

Abstract
The N-heterocyclic carbene catalyzed [4+2] cycloaddition has been shown to give γ,δ-unsaturated δ-lactones in excellent enantio- and diastereoselectivity. However, preliminary computational studies of the geometry of the intermediate enolate rendered ambiguous both the origins of selectivity and the reaction pathway. In this communication, we show that a concerted, but highly asynchronous, Diels-Alder reaction occurs rather than the stepwise Michael-type or Claisen-type pathways. In addition, two crucial interactions are identified that enable high selectivity: an oxyanion-steering mechanism and a CH-π interaction. The calculations accurately predict the enantioselectivity of a number of N-heterocyclic carbene catalysts in the hetero-Diels-Alder reaction.
Introduction
N-Heterocyclic carbenes (NHCs) are effective in a large number of organocatalytic1 and organometallic2 applications. Starting in 2006, Bode and coworkers reported an NHC-catalyzed [4+2] cycloaddition between an enolate derived from an α,β-unsaturated aldehydes or α-functionalized aldehydes and an enone as the diene.3,4 These reactions displayed remarkable diastereo- and enantioselectivity, producing γ,δ-unsaturated δ-lactones in up to 99% ee and greater than 20:1 dr, as well as near quantitative yields (eq 1).5 In this communication, we establish by computation that a concerted, but highly asynchronous Diels-Alder reaction occurs rather than the stepwise Michael-type or Claisen-type pathways. Two crucial interactions were discovered that enable the high selectivity: an oxyanion-steering mechanism and a CH-π interaction. The calculations described herein accurately predicted the selectivity of a number of NHC catalysts in the hetero-Diels-Alder reaction.
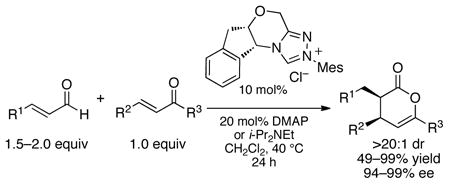 |
(1) |
The transformation in equation 1 is intriguing because the homoenolate equivalent (conjugated Breslow intermediate, II) undergoes proton transfer6 to form an enolate intermediate (Scheme 1).3,7 Alternatively, the enolate intermediate can also be reached from NHC attack on an α-chloroaldehyde7 to obtain adduct V, followed by elimination of HCl. Unexpectedly, initial calculations showed that the enolate of III lies perpendicular to the triazolium and is not stabilized by conjugation with the NHC.3b Since the enolate is blocked by the indane on one side and mesitylene on the other, the origin of the high stereochemical fidelity was ambiguous.
Scheme 1.

Proposed catalytic cycle for the NHC-catalyzed hetero-Diels-Alder reaction (only the NHC core shown).
Methods
All calculations were performed using Gaussian 09.8 All transition states were optimized with HF/6-31G(d) in the gas phase and were confirmed to have one imaginary frequency; all local minima were optimized with HF/6-31G(d) and were found to have no imaginary frequencies. Intrinsic reaction coordinate calculations were performed regularly to confirm that the calculated transition states reflected the correct reaction. Gibbs Free Energies were calculated at 1 atm and 298.15 K and are uncorrected.9
Solvation with toluene was examined for the mesitylene, para-trifluoromethyl, and pentafluoromethyl catalysts in the deprotonated Diels-Alder reaction using IEFPCM(toluene)-HF/6-31G(d)//HF/6-31G(d).
A solution of 2-chloro-3-phenylpropanal (26.0 mg, 0.4 mmol, 1.0 equiv), triethylamine (20.0 μL, 0.6 mmol, 1.5 equiv), and (E)-methyl 4-oxo-4-phenylbut-2-enoate (20.0 mg, 0.4 mmol, 1.0 equiv) was prepared using 0.5 mL PhCH3. This solution was transferred to a vial containing a chiral triazolium salt (Table 1). The reactions were carried out at room temperature and the products were isolated by preparative TLC using 7:1 hexanes: EtOAc. The identity of the product was confirmed by 1H NMR, 13C NMR, GC/MS and LC/MS.10 Percent conversion was determined by the integration of the product at 5.82–5.79 ppm against the enone starting material at 6.91–6.86 ppm. Percent enantiomeric excess was determined by SFC (AD-H, gradient 5–50% CO2: iPrOH), tr = 8.31 min and 9.61 min. The absolute configuration was assigned based on the previous literature report.
