

Abstract
Pseudomonas aeruginosa strain PA14 is an opportunistic human pathogen capable of infecting a wide range of organisms including the nematode Caenorhabditis elegans. We used a non-redundant transposon mutant library consisting of 5,850 clones corresponding to 75% of the total and approximately 80% of the non-essential PA14 ORFs to carry out a genome-wide screen for attenuation of PA14 virulence in C. elegans. We defined a functionally diverse 180 mutant set (representing 170 unique genes) necessary for normal levels of virulence that included both known and novel virulence factors. Seven previously uncharacterized virulence genes (ABC transporters PchH and PchI, aminopeptidase PepP, ATPase/molecular chaperone ClpA, cold shock domain protein PA0456, putative enoyl-CoA hydratase/isomerase PA0745, and putative transcriptional regulator PA14_27700) were characterized with respect to pigment production and motility and all but one of these mutants exhibited pleiotropic defects in addition to their avirulent phenotype. We examined the collection of genes required for normal levels of PA14 virulence with respect to occurrence in P. aeruginosa strain-specific genomic regions, location on putative and known genomic islands, and phylogenetic distribution across prokaryotes. Genes predominantly contributing to virulence in C. elegans showed neither a bias for strain-specific regions of the P. aeruginosa genome nor for putatively horizontally transferred genomic islands. Instead, within the collection of virulence-related PA14 genes, there was an overrepresentation of genes with a broad phylogenetic distribution that also occur with high frequency in many prokaryotic clades, suggesting that in aggregate the genes required for PA14 virulence in C. elegans are biased towards evolutionarily conserved genes.
Author Summary
Pseudomonas aeruginosa is an opportunistic human pathogen that can also infect a wide range of model organisms, including the nematode Caenorhabditis elegans. To identify P. aeruginosa genes that play key roles in the pathogenic process, we performed a screen for mutants that exhibited reduced ability to kill C. elegans using a previously constructed non-redundant library representing approximately 80% of the non-essential P. aeruginosa PA14 genes. We defined a functionally diverse set of 180 P. aeruginosa mutants (representing 170 unique genes) necessary for normal levels of virulence that included both known and novel virulence factors. The major contributors to P. aeruginosa virulence in the C. elegans infection model were not secretion systems or their corresponding effectors, but rather regulators (particularly ones that are involved in quorum sensing) and genes likely to play key roles in survival of P. aeruginosa within the host intestine. Moreover, these putative P. aeruginosa virulence genes are neither overrepresented in strain-specific regions nor in horizontally acquired genomic islands and furthermore tend to have orthologs that are widely distributed across sequenced prokaryotic species. These data underscore the diversity of pathways involved in virulence, and especially the importance of highly conserved genes for P. aeruginosa virulence in the C. elegans host model.
Introduction
Pseudomonas aeruginosa, an opportunistic Gram-negative human pathogen, is one of the leading causes of hospital-acquired infections. In the context of a breakdown in host defenses, it is capable of infecting a plethora of tissue types, causing both acute and chronic infections. Burn victims as well as immunocompromised, mechanically ventilated, and cystic fibrosis (CF) patients are particularly susceptible to P. aeruginosa infection [1]. Over the last few decades, a steady increase in drug resistant P. aeruginosa strains has made antibiotic treatment more difficult [2]. In part because no new antibiotics effective against P. aeruginosa are imminently available as treatment options, the pressing need for drugs to fight this pathogen has focused study on its virulence factors as potential drug targets, and more generally energized a search for novel anti-infectives [3]–[5].
One likely reason that P. aeruginosa is a common nosocomial pathogen is because it is capable of thriving in a wide variety of environmental niches, including surfaces in hospital rooms, water, soil and plants [6]. Consistent with its ecological ubiquity, P. aeruginosa has a relatively large genome that presumably promotes survival in diverse habitats. In addition to inhabiting a wide variety of ecological niches, P. aeruginosa is also a multi-host pathogen, capable of infecting hosts as divergent as amoebae, plants, insects, flies, nematodes, and mice [7]–[13]. Progress in fighting P. aeruginosa infections will be aided by a fundamental understanding of the myriad ways that P. aeruginosa can survive in different environments and cause disease in diverse hosts.
Our laboratory has developed a Pseudomonas aeruginosa - Caenorhabditis elegans infection-based model for studying host-pathogen interactions that is genetically tractable from both the perspectives of the host and the pathogen. This model (referred to as “slow-killing” or SK), which primarily utilizes P. aeruginosa strain PA14 [8], requires live bacteria and a set of bacterial virulence factors that distinguish it from rapid toxin-mediated PA14 killing of C. elegans (“fast killing” or FK) that occurs on high osmolarity media [12], [14]. Under standard laboratory conditions [15], C. elegans animals have a normal lifespan of approximately two weeks when feeding on non-pathogenic Escherichia coli strain OP50, and OP50 does not accumulate in the C. elegans intestine during the first few days of life. In contrast, when C. elegans are transferred at the L4 larval stage from E. coli OP50 to a lawn of P. aeruginosa strain PA14, the animals die in two-three days [12]. A few PA14 cells initially accumulate in the anterior and posterior portions of the nematode intestine, then over the course of one to two days bacteria spread throughout the intestine and the intestinal lumen becomes severely distended with a corresponding reduction in volume of the intestinal epithelial cells. Ultimately, PA14 cells move across the intestinal epithelial barrier destroying tissue, but it is not known whether tissue invasion is required for killing [16].
C. elegans rapidly responds to the presence of pathogenic PA14 by enhancing the transcription of hundreds of genes including a number of predicted secreted proteins (C-type lectins, CUB-domain containing proteins, ShK toxins) that may have antimicrobial or detoxifying activity [17], [18]. Two of the major classes of PA14 response genes, C-type lectins and CUB-like domain containing proteins, also play a role in mammalian innate immunity [19]–[21]. In C. elegans, many immune response genes are regulated by the PMK-1 p38 mitogen-activated protein kinase (MAPK), the terminal MAPK in an evolutionary conserved signaling cassette required for defense against pathogens in both nematodes and mammals. Approximately 25% of the C. elegans genes regulated by PMK-1 are also induced in response to P. aeruginosa PA14 [18] and C. elegans p38 MAPK pathway mutants exhibit enhanced sensitivity to PA14 as well as a variety of other bacterial and fungal pathogens [22]–[24].
Several hundred genes have been implicated in P. aeruginosa virulence based on data obtained from a wide variety of host infection models (Pseudomonas.com). Many of the well-studied P. aeruginosa virulence-related factors participate directly or indirectly in physical interactions with the host cell and/or host proteins, including secretion systems (type II, type III, type VI) and associated effectors (including ExoT, ExoU, ExoS, ToxA, phospholipase C, and alkaline protease), flagella, and structures involved in attachment to host cells such as type IV pili. Other recognized virulence factors include those involved in quorum sensing (AHL and PQS systems), iron acquisition (pyochelin, pyoverdine), small molecule/toxin synthesis (phenazines, hydrogen cyanide), alginate, LPS, and biofilm. Not all of these classes of virulence-related factors play a significant role in P. aeruginosa strain PA14 virulence in C. elegans; for example, the Type III secretion system and its associated virulence effectors have been shown to play no detectable role in nematode killing, in contrast to playing key roles in pathogenesis in mammals and insects [25]. However, a variety of P. aeruginosa PA14 virulence factors required for killing C. elegans in the SK infection model are also required for full pathogenesis in mammalian models, including the quorum sensing regulators LasR and RhlR, the two component regulator GacA, the alternate sigma factor RpoN, the periplasmic protease MucD, and the phosphoenolpyruvate-protein phosphotransferase and transcriptional regulator PtsP [13], [26]–[29]. Additionally P. aeruginosa virulence-related factors involved in LPS biogenesis and type IV pilus assembly and function also play a role in both mammalian and C. elegans hosts [30]–[33].
A common theme that has emerged from the study of bacterial virulence in a wide variety of pathogens and hosts is an association linking virulence-related genes with regions of genomic plasticity, including genomic pathogenicity islands (PAIs) [34], so-called “replacement islands” harboring the pyoverdine [35] and O-antigen biosynthetic loci [36], and plasmids [37]. These findings indicate that horizontal gene transfer has played an important role in the evolution of virulence. For example, phylogenetic analysis of three sub-families of the type III effector HopZ in the plant pathogen Pseudomonas syringae, suggested that at least two were acquired by P. syringae from disparate donors [38]. Analysis of the occurrence of virulence factors across many pathogen genomes has suggested that there is an overrepresentation of virulence factors on genomic islands [39], and two virulence factor- containing pathogenicity islands, PAPI-I and PAPI-II, have been identified in P. aeruginosa [40].
Although there are many published examples linking virulence-related factors to putative pathogenicity islands, a preliminary study from our laboratory showed that the presence of genes occurring in the highly virulent strain PA14, but not in the less virulent strain PAO1, could not be correlated with increased virulence across a wider sampling of strains, suggesting that virulence is a combinatorial and multifactorial product of the interactions of many potential virulence factors [32]. These data were seemingly at odds with the expected over-representation of virulence factors in strain-specific regions such as genomic islands, but were not definitive because only a limited set of virulence factors were available for analysis. Further, comparison of the sequences of five pathogenic P. aeruginosa strains suggested that virulence was primarily encoded by a core P. aeruginosa genome [41], a set of genes shared by all strains, and not the auxiliary genome defined by regions of genomic plasticity that are strain-specific. An unbiased comprehensive list of P. aeruginosa virulence factors required to cause disease in C. elegans would allow us to better understand what genes are the major contributors to virulence and whether these genes are primarily located in regions of genome plasticity or not. We considered this question worthy of investigation because it seemed likely to us that the virulence factors of an opportunistic multi-host pathogen might as a group be distinct from the virulence factors of host-specific pathogens.
We report here the results of a genome-wide screen using a previously constructed non-redundant PA14 transposon library consisting of 5850 members that represents insertions in approximately 80% of the non-essential ORFs in P. aeruginosa strain PA14 [42]. Previous studies to identify P. aeruginosa virulence factors in vivo using a number of different technologies and infection models have been limited by the complexity and redundancy of mutant collections or screening procedures [13], [26], [27], [43]–[51]. We examined the genes identified in this genome-wide screen for their functions, presence on putative and characterized genomic islands, and their phylogenetic distribution across prokaryotes. We demonstrate that the major genes contributing to PA14 virulence in C. elegans are not enriched on genomic islands, are not PA14 or P. aeruginosa specific genes, and may in fact be biased for ancient genes common to many other prokaryotic species.
Results
Genome-Wide Screen for PA14 Mutants Attenuated in C. elegans Killing
Primary screen
A non-redundant (NR) library of MAR2xT7 transposon insertion mutants representing approximately 75% of the total and 80% of the non-essential ORFs in P. aeruginosa PA14 was screened for attenuation of virulence in an infection-based model of C. elegans killing (see Figure 1, Figure S1 and Table S1 for an overall scheme of the screen). In this infection assay, called “slow killing” (SK), nematode death requires live bacteria and correlates with accumulation of bacteria in the nematode intestine [12], [16]. Ideally, we would have screened the entire non-redundant transposon library using the SK assay, but the relatively large number of mutants (5850) in the library made direct quantitation of killing kinetics impractical for a primary screen. In previous work [13] and in new experiments with known virulence-attenuated mutants (Figure S2), we observed that the quantity and maturity of the brood size of an infected hermaphroditic worm is generally inversely correlated with the virulence of the infecting PA14 strain. Under our standard assay conditions, wild-type PA14 kills C. elegans rapidly enough to dramatically diminish the number of progeny produced, and the few larvae that hatch do not reach maturity. However, PA14 mutants with reduced virulence (such as gacA, lasR, mucD; Figure S2) allow the C. elegans hermaphrodites to produce a significant brood that is able to reach adulthood and in turn become gravid, often consuming the entire bacterial lawn. Based on these data, we concluded that we could use PA14-mediated nematode brood size reduction, a much less time consuming assay than monitoring death of a population of worms, as a proxy for PA14-mediated killing.
Figure 1. Pipeline of screen for PA14 virulence-attenuated mutants in C. elegans.

The three screening steps for identification of P. aeruginosa PA14 virulence-attenuated mutants are outlined; details of the screens are presented in the Materials and Methods and the text. The number of mutants obtained after each round of screening, as well as those removed from the pool for various reasons, is shown. Note that the 313 mutants identified in the primary screen and the 180 from the secondary screen represent 294 and 170 unique genes respectively because some genes were represented by multiple mutants, and a small fraction of mutants were in intergenic regions (see text). In the tertiary screen a single mutant defined each gene.
A total of 5,754 mutants (the complete 5,850 PA14 NR library minus one 96-well plate consisting of previously characterized slow-growing mutants) was screened twice on SK agar in standard 6-well assay plates for mutants that permitted an increase in C. elegans progeny number or allowed the brood to mature to adulthood when compared to wild-type PA14. In this primary screen, 399 mutants (corresponding to 368 genes and three mutants in intergenic regions) were identified as potentially attenuated in virulence (Figure 1, Figure S1 and Table S1). From the set of 399 putative virulence-attenuated mutants, 86 auxotrophic or growth-defective mutants corresponding to 74 genes (Table S2) were identified by replica plating on minimal medium. Some but not all of the 86 auxotrophic mutants formed noticeably thin lawns on the SK agar plates. We reasoned that the 86 mutants with fundamental growth defects were not relevant to our study of virulence and we eliminated them from future studies. This left 313 putative virulence-related mutants corresponding to 294 distinct genes and three mutants in intergenic regions (Table S3).
Secondary screen
The 313 putative PA14 virulence-related mutants that did not appear to be dependent on the addition of amino acids or nucleotides for growth on minimal media were re-tested using C. elegans slow killing assays. Specifically, the survival of 60–80 wild-type N2 Bristol worms was quantified at two or three different time points following manual transfer of individual worms from E. coli OP50 to a lawn of each PA14 mutant. In addition, the number of progeny produced was scored after four days. Each batch of mutants tested in the secondary screen included two known attenuated mutants, the strongly avirulent quorum sensing regulatory mutant lasR and the moderately attenuated type IV pilus protein mutant pilA. The mutants from the secondary screen were ranked with respect to these controls. About 63% (198) of the 313 non-auxotrophic mutants from the primary screen exhibited an attenuated killing phenotype or increased brood size in the secondary screen (Figure 1, Table S1). The insertion sites of MAR2xT7 in the attenuated mutants from the secondary screen were re-sequenced to verify their identity and the mutants were rescored for readily apparent growth defects in overnight cultures or on plates. As a result, 18 mutants that had either incorrectly annotated sequence or were slow growing were removed from the list. Of the 180 remaining virulence-attenuated mutants from the secondary screen (representing 170 genes and one mutant in an intergenic region), 34 were strongly attenuated (similar to lasR), 76 were moderately attenuated (greater than or equal to pilA), and 70 were weakly attenuated (less than pilA but allowing greater parental or progeny survival than wild-type PA14) (Table S4).
Previously in our laboratory, 17 PA14 mutants were shown to exhibit an attenuated phenotype in a standard C. elegans SK assay. Of these 17, 15 were identified in relatively small-scale forward genetic screens (aefA, lasR, mtrR, ptsP, gacA, gacS, PA14_03370, ORF_10 (PA14_23420), ORF_11 (PA14_23430), PA14_27680 (GID6172), PA14_27700 (GID6170), pilC, PA14_59010, PA14_59070 and pilW) and two by a candidate gene approach testing predicted virulence factors (rpoN, mucD). Of these 17 genes, 16 are represented by mutants in the PA14 non-redundant library (lasR is absent). One of these 16, rpoN, grows very slowly under our conditions and was not assayed. Thus 15 previously identified mutants could potentially have been recovered in the screen of the non-redundant library. In fact, nine of these 15 previously identified mutants were identified in the primary screen and eight of these nine also scored as positives in the secondary screen (strongly attenuated ptsP, gacA, gacS, and PA14_27700 (GID6170), moderately attenuated i.e. close to the attenuation of pilA, ORF_11, mucD, and PA14_27680 (GID6172), and very weakly attenuated PA14_03370). At least four of the remaining seven previously identified virulence-related genes (ORF_10, pilC, PA14_59010, PA14_59070) that we did not isolate in the secondary screen exhibited virulence-related phenotypes approximately equal to pilA, which has a phenotype at the lower limit of sensitivity for recovery in the progeny-based screen (note an ORF-10 mutant was recovered in the primary screen but not the secondary). Although pilC and ORF_10 were not recovered in the secondary screen, other type IV pilus and O-antigen synthesis mutants (pilF, pilW, pilU and ORF_11) were identified. Based on these data, we conclude that the genome-wide screen that we carried out to identify PA14 virulence-related factors was successful in identifying the majority of the virulence-related mutants in the non-redundant library with strong killing-attenuated phenotypes, but that many potential mutants with relatively weak attenuated phenotypes were probably overlooked.
We compared the genes identified in our screen to a set of 241 P. aeruginosa strain PA14 virulence-related genes downloaded from the Virulence Factor Database (VFDB). VFDB is, a compilation of virulence factors from a wide variety of pathogens in numerous host systems that includes virulence factor sets for PA14 and for three other P. aeruginosa isolates [52] (http://www.mgc.ac.cn/VFs/). The VFDB set for PA14 incorporates well-studied virulence factor classes, most abundantly those involved in adherence/motility (flagella, type IV pilus, LPS), alginate, rhamnolipids, iron uptake (pyochelin and pyoverdine), quorum sensing, global regulators (GacA/GacS), proteases (alkaline protease, LasA, LasB), lipases (PlcH, PlcN), secretion systems and associated effectors (type III, type VI), pyocyanin pigment and toxins (ToxA, hydrogen cyanide). The degree of overlap between the VFDB genes and those identified in our screen increased between the primary and secondary round of screening, with VFDB genes constituting 10.2% (30 of 294) of the genes identified in the primary screen and 11.8% (20 of 170) of the genes identified in the secondary screen (Figure S3).
The collection of avirulent secondary positive hits does not exhibit a strong functional bias
The 170 genes (represented by 180 mutants) from the secondary screen were grouped by the 27 functional protein classes used in the annotation of the PA14 genome (http://ausubellab.mgh.harvard.edu/cgi-bin/pa14/annotation/statistics.cgi) and the fraction of genes in each functional class was compared to that of the total PA14 genome (Table S4). Only one functional class, LPS and capsule biosynthesis, was marginally statistically overrepresented (p-value = 0.013) after FDR correction. In addition, the 170 genes were mapped onto the KEGG pathway database to determine whether particular biochemical pathways were enriched and were categorized by GO term with DAVID. Neither the KEGG pathway nor the GO term analysis revealed any significant overrepresentation of pathways or GO terms.
