at all stages 15 while some dose-related adverse events have been observed in some patients during Gleevec therapy, such as nausea, vomiting, diarrhea, and fluid retention etc. 16–17. In our study, we observed a dose-dependent effect of Gleevec treatment on neurological function improvement after ICH. The medium dose (60 mg/kg) significantly improved neurological function while the low dose (30 mg/kg) or high dose (120 mg/kg) did not.
Increased BBB permeability following PDGF administration is not a new concept. Previous study led by Su and colleagues has suggested just that – specifically showing that PDGF injections into the CSF of naïve mice could increase the Evans blue extravasation compared to just PBS injections 6. Yet another study led by Yao and colleagues recently found that cocaine-induced PDGF could increase vascular permeability and that administration of a PDGF neutralizing antibody could abolish this effect 7. Similar to these studies, we found that ICH injury resulted in a transient increase in PDGFR-α/PDGF-AA levels, peaking at 6 hours and returning to baseline by 72 hours. This resulted in a significant increase in brain edema accumulation and BBB disruption which we measured at 24 hours. To our surprise, there was no simultaneous decline in neurological functions with further brain edema accumulation in the bICH with exogenous PDGFR-α agonist group compared to the bICH vehicle group. We attributed this unexpected outcome to the inability of neurological function test to pick up subtle changes in edema accumulation that occurred between the vehicle and agonist group.
With regards to mechanics, we now discuss the potential downstream signaling of PDGF-AA/ PDGFR-α which we hope will explain the mediation of the BBB disruption. MAPK pathway has been established as one of the downstream effectors of PDGFR-α signaling 18. There are several subfamilies of MAPKs including the extracellular signal-regulated kinases (ERK1/2), ERK5, the Jun amino-terminal kinases (JNK1–3) and the p38 kinases 19. Generally, p38 and JNK are detrimental in stroke models, with previous research showing that p38/MAPK2 is involved in control of the tight junctional closures among astrocytes 20 and plays a key role in orchestrating BBB disruption and vasogenic edema formation following focal cerebral ischemia and reperfusion 21. In our study, we found that p38 MAPK level, and not Erk or JNK level, were significantly decreased following Gleevec treatment - and that phosphorylated ATF2 (activating transcription factor 2), a substrate of p38 and JNK, was also markedly decreased. Moreover, the p38 MAPK inhibitor, SB 203580 hydrochloride, which was administered with PDGF-AA in naïve animals, was found to reverse the PDGF-AA induced BBB impairment. These findings suggest that PDGF-AA/ PDGFR-α system may orchestrate the damage to the BBB integrity through a p38 MAPK signaling pathway.
The detrimental role of matrix metalloproteinases, especially MMP-9 and MMP-2 has been well documented in the literature with regards to their effects on BBB integrity following ICH injury 22–24. Current studies suggested that MMP-10 and 13 were upregulated in both animal and human brain infarcted tissue following ischemic stroke damage 25–26. Similar to other members in the MMP family, MMP-13 (collagenase-3) can breakdown collagen and gelatin structures and in previous in vitro studies has been shown to cleave pro-MMP-9 to active MMP-9 27 –which can occur following MMP-10 as well28. With regards to this study, we found that PDGFR-α suppression significantly reduced MMP-9 activity but not MMP-2. We also observed that the expression of MMP-10 and MMP-13 were significantly decreased following PDGFR-α suppression. All of which resulted in preservation of the BBB integrity. In all, taken together MMPs may be the direct downstream proteins of PDGFR-α/p38 pathway and direct mediators of BBB impairment following ICH.
Now that we’ve discussed downstream orchestrators of PDGFR-α induced BBB damage, we wanted to investigate who was responsible for the upstream regulation of PDGFR-α signaling following ICH injury. Previous literature has alluded to the notion that thrombin regulates the expression of PDGF-AA through a PAR-1 receptor found in endothelial cells 29. Therefore in the present study, two different mice models were conducted to investigate the potential relationship between thrombin and PDGF-AA. First in the autologous arterial blood-induced ICH model, we found that PDGF-AA expression was significantly down-regulated following the delivery of hirudin, a thrombin specific inhibitor. We also found that the effects of hirudin on BBB preservation were reversed by exogenous PDGF-AA injection. In the thrombin injection model, we first found the increase of phosphorylated PDGFR-α level as well as its downstream signals, p38 MAPK and MMPs, and the diminishment following PDGFR-α suppression by Gleevec treatment. In this case, we also found that the PDGF-AA level was significantly upregulated in the ipsilateral hemisphere. Additionally, a PDGF-AA neutralizing antibody given with thrombin markedly reduced BBB permeability. Taken together, these findings demonstrated that thrombin is an essential upstream regulator of PDGF-AA/PDGFR-α system.