Table 1.
Prediction of reaction selectivities using the computational model.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl | Calculated ee (%)a | Conversion (%) | Experimental ee (%)b | ||
| 1 | 2,4,6-(CH3)-C6H2 | 99.8 (100) | 100 | 99 | ||
| 2 | 2-Me-4-OMe-C6H3 | 96.4 | 100 | 99 | ||
| 3 | 2-Me-C6H4 | 97.7 | 100 | 98 | ||
| 4 | 2,4,6-Cl-C6H2 | 97.3 | 95 | 99 | ||
| 5 | 2,4,6-Cl-C6F2 | 86.2 (99.6) | 93 | 97 | ||
| 6 | C6F5 | 81.5 (83.2) | 47 | 76 | ||
| 7 | 3,5-(CF3)-C6H3 | 84.1 | 34 | 89 | ||
| 8 | 4-CF3-C6H4 | 87.5 | 15 | 95 | ||
| 9 | C6H5 | 93.0 | 50 | 97 | ||
| 10 | 4-OMe-C6H4 | 95.0 | 75 | 98 | ||

Results & Discussion
To understand the stereochemical factors in this system, we examined a model system with acetaldehyde enolate as the dienophile and acrolein as the diene (R1 = R2 = R3 = H, eq 1 and Scheme 1) using HF/6-31G(d) in the gas phase. The entire NHC catalyst from equation 1 was employed to accurately determine its role.11,12
Analysis of the enantioselectivity determining step revealed three distinct plausible reaction pathways (Figure 1): 1) a concerted Diels-Alder reaction, 2) a Michael type addition of the NHC-enolate to the enone followed by addition/elimination of the enolate intermediate to cyclize the ring and expel the NHC catalyst, or 3) an initial addition of the NHC-enolate to the enone carbonyl followed by Claisen rearrangement and collapse of the intermediate following the same pathway as the Michael-type reaction. Examination of options 1 and 2 rapidly revealed that the charge separation induced in the initial Michael-type reaction gave rise to much higher energy transition states relative to the Diels-Alder reaction.
Figure 1.

Possible reaction pathways.
For the Diels-Alder pathway, three constraints were identified: 1) the acrolein adopts a cisoid geometry to avoid undesirable charge separation in the transition state, 2) the acrolein does not approach endo to the triazolium, which creates prohibitive steric interactions between the diene and the mesitylene ring and gives rise to the incorrect diastereomer, and 3) the enolate substituent (R1, Figure 1) is cis to the enolate oxygen to avoid steric interactions with the catalyst and to give rise to the correct diastereomer. Upon examination of conformations and optimization, transition states were located for the Diels-Alder pathway in which the enolates adopt a planar conformation with respect to the triazolium ring, a significant departure compared to the ground state enolates,3b which are perpendicular to the plane of the triazolium. This planarization of the enolate increases the positive charge on the enolate C1-carbon (Figure 1, top, and Figure 2, bottom) thereby optimizing the developing interaction with the negative charge on the diene oxygen.
Figure 2.

Sample Diels-Alder and Claisen transition states (one Claisen protonated, one DA protonated, and one zwitterionic DA) leading to the dihydropyranone products.