The PA14 secondary virulence screen did not enrich for known secretion systems or secreted effectors
Canonical virulence factors such as those present in VFDB are enriched in extracellular proteins (10% in VFDB vs. a little over 1% in the PA14 genome and NR mutant set) and in various secretion systems (22% in VFDB vs. approximately 2% in the PA14 genome). In contrast, mutations with consistent or strong phenotypes in any of the known secretion systems or effectors were significantly underrepresented in our genome-wide screen. Among 62 documented PA14 secreted proteins and their chaperones [53] (PA14 database at http://ausubellab.mgh.harvard.edu/cgi-bin/pa14/annotation/start.cgi), 55 of which correspond to mutants in the PA14 NR Set, only one was also found in the 180 virulence-attenuated mutant set from the secondary screen. Similarly, although our primary screen identified 15 of 97 NR set mutants annotated to be secretion apparatus proteins or their chaperones (type II, III, V and VI), only six of these were retained after the secondary screen. Three of the secretion apparatus loci mutants isolated in the secondary screen (type II secretion loci xcpT, xcpZ and secB) exhibited only slight attenuation of virulence and three had moderate attenuation of virulence roughly equivalent to the pilA mutant control (typeII tatC and type VI HSI-I locus clpVI and type VI HIS-II fha2). Although we isolated two type VI mutants in our screen it is unclear what this means as many of the type VI structural loci were not identified, and two large deletions of type VI HSI-II and HIS-III had little effect on virulence (data not shown). These data do not strongly implicate a particular secretion system as predominant in PA14 virulence in C. elegans and are consistent with previous work from our lab, which showed that the type III secretion system mutants had no detectable attenuation of virulence in C. elegans [25]. On the other hand, the failure to observe a phenotype with the secretion defective mutants may be a consequence of built-in redundancy.
Comparison of the 170 P. aeruginosa putative virulence genes identified in C. elegans with genes required for virulence in a rat chronic infection model
In order to evaluate the degree to which virulence genes from our screen represent putative conserved virulence determinants, we compared the 170 genes identified in our secondary screen with the largest available set of genes identified in another unbiased screen, a negative signature tagged mutagenesis (STM) selection for P. aeruginosa mutants defective in virulence in a rat chronic respiratory infection model [46]. It is notable that like the 170 genes identified in our screen, and in contrast with the VFDB set, the 148 P. aeruginosa genes identified by Potvin et al (2003) by STM also appear as a group to possess a broad distribution across all functional classes. The P. aeruginosa mutant set identified in the rat chronic infection model exhibits an underrepresentation of secreted proteins and secretion systems and includes a number of auxotrophic mutants and many mutants in genes with enzymatic functions not previously linked to pathogenesis, reminiscent of the mutants identified in C. elegans. Only five genes identified in the rat infection model were also found in the 170 gene set from our secondary screen and only one of these was also found in the VFDB (Figure S4). A number of well-studied P. aeruginosa virulence factors, required for slow killing in C. elegans and in some mammalian infection models, including gacA, lasR, rhlR, ptsP, mucD, and rpoN, are absent from the rat chronic infection set. We do not know if this reflects a difference between chronic and acute infections, nor whether the C. elegans model is more analogous to acute or chronic infection in mammals. These data suggest that genes required for virulence under a particular set of circumstances are highly dependent on host model, infection site, and most likely phase of infection.
Tertiary screen
The relatively large number of positive mutants (180 corresponding to 170 genes) from the secondary screen made it necessary to prioritize mutants for further characterization. A subset of 58 mutants/genes was selected based on strength of attenuation, annotation (biased toward regulators and mutants with functions previously implicated in virulence), whether they were in putative operons or functionally grouped with other attenuated mutants, and whether they exhibited normal doubling times (see Materials and Methods). These mutants (Table S5) included 31 mutants with strongly attenuated virulence similar to that of the lasR quorum sensing mutant (34 such mutants were isolated but gacA, ptsP and pepP were represented by two mutants each and a single allele of these three genes was carried through the tertiary screen), 22 mutants with moderately attenuated virulence (less attenuated than lasR but equal to or more attenuated than pilA), and five weakly attenuated mutants (less attenuated than pilA).
In the tertiary screen, each of the 58 selected mutants was subjected to SK assays using 100–150 fer-15(b26)II;fem-1(hc17)IV worms and the time to 50% survival was determined. Sterile fer-15;fem-1 worms were used in these assays to avoid production of a brood that would complicate the scoring of death of the parental worms. fer-15;fem-1 animals, while slightly more resistant to PA14 than N2 worms, have previously been used to study the transcriptional response of C. elegans to PA14 [18]. Importantly, wild-type N2 worms and fer-15;fem-1 worms exhibit comparable relative susceptibilities to a gacA mutant [18]. Four mutants (PA4296, rcsC, bkdA1, and norB) did not re-test as avirulent in the tertiary assay, three (in ORFs PA2658, PA14_4560 and PA4745) exhibited impaired growth in LB overnight cultures that had been previously overlooked and two mutants were removed based on ambiguity as to which ORF they were inactivating (mutants #38436 and #5691) (Table S5). Thus, the tertiary screen substantiated the avirulent phenotype of 49 of the 58 mutants.
Use of the Master Transposon Library to Verify Phenotypes and Examine the Genomic or Functional Context of Putative Virulence-Attenuated Mutants
Multiple transposon alleles confirm avirulent phenotypes
One of the bottlenecks in a large-scale screen such as the one described in this paper is verification of the mutant phenotypes. In general, the gold standard to correlate phenotype with genotype is to generate an in-frame deletion mutant and then complement the mutant in trans to establish that the phenotype observed is due to the defect in the identified gene. Because the goal of our screen was to obtain an overview of the major factors utilized by PA14 to infect and kill C. elegans, we were interested in characterizing a large collection of mutants. Generation of in-frame deletions for all the genes of interest was prohibitively time-consuming. As an alternative, we reasoned that by examining multiple independent MAR2xT7 insertion mutations in candidate virulence-related genes, as well as insertions in adjacent genes within putative operons, we could: a) verify the avirulent phenotype of the original mutation, b) reduce the likelihood that the avirulent phenotype was due to polar effects of the original transposon insertion on downstream ORFs, and c) eliminate the possibility that second-site mutations were responsible for the less virulent phenotype. The NR PA14 library of 5,850 mutants was selected from more that 24,000 sequenced MAR2xT7 transposon insertion mutations and on average each gene in the NR library is represented by 4.3 transposon insertions in this “master” library of 24,000 mutants [42].
Among the mutations in the 49 genes that re-tested as avirulent in the tertiary screen, there were 44 genes for which there was more than one corresponding transposon insertion. (However, three genes for which multiple insertion alleles existed, gacS, pilF and aruD, were only tested with single alleles. gacS and pilF had been previously implicated in virulence in C. elegans and in the case of aruD, the entire aru operon was shown to be virulence-attenuated (Figure 2 and Figure 3). A total of 72 additional insertion mutants corresponding to 41 of the 49 genes were tested in SK assays. These assays resulted in the elimination of eight genes (PA2089, PA0902, PA1032, PA0533, PA4016, clpS, sltB1 and pvdD) from further consideration because the additional transposon insertions in these genes did not cause an attenuated killing phenotype (representative data are shown for clpS (Figure 4) and PA4016 (Figure 5B). In the case of clpS, for example, in contrast to the virulence-attenuated clpS NR allele mutant ID#34203 isolated in the screen (LT50 mutant/LT50 wild-type (WT) PA14 = 1.66), three additional independent alleles of clpS all exhibited killing kinetics similar to WT PA14 (LT50 mutant/LT50 WT PA14 = 1.11, 1.10 and 0.98 respectively) suggesting that the phenotype of mutant #34203 was aberrant.
Figure 2. 41 PA14 genes required for virulence in a C. elegans infection based killing model.

The ratio of nematode survival on mutant PA14 to that on wild-type PA14 (mutant LT50/WT LT50) is presented for 41 mutants identified after three rounds of screening as well as for the known virulence-attenuated mutants, lasR and pilA. The time to 50% death (LT50) was calculated using a non-linear regression based on the Hill equation (Prism 5.0). 100–150 animals were tested in each experiment. Error bars represent the SEM of the ratios derived from at least two different experiments (lack of error bars indicates that the mutants for known virulence factors gacA, ptsP and vfr were tested only once). Red bars depict the ratio of the LT50 of lasR or pilA to WT PA14. The lasR and pilA mutants were generated previously (see Materials and Methods); there are no alleles of lasR or pilA in the NR library. The number of alleles tested with an avirulent phenotype is indicated by a number below the graph: 1 indicates that a single allele was tested but that there exist multiple alleles in the master transposon library, S indicates only a single allele was available in the library. Genes that are predicted to be in operons are indicated (Y = yes, N = no). Genes in a single operon are represented in the same color and an underline designates that other genes within the same operon were tested for their role in virulence.
Figure 3. The catabolic arginine succinyltransferase (aru) operon is required for normal virulence in C. elegans.
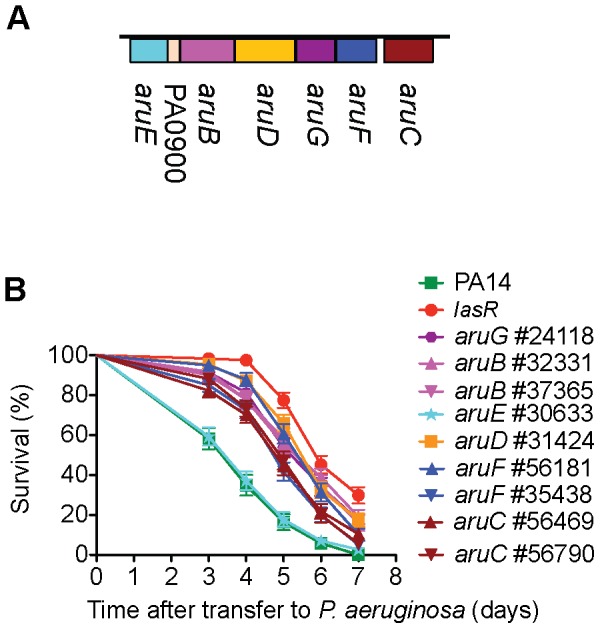
A) aruFGDB is transcribed as a unit; the transcriptional regulator aruC is transcribed separately. The aruFGDB operon encodes enzymes for the major aerobic route of arginine utilization as an energy, carbon and nitrogen source [56], [57]. In P. aeruginosa PA01, aruE belongs to a separate transcription unit [57]. B) MAR2xT7 insertions in aruC, aruF, aruG, aruD and aruB all reduce the virulence of PA14. The single mutation in aruE has normal virulence.
Figure 4. Multiple transposon alleles of clpA, but not clpS, are virulence-attenuated.
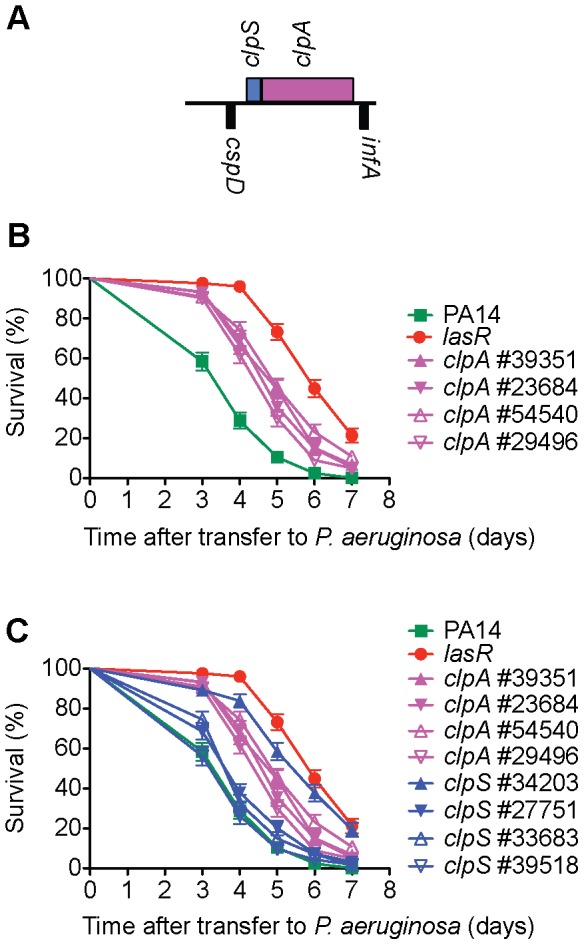
ClpA is the chaperone subunit responsible for substrate recognition of the ClpAP ATP dependent protease common to Gram-negative proteobacteria [96]. A) clpA (PA2620) is the second gene of a two gene operon; it is preceded by clpS (PA2621), encoding a ClpAP adaptor protein that has been shown to bind to the N-terminus of ClpA and inhibit ClpAP degradation of some substrates while enhancing the degradation of others [97]. B) Four different MAR2xT7 transposon insertion alleles of clpA are decreased in virulence in C. elegans. C) Three of four MAR2xT7 insertion alleles of clpS exhibit wild-type levels of virulence. Only the clpS mutant (#34203) identified in the primary and secondary screens has a virulence-attenuated phenotype. A mutant in the ClpP proteolytic subunit (#52957) was also identified in our primary screen for virulence-attenuated mutants, but this mutant was defective in growth on minimal media and therefore was not analyzed further.
Figure 5. ΔexoU may be a sensitized background that can reveal virulence-associated genes.
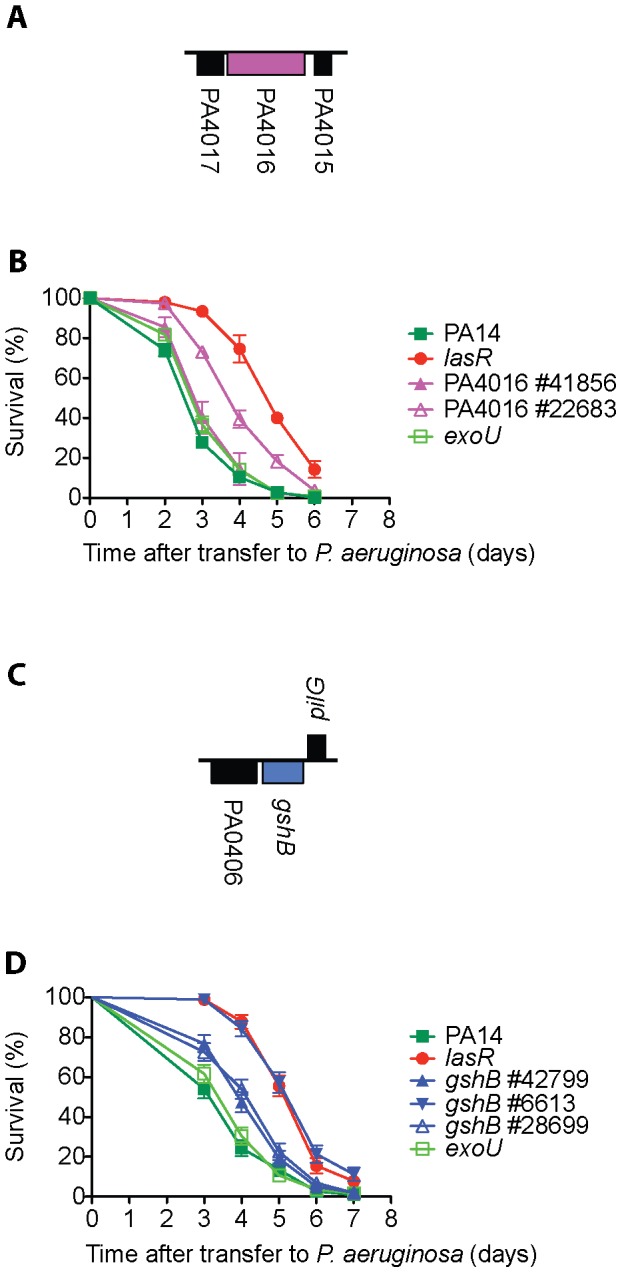
Deletion of the Type III effector protein ExoU has no statistically significant impact on PA14 virulence in C. elegans (B and D and [25]). A) Hypothetical protein PA4016 and adjacent loci PA4017 and PA4015; PA4016 is most likely a single gene transcription unit. B) A PA4016 MAR2xT7 insertion mutant (#22683) in the ΔexoU strain background has attenuated virulence in C. elegans, but a second PA4016 insertion allele (#41856) in the WT strain does not. C) Glutathione synthetase, gshB (PA0407), is a single gene transcription unit. D) Multiple alleles of gshB exhibit reduced virulence in C. elegans but the gshB #6613 allele in the ΔexoU strain background is more attenuated.
ΔexoU, a potential sensitized genetic background
We noticed that in 4/8 cases where the phenotype of the mutant identified in the screen was not recapitulated by additional alleles (putative transcriptional regulator PA0533 mutant #22525, probable penicillin amidase PA1032 mutant #6114, hypothetical protein PA4016 mutant #22683, and pvdD PA2399 mutant #5205), the original allele was in a ΔexoU background, which carries an in-frame deletion of the type III effector ExoU [42]. The ΔexoU mutant behaved similarly to wild-type PA14 in nematode killing assays (Figure 5B, D and [25]). The observation that 50% of the mutants in which the original phenotype did not recapitulate with multiple alleles also contained a ΔexoU mutation despite the fact that less than 4% of the transposon insertions in the NR library are in the ΔexoU background suggests that the mutation in exoU might create a sensitized genetic background to identify other putative virulence factors. In support of this conclusion, two alleles of gshB (#42799 identified in our screen and #28669 from the master library) exhibited modest virulence attenuation (LT50 mutant/LT50 WT PA14 = 1.25 and 1.30 respectively), whereas a third gshB insertion allele in the ΔexoU background (#6613) showed a considerably stronger attenuated phenotype (LT50 mutant/LT50 WT PA14 = 1.68) (Figure 5D). Construction of a series of double mutants carrying ΔexoU (or perhaps other mutations in the type III secretion apparatus or effectors) and mutations in other virulence loci would help to clarify the role of PA14 ExoU in pathogenesis of C. elegans.