Why not just block thrombin? The dual role of thrombin in ICH has been well described in previous studies. On one hand, thrombin itself can directly damage the BBB and cause brain edema formation following ICH; while on the other hand, it can act as an essential element in the coagulation cascade to stop bleeding. The concentration of thrombin generated in the brain following ICH has been calculated. Normally, 1 ml of whole blood can provide roughly 260 to 360 units of thrombin from prothrombin. That means that about 15 U of thrombin is generated following a 50 μl blood injection (about 30 μl plasma) 30. A number of studies have regarded thrombin generated during blood clotting and hematoma formation as a major cause of brain edema formation 31–32. One study led by Xi et al. revealed that thrombin is also responsible for prolonged brain edema following ICH 32.
Mounting evidence that thrombin infusion into the brain produces the same amount of BBB disruption suggested that thrombin could be directly responsible for the breakdown 33. Moreover, thrombin can cleave its receptors and induce downstream protein production, such as vascular endothelial growth factor (VEGF) which can lead to increased endothelial cell permeability 34. Therefore, antithrombin therapy using intravenous is considered as a way to prevent brain tissue damage during invasive procedure including surgical removal of hematomas and possibly even direct infusion of thrombin inhibitors into hematomas 35. Unfortunately, a series of studies showed that the thrombin inhibitors, such as argatroban and hirudins, can provide protective effects in animal models 36–37 but, while in phase 1 clinical trials hemorrhagic transformations and increased hemorrhages were major adverse effects that occurred in patients 35, 38. As a result, it is very reasonable to develop a therapy strategy that can disrupt downstream thrombin mediators following ICH because they provide fewer side effects than direct thrombin inhibition.
It is also important to note that in addition to PDGF-AA, thrombin also regulates the expression of other PDGFs, such as PDGF-BB 39 which has led to BBB disruption in previous study 6. Although in our study we hypothesized that PDGFR-α activation may be responsible for BBB impairment, our study cannot rule out the possibility that other PDGFs may be involved in BBB disruption and thus remains one of the main limitations of this study.
Since the PDGF signals expressed transiently and peaked 6 hours after ICH, one of the limitations of our study was the potential narrow therapeutic time window. Previous study showed that BBB permeability in the perihematomal region increased markedly 8 to 12 hours after ICH, and continued to rise for 48 hours33. And the early BBB disruption is associated with the thrombin which is generated by the hematoma31. Our study was based on the pathophysiology of intracerebral hemorrhage and brain edema formation and may provide insight in understanding the mechanism of BBB disruption and clue on brain edema therapy. In the present study, Gleevec was administered 1 hour after ICH. However, the profile of PDGFR-α expression showed that at 12 hours and 24 hours after ICH, the PDGFR-α level was still 3.08 times and 1.76 times higher than that of sham animals respectively, therefore, a delayed treatment will be conducted in our future study to further establish the therapeutic time window.
In conclusion, our findings suggest that PDGFR-α may contribute to BBB impairment and brain edema formation induced by ICH. Thrombin may in fact be the upstream regulator of PDGFR-α signaling that regulates PDGF-AA expression (potential mechanisms see Supplemental Fig 5). Targeting the PDGFR-α signaling may provide an alternative treatment to thrombin-induced BBB injury following ICH.
