

Abstract
Mangroves are complex ecosystems that regulate nutrient and sediment fluxes to the open sea. The importance of bacteria and fungi in regulating nutrient cycles has led to an interest in their diversity and composition in mangroves. However, very few studies have assessed Archaea in mangroves, and virtually nothing is known about whether mangrove rhizospheres affect archaeal diversity and composition. Here, we studied the diversity and composition of Archaea in mangrove bulk sediment and the rhizospheres of two mangrove trees, Rhizophora mangle and Laguncularia racemosa, using denaturing gradient gel electrophoresis (DGGE) and pyrosequencing of archaeal 16S rRNA genes with a nested-amplification approach. DGGE profiles revealed significant structural differences between bulk sediment and rhizosphere samples, suggesting that roots of both mangrove species influence the sediment archaeal community. Nearly all of the detected sequences obtained with pyrosequencing were identified as Archaea, but most were unclassified at the level of phylum or below. Archaeal richness was, furthermore, the highest in the L. racemosa rhizosphere, intermediate in bulk sediment, and the lowest in the R. mangle rhizosphere. This study shows that rhizosphere microhabitats of R. mangle and L. racemosa, common plants in subtropical mangroves located in Rio de Janeiro, Brazil, hosted distinct archaeal assemblages.
INTRODUCTION
Mangroves are coastal forests that represent an important ecotone between terrestrial and marine environments. These forests are ecologically and economically important, since they limit erosion in coastal areas, reduce the impact of waves and tsunamis, and are used by several aquatic animals during some part of their life cycle (1, 16, 27). Mangroves also harbor microorganisms that are important in nutrient cycling (31, 40). Many species of terrestrial plants influence microbial communities inhabiting their roots; this phenomenon is known as the rhizosphere effect (33, 39). Microorganisms in turn are essential for plant growth and organic matter turnover, and they use plant root exudates as a nutrient source (39, 70). Little is known, however, about the impact of mangrove plants on microbial rhizosphere communities (31, 32, 72). The mangrove environment differs radically in many ways from typical terrestrial environments due to regular inundation with seawater and sometimes freshwater flooding (40). Due to the rapid development of novel molecular tools, we are now able to perform more thorough investigations of the highly complex microbial communities inhabiting mangrove forests and can start to unravel how plant-microbe interactions help to maintain this endangered ecosystem.
Molecular techniques, including nucleic acid extraction, PCR, DNA cloning and sequencing, and denaturing gradient gel electrophoresis (DGGE), have enabled the study of microbial communities at the genetic level, without isolating and culturing microorganisms (53, 62). In turn, recent developments in DNA sequencing technologies, such as pyrosequencing, have greatly increased our ability to study microbial communities compared to that possible by other techniques (44). The combination of PCR, more classical molecular tools, and pyrosequencing allows us to study microorganisms at an unprecedented level of detail (22, 63, 64). In previous studies, we applied classical molecular tools and in-depth functional microarray (32) and structural barcoded pyrosequencing (31) analyses to study the structure and functions of bacterial communities inhabiting mangrove roots. These studies suggest an important ecological role of mangrove plants in the process of selective enhancement of degrading genes and functional bacterial guilds in their complex root systems.
Here, we investigate the effect of the roots of different mangrove plant species on sediment archaeal populations as a follow-up to previous studies (31, 32). Hitherto, most of the research on mangrove environments has focused on bacteria and fungi, and very little is known about archaeal assemblages in mangroves (51, 72). Several studies have, furthermore, shown that Archaea are more diverse and ubiquitous than previously thought (7, 48, 52). However, information on the influence of environmental and biological factors that contribute to their abundance, diversity, and spatial distribution in different environments is still scarce. In the first part of this study, we focus on the development of a nested primer system which generates PCR amplicons of 16S rRNA gene fragments suitable for both PCR-DGGE and barcoded pyrosequencing analyses of the Archaea domain on the basis of novel sequences recently submitted to public data banks (61). After standardization of this new methodology, we investigated if the rhizosphere of two different plant species (Rhizophora mangle and Laguncularia racemosa) hosted distinct archaeal assemblages compared to bulk mangrove sediment (here known as Sed), following a similar trend observed for bacterial assemblages in previous studies (31, 32).
MATERIALS AND METHODS
Sampling and total community DNA extraction.