Features of 41 PA14 tertiary set genes required for virulence in C. elegans
Figure 2 shows the relative virulence of the 41 mutants (corresponding to 41 genes) out of the 58 tested whose phenotypes were confirmed in the tertiary screen (Table S5, Table S6). Of the 41 genes, 21 have been previously implicated in P. aeruginosa virulence in at least one host and an additional 4 genes have been identified as virulence-related in other pathogens (Table 1). Thus 20 of the 41 (49%) genes identified in our screen are novel P. aeruginosa virulence-related genes. None of these 41 mutants exhibited significant growth defects and in 33 cases, the virulence-related phenotypes were verified by two or more independent MAR2xT7 transposon insertions. In Figure 2, the ratio of the time to 50% nematode survival on each mutant to the 50% survival time on wild-type PA14 is presented (the average value of the ratio from multiple experiments is shown in most cases). The mutants for which killing assays were not repeated (gacA, ptsP, vfR) had been previously demonstrated to be virulence-attenuated in C. elegans in published studies [13], [54]. The ratios of 50% survival time of the positive lasR and pilA controls to WT PA14 were 1.63 (SEM = 0.08 for 10 independent experiments) and 1.22 (SEM = 0.04 for 10 independent experiments), respectively, and are shown as red bars in Figure 2. Among these 41 genes, five (indicated by “S” in Figure 2; pchH, lysC, PA1592, aruG, fha2) were represented by a single allele in the master library and the avirulent phenotypes corresponding to these insertions could not be verified by an independent insertion allele. The data shown in Figure 2 are for the mutant allele identified in the initial screen from the NR library (in the case where two alleles were identified a single allele was chosen). Examples of C. elegans survival curves (representative curves of assays repeated at least twice) from which the data in Figure 2 are derived are shown for clpA (Figure 4B), gshB (Figure 5D), pchH and pchI (Figure 6B), and aruC, aruG, aruB, aruD (Figure 3B). Additional survival curves are shown in the Supporting Information for pepP (Figure S5B), PA0456 (Figure S6B), kinB (Figure S7B), PA14_27700 (Figure S8B), PA0745 (Figure S9B), vqsR (Figure S10B) and gshA (Figure S11B).
Table 1. P. aeruginosa PA14 virulence-attenuated genes identified in the C. elegans infection model.
| Locus | Description | P. aeruginosa host | Virulence-attenuated |
| gacA | two-component regulator | nematode, mouse[13] | |
| Vfr | cAMP dependent transcriptional regulator | mouse (PAK)[109] | |
| pchH | ABC transporter | slime mold, fly, mouse (22D10)[99] | |
| PA4005 | conserved hypothetical protein | ||
| PA14_27700 | putative transcriptional regulator | nematode[32] | |
| pepP | proline aminopeptidase | ||
| hemK | probable translation release factor methyltransferase | ||
| lysC | aspartokinase | ||
| vqsR | transcriptional regulator | nematode (TB toxin killing)[108] | |
| kinB | two-component sensor | zebrafish, mouse (PA01)[86], [103] | |
| ptsP | Phosphoenolpyruvate-protein phosphotransferase | nematode, mouse[13] | |
| lasR | HSL quorum sensing regulator | nematode, mouse[13], [110], [111] | |
| PA0745 | putative enoyl-CoA hydratase isomerase | nematode (PA01 cyanide)[54] | |
| rhlR | HSL quorum sensing regulator | nematode[111] | |
| PA2550 | putative acyl-CoA dehydrogenase | ||
| Mind | cell division inhibitory membrane ATPase | F. tularensis [112] | |
| PA1592 | hypothetical protein | ||
| glnK | nitrogen assimilation signal transduction protein | S. typhimurium [76] | |
| aruD | arginine catabolism | ||
| gshA | glutathione biosynthesis | ||
| PA2015 | putative isovaleryl-CoA dehydrogenase | ||
| PA0456 | cold shock domain protein | ||
| prpC | methylcitrate cycle propionate metabolism | nematode (PA01 cyanide)[54] | M. tuberculosis [113] |
| gacS | two-component sensor kinase | nematode, mouse[13] , [54] | |
| lasI | quorum sensing HSL production | mouse (PA01)[28] | |
| pchI | ABC transporter | slime mold, fly, mouse[99] | |
| aruG | arginine catabolism | ||
| fabF1 | fatty acid biosynthesis | fly, mouse[45] | |
| clpA | ClpP protease chaperone & ATPase | H. pylori [114] | |
| aruB | arginine catabolism | ||
| PA1766 | conserved hypothetical protein | ||
| PA1216 | hypothetical protein | ||
| pqsE | quorum sensing regulation | mouse[115] | |
| kdpD | two-component sensor kinase | S. typhimurium [76] | |
| PA1767 | hypothetical cytoplasmic membrane protein | ||
| prpB | methylcitrate cycle propionate metabolism | nematode (PA01 cyanide)[54] | |
| fha2 | type VI secretion protein | ||
| gshB | glutathione biosynthesis | ||
| aruC | arginine catabolism | ||
| wbpL | LPS O antigen synthesis | nematode, mouse[32] , [33] | |
| pilA | type IV pilus protein | nematode, mouse, human cells[32] , [116], [117] | |
| pilF | type IV pilus protein | nematode, mouse, human cells[32] , [116], [117] | |
| ORF_11 | LPS O antigen synthesis | nematode, mouse[32] , [33] |
Genes are listed in descending order of contribution to virulence (according to the ratio of mutant LT50/wild-type LT50) using the data from Figure 2. Genes previously identified as required for normal levels of P. aeruginosa virulence in various model systems are indicated. In some cases only P. aeruginosa strains other than PA14 were examined and the strain and mode of killing is indicated in parentheses. The other pathogens, in which orthologs of these genes have been implicated in virulence, are noted in the last column.
Figure 6. PchH and PchI but not pyochelin are required for normal virulence of PA14 in C. elegans.
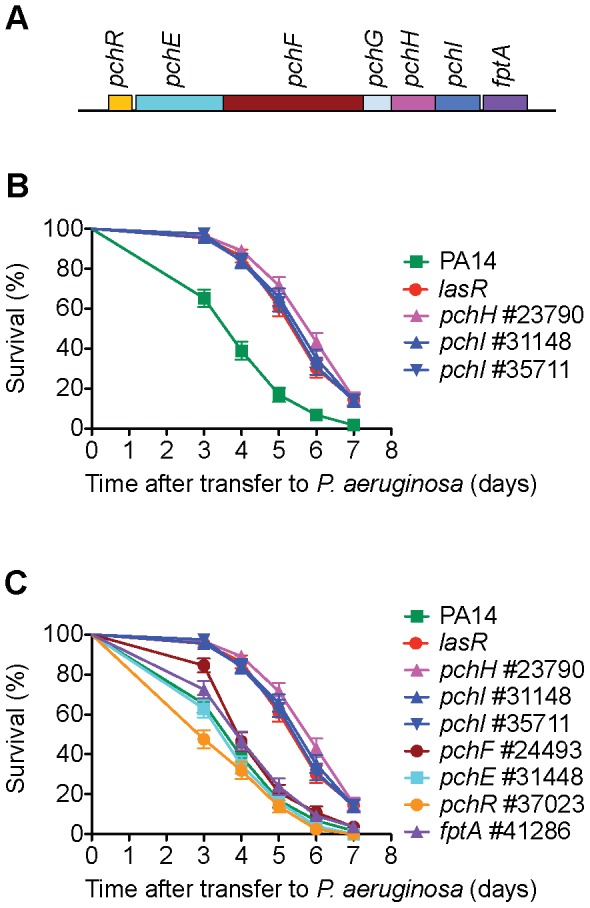
A) pchH and pchI encode putative ABC transporters with potential export functions and are the two terminal genes in a pyochelin biosynthetic operon [98]. B) Two MAR2xT7 transposon insertions in pchI and the single available MAR2xT7 allele of pchH are virulence-attenuated. C) Transposon insertion mutations in either the pyochelin biosynthetic genes pchE and pchF or the outer membrane transporter fptA, which transports pyochelin complexed with iron into the cell, have little effect on virulence. Mutations in pchH and pchI have been previously shown to produce wild-type levels of pyochelin in culture supernatant [98] and exhibit attenuated virulence in a neutropenic mouse model [99].
Additional transposon mutants used to examine the role of identified operons
In bacteria, genes that function in common processes are frequently co-regulated in operons or clustered in the genome. We utilized the public PA14 database constructed in our laboratory [http://ausubellab.mgh.harvard.edu/cgi-bin/pa14] and BIOCYC [http://biocyc.org/PAER208963/NEW-IMAGE?object=Transcription-Units] to predict whether the genes corresponding to the avirulent mutants identified were in operons, based on annotation and the proximity and direction of transcription of adjacent genes. Of the 41 genes shown in Figure 2, our analysis suggested that 27 are located in 22 putative operons (see Figure 2). Multiple insertion mutations in three operons were identified in our screen, four in the aru operon (aruC, aruG, aruD, aruB), two in a pyochelin biosynthetic operon (pchH, pchI), and two (PA1766, PA1767) in an operon of unknown function. Utilizing the master insertion library of 24,000 mutants, we tested an additional 38 mutants (corresponding to 24 genes) in 11/22 of the putative operons and thereby identified six more genes in five operons that resulted in virulence attenuation when mutated: aruF, which is part of the aru operon; ORF_10, which had been isolated in a previous screen and together with ORF_11 forms a predicted transcription unit involved in O-antigen biosynthesis [32]; pqsA, which is required for synthesis of the quinolone quorum sensing molecule located upstream in the pqsE operon [55]; PA1218 and PA1221, which are upstream of PA1216 in an operon of unknown function; and PA4000 at the end of the operon containing PA4005. Note that mutations in proA and nadD directly upstream of PA4005 in the operon were identified in our primary screen but discarded because they are auxotrophs. Interestingly, in the case of 5/11 putative operons (containing pepP, kinB, PA0745, clpA and fha2) for which mutations in multiple genes were analyzed, only mutation of a single gene in the operon resulted in an attenuated phenotype. Two genes in the final operon examined (pchH, pchI) were identified in the primary and secondary screens, but mutations in the other genes in this pyochelin operon had no significant effect on virulence (Figure 6C). It is important to keep in mind that not all the genes in each of the operons examined were represented by MAR2xT7 insertions in the master library.
Our finding that mutation of some but not all genes in an operon affect virulence may indicate redundant functions (perhaps located elsewhere in the genome) for those with no mutant phenotype or unique roles in virulence for specific genes within the operon. The aru operon, which encodes the catabolic enzymes for aerobic utilization of arginine as a carbon, nitrogen and energy source [56], [57], represented an unusual case where 5/5 genes in the aruCFDGB operon all exhibited a similar moderate attenuation of virulence when mutated. The aru insertion mutations did not observably affect growth of the bacteria on SK plates (although a single mutant in aruF #56181 had somewhat reduced growth on minimal media in the absence of nucleotides and amino acids) suggesting that the catabolism of arginine may be important for growth or virulence of the bacteria within the nematode. Interestingly, mutation of aruE, which is located downstream and adjacent to the aruFGDB operon, did not result in an avirulent phenotype. In the P. aeruginosa strain PA01 aruE is reported to be a separate transcription unit [57] (Figure 3).
Virulence-Related Phenotypes of Selected Mutants
Seven putative virulence-related factors, cold shock domain protein PA0456, ABC transporters PchH and PchI, aminopeptidase PepP, putative enoyl-CoA hydratase/isomerase PA0745, ATPase/molecular chaperone ClpA, and putative transcriptional regulator PA14_27700 were chosen for further study. Mutants corresponding to these factors (clpA, Figure 4B; pchH and pchI, Figure 6B; pepP, Figure S5B; PA0456, Figure S6B; PA14_27700, Figure S8B; and PA0745, Figure S9B, C) all have a strong avirulent phenotype in C. elegans, exhibit normal growth kinetics in vitro (Figure S12), and represent genes whose role in P. aeruginosa virulence has not been previously characterized. The avirulent phenotype of all these mutants was confirmed with multiple transposon alleles except for pchH for which there is only a single allele available. In addition, in the case of PA0745, an in-frame deletion mutant was generated that was severely impaired in virulence, similar to the transposon allele #37629 isolated in the screen (Figure S9C).
Many of the genes previously identified as necessary for virulence of PA14 in C. elegans, for example those coding for the quorum sensing regulators RhlR and LasR, are known regulators of multiple virulence factors or virulence associated pathways [58], [59]. Both lasR and rhlR mutants have a spectrum of pigment and motility defects. lasR and rhlR mutants produce reduced levels of the blue-green pigment pyocyanin, rhlR produces no pyocyanin, and lasR mutants produce varying amounts dependent on conditions and growth phase [60]. Under certain growth conditions, rhlR and lasR mutants have been reported to produce less of the fluorescent siderophore pyoverdine [61]. lasR and rhlR mutants also exhibit dramatically reduced swarming motility [62], which is dependent on both the type IV pilus and the flagella and regulated by quorum sensing and a host of transcription factors [63].
We tested the seven selected virulence-related mutants for defects in motility (twitching, swimming and swarming assays) as well as for pyocyanin and pyoverdine production in comparison to lasR and rhlR mutants to determine whether they had a similar spectrum of defects and/or could be classified into groups based on common pigment or motility phenotypes (Table 2). It should be noted that of these phenotypes, only mutants in which type IV pilus function is affected have been shown to exhibit reduced virulence in the C. elegans infection model; pyocyanin does not appear to be necessary for virulence in the SK model and the roles of pyoverdine production, swimming and swarming have not been directly tested [12], [14]. Significantly, with the exception of PA14_27700, all of the mutants exhibited defects in some aspect of motility or pigment production. Mutation of putative cold shock protein PA0456 diminished pyocyanin production as did the quorum sensing regulators lasR and rhlR, whereas a pepP mutant had elevated pyocyanin levels. The putative enoyl-CoA hydratase/isomerase PA0745 produced reduced levels of pyoverdine. Among the tested mutants, 4/7 had clear swarming defects, but exhibited normal levels of swimming and twitching motility, suggesting that neither flagella nor type IV pili function was compromised. The clpA mutant was slightly attenuated for swarming and twitching, implying that there might be a type IV pilus defect in this mutant.
Table 2. Pigment and motility phenotypes of seven novel virulence mutants.
| Strain | Pyocyanin | Pyoverdine | Twitching (cm) | Swimming (cm) | Swarming (SK) | Swarming (LB) |
| lasR (deletion) | 0.06±0.01 | 1.10±0.18 | 0.49±0.02 | 2.30±0.12 | ± | − |
| rhlR (deletion) | 0.00 | 1.11±0.17 | 0.53±0.01 | 2.53±0.09 | − | − |
| PA0456 (#36116) | 0.20±0.11 | 1.07±0.04 | 0.53±0.02 | 2.43±0.03 | + | ± |
| PA0745 (deletion) | 1.05±0.04 | 0.33±0.04 | 0.56±0.02 | 2.32±0.05 | ± | ± |
| pchH (#23790) | 0.89±0.12 | 0.99±0.04 | 0.53±0.02 | 2.40±0.03 | + | ± |
| pchI (#35711) | 1.12±0.05 | 1.09±0.07 | 0.60±0.01 | 2.30±0.10 | + | ± |
| pepP (#31907) | 1.46±0.13 | 0.96±0.06 | 0.58±0.02 | 1.93±0.03 | +++ | +++ |
| clpA (#39351) | 0.92±0.01 | 0.88±0.05 | 0.48±0.02 | 2.30±0.12 | + | +++ |
| PA14_27700 (#32578) | 0.98±0.03 | 0.92±0.06 | 0.58±0.02 | 2.40±0.00 | +++ | +++ |
| PA14 WT | 1.00 | 1.00 | 0.59±0.01 | 2.31±0.02 | +++ | +++ |
The average ratio of mutant to wild-type pyocyanin levels from four samples and the SEM is shown. The average ratio of mutant to wild-type pyoverdine levels from four samples and the SEM is shown. Twitching motility (1.5% LB agar) was measured as the radius of growth at the interface of the medium and the polystyrene plate and average radius and SEM from three inoculations is presented. Swimming motility was determined by the diameter of the turbid zone in semi-solid LB agar (0.35%) and average radius and SEM from three inoculations is presented. Swarming levels on the surface of 0.5% agar medium were qualitatively evaluated with number of and length of tendrils taken into account.
Characterization of Genomic and Phylogenetic Distribution of Virulence-Attenuated Genes
The list of PA14 genes identified as being required for full virulence in C. elegans from the genome-wide screen provided the opportunity to examine the distribution within a species and conservation across bacterial species of a large set of genes required for virulence in a single host. We determined whether this set of virulence associated genes was biased towards Pseudomonas core or strain-specific (auxiliary) regions of the P. aeruginosa genome (as defined by Mathee et al. [41] and/or whether these virulence genes were preferentially located on genomic islands, as previously suggested for Pseudomonas virulence factors [39], [40]. In addition, we examined whether the PA14 virulence genes had a narrow phylogenetic distribution (unique to PA14, P. aeruginosa, Pseudomonas, or closely-related organisms) or were broadly distributed across prokaryotic phylogeny.
We used four sets of genes identified in our screen and a set of previously defined PA14 virulence genes downloaded from the Virulence Factor Database (VFDB) for all analyses. The sets of unique genes identified in the primary (294), secondary (170) and tertiary (41) virulence-attenuated screens outlined above and the auxotrophic genes identified in the primary screen but subsequently discarded (76) were used and for simplicity are referred to below as primary, secondary, tertiary, and auxotroph sets. All statistical analyses of the virulence genes identified in the C. elegans screen were done in comparison to the genes represented in the non-redundant (NR) library, as opposed to the entire PA14 genome, because this was the starting set for the screen. We used all four sets of genes in our comparisons to gain statistical power because the final set of 41 verified virulence-related genes was so small that most analyses did not make statistical cutoffs. In addition, using sets of genes from subsequent rounds of screening allowed us to look for enrichment that correlated with the refinement of the screen. To compare the virulence-related genes identified in our screen to previously identified P. aeruginosa genes, we made use of a set of 241 P. aeruginosa strain PA14 virulence genes downloaded from the Virulence Factor Database (VFDB) [52].
PA14 virulence-attenuated genes in the P. aeruginosa core and auxiliary (strain-specific) genome
Among the 5,893 annotated PA14 genes, 5,016 (85%) are core genes in that they are also present in P. aeruginosa strains PA01, PACS2, PA2192 and C3719, four P. aeruginosa clinical isolates [41]. The remaining 15% of the PA14 genes are designated as the auxiliary PA14 genome because they are genes that are missing in at least one of these five sequenced P. aeruginosa strains. The auxiliary genome is dispersed across the chromosome with regions of genomic plasticity containing strain-specific segments bordered by conserved genes. It should be noted that approximately 66% of the auxiliary genome is found in species outside of the Pseudomonas genus [41] and that the designated auxiliary and core genes may change somewhat over time as more P. aeruginosa genomes are sequenced. The distribution of the primary, secondary, and tertiary genes in the core versus auxiliary genome was statistically identical to the distribution of genes in the NR set. That is, roughly 15% of the virulence-related genes were part of the auxiliary genome (Figure 7A, Table S7). By comparison, we performed the same analysis with the auxotrophic genes and as expected for genes necessary for fundamental cellular metabolism, this set was significantly over-represented in the core genome (p-value = 0.009). Therefore, despite the fact that PA14 is much more virulent than strain PA01 in the C. elegans killing assay [32], our set of functionally defined PA14 virulence factors show no bias for the set of genes that is present in PA14 but absent in PAO1, consistent with our previous preliminary conclusion based on a much smaller data set [32]. Likewise, the representation of the VFDB genes in the core and auxiliary genomes was not significantly different from that of the NR set.