Supplementary Material
Acknowledgments
This study was supported by NIH R01 NS060936-01A2 to Jiping Tang and NIH R01 NS053407-01A to John H. Zhang
References
- 1.Ribo M, Grotta JC. Latest advances in intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2006 Jan;6(1):17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- 2.He Y, Hua Y, Lee JY, et al. Brain alpha- and beta-globin expression after intracerebral hemorrhage. Transl Stroke Res. 2010 Mar;1(1):48–56. doi: 10.1007/s12975-009-0004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keep RF, Xiang J, Ennis SR, et al. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–7. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- 4.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999 Oct;79(4):1283–316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 5.Marx M, Perlmutter RA, Madri JA. Modulation of platelet-derived growth factor receptor expression in microvascular endothelial cells during in vitro angiogenesis. J Clin Invest. 1994 Jan;93(1):131–9. doi: 10.1172/JCI116936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su EJ, Fredriksson L, Geyer M, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008 Jul;14(7):731–7. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao H, Duan M, Buch S. Cocaine-mediated induction of platelet-derived growth factor: implication for increased vascular permeability. Blood. 2010 Dec 8; doi: 10.1182/blood-2010-10-313593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rynkowski MA, Kim GH, Komotar RJ, et al. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3(1):122–8. doi: 10.1038/nprot.2007.513. [DOI] [PubMed] [Google Scholar]
- 9.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation Stroke. 1995 Apr;26(4):627–34. doi: 10.1161/01.str.26.4.627. discussion 35. [DOI] [PubMed] [Google Scholar]
- 10.Wu B, Ma Q, Khatibi N, et al. Ac-YVAD-CMK Decreases Blood-Brain Barrier Degradation by Inhibiting Caspase-1 Activation of Interleukin-1beta in Intracerebral Hemorrhage Mouse Model. Transl Stroke Res. 2010 Mar 1;1(1):57–64. doi: 10.1007/s12975-009-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002 Oct;33(10):2478–84. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 12.Seiffert E, Dreier JP, Ivens S, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004 Sep 8;24(36):7829–36. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Hartman R, Ayer R, et al. Matrix metalloproteinases inhibition provides neuroprotection against hypoxia-ischemia in the developing brain. J Neurochem. 2009 Nov;111(3):726–36. doi: 10.1111/j.1471-4159.2009.06362.x. [DOI] [PubMed] [Google Scholar]
- 14.Strbian D, Durukan A, Tatlisumak T. Rodent models of hemorrhagic stroke. Curr Pharm Des. 2008;14(4):352–8. doi: 10.2174/138161208783497723. [DOI] [PubMed] [Google Scholar]
- 15.Peggs K, Mackinnon S. Imatinib mesylate--the new gold standard for treatment of chronic myeloid leukemia. N Engl J Med. 2003 Mar 13;348(11):1048–50. doi: 10.1056/NEJMe030009. [DOI] [PubMed] [Google Scholar]
- 16.Deininger MW, O’Brien SG, Ford JM, Druker BJ. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol. 2003 Apr 15;21(8):1637–47. doi: 10.1200/JCO.2003.11.143. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003 Mar 13;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 18.Dibb NJ, Dilworth SM, Mol CD. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer. 2004 Sep;4(9):718–27. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- 19.Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep. 2010 Nov;11(11):834–40. doi: 10.1038/embor.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zvalova D, Cordier J, Mesnil M, Junier MP, Chneiweiss H. p38/SAPK2 controls gap junction closure in astrocytes. Glia. 2004 May;46(3):323–33. doi: 10.1002/glia.10334. [DOI] [PubMed] [Google Scholar]
- 21.Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008 Oct;28(10):1686–96. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg GA, Navratil M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology. 1997 Apr;48(4):921–6. doi: 10.1212/wnl.48.4.921. [DOI] [PubMed] [Google Scholar]