Four composite replicates of bulk sediment samples (∼20 cm of top sediment with 4-cm diameter) and roots of individual mangrove native plants (4 replicates each of R. mangle and L. racemosa) were sampled from a fringe mangrove forest in Guanabara Bay (Rio de Janeiro, Brazil) (22°46′53.50″S, 43°04′16.16″W). Young plants of similar height (R. mangle, 50 cm; L. racemosa, 35 cm) were collected haphazardly. Total community DNA (TC-DNA) extraction from microbial cell pellets retrieved from sediment and rhizosphere samples was performed as previously described (30).
Primer design.
A nested PCR system for the analysis of archaeal communities suitable for both DGGE fingerprinting and barcoded pyrosequencing was developed. This consisted of a first PCR with Archaea-specific outer primers (fragment size, ∼624 bp) previously published by DeLong (19) and Casamayor et al. (6a), with modifications (primers ARC344f-mod and Arch958R-mod) (Table 1). The original primers were modified in order to cover, with high specificity, the V4 and V5 regions of the 16S rRNA gene of Archaea and most archaeal sequences present in the Silva ARB database (release 102, containing high-quality sequences; http://www.arb-silva.de/) (61). A second PCR using the amplicons from the first PCR as the template was subsequently carried out to amplify the inner fragments of the archaeal 16S rRNA sequences. This approach generated PCR amplicons suitable for DGGE or barcoded pyrosequencing analyses. For DGGE analyses, the second PCR used the miniprimer 524F-10 (45) and Arch958R-mod (fragment size, ∼425 bp) with a GC clamp attached to the 5′ end to prevent complete melting of double-stranded DNA during DGGE.
Table 1.
Archaea-specific 16S rRNA gene primersa
| Primer | Sequence (5′–3′) | Bacteria |
Archaea |
Reference or source | ||||
|---|---|---|---|---|---|---|---|---|
| PM | M | % M | PM | M | % M | |||
| ARC344f-mod | ACGGGGYGCASSAGKCGVGA | 1,244,196 | 52,937 | 4.25 | 52,211 | 47,911 | 91.76 | This study |
| Arch958R-mod | YCCGGCGTTGAVTCCAATT | 957,411 | 402 | 0.04 | 22,329 | 19,543 | 87.52 | This study |
| Arch958R-mod (GC)b | CCGGCGTTGAVTCCAATT | |||||||
| 524F-10-ext | TGYCAGCCGCCGCGGTAA | 1,197,614 | 1,129,558 | 94.32 | 58,408 | 56,762 | 97.18 | This study |
| Arch958R | YCCGGCGTTGAMTCCAATT | 957,411 | 365 | 0.04 | 22,329 | 13,730 | 61.49 | 19 |
| 524F-10 | GCCGCGGTAA | 1,197,614 | 1,129,558 | 94.32 | 58,408 | 56,762 | 97.18 | 45 |
| ARC344f | ACGGGGYGCAGCAGGCGCGA | 1,244,196 | 179 | 0.01 | 52,211 | 34,460 | 66.00 | 6a |
M, number of matches; PM, number of possible matches; % M, percentage of matches. Primers were submitted to the check probe facility of the Ribosomal Database Project (http://www.rdp.cme.msu.edu/) to check for archaeal specificity. All forward primers were submitted as target sequence, and reverse primers were submitted as probes.
Primer Arch958R-mod (GC) had the degeneration Y removed and a GC clamp attached to the 5′ end used for DGGE analyses; GC clamp sequence: 5′-CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGG-3′ (57).
The primers described above for PCR-DGGE were adapted for pyrosequencing using the pyrosequencing platform 454 Life Sciences (Roche Diagnostics). Briefly, the miniprimer 524F-10 was extended 8 nucleotides toward the 5′ end (524F-10-ext; Table 1) and used in combination with Arch958R-mod (without the GC clamp). In addition to this, fusion nucleotide sequences A and B were added at the 5′ ends of primers 524F-10-ext and Arch958R-mod, respectively (Table 1). Tag sequences were added between fusion A and forward primer 524F-10-ext.