Figure 7. The distribution of P. aeruginosa PA14 genes required for virulence in C. elegans in the Core vs. Auxiliary genome and on both predicted and known genomic islands.
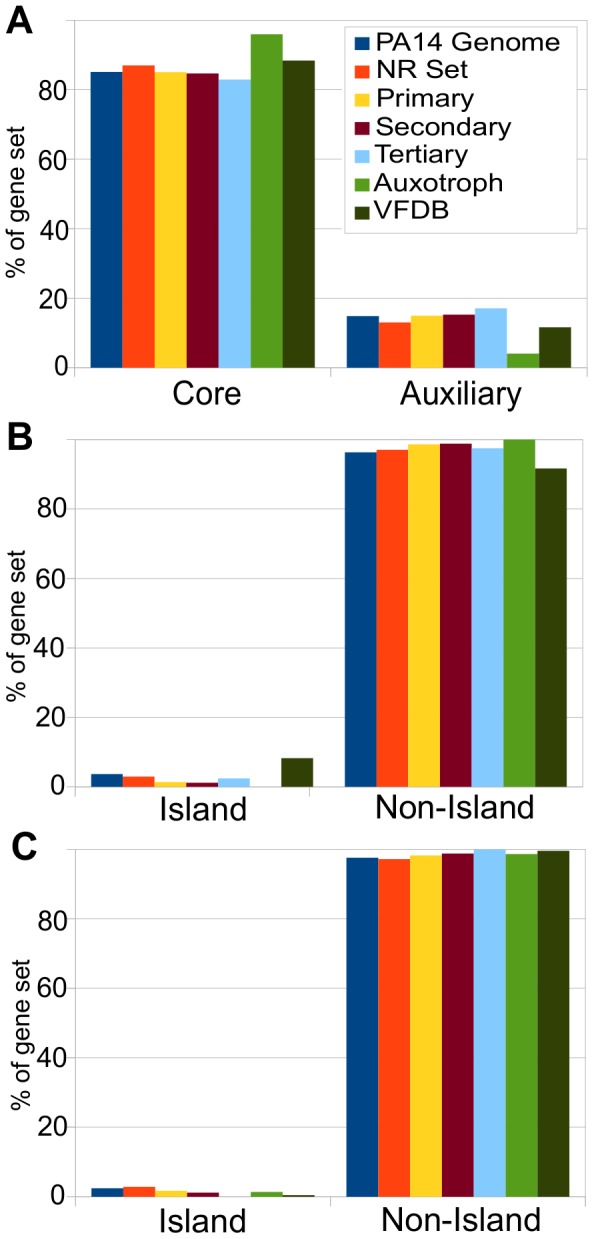
A) The percentages of P. aeruginosa PA14 genomic genes, PA14-NR Set mutants, and primary, secondary, tertiary, auxotroph, and VFDB set genes that are part of the P. aeruginosa core and auxiliary genome as defined by Mathee et al. [41]. The auxotroph set is disproportionately part of the core genome. Genes in the primary, secondary, tertiary, and VFDB sets have proportions in the core and auxiliary genes that are statistically indistinguishable from the PA14 NR set and from the genome as a whole. B) The percentages of genes from the PA14 genome, the PA14-NR Set, and from the primary, secondary, tertiary, auxotroph, and VFDB sets that are located on genomic islands predicted by IslandViewer. Representation of primary, secondary, and tertiary gene sets on predicted islands was statistically representative of the genome as a whole and of the NR-set, whereas VFDB genes had a statistical overrepresentation of genes located on predicted genomic islands (p = 0.0005). C) The percentages of genes from the PA14 genomic, the PA14-NR Set, and from the primary, secondary, tertiary, auxotroph, and VFDB sets located on the known PAPI-1, PAPI-2, and PAGI-1 genomic islands. Representation of primary, secondary, and tertiary set genes was statistically identical to the genome as a whole and the NR-set. VFDB genes were statistically underrepresented on the known islands (p = 0.007). Refer to Table S7 for statistics.
Location of PA14 virulence-attenuated genes on genomic island versus non-islands
We retrieved a list of predicted genomic islands of P. aeruginosa strain PA14 from the IslandViewer website [64] at (http://www.pathogenomics.sfu.ca/islandviewer/query.php). IslandViewer combines multiple methods of genomic island prediction. A list of PA14 genes located on the predicted islands was compiled. However, we noticed that the set of genes predicted to be on islands by IslandViewer only included 20 of the 143 genes located on the previously defined genomic islands PAPI-1, PAPI-2 and PAGI-1 [40], [65]. Therefore, an additional set of genes was compiled consisting of the 143 genes located on these known islands. We determined whether PA14 virulence genes identified in the C. elegans screen were associated with predicted genomic islands or with previously defined islands. Among the PA14 NR mutant set (3.0%; 131) of the genes represented are located on predicted genomic islands (similar to the 3.6%; 215 in entire PA14 genome). None of the identified auxotroph genes were located on predicted genomic islands and only one was located on a known genomic island. The virulence-related genes identified in our screen exhibited no bias for incorporation on the genomic islands predicted by IslandViewer nor on known genomic islands (PAPI-1, PAPI-2, and PAGI-1); in fact a non-statistically significant skew towards non-island regions of the genome was observed for the primary, secondary and tertiary sets in both comparisons. (Figure 7B,C, Table S7). This is in contrast to the significant overrepresentation of VFDB genes located in predicted genomic islands; i.e., 8.3% of total VFDB genes compared to 3.6% of genes in the genome as a whole (Figure 7B). However, the VFDB virulence factors were also marginally underrepresented (1 gene, 0.4%, p-value = 0.017) on the known islands PAPI-1, PAPI-2 and PAGI-1 (Figure 7C, Table S7). The data above suggest that the virulence-related genes identified in the C. elegans SK model are not preferentially located in plastic regions of the P. aeruginosa genome.
Breadth of phylogenetic distribution of PA14 virulence genes across prokaryotes
Are the predominant contributors to P. aeruginosa PA14 virulence in C. elegans genes that are unique to PA14 or P. aeruginosa or are they common to many bacteria? The comparisons above suggest that they are not PA14 specific genes, but to ask the question more generally we used a variation of the phylostratigraphy approach [66] in which we evaluated the breadth of the phylogenetic distribution of each PA14 gene by examining 727 bacterial genomes for orthologs of these genes. Each gene was designated as belonging to one of seven breadth categories or phylostrata (0–6) depending on its distribution across the bacterial kingdom, with “0” representing genes that are only present in P. aeruginosa PA14, “1” for genes present in other P. aeruginosa strains but absent outside the species, “2” for genes occurring in other species in the genus Pseudomonas but absent outside the genus, and so forth, with the most broadly-distributed category “6” found in Archaea as well as bacteria (Figure S13; see Materials and Methods). It should be noted that each ORF is given a “breadth of phylogenetic distribution” designation based on the presence of a putative ortholog in the most distantly related organism and does not necessarily imply that orthologs for the given ORF are present in all groups less divergent than this most distantly related organism.
Whereas “breadth of phylogenetic distribution” may be a meaningful surrogate for the age of a gene in the case of eukaryotes, there is a significant pitfall to using it as such in prokaryotes, because horizontal gene transfer between unrelated prokaryotic lineages could distort the apparent age of a gene. An apparently young, narrowly distributed gene could be older than it seems if horizontally transferred from a previously unrelated unsequenced organism, but it would appear to be a new within the P. aeruginosa lineage. A more serious problem is presented by genes that appear older than they are, as a result of a limited number of horizontal gene transfer events between unrelated lineages. We reasoned that we could mitigate this latter complication by including information about the frequency of occurrence across the clades in which genes are represented. Broadly distributed genes that occur with high frequency across their phylogenetic breadths, have, we believe, a greater likelihood of being genuinely “old” genes. We therefore created a subset of our most broadly distributed genes, categories 4, 5, and 6, that are also represented in 50% or more of the sequenced genomes in the clades in which they occur. We dubbed this subset “high-frequency-broad-phylogeny” or HFBP genes, which comprise 6.9% of the NR Set. Since we were also interested in examining the representation of the newest or most recently acquired genes in P. aeruginosa strain PA14, we also considered genes that were Pseudomonas-genus-specific, which we dubbed “PGS” genes. For this set, we binned genes of breadths 0, 1, and 2, which total 9.6% of the NR set, reasoning that such a set would represent relatively new genes, while at the same time being a large enough set to provide some statistical power.
The distribution of phylogenetic breadths of all the PA14 genes, all the PA14 NR set mutants, and the primary, secondary, tertiary and auxotrophic set mutants is shown in Figure S13, with statistical analysis presented in Table S7. The virulence-attenuated primary, secondary, and tertiary sets of genes identified in the C. elegans screen show a trend of underrepresentation in the narrowest phylogenetic breadth classes, with the tertiary positives, the set that predominantly includes highly virulence-attenuated mutants, being most underrepresented in the narrow phylogenetic breadth classes. However, taken individually, these underrepresentations are not statistically significant, due to the small number of mutants in each set.
When the same analysis was performed examining the HFBP and PGS gene sets in comparison with all the other PA14 genes (Figure 8, Table S7), we observed that there was a significant overrepresentation of HFBP genes among auxotroph set (p-value = 5.47×10−28) and in the primary, secondary and tertiary mutant sets (p-values of 0.00004, 0.0005, 0.006, respectively). At the same time, there was an underrepresentation of Pseudomonas-genus-specific (PGS) genes among the primary set (5.8% vs 9.6% in the NR Set, with p-value = 0.01), and the proportion of PGS genes appeared even more depleted among secondary and tertiary gene sets (5.3% and 2.4%), although the smaller size of these sets rendered these underrepresentations not statistically significant. However, we believe the statistically significant underrepresentation of PGS genes in the primary set, combined with the successive, increasing depletion of that category in the secondary and tertiary positives, suggests that those depletions are not spurious. In contrast to the gene sets from our screen, HFBP genes were underrepresented in the VFDB P. aeruginosa strain PA14 gene set (p-value = 0.00013). The apparent underrepresentation of Pseudomonas-genus-specific genes, most likely the youngest genes, and the overrepresentation of high-frequency-broad-phylogeny genes that likely represent older genes, point to a skew in the primary, secondary, and tertiary mutant sets toward older genes, and away from young, newly-acquired, or fast-evolving genes.
Figure 8. Among the PA14 genes required for virulence in C. elegans, “Pseudomonas-genus-specific” (PGS) genes are underrepresented, whereas “high-frequency-broad-phylogeny” (HFBP) genes are overrepresented.
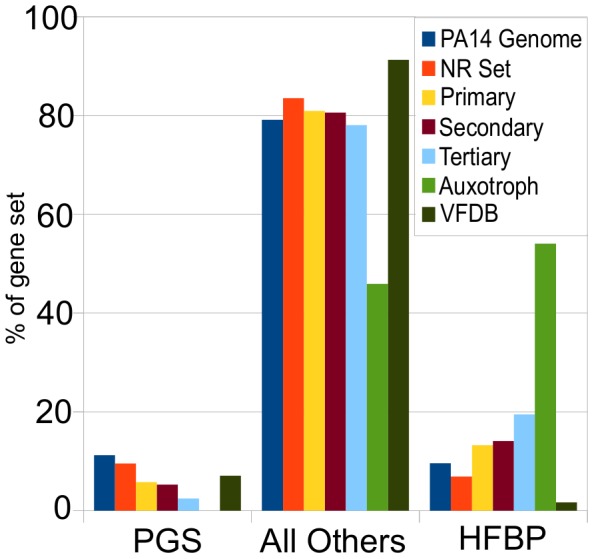
Based on phylostratigraphic analysis, PA14 genes required for virulence in C. elegans were classified as either “Pseudomonas-genus-specific” (PGS), presumably representing the newest genes in PA14, “high-frequency-broad-phylogeny” (HFBP), representing the oldest, most conserved genes in PA14, or “all others”. The percentage of each gene set, including the PA14 genome genes, the PA14-NR, primary, secondary, tertiary, auxotroph, and VFDB gene sets that are classified as PGS genes, HFBP genes, or all others genes, are shown. HFBP genes comprise 10% of the PA14 genome, and about 7% of the NR set genes. Furthermore, HFBP genes are increasingly overrepresented with successive iterations of the screen accounting for 13% of the primary set (p-value = 0.00004), 14% of the secondary set (p-value = 0.0005) and 19% of the tertiary set (p-value = 0.006). HFBP genes make up greater than 50% of the auxotroph set with a (p-value = 5.47×10−28) relative to the NR set. The PA14 VFDB set contains an underrepresentation of HFBP genes (1.6%, p-value = 0.0001). PGS genes make up 11% and 9.6% of the PA14 genome and NR set respectively. Over successive iterations of the screen, PGS genes become numerically more underrepresented relative to the NR set, comprising 5.7% of the primary set (5.7%, p-value = 0.01), 5.2% of the secondary set (p-value = 0.03, not statistically significant), and 2.4% of the tertiary set (p-value = 0.08, not statistically significant). Due to the small numbers of genes in the secondary and tertiary sets, only the underrepresentation in the primary set is significant after application of multiple comparison correction (FDR, q< = 0.05). PGS genes are underrepresented in the auxotroph set (0%, p-value = 0.0006). Statistical data for this figure are presented in supplemental Table S7.
Taken together, these analyses of the genomic and phylogenetic distribution of virulence-attenuated genes demonstrate that the genes required for PA14 virulence in C. elegans are distributed throughout the PA14 genome on both predicted genomic island and non-island regions, are not unique to a particular P. aeruginosa strain and in fact are disproportionately potentially old genes with identifiable orthologs across a wide breadth of prokaryotic species.
Discussion
P. aeruginosa PA14 Genes Required for C. elegans Killing
We set out to define the spectrum of genes required for P. aeruginosa PA14 infection in a single host organism with the ultimate goal of elucidating the mechanisms underlying pathogenesis in this multi-host opportunistic pathogen. A genome-wide unbiased screen for P. aeruginosa strain PA14 mutants defective in killing C. elegans identified a set of 180 putative virulence-related mutants (corresponding to 170 genes) after two rounds of screening. The screen was validated by the isolation of mutants previously shown to be required for P. aeruginosa virulence in both nematodes and mammals or known to regulate processes or pathways linked to pathogenesis including, but not limited to, genes involved in quorum sensing, two component regulators of virulence, transcriptional regulators, genes involved in type IV pilus production, and O-antigen biosynthesis. Twenty genes in the 170 gene set overlapped with a set of previously defined virulence factors in VFDB, a database of Pseudomonas-related virulence factors. Overall, the PA14 genes identified in the genome-wide screen have an overrepresentation of highly conserved genes present in many bacterial phyla and are part of the stable P. aeruginosa genome, rather than being located on pathogenicity islands.
The set of 170 virulence-related genes is broadly distributed across 27 defined functional classes and DAVID GO term analysis and mapping onto KEGG pathways did not reveal any interpretable enrichment for particular functions or pathways. This breadth of functional classes parallels the virulence-attenuated mutants identified in an independent unbiased screen using signature tagged mutagenesis carried out by Potvin and coworkers in a rat chronic infection model [46]. Unlike the genes identified in our screen and by Potvin et al., virulence-related factors in the VFDB are enriched for secretion- and adherence-related proteins. A major difference between the virulence-related genes in VFDB and the genes identified in our unbiased screen is that many of the genes in VFDB were included because they encode secreted toxins, secretion systems, or cell surface structures [52], [67]. However, the sensitivity of our screen favored identification of mutants with strongly attenuated virulence. This was expected given the nature of the primary screen that required that both the parent nematodes live long enough to produce a significant brood and that the nematode brood mature on the mutant bacterial lawn. It is possible, therefore that mutants with a weak virulence-attenuated phenotype were not detected and this could potentially skew the collective analysis of virulence factors.
One explanation for why so few mutants identified in our screen correspond to secretion pathways or to secreted effectors is that many of P. aeruginosa virulence effectors appear to function redundantly in the C. elegans killing assay. In support of this conclusion, disruption of the ExoU cytotoxic phospholipase had no statistically significant impact on virulence, but appeared to create a sensitized background that allowed the detection of other relatively weak virulence factors. Further, McEwan et al. have shown that whereas PA14 exotoxin A (toxA) mutants have no or little defect in virulence, overexpression of ToxA in E. coli activates the worm immune system and ultimately kills an immune-compromised animal, suggesting that ToxA may play an active, but to date undetected, role in PA14 pathogenesis in the nematode host [68]. Similarly, Dunbar et al. [69] have shown that ToxA inhibits protein synthesis in C. elegans intestinal cells during an infection. ExoU and ToxA are secreted by distinct systems and the weak virulence attenuation of secretion system mutants in C. elegans implies that multiple secretion systems and effectors may contribute to virulence in C. elegans with no single system being paramount.
An alternative explanation for the identification of a limited number of secretion-related mutants in our screen may be linked to the fitness costs of maintaining a large set of effectors targeting a wide range of potential hosts. Only four Type III effectors have been identified in P. aeruginosa, whereas 46 families of effector proteins have been identified in various strains of the related plant pathogen P. syringae [70] and a typical P. syringae strain has 20 to 30 effectors [71]. In P. syringae, which has a much more limited host range than P. aeruginosa, type III effectors mostly target host defense signaling pathways and both enhance virulence in particular host plants, while eliciting a strong immune response in others [72]. This fact, combined with the observations that the genes encoding secreted effectors are often under diversifying selection [38], [73] and are typically located in plastic regions of the genome [70], [74], suggests that P. syringae strains actively co-evolve with a limited number of hosts. In contrast, from first principles, it seems highly unlikely that a broad host-range pathogen like P. aeruginosa PA14 can be simultaneously co-evolving with all of its multiple hosts. Therefore P. aeruginosa might employ a broader set of strategies to ensure survival in diverse hosts instead of maintaining large sets of host-specific virulence-related effectors.
In this context, a number of the strongly virulence-attenuated PA14 mutants identified in our screen may correspond to factors that enable survival of P. aeruginosa in the hostile environment of the C. elegans intestinal tract, which is acidic and filled with enzymes such as proteases, lipases, and DNAse that potentially disrupt bacteria [75]. Moreover, in response to pathogens, the nematode specifically upregulates transcription of many putative antimicrobial genes [18]. In order for P. aeruginosa to proliferate in the intestine and cause disease, it first has to survive. Both the two component potassium sensor KpdD and the nitrogen assimilation regulatory protein GlnK, identified in our screen, have recently been shown to play a role in the persistence of S. typhimurium in the C. elegans intestine and defects in outer membrane integrity may reduce the survival of kdpD and glnK mutants in the host [76]. In addition, the identification of two PA14 genes required for biosynthesis of glutathione, gshA and gshB, may be related to the role of glutathione as a protectant against stresses encountered in the worm intestine including reactive oxygen species and low pH [77]. Cold shock domain proteins, like PA0465 identified in our screen are another class of molecule that are induced by environmental stress and are generally thought to play a protective role in the cell [78].