- 23.Power C, Henry S, Del Bigio MR, et al. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003 Jun;53(6):731–42. doi: 10.1002/ana.10553. [DOI] [PubMed] [Google Scholar]
- 24.Tang J, Liu J, Zhou C, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutantmice. J Cereb Blood Flow Metab. 2004 Oct;24(10):1133–45. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 25.Rosell A, Alvarez-Sabin J, Arenillas JF, et al. A matrix metalloproteinase protein array reveals a strong relation between MMP-9 and MMP-13 with diffusion-weighted image lesion increase in human stroke. Stroke. 2005 Jul;36(7):1415–20. doi: 10.1161/01.STR.0000170641.01047.cc. [DOI] [PubMed] [Google Scholar]
- 26.Cuadrado E, Rosell A, Penalba A, et al. Vascular MMP-9/TIMP-2 and neuronal MMP-10 up-regulation in human brain after stroke: a combined laser microdissection and protein array study. J Proteome Res. 2009 Jun;8(6):3191–7. doi: 10.1021/pr801012x. [DOI] [PubMed] [Google Scholar]
- 27.Knauper V, Smith B, Lopez-Otin C, Murphy G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13) Eur J Biochem. 1997 Sep 1;248(2):369–73. doi: 10.1111/j.1432-1033.1997.00369.x. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura H, Fujii Y, Ohuchi E, Yamamoto E, Okada Y. Activation of the precursor of human stromelysin 2 and its interactions with other matrix metalloproteinases. Eur J Biochem. 1998 Apr 1;253(1):67–75. doi: 10.1046/j.1432-1327.1998.2530067.x. [DOI] [PubMed] [Google Scholar]
- 29.Chandrasekharan UM, Yang L, Walters A, Howe P, DiCorleto PE. Role of CL-100, a dual specificity phosphatase, in thrombin-inducedendothelial cell activation. J Biol Chem. 2004 Nov 5;279(45):46678–85. doi: 10.1074/jbc.M406441200. [DOI] [PubMed] [Google Scholar]
- 30.Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996 Jan;84(1):91–6. doi: 10.3171/jns.1996.84.1.0091. [DOI] [PubMed] [Google Scholar]
- 31.Lee KR, Kawai N, Kim S, Sagher O, Hoff JT. Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J Neurosurg. 1997 Feb;86(2):272–8. doi: 10.3171/jns.1997.86.2.0272. [DOI] [PubMed] [Google Scholar]
- 32.Xi G, Wagner KR, Keep RF, et al. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke. 1998 Dec;29(12):2580–6. doi: 10.1161/01.str.29.12.2580. [DOI] [PubMed] [Google Scholar]
- 33.Yang GY, Betz AL, Chenevert TL, Brunberg JA, Hoff JT. Experimental intracerebral hemorrhage: relationship between brain edema, blood flow, and blood-brain barrier permeability in rats. J Neurosurg. 1994 Jul;81(1):93–102. doi: 10.3171/jns.1994.81.1.0093. [DOI] [PubMed] [Google Scholar]
- 34.Sarker KP, Yamahata H, Nakata M, et al. Recombinant thrombomodulin inhibits thrombin-induced vascular endothelial growth factor production in neuronal cells. Haemostasis. 1999 Nov-Dec;29(6):343–52. doi: 10.1159/000022522. [DOI] [PubMed] [Google Scholar]
- 35.Matsuoka H, Hamada R. Role of thrombin in CNS damage associated with intracerebral haemorrhage: opportunity for pharmacological intervention? CNS Drugs. 2002;16(8):509–16. doi: 10.2165/00023210-200216080-00001. [DOI] [PubMed] [Google Scholar]
- 36.Kitaoka T, Hua Y, Xi G, Hoff JT, Keep RF. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke. 2002 Dec;33(12):3012–8. doi: 10.1161/01.str.0000037673.17260.1b. [DOI] [PubMed] [Google Scholar]
- 37.Xue M, Fan Y, Liu S, Zygun DA, Demchuk A, Yong VW. Contributions of multiple proteases to neurotoxicity in a mouse model of intracerebral haemorrhage. Brain. 2009 Jan;132(Pt 1):26–36. doi: 10.1093/brain/awn215. [DOI] [PubMed] [Google Scholar]
- 38.Hursting MJ, Alford KL, Becker JC, et al. Novastan (brand of argatroban): a small-molecule, direct thrombin inhibitor. Semin Thromb Hemost. 1997;23(6):503–16. doi: 10.1055/s-2007-996128. [DOI] [PubMed] [Google Scholar]
- 39.Stenina OI, Shaneyfelt KM, DiCorleto PE. Thrombin induces the release of the Y-box protein dbpB from mRNA: a mechanism of transcriptional activation. Proc Natl Acad Sci U S A. 2001 Jun 19;98(13):7277–82. doi: 10.1073/pnas.121592298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.