The modified primers listed above were redesigned with the Probe Design and Match Probe subroutines in the ARB software (50), optimized using the program Oligo (version 4.0; National Biosciences Inc.), and empirically tested against environmental samples. The Probe Match function of Ribosomal Database Project II (RDP II; http://rdp.cme.msu.edu/) was used for in silico analysis of primer specificity, based on the last 10 3′-end nucleotides (Table 1).
Nested PCR conditions.
The nested PCR for DGGE and barcoded pyrosequencing analyses consisted of only one first PCR with Archaea-specific primers (ARC344f-mod/Arch958R-mod). A PCR mix of 25 μl containing 1× PCR buffer (Fermentas, Vilnius, Lithuania), 0.2 mM deoxynucleoside triphosphates (dNTPs), 2.75 mM MgCl2, 2.5 μg bovine serum albumin (BSA), 1% (vol/vol) formamide, 0.2 μM primers 344f-mod and Arch958R-mod, 2.5 U Dream Taq polymerase (Fermentas), and template DNA (ca. 10 ng) was prepared. After 5 min denaturation at 94°C, 30 thermal cycles of 1 min at 94°C, 1 min at 56°C, and 1 min at 72°C were carried out. A final extension step at 72°C for 7 min was performed to finish the reaction.
DGGE analyses of archaeal 16S rRNA gene fragments.
DGGE fingerprinting was used prior to pyrosequencing, to compare archaeal community compositions among samples, and later, the data were complemented with a more-in-depth barcoded pyrosequencing analysis of composite samples (13).
PCR-DGGE mixtures (25 μl) consisted of 1 μl of the first nested PCR product (ARC344f-mod/Arch958R-mod), 1× PCR buffer (Fermentas, Vilnius, Lithuania), 0.2 mM dNTPs, 2.75 mM MgCl2, 8% (vol/vol) acetamide, 0.2 μM primers 524F-10 and Arch958R-mod (GC), and 2.5 U Dream Taq polymerase (Fermentas). After 5 min denaturation at 94°C, 35 thermal cycles of 1 min at 94°C, 1 min at 50°C, and 1 min at 72°C were carried out. A final extension step at 72°C for 7 min was performed to finish the reaction.
DGGE gels of the amplified 16S rRNA gene sequences of each replicate sample were made using a CBS system (CBS Scientific Company, Del Mar, CA). The run was performed in 1× Tris-acetate-EDTA buffer with a denaturant gradient of 22 to 57% at 60°C and a constant voltage of 220 V for 8 h; 8 μl of each PCR product was loaded with 5 μl of loading buffer for DGGE. The DGGE gels were silver stained as described by Heuer et al. (38), with slight differences. Gels were scanned using a Molecular Image FX apparatus (Bio-Rad, Hercules, CA).
The digitalized DGGE gels were analyzed with the software package GelCompar (version 4.0; Applied Maths), as described by Smalla et al. (65). Briefly, both band position and intensity were processed in a spreadsheet. The data matrix of band abundance (band positions and their corresponding intensities) per sample was log10 (x + 1) transformed, and a distance matrix was constructed using the Bray-Curtis index with the vegdist() function in the vegan package (58) in R (version 2.11.1; http://www.r-project.org/; checked 13 September 2010). The Bray-Curtis index is one of the most frequently applied (dis)similarity indices used in ecological work (10–12, 24, 31, 47). Variation in archaeal composition among microhabitats (bulk sediment and rhizosphere sediment from both mangrove tree species) was visually assessed with principal coordinates analysis (PCO) using the cmdscale() function in R using the Bray-Curtis distance matrix as input. We tested whether the compositions differed significantly among microhabitats using the adonis() function in vegan. This function performs an analysis of variance with distance matrices using permutations that partition the distance matrices among sources of variation, in this case, microhabitats. In the adonis() analysis, the Bray-Curtis distance matrix of band composition was the response variable, with microhabitat being the independent variable. The number of permutations was set at 999; all other arguments used the default values set in the function.
16S rRNA gene barcoded pyrosequencing.