Identification of a number of PA14 mutants corresponding to metabolic genes illustrates the importance of nutrient acquisition in virulence. Without specific biosynthetic or metabolic capabilities a pathogen may be unable to colonize or grow within a host. For example, P. aeruginosa mutants defective in purine biosynthesis are unable to replicate in neutropenic mice, presumably because the in vivo environment is deficient in purine [48]. In the cases of intercellular pathogens such as Listeria monocytogenes and Mycobacterium tuberculosis, specific amino acid and nucleotide auxotrophs are reduced in growth in vivo [79], [80]. We identified a number of metabolic genes including prpB and prpC and several aru genes that may be important for bacterial metabolism and growth under the nutrient conditions within the nematode intestine. Further, some of the putative virulence-attenuated mutants identified in the primary screen that were set aside for further study because they were determined to be auxotrophs (a typical step in many screens) might specifically reflect nutrient availability in the nematode intestine. In this regard it is notable that mutations in nine purine, five pyrimidine, and six tryptophan biosynthetic genes were identified in the primary screen for virulence-attenuated mutants, and although some of these mutants exhibited reduced growth on the killing assay medium, many did not have any observable difference from wild-type PA14, suggesting that the reduction in virulence might be due to aberrant growth of these auxotrophs in vivo.
The predominant contributors to PA14 virulence in our C. elegans infection based assay appear not to be individual effectors, but genes that regulate numerous effectors (like the quorum sensing regulators lasR and rhlR), genes that are vital for protecting the bacteria from the host defense onslaught, and genes that help P. aeruginosa obtain the necessary nutrients to survive in the host. Therefore the strategy of a broad host range opportunistic pathogen might fundamentally differ from a pathogen that targets specific hosts, relying more on multiple partially redundant secretion systems and their cognate effectors and strategies for survival under a wide-variety of metabolic and environmental conditions.
Location of P. aeruginosa Virulence Factors in the Core P. aeruginosa Genome
The long-recognized association between virulence genes and regions of genomic plasticity, particularly genomic islands acquired by lateral transfer of genetic material [81], has been attributed to the competitive advantage conferred by horizontally-acquired virulence factors in an ongoing co-evolutionary struggle between a host and pathogen. Two pathogenicity islands carrying plant and animal virulence-related genes have been identified and characterized in P. aeruginosa PA14 (PAPI-1 and PAPI-2). Of the 11 genes located on PAPI-1 previously shown to be required for normal levels of virulence in plants and mice [40], only one (rcsC) was identified in our secondary screen as a weak mutant and it did not re-test in the tertiary screen. Overall our results showed no enrichment of virulence-associated genes on predicted genomic islands, or on the known genomic islands PAPI-1, PAPI-2, or PAGI-1. More generally, whether or not virulence genes in aggregate are preponderantly associated with genomic islands in P. aeruginosa has not been experimentally demonstrated. The statistical power of analysis with respect to the genomic locations of genes is limited in part by the fact that not all genes are expected to segregate independently. In this regard, it is worth noting that the apparent enrichment of P. aeruginosa VFDB genes on predicted genomic islands is primarily due to the cluster of functionally interdependent Type III secretion apparatus genes located between gene loci PA14_42440 and PA14_42660. In summary, our data and analysis of existing data suggest that neither P. aeruginosa VFDB genes nor the virulence-attenuated genes identified in our screen are preferentially found on genomic islands.
Roughly paralleling our observations with genomic islands, our analysis of the frequency of PA14 virulence genes in the core and auxiliary genomes (both from our screen and the VFDB) showed no statistically significant over or underrepresentation. The main difference between the core versus auxiliary genome distinction, and that of genomic islands, is that auxiliary genes include both genes that are lost from the genomes of some isolates, as well as those genes that are newly acquired. Genomic islands by contrast, specifically include only newly acquired genes. Taken together, these results suggest that there is no specific enrichment of P. aeruginosa virulence-related genes on islands or in strain-specific regions of the P. aeruginosa genome, at least with respect to those that are involved in the C. elegans slow killing assay.
To further test the hypothesis that the arsenal of P. aeruginosa virulence factors includes newly evolved and or newly acquired genes we investigated the phylogenetic breadth of distribution of the PA14 virulence genes, as well as the degree to which they belong to a set of putative old conserved genes, the so-called high-frequency-broad-phylogeny (HFBP) set, and probable newer genes, the Pseudomonas-genus-specific (PGS) set. We found that consistent with the result that virulence genes are not located on islands, that PA14 virulence genes involved in C. elegans killing are enriched in HFBP genes, which as a group are likely to be the most ancient and conserved prokaryotic genes. The vast majority of PA14 virulence genes appear not to be specific to the Pseudomonas genus, and in fact, such recently acquired or novel genes are underrepresented among our putative virulence genes. These observations underscore the point that many virulence factors with the most significant contribution to virulence can be old, highly conserved genes.
Assessing the Generality of the P. aeruginosa C. elegans Infection Model
Although the C. elegans model, in which nematodes are “force-fed” a monoculture of P. aeruginosa [13], [43], [44], is somewhat artificial, so are other laboratory models of P. aeruginosa infection that require pricking of the body of a fly [45], lung inoculation with agarose beads in rats [46] and mice [47], and non-lethal cutaneous burns in mice [82]. The artificiality of these models and the need for a compromised host is in part dictated by the opportunistic nature of the pathogen. The fact that conserved immune defense pathways are activated in the C. elegans host by P. aeruginosa strongly supports the view that the nematode is responding to P. aeruginosa as a pathogen [83]. Although we don't yet know whether P. aeruginosa is a natural pathogen of C. elegans, since both organisms live in the soil, it is likely that they encounter one another, and it is not inconceivable that the pathogenicity interactions that we observe may approximate the interactions of P. aeruginosa with weakened individual C. elegans animals in the wild.
Although some putative P. aeruginosa virulence factors are common to both mammalian and C. elegans host models, the degree to which P. aeruginosa virulence factors are shared, and implicitly, the degree to which P. aeruginosa virulence strategies are common to C. elegans and mammalian hosts is not yet clear. That only a single putative virulence-related gene is shared between the 170 virulence-attenuated genes from our secondary set, the rat chronic infection set defined by Potvin et al., and the VFDB set, argues against the idea of a core set of virulence factors common to all infection models. The spectrum of virulence factors that play a role in a given host model is likely to depend on a wide variety of factors including the characteristics of the site of infection, such as pH, ionic strength, nutrient availability, and temperature, the type of immune compromise, the phase of infection, and the particulars of the immune response, including the presence of host factors, and even host behavior. Indeed, two additional PA14 pathogenicity assays in C. elegans have been developed in our laboratory, a toxin-mediated killing model [12], [14] and a liquid killing assay (N. Kirienko and F. Ausubel, unpublished). PA14 appears to utilize distinct mostly non-overlapping sets of virulence-related genes to kill nematodes in the three different models. Nevertheless, all three of these assays have identified virulence factors that play important roles in various aspects of mammalian pathogenesis. The implication for human pathology is that the predominant virulence factors that play a role in different types of P. aeruginosa infection in humans may be somewhat distinct.
Implications for Multi-host Opportunistic Pathogens
In summary, the data from our unbiased genome-wide screen for P. aeruginosa virulence factors involved in C. elegans killing in a specific infection model, suggest that in comparison to host-specific pathogens, P. aeruginosa may employ a smaller arsenal of host-specific effectors, and rely more on conserved, generic virulence factors and on its ability to endure host defense responses. While it is not yet clear that this same strategy is employed by multi-host pathogens beyond P. aeruginosa that are capable of infecting organisms from multiple phylogenetic kingdoms, this may explain why the major genes contributing to PA14 virulence in C. elegans are not overrepresented on genomic islands, are not PA14 or P. aeruginosa specific genes, and may in fact be biased for ancient genes common to many other prokaryotic species. These observations are consistent with the view of P. aeruginosa PA14 as a generalist pathogen for which the relationship with C. elegans is opportunistic rather than co-evolved. Indeed it is likely that no significant co-evolution occurs between P. aeruginosa and any of the hosts for which it is an opportunistic pathogen, both because of the rarity of pathogenic interaction, and because of the likelihood that co-evolution with multiple hosts would necessitate balancing opposing evolutionary pressures from those hosts. From a clinical perspective, the multiplicity and apparent combinatorial nature of P. aeruginosa's virulence factors may pose a challenge for the development of new therapeutics to fight Pseudomonas infection. This work does not settle the question of whether the profile of virulence factors of a multi-host pathogen is likely to be different from that of host-specific pathogens in terms of the reliance on conserved effectors that target highly conserved features of eukaryotic biology, but it is a question that deserves further inquiry.
Materials and Methods
Bacterial Strains
P. aeruginosa strain PA14 mutants are gentamycin resistant MAR2xT7 transposon insertion mutants unless otherwise stated [42]. The lasR mutant used as a control was a deletion that removes the lasR ATG and carries a gentamycin cassette [84]. Similarly, the rhlR mutant used as a control in the pigment and motility assays was a deletion mutant that carries a gentamycin cassette [84]. These mutants have an identical avirulent phenotype to subsequently generated clean in-frame PA14 ΔlasR and ΔrhlR mutants (data not shown). The pilA mutant is a tetracycline resistant Tn5-B30 transposon insertion mutant provided by G. O'Toole [85]. ΔPA0745 is a complete in-frame deletion of the ORF with a concomitant insertion of a PacI restriction site generated by the method previously described in Chand et. al. [86]. Unless otherwise indicated, MAR2xT7 mutant bacteria were streaked from frozen stocks onto LB agar containing 15 µg/ml gentamycin to isolate single colonies used for inoculation. WT PA14 (as well as lasR and pilA mutants) was streaked on LB agar containing 100 µg/ml rifampicin.
Nematode Strains
N2 Bristol L4 animals were used for the primary and secondary screens. Tertiary tests were done with CF512 fer-15(b26)II;fem-1(hc17)IV temperature sensitive sterile nematodes. CF512 worms were propagated at 15°C, egg-prepped and hatched overnight at 20°C in M9 liquid media in the absence of food. Starved L1 animals were dropped onto NGM plates seeded with standard E. coli OP50 and grown for approximately 20 hours at 15°C and then 20 more hours at 25°C to generate staged L4 sterile animals for experimentation.
Primary Screen
We used the non-redundant PA14 MAR2xT7 transposon library of ordered mutants to screen for avirulent mutants [42]. The entire library was screened twice with the exception of a single 96-well plate of mutants (plate 14.3) that was not screened because it contained mostly known slow-growing mutants. Bacteria were inoculated from the frozen 96-well stock plates into 150 µl of LB in 0.5 ml 96-well Masterblocks (Greiner) and grown overnight with shaking for 16 hours at 37°C. 10 µl of each culture was spotted onto slow killing (SK) agar (standard nematode growth media, NGM, containing 0.35% instead of 0.25% peptone) in each of two wells of a 6-well culture plate (Falcon) [12]. Two previously identified virulence-attenuated mutants, quorum sensing regulator lasR (highly attenuated) and typeIV pilin pilA (moderately attenuated) were included as positive controls in the screen. Plates were scored qualitatively after 4 days at 25°C and designated as either “strong”, indicating large numbers of gravid animals (generally on lasR mutant bacteria hundreds of gravid nematodes were present and the bacterial lawn was mostly or completely consumed) or “weak”, indicating an observable increase in the number of worms or gravid adults present compared to PA14 (pilA usually resulted in an increase in the total brood size with a bias towards older stage larvae and gravid adults; the pilA control wells exhibited variability in the number of progeny alive and were near the limit of sensitivity in the screen). Each mutant was screened blind on two separate days for a total of four wells screened per mutant. Mutants that were scored as strong in either one or both repetitions or weak in both were retained for a total of 399 primary mutants. Approximately half way through the primary screen it became clear that lawn growth phenotype was relevant to our study, and from then on we scored for lawn growth. We therefore have a crude assessment of growth for about 50% of the mutants identified in the primary screen.
Auxotroph Screen
All 399 of the mutants from the primary screen were streaked from the frozen library stock onto LB agar plates containing 15 µg/ml gentamycin and grown overnight at 37°C. To quickly test for potential auxotrophs, a single colony from each mutant was picked and streaked on 1) Neidhardt supplemented MOPS defined media without amino acids or nucleotides (TEKNOVA EZ Rich Media), 2) Neidhardt supplemented MOPS defined media plus amino acids and nucleotides, and 3) LB containing 15 µg/ml gentamycin agar plates. Plates were incubated overnight at 37°C. 86 mutants that either exhibited no growth on minimal media minus supplements or significantly reduced growth were removed from the pool of putative avirulent mutants and not examined in further rounds of screening (Table S2). 15/56 of the auxotroph mutants for which a plate growth phenotype had been annotated (see above) had obvious growth defects on SK agar plates.
Secondary Screen
The 313 mutants from the primary set that exhibited normal growth on minimal media without addition of amino acids and nucleotides (Table S3) were screened for attenuation of virulence in standard SK assays and scored for both killing of parental (P0) animals and the number and maturity of worm progeny produced [13]. A single bacterial colony was inoculated into 5 ml of LB media and grown for 14.5 hours at 37°C with aeration on a rotating wheel. 10 µl of this overnight culture was spread onto each of two SK agar plates (3.5 cm culture plates (Falcon), plates were incubated for 24 hours at 37°C, and then 24 hours at 25°C. 30–40 N2 L4 animals (raised on standard NGM plates with E. coli OP50 as food) were picked to each SK plate and the plates were incubated at 25°C for a total of 60–80 animals per assay. Live and dead animals were counted 2–3 times over 1–3 days after transfer to the pathogen. Animals were considered dead if they did not respond to a gentle touch and were removed from the plate. After 4 days at 25°C, the number and age of the nematode progeny on each plate was qualitatively assessed as compared to those on PA14 WT. Secondary positives were ranked as “strong” (strongly virulence-attenuated similar to the lasR mutant), “moderate” (less attenuated than lasR but greater than or equal to pilA) and “weak” (less attenuated than pilA for parental killing but clearly exhibiting an increase in progeny number and age over WT PA14). 180 MAR2xT7 mutants exhibited either attenuation of P0 parental killing or allowed increased production and development of nematode progeny (Table S4). The identity of the MAR2xT7 mutants was confirmed by sequencing of arbitrary PCR products as previously described [42], see Table S4.
Tertiary Screen
58 MAR2xT7 mutants were re-screened in standard SK assays using sterile fer-15(b26)I:fem-1(hc17)IV animals (Table S5). Bacteria were grown as indicated above for the secondary screen. Three 3.5 cm SK agar plates were seeded with each bacterial strain and 35–50 L4 worms were transferred to each plate for a total of 100–150 animals per assay. Live and dead animals were counted every day over approximately 7 days. PA14 WT and lasR were included in each assay as controls. The resulting 41 virulence-attenuated mutants shown in Figure 2 were all tested in at least two separate experiments unless otherwise indicated. Statistical analysis of the curves summarized in Figure 2 was done using Prism 5 Log-rank (Mantel-Cox) test and the difference between the mutant and wild-type curves was highly significant in all cases with a p-value<0.0001. Time to 50% survival was calculated (Prism 5.0 linear regression Hill equation LogEC50). All killing assays presented in the manuscript (Figures 3, 4, 5, 6, S5, S6, S7, S8, S9, S10, and S11) are a representative example of two or more experiments that resulted in the summary shown in Figure 2.
Assessment of Growth
Growth of bacterial mutants was evaluated by four methods: 1) Overnight cultures inoculated from a single colony into 5 ml of LB and grown for 14.5 hours at 37°C with shaking were visually compared to wild-type PA14 grown under the same conditions. Mutants that were observably less turbid were considered slow growers (in the case of PA14_45650 mutant #54246 the cells appeared to be lysed). 2) Slow killing agar plates spread with each mutant were examined prior to transfer of worms. Bacterial lawns that were thin or exhibited other aberrant phenotypes (nusA mutant #55834 had large colonies that emerged on top of the lawn) were removed from the mutant pool. 3) Many of the secondary and tertiary positive mutants were grown overnight in LB and M63 minimal media in a 96-well plate without shaking and the OD600 was measured every 15 minutes for 15 hours at 37°C in a Molecular Devices Spectra Max M5. The rate of growth during two hours of maximal growth was compared to WT (Table S5). 4) The growth of nine mutants (pchH, pchI, PA14_27700, PA2550, PA0456, PA0745, clpA, PA1216, vqsR) in 5 ml of M63 minimal in a standard culture tube on a rotation wheel at 37°C was measured by counting colony forming units. All nine of these mutants grew as well as WT (Figure S12).
Pyocyanin Assay
A pyocyanin assay was modified from Essar et. al. 1990 [87]. A single colony of WT or mutant bacteria was inoculated into 5 ml of LB media and grown for 16 hours at 37°C on a rotating wheel. 1 ml of saturated overnight culture was transferred to a microfuge tube and cells were pelleted by centrifugation at 14,000 RPM for 2 minutes. 800 µl of the supernatant was transferred to a new tube, extracted with 600 µl of chloroform and the phases were separated by centrifugation for 5 minutes at 14,000 RPM. The chloroform phase was then re-extracted with 0.3 ml of 0.2 N hydrochloric acid. The pyocyanin content of 100 µl of the aqueous acidic phase was quantitated based on absorbance at 520 nm. The A520 was normalized to cell number (A600 of the original overnight culture). A ΔphzA1-G1/ΔphzA2-G2 [88] mutant that does not produce any phenazines had no detectable A520. The pyocyanin produced by each bacterial strain was measured from the growth of two individual colonies on two separate days. For each of the four cultures, the ratio of A520 (normalized to cell number as measured by A600) of mutant to WT was calculated and the average of these four ratios is shown in Table 2. The error presented is the SEM of the four mutant/WT ratios.
Pyoverdine Assay
A single colony of WT or mutant bacteria was inoculated into 5 ml of M9 media and grown overnight for 18 hours at 37°C on a rotating wheel. Cells were pelleted by centrifugation at 14,000 RPM for 2 minutes in a microfuge tube and the supernatant was diluted 10 fold in 10 mM Tris pH 7.4. Pyoverdine content was determined by measurement of fluorescence (400 nm excitation, 460 nm emission) [89], [90]. No fluorescence above background was detected in pyoverdine biosynthetic mutants, pvdD (#40342) and pvdA (#30448). The pyoverdine produced by each bacterial strain was measured from the outgrowth of two individual colonies on two separate days. For each of the four cultures, the ratio of fluorescence (normalized to cell number as measured by A600) of mutant to WT was calculated and the average of these four ratios is shown in Table 2. The error presented is the SEM of the four mutant/WT ratios.