The transition state geometries indicate a very asynchronous Diels-Alder reaction. In the lowest energy model system transition state, the forming carbon-carbon bond is 2.109 Å while the forming carbon-oxygen bond is 3.049 Å. The atom-atom net linear natural localized molecular orbital natural population analysis (NLMO/NPA)13 bond orders14 are 0.435 and 0.0550 respectively. Similar values were seen for all the Diels-Alder transition states.9
Intrinsic reaction coordinate (IRC) calculations show that the enolate commences nearly perpendicular to the triazolium ring and flattens as the enone approaches. Notably, the starting geometry with the enolate oxygen up always leads to enone approach from the top face; the starting geometry with the enolate oxygen down always leads to enone approach from the bottom face (seen in IRCs of all the 7 transition states located, see Figure 4). This trend suggests a secondary orbital interaction between the enone carbonyl π* and an enolate lone pair (Figure 3), which guides the enone in its approach and ultimately leads to the Diels-Alder reaction.
Figure 4.

The eight possible transition states and relative free energies for the NHC-catalyzed Diels-Alder reaction (relative ΔG values in kcal/mol). Labels in red indicate transition states that lead to the major enantiomer; blue labels lead to the minor enantiomer.
Figure 3.

The oxyanion guiding interaction as seen in the intrinsic reaction coordinate calculation (NHC indane and mesitylene rings removed for clarity).
The presence of this interaction also suggests an alternative reaction pathway (Claisen-type, Figure 1) in which the enolate oxygen first adds to the enone carbonyl. Subsequent Claisen rearrangement of this intermediate would lead to the product.15 When a protonated enolate was used in the calculation, this Claisen transition state was 7.0 kcal/mol lower in energy than the Diels-Alder reaction, but when the deprotonated enolate was used the model converged to the asynchronous Diels-Alder transition state. This suggests two possibilities: 1) if the reaction proceeds through the enolate, it possesses both Diels-Alder and Claisen character, and 2) if the complex reacts as the enol, it undergoes the Claisen rearrangement.15a The reaction is performed with catalytic DMAP or i-Pr2NEt (water pKa = 9.2 and 10.8, respectively), so the enol will dominate if its pKa is above 12.
To calculate the pKa values16 of the relevant enolates, the method that Pulay17 used with phenols was implemented except that the integral equation formalism variant of the polarization continuum model (IEFPCM) solvation method was employed instead of the COSMO model.18 Each of the four enolate conformations was calculated and the overall pKa was generated from a weighted average of the four resultant values using a Boltzmann distribution at 25 °C. The resulting theoretical pKa of the enolate in water is 5.5,9 much less than that of the bases in water. Since the reactions under discussion here are conducted in toluene or dichloromethane, this estimate needs to be used with caution. However, analysis of the conjugate acids of other zwitterions, such as pyridine N-oxide, is informative. Here, the acid is a resonance-stabilized cation and the conjugate base is a zwitterion in which the negative charge is not stabilized by resonance. The pKa of pyridine N-oxide is 0.79 in water and 1.63 in DMSO. On this basis we expect the pKa of the triazolium enolate to be ~6.5 in DMSO (vs 9.00 for Et3NH+ in DMSO). Therefore, the main species in solution is most likely the enolate for which the Claisen-type pathway does not occur.
In the Diels-Alder pathway there are the eight possible transition states differing by the ring conformation of the morpholine, the orientation of the enolate, and the facial approach of the enone. Upon examination of all the combinations, only seven of the eight transition states could be located (Figure 4); TS5, in which the morpholine oxygen orients cis to the indane, the enolate oxygen points toward the indane, and the acrolein approaches from the bottom, converged instead to TS6.
For the morpholine ring, interconversion between two different half-chair conformations causes a large distortion of the catalyst. When the morpholine oxygen orients down (cis to the indane), the indane ring extends in front of the triazolium alleviating steric interactions with the mesityl and resulting in the lowest energy conformation in the absence of enone. In this conformation, the indane more effectively blocks the approach of the substrate from the bottom face as seen in the energy difference of 6.65 kcal/mol between TS3 and TS7, which vary only in the enone facial approach. In addition, this steric hindrance blocks enone approach in the counterpart of TS1 (i.e. TS5) so effectively that it could not be located. In all cases where the enone approaches from the top, the lower energy conformation has the indane extended forward (TS1 vs TS2 and TS3 vs TS4).