A barcoded pyrosequencing approach was used for the compositional analyses of archaeal communities. Prior to pyrosequencing, the amplicons of the first nested PCR (ARC344f-mod/Arch958R-mod) of all four replicates of each microhabitat were combined, forming one DNA library per microhabitat (Sed, R. mangle, and L. racemosa). Pyrosequencing libraries were obtained using the 454 Genome Sequencer FLX platform (Roche Diagnostics Ltd., West Sussex, United Kingdom). Archaeal 16S rRNA gene amplicons (nested PCR) were amplified using barcoded fusion primers with the Roche 454 titanium sequencing adapters. The forward fusion primers used for each sample were 5′-CGTATCGCCTCCCTCGCGCCATCAG (fusion) ATCATC/ATGATG/AGAGAG (tags for samples Sed, R. mangle, and L. racemosa, respectively) TGYCAGCCGCCGCGGTAA-3′ (primer 524F-10-ext). The reverse fusion primer was 5′-CTATGCGCCTTGCCAGCCCGCTCAG (fusion) CCGGCGTTGAVTCCAATT-3′ (primer Arch958R-mod). The PCR mixture (50 μl) consisted of 1 μl of the first nested PCR product, 1× FastStart high-fidelity reaction buffer (Roche), 0.2 mM dNTPs, 2 mM MgCl2, 5% (vol/vol) dimethyl sulfoxide (DMSO), 0.2 μM pyrosequencing primers (Table 1), and 5 U FastStart high-fidelity reaction buffer (Roche). After 3 min denaturation at 94°C, 20 thermal cycles of 30 s at 94°C, 45 s at 50°C, and 1 min at 72°C were carried out. A final extension step at 72°C for 7 min was performed to finish the reaction. The PCR product was quantified by fluorimetry with PicoGreen (Invitrogen, CA), and PCR products were pooled at equimolar concentrations and sequenced in the A direction with GS 454 FLX titanium chemistry, according to the manufacturer's instructions (Roche, 454 Life Sciences, Bradford, CT).
Barcoded pyrosequencing analyses.
The initial quality check of pyrosequencing libraries consisted of the removal of sequences with ambiguous bases, wrong primer sequences and barcoded tags, and sequences with reads below 300 bp. Trimmed sequences were submitted to Greengenes alignment using a minimum length of 100 bp and subsequently to analysis on the Greengenes server using the Bellerophon program for the detection and elimination of chimeric sequences with a window size of 200 bp and a parent-to-fragment threshold of 80% (21, 43). Some sequences tested with the Greengenes chimera check returned as “not tested,” as described by Gontcharova et al. (34). These sequences were submitted to a Greengenes alignment and chimera check a second time; the sequences returned as not tested retrieved were discarded from further analyses. Since Greengenes allows the analysis only of sequences with lengths of ≥400 bp (34, 43), all the sequences with less than 400 bp were also rejected. The relative abundance of the archaeal groups in each microhabitat and the representative sequences of the most dominant operational taxonomic units (OTUs) (≥20 sequences) were determined according to the Naive Bayesian rRNA classifier (version 1.0) of the RDP (release 10, update 20), with 80% used as the bootstrap cutoff. Only a few sequences (nine) were not classified into the Archaea domain and were discarded.
The selected pyrosequencing reads were aligned online using the Infernal aligner algorithm (55). Aligned sequences were assigned (97% identity) to OTUs (phylotype clusters) using the complete linkage clustering application of the RDP pyrosequencing pipeline (9). The complete linkage cluster file was then converted into a square matrix containing the presence and abundance of OTUs per sample using a self-written function in R (31).
OTU richness was assessed using rarefaction; a rarefaction curve for each microhabitat was computed using a self-written function in R (31). In addition to this, we used nonparametric richness estimators to estimate true richness (4, 15). An advantage of nonparametric richness estimators is that no assumptions about community structure need to be made, as opposed to the case in parametric models. Some nonparametric estimators also outperform other nonparametric and parametric estimators (14). Here we used the Chao1 species richness estimator (8) to estimate the total number of archaeal OTUs in rhizosphere and bulk sediment samples.
Sequences from selected dominant OTUs (≥20 reads), along with their closest relatives retrieved from GenBank (http://www.ncbi.nlm.nih.gov/), were aligned, and a phylogenetic tree was built with the Mega program (http://www.megasoftware.net/; last checked 11 December 2011) using the neighbor-joining algorithm with Jukes-Cantor corrections with values generated from 1,000 replicates. Bootstrap values lower than 50% were omitted.
Nucleotide sequence accession numbers.
The selected sequences obtained after quality check were submitted to the GenBank database under accession numbers JN874931 to JN880408.
RESULTS
Primer design.