Swarming Assay
The swarming assay was modified from Overhage (2008) [91]. A single colony of WT or mutant bacteria was inoculated into 5 ml of LB media and grown for 15 hours at 37°C with aeration. 2 µl of each overnight culture was spotted onto the surface of LB 0.5% agar and SK 0.5% agar plates and then incubated at 37°C overnight. Each mutant was tested in triplicate on two separate days. As expected, the rhlR mutant (deficient in rhamnolipid production) and pilA mutant (typeIV pilin) were defective in swarming [62]. Mutants were visually compared to WT PA14 and were qualitatively evaluated for swarming radius and number of tendrils (Table 2).
Swimming Motility
The swimming motility assay was based on Darzins (1993) [92]. A single bacterial colony was picked with a straight end loop and inoculated into LB swim agar (0.35% agar). Plates were incubated 8–12 hours at 37°C. The diameter of the flagellum-mediated motility generated turbid zone was measured. Each mutant was tested in triplicate and the average with SEM is presented in Table 2.
Twitching Motility Assay
The twitching motility assay was based on O'Toole 1998 [85]. A portion of a single bacterial colony was picked with a straight end inoculation loop and stabbed to the bottom of a LB agar plate (1.5% agar). Plates were incubated overnight at 37°C and then 2 days at room temperature. The growth at the interface between the agar and the polystyrene plate (radius from the inoculation point) was measured. The pilA mutant exhibited no twitching motility. Each mutant was tested in triplicate and the average with SEM is presented in Table 2.
Breadth of Phylogenetic Distribution of P. aeruginosa strain PA14 Genes Using a Variation of the Method of Phylostratigraphy [66]
A database of orthologs of P. aeruginosa strain PA14 genes across 727 sequenced prokaryotic genomes (including PA14) was created. Finished microbial genome sequences were obtained as downloaded packages from the NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/) on August 25, 2008. PA14 proteins were used as BLASTP queries against each bacterial genome. Putative orthologs were reciprocal best hits against the corresponding proteins in the subject genomes. Blast results against subject genomes were required to have an e-value equal to or less than 0.0001. Reciprocal blasts against the PA14 genome were required to have e-values of 0.001 or less. Putative orthologs were required to align for at least 80 percent of their length and have less than 20% difference in protein sequence lengths, thereby conserving overall domain structure. The e-value constraint was permissive to allow detection of distant orthologs, but the requirement for alignment length was fairly stringent. Breadth of phylogenetic distribution, also called phylostratum, was a measure of the maximal phylogenetic distance at which an ortholog occurs. Breadth was defined as: 0 for proteins specific to PA14, 1 for proteins that occur in multiple strains of Pseudomonas aeruginosa, but not in other species, 2 for proteins that occur in multiple Pseudomonas species but not in other genera, 3 if across gamma and beta proteobacteria, 4 if across proteobacteria, 5 if across eubacteria, and 6 if across eubacteria and archaea (Figure S13).
Statistics
P-values for overrepresentation and underrepresentation were calculated as Fisher Exact Test right and left probabilities, respectively, using version 1.21 of the Text::NSP::Measures::2D::Fisher Perl module, available from CPAN.org [93]. Multiple comparison correction using False Discovery Rate (FDR) was performed where indicated [94]. The maximal value of q was 0.05.
KEGG Pathway and GO Term Analysis
Genes of P. aeruginosa strain PA14 were mapped to KEGG pathways using the KEGG Mapper program (http://www.genome.jp/kegg/tool/map_pathway1.html). The total number of mutants in each pathway was summed for those genes in the PA14 NR set and for each mutant set analyzed. P-values for overrepresentation of each pathway were calculated using Fisher exact test right-probabilities, and their significance was assessed using the Bonferroni correction [94], [95]. GO term analysis was performed using DAVID (http://david.abcc.ncifcrf.gov/home.jsp) using the genes in the NR set as a background, and the genes in the sets of analyzed positives as gene-lists. The DAVID software package calculates p-values for each GO term automatically, and also gives an EASE score, which was used to assess the significance of the overrepresentation of any given GO term.
Supporting Information
Flow chart of screening procedure for virulence-attenuated PA14 transposon mutants in C. elegans .
(TIF)
Correlation of virulence attenuation in standard SK and progeny survival assays. A) lasR, gacA, ptsP, mucD, rhlR and pilA mutants all have attenuated virulence in a standard slow killing (SK) assay (60–80 N2 worms tested). B) The number and developmental stage of the progeny on the SK plates in panel (A) four days after transfer of the parent worms to pathogen at 25°C. On PA14, there were few progeny and no gravid adults were observed whereas gravid adult progeny were found on the virulence-attenuated P. aeruginosa strains. The number and age of progeny on the mutant plates was qualitatively scored in comparison to the PA14 WT plates. Plates seeded with gacA and ptsP were overrun with hundreds of gravid adult worms that consumed the bacterial lawn. lasR, mucD, rhlR and pilA plates contained fewer gravid adult worms but all were observably greater than plates seeded with WT PA14.
(TIF)
Percentage of mutants that are VFDB genes. The percentage of primary, secondary, and tertiary, and auxotroph set virulence-attenuated genes that are VFDB and non-VFDB genes are indicated. VFDB genes are significantly overrepresented in the primary, secondary, and tertiary sets, with p-values of 5.5×10−6, 2.1×10−5, and 3.7×10−5, respectively. Furthermore, their overrepresentation increases with successive screen iterations.
(TIF)
Venn diagram showing the overlaps between the 170 virulence-attenuated genes obtained in the secondary screen, the Potvin set, and the PA14 VFDB set.
(TIF)
Multiple pepP alleles but not alleles of other genes in the operon have attenuated virulence. A) PepP is a cytoplasmic aminopeptidase that cleaves aminoacyl proline dipeptides from the N terminus of polypeptides. In PA14 pepP appears to be the second gene in a 5 gene operon comprised of PA5225, a hypothetical protein, pepP, ubiH a ubiquinone biosynthetic enzyme, PA5225 another hypothetical protein, and terminating with PA5221, an ORF with homology to E. coli visC a pyridine nucleotide-disulphide oxidoreductase in the ubiH family. B) Four MAR2xT7 alleles of pepP display highly attenuated C. elegans killing. C) Transposon mutants in 4/5 genes in the pepP operon were tested for their effect on virulence (no mutant was available in PA5221). Mutants in the two downstream genes tested, ubiH and PA5222 exhibited wild-type levels of virulence whereas the single mutant in the upstream PA5225 had a modest attenuation of virulence (compared to pepP) that might be due to effects on pepP expression. pepP mutant #31097 produced elevated levels of pyocyanin and had somewhat reduced swimming ability, although swarming was normal on both SK and LB (Table 2). The PepP proline aminopeptidase could function in utilization of exogenous peptides as nutrients, degradation of proteins or protein maturation, perhaps of a specific substrate relevant to virulence. There is a precedent for aminopeptidase function being linked to virulence associated phenotypes. P. aeruginosa PepA was shown to be involved in the regulation of alginate biosynthesis [100].
(TIF)
Mutations in cold shock domain (CSD) protein PA0456 are virulence-attenuated in C. elegans but mutations in four other CSD proteins do not affect virulence. A) PA0456 is most likely a single gene transcription unit. PA0456 (previously annotated as cspB in the PA14 genome) contains a canonical cold shock domain (CSD) and is homologous to other CSD proteins (57% identical to the major E. coli cold shock protein, CspA, and 65% to B. subtilis CspB as determined by BLASTP). Although the first cold shock proteins (Csps), a conserved family of small mostly acidic proteins that bind single stranded DNA and RNA, were identified as major proteins induced upon temperature downshift, some members of the family are not induced upon cold shock and many are implicated in other cellular functions [101]. B) Three independent MAR2xT7 transposon insertions in PA0456 have attenuated virulence in C. elegans. C) Transposon insertion mutants in four additional CSD containing genes (capB PA3266, PA0961, cspD PA2622 and PA1960) have wild-type virulence in C. elegans. Five additional CSD-containing homologs of PA0456 (PA3266, PA1159, PA0961, PA2622, PA1960) were identified by BLASTP against the PA14 protein database with 76, 66, 62, 54 and 41% identity to PA0456 respectively; all 5 contain a CSD as analyzed by Prosite and transposon insertion mutants were available in 4/5 of these genes. Among the CSD containing proteins tested, PA0456 appears to be unique in its role in virulence, suggesting that the cold shock response per se is most likely not required for virulence of PA14. PA14 PA0456 has been reported to be regulated by quorum sensing [102], and in keeping with these findings PA0456 exhibited defects in quorum sensing regulated phenotypes; the PA0456 mutant had reduced pyocyanin production and swarming motility (Table 2).
(TIF)
KinB (PA5484) sensor kinase is required for PA14 virulence in C. elegans . KinB negatively regulates alginate production and has been recently shown to be required for virulence in zebrafish embryos and mice [86], [103]. A) kinB and its cognate response regulator algB form a two gene operon. B) Two MAR2xT7 transposon alleles in kinB (as well as an in-frame deletion of kinB described in Chand et al., data not shown) are reduced in virulence. C) Transposon mutants in algB (and an in-frame deletion of algB described in Chand el al., data not shown) exhibited wild-type virulence in C. elegans, paralleling their lack of phenotype in zebrafish [86].
(TIF)
Mutations in putative transcriptional regulator PA14_27700 and possible ECF sigma factor PA14_27690 are virulence-attenuated in C. elegans . A) PA14_27700, a Crp/FNR-type transcriptional regulator that appears to have no homologue in PA01 [32], is located 264 bp downstream of PA2817 and 66 upstream of PA14_26790, a FecI-like extracytoplasmic function (ECF) sigma that also has no PA01 homologue. It is unclear whether PA14_27700 and PA14_26790 form an operon. B) Multiple MAR2xT7 insertions in both PA14_27700 and PA14_27690 are attenuated in virulence. Although not initially identified in our screen, mutants in PA14_26790 have a strong virulence-attenuated phenotype like PA14_27700. ECF sigma factors, to which PA14_26790 has homology, are commonly co-transcribed with a regulatory anti-sigma factor and the ECF sigma factor and its regulatory anti-sigma factor frequently play an important role in adaptation to the external environment [104]. Canonical transmembrane anti-sigma factors have a small cytoplasmic regulatory/inhibitory domain linked by a transmembrane domain to a C-terminus sensory domain that resides in the periplasm. PA14_27700 has neither a signal sequence nor a transmembrane domain suggesting that it functions in the cytoplasm. However, most anti-sigma factors are poorly conserved at the sequence level and there are anti-sigma factors that sense cytoplasmic stimuli. PA14_27700 #32578, although attenuated in virulence, does not exhibit any obvious defects in pigment production or motility in our assays (Table 2).
(TIF)
The virulence-attenuated phenotype of MAR2xT7 insertions in putative enoyl-CoA hydratase isomerase PA0745 is recapitulated by an in-frame deletion and is not dependent on pyoverdine production. A) PA0745, a putative enoyl-CoA hydratase isomerase, is the third gene of what is most likely a 5 gene operon: PA0747 a probable aldehyde dehydrogenase, PA0746 a putative acyl-CoA dehydrogenase, PA0745, PA0744 another putative enoyl-CoA hydratase/isomerase, and PA0743 a probable 3-hydroxyisobutyrate dehydrogenase. B) Insertions in PA0745 exhibit a virulence-attenuated phenotype but mutants of PA0746, the only other gene in the putative operon for which MAR2xT7 mutants were available, exhibited wild-type levels of virulence. C) The virulence-attenuated phenotype of an in-frame deletion mutant of PA0745 is complemented by expression of the entire operon (PA0747-PA0743) in trans. The operon was expressed under its own promoter; PA14 DNA from genome position 4851671–4848075 was cloned in Pseudomonas vector pucP19 [105]. D) A defect in pyoverdine production in PA0745 mutants (Table 2) is not the cause of the PA0745 avirulent phenotype. Pyoverdine biosynthetic mutants pvdA and pvdD show minimal to no attenuation of virulence. It has been suggested that PA0745 enoyl-CoA hydratase isomerase may function in the production of cis-2-decenoic acid, a fatty acid signal responsible for inducing biofilm dispersal, due to its weak homology to RpfF which produces cis-11-methyl-2-dodecenoic acid, the diffusible soluble factor (DSF) required for virulence in Xanthomonas campestris [106], [107]. However, PA0745 and X. campestris paaF (required for the breakdown of phenlyacetic acid) are reciprocal top BLASTP hits with 65% identity over 95% of the protein. The putative enzymatic functions of the genes in the PA0745 operon suggests that this operon may collectively synthesize or degrade an as yet unidentified product/products that affect siderophore production, control of swarming, and virulence in P. aeruginosa.
(TIF)
Transposon mutants in transcriptional regulator vqsR , but not genes in the adjacent downstream operon, are attenuated in virulence. VqsR (PA2591) is a LuxR type transcriptional regulator that controls expression of quorum sensing and virulence genes. A vqsR mutant of P. aeruginosa strain TB has been shown to be impaired in homoserine lactone production and is attenuated in a liquid C. elegans killing assay that occurs over a period of hours, similar in kinetics to toxin mediated PA14 “fast-killing” [108]. A) P. aeruginosa PA14 vqsR appears to be transcribed as an individual ORF; it is located 120 bp upstream of an operon that encodes two conserved hypothetical proteins, PA2590 a putative outer membrane receptor protein and PA2589 a possible permease. B) Two MAR2xT7 vqsR transposon mutants have reduced virulence in C. elegans. C) MAR2xT7 mutants in the operon adjacent to and downstream of vqsR (PA2590 and PA2589) are as virulent as WT PA14. In keeping with its role in quorum sensing, the spectrum of pigment and motility defects of a PA14 vqsR mutant paralleled those of lasR in our assays; the vqsR mutant was defective in both pyocyanin production and swarming, and displayed slightly reduced twitching (data not shown).
(TIF)
Glutamate-cysteine ligase ( gshA ) is required for full virulence of PA14. A) gshA (PA5203) is a single gene transcription unit. B) Four MAR2xT7 transposon insertions in gshA are virulence-attenuated. Mutations in gshA and gshB (PA0407) exhibit a similar degree of attenuation suggesting that production of glutathione may be required for WT levels of virulence (see Figure 5D).
(TIF)
Nine virulence-attenuated mutants exhibit normal growth. Cultures were diluted from overnight saturated cultures to 104 CFU/ml in M63 minimal media and grown at 37°C on a rotating wheel. At the indicated times, samples were taken of each culture, diluted and plated on LB plates. Plates were incubated overnight and colonies were counted the next day. A) Growth of virulence-attenuated MAR2xT7 mutants pchH #23790, PA14_27700 #32578, PA2550 #34827, pchI #35711, PA0456 #36116, clpA #39351, PA1216 #47923, vqsR #52787. B) Growth of PA0745 in-frame deletion mutant.
(TIF)
PA14 genes required for virulence in C. elegans were classified according to the breadth of their phylogenetic distribution across sequenced prokaryotes. A) Phylogenetic tree indicating breadths of phylogenetic distribution (or phylostrata) from 0, the narrowest breadth corresponding to PA14-specific genes, and 1 corresponding to genes shared found only in other strains of P. aeruginosa, to 6, the root of the tree and the broadest breadth, corresponding to genes distributed across eubacteria and archea. Phylogenetic breadths were assigned to PA14 genes corresponding to the parent node uniting all the child taxa to which all orthologs of a particular gene are found. B) The percentages of P. aeruginosa strain PA14 genes, PA14-NR set genes, and primary, secondary, tertiary, auxotroph, and VFDB set genes within each phylogenetic breadth is shown. The primary, secondary, tertiary, and auxotroph gene sets were apparently underrepresented in the breadths 0, 1, 2, and 3, but without statistical significance after multiple comparison correction using false discovery rate (q< = 0.05) due to the small number of genes involved. Among the auxotrophs, genes of breadth 4 were underrepresented (p-value = 0.0027) and 6 overrepresented (p-value = 1.2×10−8). Among VFDB genes, genes of breadth 3 were overrepresented (p-value = 1.22×10−8) and breadth 6 were underrepresented (p-value = 7.92×10−11).
(TIF)
Numerical accounting of virulence-attenuated PA14 transposon mutants identified in the primary, secondary and tertiary screens. (*) The 5754 mutants screened was the NR library (5850 insertion mutants) minus one 96-well plate of known slow growing mutants. Note that for the primary and secondary screen, the number of mutants obtained is presented; some genes were represented by multiple mutants and a few mutants were in intergenic regions (see text). In the tertiary screen, the number of genes is designated.
(DOCX)
Auxotrophic and slow-growing mutants identified in the primary screen for virulence-attenuated mutants.
(XLSX)
Virulence-attenuated mutants identified in the primary screen minus auxotrophs from Table S2.
(XLSX)
PA14 virulence-attenuated mutants identified in the secondary screen.
(XLSX)
Mutants in 58 genes tested in the tertiary screen for attenuation of virulence.
(XLSX)
Contains the following sheets showing totals and statistics for Figures 7 , 8 , and S13. This spreadsheet contains total counts, and percentages for comparisons across the various gene sets: the PA14 genome, the NR set, and the primary, secondary, tertiary, auxotroph, and VFDB sets. Fisher exact tests were performed relative to gene totals the NR set, and left, right, and two-tailed probabilities (p-values) indicating underrepresentation, overrepresentation, or extreme value, respectively, are shown for the primary, secondary, tertiary, auxotroph, and VFDB sets. FDR q-values are calculated only for the left and right probabilities, and are shown below the corresponding left and right probabilities. Results with statistical significance after FDR correction are shown in red font. Cells are colored according to the coloring of the corresponding legends in Figure 7 and Figure 8. Sheet “A. Core vs Auxiliary Genes”. This sheet contains the total counts and percentages of Core and Auxiliary genes across the various gene sets (see above). These data are presented graphically in Figure 7A with corresponding coloring of bars. Sheet “B. Predicted vs Non-Island”. This sheet contains the total counts and percentages of “predicted island” and “non-island” genes across the various gene sets. These data are presented graphically in Figure 7B. Sheet “C. Known Island vs Non-Island”. This sheet contains the total counts and percentages of “known island” and “non-island” genes across the various gene sets. These data are presented graphically in Figure 7C. Sheet “D. Phylogenetic Breadths”. This sheet contains the total counts and percentages of genes with various breadths of phylogenetic distribution, or phylostrata across the various gene sets. These data are presented graphically in Figure S13. Sheet “E. PGS and HFBP”. This sheet contains the total counts and percentages of genes within the Pseudomonas-genus-specific (PGS), in the HFBP set, and in non-PGS non-HFBP (“All Others”), across the various gene sets. These data are presented graphically in Figure 8.