When the morpholine oxygen points up (trans to the indane), the indane ring tucks under the triazolium (e.g. TS2) incurring greater steric interactions with the mesityl as compared to the other half-chair conformation (cf. TS1). On the other hand, this conformation does open up approach from the bottom face of the enolate such that TS6 is lower than TS5 and TS8 is lower than TS7. Even so, considerable steric blocking remains on the bottom face and all the transitions states are higher in energy (TS5-TS8).
On the whole, the transition states with C2 of the enolate pointing toward the mesitylene are lower in energy than when it is pointing toward the indane due to a CH–π interaction between the terminal CH2 of the enolate and the aromatic ring (5).19,20,21 Specifically, the inner hydrogen on the enolate carbon is just 2.9 Å from C1 of the mesitylene, well within the combined van der Waals distance of 3.4 Å. Similar CH–π interactions have been observed in the transition states of Diels Alder reactions,22 sulfide oxidations,23 and hydride reductions.24 This effect can be increased with electron donating groups and diminished with withdrawing groups (see below).
To test the importance of the CH–π interaction, the eight transition states of the model system were optimized for a series of catalysts with varied aryl substitution and enantioselectivities were calculated using Boltzmann distributions (Table 1).26,9 The calculations were performed concurrently with the reaction in equation 2. In principle, the stereoselectivity predictions provided are for both the α-haloaldehydes or the enals since the both yield the same enolate that precedes the stereoselectivity determining step calculated here (Scheme 1). In practice, the enals are unreactive with catalysts lacking ortho substitution on the N-aryl ring due to reversibility in the formation of the initial adduct between the aldehyde and the NHC.7
Overall, the calculations were highly effective in anticipating the experimental selectivity, with the exception of entry 5. Further calculations revealed that M06-2X/6-311+G(d,p)27 single point calculations of the full system were necessary to reproduce the experimental results in this case, which is attributed to the combination of five halogen substituents. Note that Table 1 contains all the catalysts that were examined computationally; none were excluded. Solvation with toluene was examined for the mesitylene, para-trifluoromethyl, and pentafluoromethyl systems using IEFPCM(toluene)-HF/6-31G(d)//HF/6-31G(d) of all the tranisition states; the computed selectivities did not change significantly, indicating that the gas phase calculations are sufficient for this reaction in this nonpolar solvent.28
A significant drop in selectivity was expected with electron-poor catalysts, which was confirmed in the experiments (entries 6–8). Analysis of the computational data shows that when Ar = mesitylene, the relative energy between TS1 and TS3, which differ only in orientation of the enolate, is 4.83 kcal/mol. When Ar = C6F5, that energy difference drops to 1.35 kcal/mol. The electron-poor arene cannot support the CH–π interaction, destabilizing TS1, TS2, TS5, and TS6 relative to TS3, TS4, TS6, and TS7, and eroding the theoretical selectivity.
A Hammett analysis of the experimental results for aryls without ortho-substitution shows a strong correlation between enantiomeric ratio and the electron density of the aryl ring (ρ = −0.63, R2 = 0.96).9 Electron-withdrawing groups attenuate the CH-π interaction, decreasing the enantiomeric ratio. On the other hand, ortho-substitution rescues even highly electron deficient catalysts (entries 4–5). Apparently, decreased rotational freedom of the N-aryl bond is a key factor in these transformations and a further analysis of this contribution is underway.
Conclusions
In summary, a computational model has been developed that successfully estimates the enantioselectivity of various catalysts in the NHC-catalyzed hetero-Diels-Alder reaction. Two effects account for the selectivity in these reactions: an oxyanion guiding interaction which delivers the substrate and a CH-π interaction. This study provides a basis for using these two elements in future development of catalyst systems.
Supplementary Material
Figure 5.

The CH-π interaction from the front (top) and from overhead (bottom) for TS1. Enone and indane ring removed for clarity.