In this study, primers ARC344f and ARCH958R were modified in order to cover the V4 and V5 regions of the 16S rRNA gene of most Archaea present in the RDP database. The in silico primer analyses showed that the primer modifications drastically increased the number of Archaea sequences with a perfect match to the last 10 3′-end nucleotides of primers 344f-mod and Arch958R-mod (Table 1). However, in contrast to primer Arch958R-mod, primer 344f-mod had unspecific matches to several bacterial groups. The in silico analysis of the primers used in the subsequent nested PCRs revealed that primer 524F-10 and its extended version (524F-10-ext) were highly generalist (Table 1). The Archaea-specific primer Arch958R-mod was used in combination with 524F-10 (with and without extension) prior to Archaea community profiling.
DGGE analyses of archaeal 16S rRNA gene fragments.
DGGE profiles revealed significant differences (Adonis F2,11 = 3.60, P < 0.001, R2 = 0.444) in archaeal composition among microhabitats (Fig. 1). This is also apparent in the PCO ordination presented in Fig. 2, which shows the first two PCO axes and explains 58% of the variation in the data set; samples from each microhabitat clearly cluster together (Fig. 2). Some ribotypes were, furthermore, present in both rhizosphere samples (indicated with arrows in Fig. 1) but not in bulk sediment samples.
Fig 1.
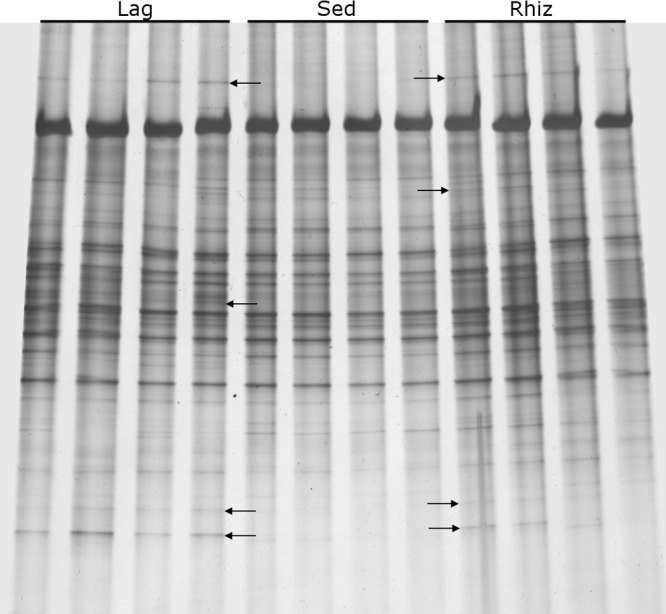
DGGE profiles of 16S rRNA fragments of Archaea amplified from total community DNA extracted from four replicates of bulk (Sed) and rhizosphere (Lag, L. racemosa; Rhiz, R. mangle) sediment samples. Short arrows show differentiating ribotypes. Some ribotypes were present in the rhizosphere of both mangrove species but absent in bulk sediment.
Fig 2.
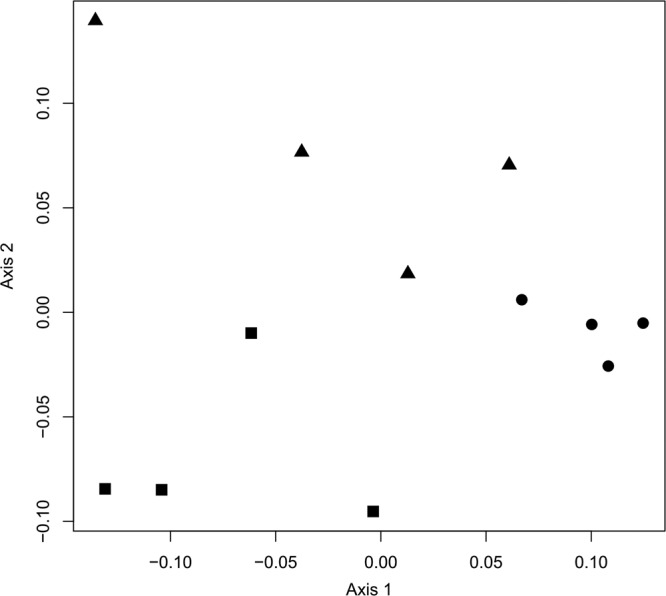
Ordination showing the first two axes of the PCO. The symbols represent microhabitats: squares, L. racemosa; triangles, R. mangle; circles, bulk sediment.