(XLS)
Acknowledgments
We acknowledge M. Borowsky for help interpreting bioinformatics data and E. Drenkard for much advice on working with P. aeruginosa, particularly with regards to phenotypic assays. We thank N. Chand for providing kinB and algB deletion mutants and for help with generation of the PA0745 deletion mutant. We thank J. Bergelson (University of Chicago), D. Guttman (University of Toronto), L. Raleigh (New England Biolabs), J. Chang (University of Oregon), G. Narasimhan (Florida International University), and K. Mathee (Florida International University) for very helpful discussions and advice on the manuscript.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by NIH grants R01 AI085581, R01 AI064332, and P01 AI044220. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir and Crit Care Med. 2005;171:1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore NM, Flaws ML. Antimicrobial resistance mechanisms in Pseudomonas aeruginosa. Clin Lab Sci. 2011;24:47–51. [PubMed] [Google Scholar]
- 3.Lesic B, Lepine F, Deziel E, Zhang J, Zhang Q, et al. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog. 2007;3:1229–1239. doi: 10.1371/journal.ppat.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 5.Sintim HO, Smith JA, Wang J, Nakayama S, Yan L. Paradigm shift in discovering next-generation anti-infective agents: targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med Chem. 2010;2:1005–1035. doi: 10.4155/fmc.10.185. [DOI] [PubMed] [Google Scholar]
- 6.Romling U, Wingender J, Muller H, Tummler B. A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol. 1994;60:1734–1738. doi: 10.1128/aem.60.6.1734-1738.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pukatzki S, Kessin RH, Mekalanos JJ. The human pathogen Pseudomonas aeruginosa utilizes conserved virulence pathways to infect the social amoeba Dictyostelium discoideum. Proc Natl Acad Sci U S A. 2002;99:3159–3164. doi: 10.1073/pnas.052704399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, et al. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 9.Jander G, Rahme LG, Ausubel FM. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol. 2000;182:3843–3845. doi: 10.1128/jb.182.13.3843-3845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apidianakis Y, Rahme LG. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. Nat Protoc. 2009;4:1285–1294. doi: 10.1038/nprot.2009.124. [DOI] [PubMed] [Google Scholar]
- 11.Lau GW, Goumnerov BC, Walendziewicz CL, Hewitson J, Xiao W, et al. The Drosophila melanogaster toll pathway participates in resistance to infection by the gram-negative human pathogen Pseudomonas aeruginosa. Infect Immun. 2003;71:4059–4066. doi: 10.1128/IAI.71.7.4059-4066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A. 1999;96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci U S A. 1999;96:2408–2413. doi: 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 15.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, et al. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 2010;6:e1000982. doi: 10.1371/journal.ppat.1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol. 2012;24:3–9. doi: 10.1016/j.coi.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, et al. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2006;2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi L, Takahashi K, Dundee J, Shahroor-Karni S, Thiel S, et al. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J Exp Med. 2004;199:1379–1390. doi: 10.1084/jem.20032207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bork P, Beckmann G. The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol. 1993;231:539–545. doi: 10.1006/jmbi.1993.1305. [DOI] [PubMed] [Google Scholar]
- 22.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 23.Pukkila-Worley R, Ausubel FM, Mylonakis E. Candida albicans infection of Caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 2011;7:e1002074. doi: 10.1371/journal.ppat.1002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sifri CD, Begun J, Ausubel FM, Calderwood SB. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun. 2003;71:2208–2217. doi: 10.1128/IAI.71.4.2208-2217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyata S, Casey M, Frank DW, Ausubel FM, Drenkard E. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect Immun. 2003;71:2404–2413. doi: 10.1128/IAI.71.5.2404-2413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendrickson EL, Plotnikova J, Mahajan-Miklos S, Rahme LG, Ausubel FM. Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J Bacteriol. 2001;183:7126–7134. doi: 10.1128/JB.183.24.7126-7134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yorgey P, Rahme LG, Tan MW, Ausubel FM. The roles of mucD and alginate in the virulence of Pseudomonas aeruginosa in plants, nematodes and mice. Mol Microbiol. 2001;41:1063–1076. doi: 10.1046/j.1365-2958.2001.02580.x. [DOI] [PubMed] [Google Scholar]
- 28.Pearson JP, Feldman M, Iglewski BH, Prince A. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect Immun. 2000;68:4331–4334. doi: 10.1128/iai.68.7.4331-4334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willcox MD, Zhu H, Conibear TC, Hume EB, Givskov M, et al. Role of quorum sensing by Pseudomonas aeruginosa in microbial keratitis and cystic fibrosis. Microbiology. 2008;154:2184–2194. doi: 10.1099/mic.0.2008/019281-0. [DOI] [PubMed] [Google Scholar]
- 30.Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol. 2004;2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 31.Heiniger RW, Winther-Larsen HC, Pickles RJ, Koomey M, Wolfgang MC. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell Microbiol. 2010;12:1158–1173. doi: 10.1111/j.1462-5822.2010.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pier GB. Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol. 2007;297:277–295. doi: 10.1016/j.ijmm.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groisman EA, Ochman H. Pathogenicity islands: bacterial evolution in quantum leaps. Cell. 1996;87:791–794. doi: 10.1016/s0092-8674(00)81985-6. [DOI] [PubMed] [Google Scholar]
- 35.Smith EE, Sims EH, Spencer DH, Kaul R, Olson MV. Evidence for diversifying selection at the pyoverdine locus of Pseudomonas aeruginosa. J Bacteriol. 2005;187:2138–2147. doi: 10.1128/JB.187.6.2138-2147.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raymond CK, Sims EH, Kas A, Spencer DH, Kutyavin TV, et al. Genetic variation at the O-antigen biosynthetic locus in Pseudomonas aeruginosa. J Bacteriol. 2002;184:3614–3622. doi: 10.1128/JB.184.13.3614-3622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson RW, Athanassopoulos E, Tsiamis G, Mansfield JW, Sesma A, et al. Identification of a pathogenicity island, which contains genes for virulence and avirulence, on a large native plasmid in the bean pathogen Pseudomonas syringae pathovar phaseolicola. Proc Natl Acad Sci U S A. 1999;96:10875–10880. doi: 10.1073/pnas.96.19.10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma W, Dong FF, Stavrinides J, Guttman DS. Type III effector diversification via both pathoadaptation and horizontal transfer in response to a coevolutionary arms race. PLoS Genet. 2006;2:e209. doi: 10.1371/journal.pgen.0020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho Sui SJ, Fedynak A, Hsiao WW, Langille MG, Brinkman FS. The association of virulence factors with genomic islands. PloS One. 2009;4:e8094. doi: 10.1371/journal.pone.0008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He J, Baldini RL, Deziel E, Saucier M, Zhang Q, et al. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc Natl Acad Sci U S A. 2004;101:2530–2535. doi: 10.1073/pnas.0304622101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci U S A. 2008;105:3100–3105. doi: 10.1073/pnas.0711982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiehlmann L, Munder A, Adams T, Juhas M, Kolmar H, et al. Functional genomics of Pseudomonas aeruginosa to identify habitat-specific determinants of pathogenicity. Int J Med Microbiol. 2007;297:615–623. doi: 10.1016/j.ijmm.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Garvis S, Munder A, Ball G, de Bentzmann S, Wiehlmann L, et al. Caenorhabditis elegans semi-automated liquid screen reveals a specialized role for the chemotaxis gene cheB2 in Pseudomonas aeruginosa virulence. PLoS Pathog. 2009;5:e1000540. doi: 10.1371/journal.ppat.1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim SH, Park SY, Heo YJ, Cho YH. Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor, HudA. Infect Immun. 2008;76:4152–4162. doi: 10.1128/IAI.01637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Potvin E, Lehoux DE, Kukavica-Ibrulj I, Richard KL, Sanschagrin F, et al. In vivo functional genomics of Pseudomonas aeruginosa for high-throughput screening of new virulence factors and antibacterial targets. Environ Microbiol. 2003;5:1294–1308. doi: 10.1046/j.1462-2920.2003.00542.x. [DOI] [PubMed] [Google Scholar]
- 47.Bianconi I, Milani A, Cigana C, Paroni M, Levesque RC, et al. Positive signature-tagged mutagenesis in Pseudomonas aeruginosa: tracking patho-adaptive mutations promoting airways chronic infection. PLoS Pathog. 2011;7:e1001270. doi: 10.1371/journal.ppat.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Mushegian A, Lory S, Jin S. Large-scale isolation of candidate virulence genes of Pseudomonas aeruginosa by in vivo selection. Proc Natl Acad Sci U S A. 1996;93:10434–10439. doi: 10.1073/pnas.93.19.10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A. 2006;103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lau GW, Britigan BE, Hassett DJ. Pseudomonas aeruginosa OxyR is required for full virulence in rodent and insect models of infection and for resistance to human neutrophils. Infect Immun. 2005;73:2550–2553. doi: 10.1128/IAI.73.4.2550-2553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mulcahy H, O'Callaghan J, O'Grady EP, Macia MD, Borrell N, et al. Pseudomonas aeruginosa RsmA plays an important role during murine infection by influencing colonization, virulence, persistence, and pulmonary inflammation. Infect Immun. 2008;76:632–638. doi: 10.1128/IAI.01132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang J, Chen L, Sun L, Yu J, Jin Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008;36:D539–542. doi: 10.1093/nar/gkm951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bleves S, Viarre V, Salacha R, Michel GP, Filloux A, et al. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int J Med Microbiol. 2010;300:534–543. doi: 10.1016/j.ijmm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Gallagher LA, Manoil C. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol. 2001;183:6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deziel E, Lepine F, Milot S, He J, Mindrinos MN, et al. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A. 2004;101:1339–1344. doi: 10.1073/pnas.0307694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider BL, Kiupakis AK, Reitzer LJ. Arginine catabolism and the arginine succinyltransferase pathway in Escherichia coli. J Bacteriol. 1998;180:4278–4286. doi: 10.1128/jb.180.16.4278-4286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itoh Y. Cloning and characterization of the aru genes encoding enzymes of the catabolic arginine succinyltransferase pathway in Pseudomonas aeruginosa. J Bacteriol. 1997;179:7280–7290. doi: 10.1128/jb.179.23.7280-7290.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol. 2003;185:2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuster M, Lostroh CP, Ogi T, Greenberg EP. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol. 2003;185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dekimpe V, Deziel E. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology. 2009;155:712–723. doi: 10.1099/mic.0.022764-0. [DOI] [PubMed] [Google Scholar]
- 61.Stintzi A, Evans K, Meyer JM, Poole K. Quorum-sensing and siderophore biosynthesis in Pseudomonas aeruginosa: lasR/lasI mutants exhibit reduced pyoverdine biosynthesis. FEMS Microbiol Lett. 1998;166:341–345. doi: 10.1111/j.1574-6968.1998.tb13910.x. [DOI] [PubMed] [Google Scholar]
- 62.Kohler T, Curty LK, Barja F, van Delden C, Pechere JC. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J Bacteriol. 2000;182:5990–5996. doi: 10.1128/jb.182.21.5990-5996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeung AT, Torfs EC, Jamshidi F, Bains M, Wiegand I, et al. Swarming of Pseudomonas aeruginosa is controlled by a broad spectrum of transcriptional regulators, including MetR. J Bacteriol. 2009;191:5592–5602. doi: 10.1128/JB.00157-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langille MG, Brinkman FS. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang X, Pham XQ, Olson MV, Lory S. Identification of a genomic island present in the majority of pathogenic isolates of Pseudomonas aeruginosa. J Bacteriol. 2001;183:843–853. doi: 10.1128/JB.183.3.843-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domazet-Loso T, Brajkovic J, Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. doi: 10.1016/j.tig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 67.Chen L, Yang J, Yu J, Yao Z, Sun L, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33:D325–328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McEwan DL, Kirienko NV, Ausubel FM. Host Translational Inhibition by Pseudomonas aeruginosa Exotoxin A Triggers an Immune Response in Caenorhabditis elegans. Cell Host Microbe. 2012;11:364–374. doi: 10.1016/j.chom.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. C. elegans Detects Pathogen-Induced Translational Inhibition to Activate Immune Signaling. Cell Host Microbe. 2012;11:375–386. doi: 10.1016/j.chom.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lindeberg M, Myers CR, Collmer A, Schneider DJ. Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol Plant Microbe Interact. 2008;21:685–700. doi: 10.1094/MPMI-21-6-0685. [DOI] [PubMed] [Google Scholar]
- 71.Chang JH, Urbach JM, Law TF, Arnold LW, Hu A, et al. A high-throughput, near-saturating screen for type III effector genes from Pseudomonas syringae. Proc Natl Acad Sci U S A. 2005;102:2549–2554. doi: 10.1073/pnas.0409660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindeberg M, Cunnac S, Collmer A. The evolution of Pseudomonas syringae host specificity and type III effector repertoires. Mol Plant Pathol. 2009;10:767–775. doi: 10.1111/j.1364-3703.2009.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schirawski J, Mannhaupt G, Munch K, Brefort T, Schipper K, et al. Pathogenicity determinants in smut fungi revealed by genome comparison. Science. 2010;330:1546–1548. doi: 10.1126/science.1195330. [DOI] [PubMed] [Google Scholar]
- 74.Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, et al. The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc Natl Acad Sci U S A. 2000;97:4856–4861. doi: 10.1073/pnas.97.9.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGhee JD The C. elegans Research Community, editor. The C. elegans Intestine. WormBook. 2007. Available: http://www.wormbook.org. doi/10.1895/wormbook.1.7.1.
- 76.Alegado RA, Chin CY, Monack DM, Tan MW. The two-component sensor kinase KdpD is required for Salmonella typhimurium colonization of Caenorhabditis elegans and survival in macrophages. Cell Microbiol. 2011;13:1618–1637. doi: 10.1111/j.1462-5822.2011.01645.x. [DOI] [PubMed] [Google Scholar]
- 77.Riccillo PM, Muglia CI, de Bruijn FJ, Roe AJ, Booth IR, et al. Glutathione is involved in environmental stress responses in Rhizobium tropici, including acid tolerance. J Bacteriol. 2000;182:1748–1753. doi: 10.1128/jb.182.6.1748-1753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Phadtare S. Recent developments in bacterial cold-shock response. Curr Issues Mol Biol. 2004;6:125–136. [PubMed] [Google Scholar]
- 79.Smith DA, Parish T, Stoker NG, Bancroft GJ. Characterization of auxotrophic mutants of Mycobacterium tuberculosis and their potential as vaccine candidates. Infect Immun. 2001;69:1142–1150. doi: 10.1128/IAI.69.2.1142-1150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marquis H, Bouwer HG, Hinrichs DJ, Portnoy DA. Intracytoplasmic growth and virulence of Listeria monocytogenes auxotrophic mutants. Infect Immun. 1993;61:3756–3760. doi: 10.1128/iai.61.9.3756-3760.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kung VL, Ozer EA, Hauser AR. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2010;74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stieritz DD, Holder IA. Experimental studies of the pathogenesis of infections due to Pseudomonas aeruginosa: description of a burned mouse model. J Infect Dis. 1975;131:688–691. doi: 10.1093/infdis/131.6.688. [DOI] [PubMed] [Google Scholar]
- 83.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58. doi: 10.1038/nri2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Limmer S, Haller S, Drenkard E, Lee J, Yu S, et al. Pseudomonas aeruginosa RhlR is required to neutralize the cellular immune response in a Drosophila melanogaster oral infection model. Proc Natl Acad Sci U S A. 2011;108:17378–17383. doi: 10.1073/pnas.1114907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 86.Chand NS, Lee JS, Clatworthy AE, Golas AJ, Smith RS, et al. The sensor kinase KinB regulates virulence in acute Pseudomonas aeruginosa infection. J Bacteriol. 2011;193:2989–2999. doi: 10.1128/JB.01546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Essar DW, Eberly L, Hadero A, Crawford IP. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J Bacteriol. 1990;172:884–900. doi: 10.1128/jb.172.2.884-900.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dietrich LE, Price-Whelan A, Petersen A, Whiteley M, Newman DK. The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol Microbiol. 2006;61:1308–1321. doi: 10.1111/j.1365-2958.2006.05306.x. [DOI] [PubMed] [Google Scholar]
- 89.Kim EJ, Sabra W, Zeng AP. Iron deficiency leads to inhibition of oxygen transfer and enhanced formation of virulence factors in cultures of Pseudomonas aeruginosa PAO1. Microbiology. 2003;149:2627–2634. doi: 10.1099/mic.0.26276-0. [DOI] [PubMed] [Google Scholar]
- 90.Adonizio A, Kong KF, Mathee K. Inhibition of quorum sensing-controlled virulence factor production in Pseudomonas aeruginosa by South Florida plant extracts. Antimicrob Agents Chemother. 2008;52:198–203. doi: 10.1128/AAC.00612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Overhage J, Bains M, Brazas MD, Hancock RE. Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J Bacteriol. 2008;190:2671–2679. doi: 10.1128/JB.01659-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Darzins A. The pilG gene product, required for Pseudomonas aeruginosa pilus production and twitching motility, is homologous to the enteric, single-domain response regulator CheY. J Bacteriol. 1993;175:5934–5944. doi: 10.1128/jb.175.18.5934-5944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Banerjee S, Pedersen T. The Design, Implementation, and Use of the Ngram Statistic Package. 2003. pp. 370–381. Proceedings of the Fourth International Conference on Intelligent Text Processing and Computational Linguistics; Mexico City. CICLing'03. Available: http://cpansearch.perl.org/src/TPEDERSE/Text-NSP-371.313/doc/cicling2003.pdf.