Acknowledgments
We are grateful to the National Institutes of Health (GM–079339 and GM-087605) and the National Science Foundation (CHE-0449587) for financial support of this research. Computing resources were provided by the National Science Foundation (CRIF CHE-0131132) and XSEDE (TG-CHE110080). We thank Bill Dailey for helpful discussions.
Footnotes
Supporting Information. Full computational and experimental details and full citation of reference 9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Moore JL, Rovis T. Top Curr Chem. 2009;291:77–144. doi: 10.1007/978-3-642-02815-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chiang P-C, Bode JW. The Royal Society of Chemistry. 2011. N-Heterocyclic Carbenes; pp. 399–435. [Google Scholar]; (d) Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem Soc Rev. 2011;40:5336–5346. doi: 10.1039/c1cs15139h. [DOI] [PubMed] [Google Scholar]; (e) Bugaut X, Glorius F. Chem Soc Rev. 2012;41:3511–3522. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]
- 2.For a review of the application of NHC–Pd complexes, see: Kantchev EAB, O’Brien CJ, Organ MG. Angew Chem Int Ed. 2007;46:2768–2813. doi: 10.1002/anie.200601663.For other metals, see: Díez-González S, Marion N, Nolan SP. Chem Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m.
- 3.(a) He M, Struble JR, Bode JW. J Am Chem Soc. 2006;128:8148–8150. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (b) He M, Uc GJ, Bode JW. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]; (c) Kaeobamrung J, Kozlowski MC, Bode JW. Proc Nat Acad Sci USA. 2010;107:20661–20665. doi: 10.1073/pnas.1007469107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For an intramolecular variant, see: Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235.
- 5.For selected, related works from other groups, see: Zhang YR, Lv H, Zhou D, Ye S. Chem Eur J. 2008;14:8473–8476. doi: 10.1002/chem.200801165.Kobayashi S, Kinoshita T, Uehara H, Sudo T, Ryu I. Org Lett. 2009;11:3934–3937. doi: 10.1021/ol901544q.Lv H, Mo J, Fang X, Chi YR. Org Lett. 2011;13:5366–5369. doi: 10.1021/ol202250s.
- 6.The mechanism of this proton transfer is explored in: Verma P, Patni PA, Sunoj RB. J Org Chem. 2011;76:5606–5613. doi: 10.1021/jo200560t.Reddi Y, Sunoj RB. Org Lett. 2012;14:2810–2813. doi: 10.1021/ol301036u.
- 7.Mahatthananchai J, Bode JW. Chem Sci. 2012;3:192–197. doi: 10.1039/C1SC00397F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frisch MJ. Gaussian 09, Revision B.01. Gaussian, Inc; Wallingford, CT: 2010. [Google Scholar]
- 9.See Supporting Information for computational details.
- 10.He M, Struble JR, Bode JW. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]
- 11.Calculations of other NHC-catalyzed reactions with truncated catalysts: Tang K, Wang J, Cheng X, Hou Q, Liu Y. Eur J Org Chem. 2010:6249–6255.Zhao L, Chen XY, Ye S, Wang ZX. J Org Chem. 2011;76:2733–2743. doi: 10.1021/jo200087u.Piel I, Steinmetz M, Hirano K, Fröhlich R, Grimme S, Glorius F. Angew Chem Int Ed. 2011;50:4983–4987. doi: 10.1002/anie.201008081.Wei S, Wei XG, Su X, You J, Ren Y. Chem Eur J. 2011;17:5965–5971. doi: 10.1002/chem.201002839.Verma P, Patni PA, Sunoj RB. J Org Chem. 2011;76:5606–5613. doi: 10.1021/jo200560t.Hawkes KJ, Yates BF. Eur J Org Chem. 2008:5563–5570.