Diversity analyses of rhizosphere and bulk sediment samples.
A total of 5,487 sequences were detected, of which 99.84% were identified as archaeal sequences. After quality control, a total of 1,694, 2,087, and 1,697 sequences were associated with the L. racemosa, R. mangle, and Sed microhabitats, representing 318, 298, and 295 OTUs, respectively (using a 3% cutoff). Rarefied OTU richness was the highest in L. racemosa, intermediate in the Sed, and the lowest in R. mangle (Fig. 3). Qualitatively, the Chao1 richness estimators revealed a pattern similar to that for rarefied richness, with the highest expected number of OTUs occurring in L. racemosa, followed by Sed and R. mangle (data not shown). OTU richness, however, estimated using the Chao1 richness estimator was substantially higher than the observed richness. On the basis of 1,650 sequences, values for the Chao1-estimated richness estimator were 614.60 ± 20.72 OTUs for L. racemosa, 481.07 ± 38.42 OTUs for R. mangle, and 534.05 ± 18.13 OTUs for Sed. None of the microhabitats, however, showed signs of reaching an asymptote, thus indicating that true OTU richness was even higher.
Fig 3.
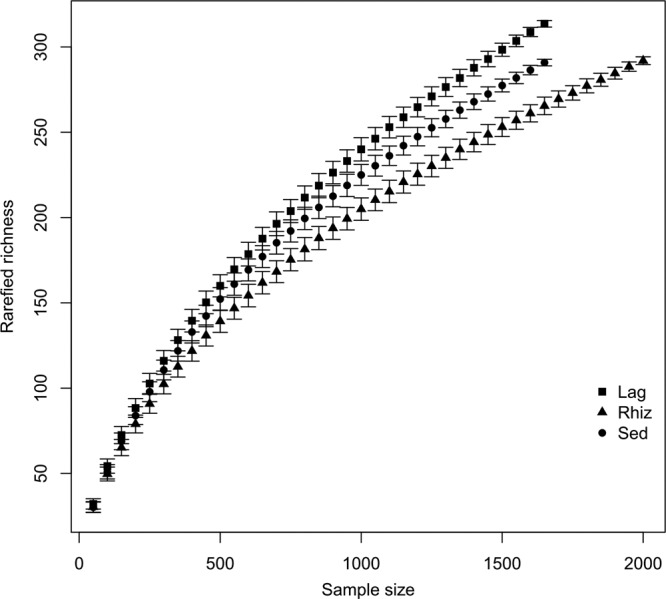
Species accumulation curves as a function of the number of sequences using resampling of archaeal 16S rRNA sequences from bulk (Sed) and rhizosphere (Lag, L. racemosa; Rhiz, R. mangle) samples.
Archaeal 16S rRNA gene sequence analyses and relative abundance of the most dominant taxon groups.
Taxonomic analyses using the RDP classifier grouped the rhizosphere-associated archaeal sequences into 2 archaeal phyla (Euryarchaeota and Crenarchaeota) and 5 classes (Halobacteria, Methanobacteria, Methanomicrobia, Thermoplasmata, and Thermoprotei). Both rhizosphere microhabitats (L. racemosa and R. mangle) had the same class distribution but with different relative abundances of the classes (Fig. 4). On the other hand, the distribution in the bulk sediment microhabitat differed from that for the rhizosphere samples; members of the euryarchaeotal class Methanobacteria were present in both rhizosphere microhabitats but not in the bulk sediment, and the euryarchaeotal class Thermoplasmata was present in the bulk sediment but absent from both rhizosphere microhabitats. Methanomicrobia were abundant in all microhabitats. All microhabitats contained high percentages of unclassified sequences at the domain, phylum, and class levels (Archaea, Euryarchaeota, and Thermoprotei, respectively) and were dominated by unknown Crenarchaeota members (Fig. 4). The Crenarchaeota group was also more abundant in both rhizosphere microhabitats than in bulk sediment, whereas the Euryarchaeota group was more abundant in the bulk sediment. No Korarchaea or Nanoarchaea were identified.