- 94.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300. Available: http://www.jstor.org/stable/2346101. [Google Scholar]
- 95.Abdi H. The Bonferonni and Sidak Corrections for Multiple Comparisons. In: Salkind N, editor. Encylopedia of Measurement and Statistics. Thousand Oaks, CA: Sage; 2007. pp. 1–9. [Google Scholar]
- 96.Kress W, Maglica Z, Weber-Ban E. Clp chaperone-proteases: structure and function. Res Microbiol. 2009;160:618–628. doi: 10.1016/j.resmic.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 97.Dougan DA, Reid BG, Horwich AL, Bukau B. ClpS, a substrate modulator of the ClpAP machine. Mol Cell. 2002;9:673–683. doi: 10.1016/s1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- 98.Reimmann C, Patel HM, Serino L, Barone M, Walsh CT, et al. Essential PchG-dependent reduction in pyochelin biosynthesis of Pseudomonas aeruginosa. J Bacteriol. 2001;183:813–820. doi: 10.1128/JB.183.3.813-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alibaud L, Kohler T, Coudray A, Prigent-Combaret C, Bergeret E, et al. Pseudomonas aeruginosa virulence genes identified in a Dictyostelium host model. Cell Microbiol. 2008;10:729–740. doi: 10.1111/j.1462-5822.2007.01080.x. [DOI] [PubMed] [Google Scholar]
- 100.Woolwine SC, Wozniak DJ. Identification of an Escherichia coli pepA homolog and its involvement in suppression of the algB phenotype in mucoid Pseudomonas aeruginosa. J Bacteriol. 1999;181:107–116. doi: 10.1128/jb.181.1.107-116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Horn G, Hofweber R, Kremer W, Kalbitzer HR. Structure and function of bacterial cold shock proteins. Cell Mol Life Sci. 2007;64:1457–1470. doi: 10.1007/s00018-007-6388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Balasubramanian D, Mathee K. Comparative transcriptome analyses of Pseudomonas aeruginosa. Hum Genomics. 2009;3:349–361. doi: 10.1186/1479-7364-3-4-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Damron FH, Owings JP, Okkotsu Y, Varga JJ, Schurr JR, et al. Analysis of the Pseudomonas aeruginosa regulon controlled by the sensor kinase KinB and sigma factor RpoN. J Bacteriol. 2011;194:1317–30. doi: 10.1128/JB.06105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Potvin E, Sanschagrin F, Levesque RC. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev. 2008;32:38–55. doi: 10.1111/j.1574-6976.2007.00092.x. [DOI] [PubMed] [Google Scholar]
- 105.Schweizer HP. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene. 1991;97:109–121. doi: 10.1016/0378-1119(91)90016-5. [DOI] [PubMed] [Google Scholar]
- 106.Davies DG, Marques CN. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J Bacteriol. 2009;191:1393–1403. doi: 10.1128/JB.01214-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang LH, He Y, Gao Y, Wu JE, Dong YH, et al. A bacterial cell-cell communication signal with cross-kingdom structural analogues. Mol Microbiol. 2004;51:903–912. doi: 10.1046/j.1365-2958.2003.03883.x. [DOI] [PubMed] [Google Scholar]
- 108.Juhas M, Wiehlmann L, Huber B, Jordan D, Lauber J, et al. Global regulation of quorum sensing and virulence by VqsR in Pseudomonas aeruginosa. Microbiology. 2004;150:831–841. doi: 10.1099/mic.0.26906-0. [DOI] [PubMed] [Google Scholar]
- 109.Smith RS, Wolfgang MC, Lory S. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun. 2004;72:1677–1684. doi: 10.1128/IAI.72.3.1677-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol. 2002;184:6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Evans EA, Kawli T, Tan MW. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 2008;4:e1000175. doi: 10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anthony LS, Cowley SC, Mdluli KE, Nano FE. Isolation of a Francisella tularensis mutant that is sensitive to serum and oxidative killing and is avirulent in mice: correlation with the loss of MinD homologue expression. FEMS Microbiol Lett. 1994;124:157–165. doi: 10.1016/0378-1097(94)90243-7. [DOI] [PubMed] [Google Scholar]
- 113.Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- 114.Loughlin MF, Arandhara V, Okolie C, Aldsworth TG, Jenks PJ. Helicobacter pylori mutants defective in the clpP ATP-dependant protease and the chaperone clpA display reduced macrophage and murine survival. Microb Pathog. 2009;46:53–57. doi: 10.1016/j.micpath.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 115.Deziel E, Gopalan S, Tampakaki AP, Lepine F, Padfield KE, et al. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol Microbiol. 2005;55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- 116.Hahn HP. The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa–a review. Gene. 1997;192:99–108. doi: 10.1016/s0378-1119(97)00116-9. [DOI] [PubMed] [Google Scholar]
- 117.Woods DE, Straus DC, Johanson WG, Jr, Berry VK, Bass JA. Role of pili in adherence of Pseudomonas aeruginosa to mammalian buccal epithelial cells. Infect Immun. 1980;29:1146–1151. doi: 10.1128/iai.29.3.1146-1151.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow chart of screening procedure for virulence-attenuated PA14 transposon mutants in C. elegans .
(TIF)
Correlation of virulence attenuation in standard SK and progeny survival assays. A) lasR, gacA, ptsP, mucD, rhlR and pilA mutants all have attenuated virulence in a standard slow killing (SK) assay (60–80 N2 worms tested). B) The number and developmental stage of the progeny on the SK plates in panel (A) four days after transfer of the parent worms to pathogen at 25°C. On PA14, there were few progeny and no gravid adults were observed whereas gravid adult progeny were found on the virulence-attenuated P. aeruginosa strains. The number and age of progeny on the mutant plates was qualitatively scored in comparison to the PA14 WT plates. Plates seeded with gacA and ptsP were overrun with hundreds of gravid adult worms that consumed the bacterial lawn. lasR, mucD, rhlR and pilA plates contained fewer gravid adult worms but all were observably greater than plates seeded with WT PA14.
(TIF)
Percentage of mutants that are VFDB genes. The percentage of primary, secondary, and tertiary, and auxotroph set virulence-attenuated genes that are VFDB and non-VFDB genes are indicated. VFDB genes are significantly overrepresented in the primary, secondary, and tertiary sets, with p-values of 5.5×10−6, 2.1×10−5, and 3.7×10−5, respectively. Furthermore, their overrepresentation increases with successive screen iterations.
(TIF)
Venn diagram showing the overlaps between the 170 virulence-attenuated genes obtained in the secondary screen, the Potvin set, and the PA14 VFDB set.
(TIF)
Multiple pepP alleles but not alleles of other genes in the operon have attenuated virulence. A) PepP is a cytoplasmic aminopeptidase that cleaves aminoacyl proline dipeptides from the N terminus of polypeptides. In PA14 pepP appears to be the second gene in a 5 gene operon comprised of PA5225, a hypothetical protein, pepP, ubiH a ubiquinone biosynthetic enzyme, PA5225 another hypothetical protein, and terminating with PA5221, an ORF with homology to E. coli visC a pyridine nucleotide-disulphide oxidoreductase in the ubiH family. B) Four MAR2xT7 alleles of pepP display highly attenuated C. elegans killing. C) Transposon mutants in 4/5 genes in the pepP operon were tested for their effect on virulence (no mutant was available in PA5221). Mutants in the two downstream genes tested, ubiH and PA5222 exhibited wild-type levels of virulence whereas the single mutant in the upstream PA5225 had a modest attenuation of virulence (compared to pepP) that might be due to effects on pepP expression. pepP mutant #31097 produced elevated levels of pyocyanin and had somewhat reduced swimming ability, although swarming was normal on both SK and LB (Table 2). The PepP proline aminopeptidase could function in utilization of exogenous peptides as nutrients, degradation of proteins or protein maturation, perhaps of a specific substrate relevant to virulence. There is a precedent for aminopeptidase function being linked to virulence associated phenotypes. P. aeruginosa PepA was shown to be involved in the regulation of alginate biosynthesis [100].
(TIF)
Mutations in cold shock domain (CSD) protein PA0456 are virulence-attenuated in C. elegans but mutations in four other CSD proteins do not affect virulence. A) PA0456 is most likely a single gene transcription unit. PA0456 (previously annotated as cspB in the PA14 genome) contains a canonical cold shock domain (CSD) and is homologous to other CSD proteins (57% identical to the major E. coli cold shock protein, CspA, and 65% to B. subtilis CspB as determined by BLASTP). Although the first cold shock proteins (Csps), a conserved family of small mostly acidic proteins that bind single stranded DNA and RNA, were identified as major proteins induced upon temperature downshift, some members of the family are not induced upon cold shock and many are implicated in other cellular functions [101]. B) Three independent MAR2xT7 transposon insertions in PA0456 have attenuated virulence in C. elegans. C) Transposon insertion mutants in four additional CSD containing genes (capB PA3266, PA0961, cspD PA2622 and PA1960) have wild-type virulence in C. elegans. Five additional CSD-containing homologs of PA0456 (PA3266, PA1159, PA0961, PA2622, PA1960) were identified by BLASTP against the PA14 protein database with 76, 66, 62, 54 and 41% identity to PA0456 respectively; all 5 contain a CSD as analyzed by Prosite and transposon insertion mutants were available in 4/5 of these genes. Among the CSD containing proteins tested, PA0456 appears to be unique in its role in virulence, suggesting that the cold shock response per se is most likely not required for virulence of PA14. PA14 PA0456 has been reported to be regulated by quorum sensing [102], and in keeping with these findings PA0456 exhibited defects in quorum sensing regulated phenotypes; the PA0456 mutant had reduced pyocyanin production and swarming motility (Table 2).
(TIF)
KinB (PA5484) sensor kinase is required for PA14 virulence in C. elegans . KinB negatively regulates alginate production and has been recently shown to be required for virulence in zebrafish embryos and mice [86], [103]. A) kinB and its cognate response regulator algB form a two gene operon. B) Two MAR2xT7 transposon alleles in kinB (as well as an in-frame deletion of kinB described in Chand et al., data not shown) are reduced in virulence. C) Transposon mutants in algB (and an in-frame deletion of algB described in Chand el al., data not shown) exhibited wild-type virulence in C. elegans, paralleling their lack of phenotype in zebrafish [86].
(TIF)
Mutations in putative transcriptional regulator PA14_27700 and possible ECF sigma factor PA14_27690 are virulence-attenuated in C. elegans . A) PA14_27700, a Crp/FNR-type transcriptional regulator that appears to have no homologue in PA01 [32], is located 264 bp downstream of PA2817 and 66 upstream of PA14_26790, a FecI-like extracytoplasmic function (ECF) sigma that also has no PA01 homologue. It is unclear whether PA14_27700 and PA14_26790 form an operon. B) Multiple MAR2xT7 insertions in both PA14_27700 and PA14_27690 are attenuated in virulence. Although not initially identified in our screen, mutants in PA14_26790 have a strong virulence-attenuated phenotype like PA14_27700. ECF sigma factors, to which PA14_26790 has homology, are commonly co-transcribed with a regulatory anti-sigma factor and the ECF sigma factor and its regulatory anti-sigma factor frequently play an important role in adaptation to the external environment [104]. Canonical transmembrane anti-sigma factors have a small cytoplasmic regulatory/inhibitory domain linked by a transmembrane domain to a C-terminus sensory domain that resides in the periplasm. PA14_27700 has neither a signal sequence nor a transmembrane domain suggesting that it functions in the cytoplasm. However, most anti-sigma factors are poorly conserved at the sequence level and there are anti-sigma factors that sense cytoplasmic stimuli. PA14_27700 #32578, although attenuated in virulence, does not exhibit any obvious defects in pigment production or motility in our assays (Table 2).
(TIF)
The virulence-attenuated phenotype of MAR2xT7 insertions in putative enoyl-CoA hydratase isomerase PA0745 is recapitulated by an in-frame deletion and is not dependent on pyoverdine production. A) PA0745, a putative enoyl-CoA hydratase isomerase, is the third gene of what is most likely a 5 gene operon: PA0747 a probable aldehyde dehydrogenase, PA0746 a putative acyl-CoA dehydrogenase, PA0745, PA0744 another putative enoyl-CoA hydratase/isomerase, and PA0743 a probable 3-hydroxyisobutyrate dehydrogenase. B) Insertions in PA0745 exhibit a virulence-attenuated phenotype but mutants of PA0746, the only other gene in the putative operon for which MAR2xT7 mutants were available, exhibited wild-type levels of virulence. C) The virulence-attenuated phenotype of an in-frame deletion mutant of PA0745 is complemented by expression of the entire operon (PA0747-PA0743) in trans. The operon was expressed under its own promoter; PA14 DNA from genome position 4851671–4848075 was cloned in Pseudomonas vector pucP19 [105]. D) A defect in pyoverdine production in PA0745 mutants (Table 2) is not the cause of the PA0745 avirulent phenotype. Pyoverdine biosynthetic mutants pvdA and pvdD show minimal to no attenuation of virulence. It has been suggested that PA0745 enoyl-CoA hydratase isomerase may function in the production of cis-2-decenoic acid, a fatty acid signal responsible for inducing biofilm dispersal, due to its weak homology to RpfF which produces cis-11-methyl-2-dodecenoic acid, the diffusible soluble factor (DSF) required for virulence in Xanthomonas campestris [106], [107]. However, PA0745 and X. campestris paaF (required for the breakdown of phenlyacetic acid) are reciprocal top BLASTP hits with 65% identity over 95% of the protein. The putative enzymatic functions of the genes in the PA0745 operon suggests that this operon may collectively synthesize or degrade an as yet unidentified product/products that affect siderophore production, control of swarming, and virulence in P. aeruginosa.
(TIF)
Transposon mutants in transcriptional regulator vqsR , but not genes in the adjacent downstream operon, are attenuated in virulence. VqsR (PA2591) is a LuxR type transcriptional regulator that controls expression of quorum sensing and virulence genes. A vqsR mutant of P. aeruginosa strain TB has been shown to be impaired in homoserine lactone production and is attenuated in a liquid C. elegans killing assay that occurs over a period of hours, similar in kinetics to toxin mediated PA14 “fast-killing” [108]. A) P. aeruginosa PA14 vqsR appears to be transcribed as an individual ORF; it is located 120 bp upstream of an operon that encodes two conserved hypothetical proteins, PA2590 a putative outer membrane receptor protein and PA2589 a possible permease. B) Two MAR2xT7 vqsR transposon mutants have reduced virulence in C. elegans. C) MAR2xT7 mutants in the operon adjacent to and downstream of vqsR (PA2590 and PA2589) are as virulent as WT PA14. In keeping with its role in quorum sensing, the spectrum of pigment and motility defects of a PA14 vqsR mutant paralleled those of lasR in our assays; the vqsR mutant was defective in both pyocyanin production and swarming, and displayed slightly reduced twitching (data not shown).
(TIF)
Glutamate-cysteine ligase ( gshA ) is required for full virulence of PA14. A) gshA (PA5203) is a single gene transcription unit. B) Four MAR2xT7 transposon insertions in gshA are virulence-attenuated. Mutations in gshA and gshB (PA0407) exhibit a similar degree of attenuation suggesting that production of glutathione may be required for WT levels of virulence (see Figure 5D).
(TIF)
Nine virulence-attenuated mutants exhibit normal growth. Cultures were diluted from overnight saturated cultures to 104 CFU/ml in M63 minimal media and grown at 37°C on a rotating wheel. At the indicated times, samples were taken of each culture, diluted and plated on LB plates. Plates were incubated overnight and colonies were counted the next day. A) Growth of virulence-attenuated MAR2xT7 mutants pchH #23790, PA14_27700 #32578, PA2550 #34827, pchI #35711, PA0456 #36116, clpA #39351, PA1216 #47923, vqsR #52787. B) Growth of PA0745 in-frame deletion mutant.
(TIF)
PA14 genes required for virulence in C. elegans were classified according to the breadth of their phylogenetic distribution across sequenced prokaryotes. A) Phylogenetic tree indicating breadths of phylogenetic distribution (or phylostrata) from 0, the narrowest breadth corresponding to PA14-specific genes, and 1 corresponding to genes shared found only in other strains of P. aeruginosa, to 6, the root of the tree and the broadest breadth, corresponding to genes distributed across eubacteria and archea. Phylogenetic breadths were assigned to PA14 genes corresponding to the parent node uniting all the child taxa to which all orthologs of a particular gene are found. B) The percentages of P. aeruginosa strain PA14 genes, PA14-NR set genes, and primary, secondary, tertiary, auxotroph, and VFDB set genes within each phylogenetic breadth is shown. The primary, secondary, tertiary, and auxotroph gene sets were apparently underrepresented in the breadths 0, 1, 2, and 3, but without statistical significance after multiple comparison correction using false discovery rate (q< = 0.05) due to the small number of genes involved. Among the auxotrophs, genes of breadth 4 were underrepresented (p-value = 0.0027) and 6 overrepresented (p-value = 1.2×10−8). Among VFDB genes, genes of breadth 3 were overrepresented (p-value = 1.22×10−8) and breadth 6 were underrepresented (p-value = 7.92×10−11).
(TIF)
Numerical accounting of virulence-attenuated PA14 transposon mutants identified in the primary, secondary and tertiary screens. (*) The 5754 mutants screened was the NR library (5850 insertion mutants) minus one 96-well plate of known slow growing mutants. Note that for the primary and secondary screen, the number of mutants obtained is presented; some genes were represented by multiple mutants and a few mutants were in intergenic regions (see text). In the tertiary screen, the number of genes is designated.
(DOCX)
Auxotrophic and slow-growing mutants identified in the primary screen for virulence-attenuated mutants.
(XLSX)
Virulence-attenuated mutants identified in the primary screen minus auxotrophs from Table S2.
(XLSX)
PA14 virulence-attenuated mutants identified in the secondary screen.
(XLSX)
Mutants in 58 genes tested in the tertiary screen for attenuation of virulence.
(XLSX)
Contains the following sheets showing totals and statistics for Figures 7 , 8 , and S13. This spreadsheet contains total counts, and percentages for comparisons across the various gene sets: the PA14 genome, the NR set, and the primary, secondary, tertiary, auxotroph, and VFDB sets. Fisher exact tests were performed relative to gene totals the NR set, and left, right, and two-tailed probabilities (p-values) indicating underrepresentation, overrepresentation, or extreme value, respectively, are shown for the primary, secondary, tertiary, auxotroph, and VFDB sets. FDR q-values are calculated only for the left and right probabilities, and are shown below the corresponding left and right probabilities. Results with statistical significance after FDR correction are shown in red font. Cells are colored according to the coloring of the corresponding legends in Figure 7 and Figure 8. Sheet “A. Core vs Auxiliary Genes”. This sheet contains the total counts and percentages of Core and Auxiliary genes across the various gene sets (see above). These data are presented graphically in Figure 7A with corresponding coloring of bars. Sheet “B. Predicted vs Non-Island”. This sheet contains the total counts and percentages of “predicted island” and “non-island” genes across the various gene sets. These data are presented graphically in Figure 7B. Sheet “C. Known Island vs Non-Island”. This sheet contains the total counts and percentages of “known island” and “non-island” genes across the various gene sets. These data are presented graphically in Figure 7C. Sheet “D. Phylogenetic Breadths”. This sheet contains the total counts and percentages of genes with various breadths of phylogenetic distribution, or phylostrata across the various gene sets. These data are presented graphically in Figure S13. Sheet “E. PGS and HFBP”. This sheet contains the total counts and percentages of genes within the Pseudomonas-genus-specific (PGS), in the HFBP set, and in non-PGS non-HFBP (“All Others”), across the various gene sets. These data are presented graphically in Figure 8.
(XLS)