- 12.Calculations of other NHC-catalyzed reactions with full catalysts: Dudding T, Houk KN. Proc Natl Acad Sci. 2004;101:5770–5775. doi: 10.1073/pnas.0307256101.Berkessel A, Elfert S, Etzenbach-Effers K, Teles JH. Angew Chem, Int Ed. 2010;49:7120–7124. doi: 10.1002/anie.200907275.Wei D, Zhu Y, Zhang C, Sun D, Zhang W, Tang M. J Mol Catal A: Chem. 2011;334:108–115.Um JM, DiRocco DA, Noey EL, Rovis T, Houk KN. J Am Chem Soc. 2011;133:11249–11254. doi: 10.1021/ja202444g.Ryan SJ, Stasch A, Paddon-Row MN, Lupton DW. J Org Chem. 2012;77:1113–1124. doi: 10.1021/jo202500k.DiRocco DA, Noey EL, Houk KN, Rovis T. Angew Chem Int Ed. 2012;51:2391–2394. doi: 10.1002/anie.201107597.
- 13.a) Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899–926. [Google Scholar]; b) Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]; c) Reed AE, Weinhold F. J Chem Phys. 1985;83:1736–1740. [Google Scholar]
- 14.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO Version 31. [Google Scholar]
- 15.(a) Kaeobamrung J, Mahatthananchai J, Zheng P, Bode JW. J Am Chem Soc. 2010;132:8810–8812. doi: 10.1021/ja103631u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wanner B, Mahatthananchai J, Bode JW. Org Lett. 2011;13:5378–5381. doi: 10.1021/ol202272t. [DOI] [PubMed] [Google Scholar]
- 16.For a review: Alongi KS, Shields GC. In: Annual Reports in Computational Chemistry. Wheeler RA, editor. Vol. 6. Elsevier; pp. 113–138.
- 17.a) Zhang S, Baker J, Pulay P. J Phys Chem A. 2010;114:425–431. doi: 10.1021/jp9067069. [DOI] [PubMed] [Google Scholar]; b) Zhang S, Baker J, Pulay P. J Phys Chem A. 2010;114:432–442. doi: 10.1021/jp9067087. [DOI] [PubMed] [Google Scholar]
- 18.For comparison of solvation models see: Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009.
- 19.For reviews on the CH/π interaction: Nishio M, Hirota M, Umezawa Y. The CH/π Interaction: Evidence, Nature, and Consequences. Wiley-VCH; New York, NY: 1998. Nishio M. Tetrahedron. 2005;61:6923–6950.Nishio M, Umezawa Y, Honda K, Tsuboyama S, Suezawa H. CyrstEngComm. 2009;11:1757–1788.
- 20.For review on the CH/π interaction in organic conformations see: Takahashi O, Kohno Y, Nishio M. Chem Rev. 2010;110:6049–6076. doi: 10.1021/cr100072x.
- 21.After this manuscript was submitted, a report (ref 6b) appeared on calculations of NHC enolates reaching some similar conclusions.
- 22.(a) Gordillo R, Houk KN. J Am Chem Soc. 2006;128:3543–3553. doi: 10.1021/ja0525859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Anderson CD, Dudding T, Gordillo R, Houk KN. Org Lett. 2008;10:2749–2752. doi: 10.1021/ol800875k. [DOI] [PubMed] [Google Scholar]
- 23.Capozzi MAM, Centrone C, Fracchiolla G, Naso F, Cardellicchio C. Eur J Org Chem. 2011:4327–4334. [Google Scholar]
- 24.Gutierrez O, Iafe RG, Houk KN. Org Lett. 2009;11:4298–4301. doi: 10.1021/ol901586t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 26.For a review on the electronics of NHC catalysts, see: Dröge T, Glorius F. Angew Chem Int Ed. 2010;49:6940–6952. doi: 10.1002/anie.201001865.
- 27.This functional reduces inflated relative energies seen in HF: Simón L, Goodman JM. Org Biomol Chem. 2011;9:689–700. doi: 10.1039/c0ob00477d.
- 28.Enantioselectivies calculated using toluene solvation (IEFPCM(toluene)-HF/6-31G(d)//HF/6-31G(d)): 2,4,6-(CH3)-C6H2, 99.8% ee; 4-CF3-C6H4, 90.7% ee; C6F5, 86.9% ee.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.