Fig 4.

Taxonomic assignment and relative abundance of archaeal partial 16S rRNA gene sequences from bulk (Sed) and rhizosphere (Lag, L. racemosa; Rhiz, R. mangle) samples, using the classifier algorithm of RDP.
The ternary plot of dominant OTUs (Fig. 5) revealed several abundant OTUs in both rhizosphere microhabitats (L. racemosa and R. mangle). From these, OTUs 60, 64, and 182 were more abundant in the R. mangle rhizosphere than in the other microhabitats; OTUs 64 and 182 were assigned to Archaea with 100% and 99% confidence, respectively, while OTU 60 was assigned to Thermoprotei with 81% confidence.
Fig 5.
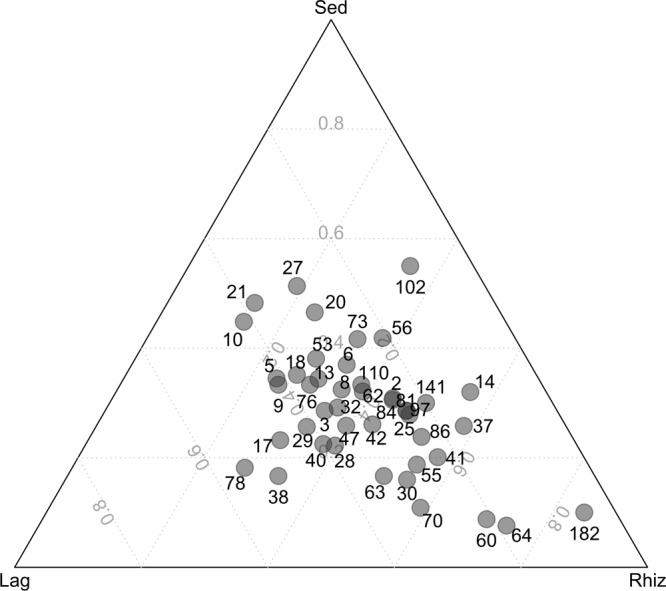
Ternary plot representing the ratio of the most abundant (≥20 reads) OTUs across treatments. Lag, rhizosphere of L. racemosa; Rhiz, rhizosphere of R. mangle; Sed, bulk sediment.
Figure 6 shows that the archaeal sequences retrieved were classified into three crenarchaeotal groups, three euryarchaeotal groups, and one thaumarchaeotal group. Crenarchaeota was represented by the miscellaneous crenarchaeotal group (MCG) (67), group C3 (20), and marine benthic group B (MBGB) (69). Euryarchaeota groups included terrestrial miscellaneous euryarchaeotal group (TMEG) (67), marine benthic group D (MBGD) (69), and marine and estuarine methane-oxidizing euryarchaeotal group (MEMOEG). Finally, the Thaumarchaeota group was represented by marine group I (MGI) (6, 19). Out of a total of 3,921 OTUs, Crenarchaeota represented the majority with 62.05%, while Euryarchaeota represented 37.44% and Thaumarchaeota represented only 0.51%; MCG was the most abundant crenarchaeotal group (89.68%), and TMEG was the most abundant euryarchaeotal group (73.71%). Interestingly, the phylogenetic analysis of archaeal groups associated with the roots of R. mangle showed that OTUs 60, 64, and 182 were closely related to Crenarchaeota in salt marsh sediments (GenBank accession no. FJ655681), an uncultured archaeon (GenBank accession no. AB177120), and a novel marine ammonia-oxidizing archaea isolated from gravel collected from a tropical marine tank (GenBank accession no. DQ085097).
Fig 6.

Phylogenetic tree of the archaeal 16S rRNA gene sequences recovered from bulk sediment and mangrove rhizospheres of L. racemosa and R. mangle. Sequences from different environments retrieved from BLAST searches were used to define the different archaeal phylogenetic groups (see accession number). The tree was constructed using the neighbor-joining method with bootstrap values generated from 1,000 replicates; bootstrap values lower than 50% were omitted. The number of each OTU is in parentheses.
DISCUSSION
Cloning, community fingerprinting, and barcoded pyrosequencing analyses of PCR amplicons are powerful tools which have greatly contributed to unraveling microbial diversity in saltwater environments (41, 60, 66). PCR-based techniques have been used as a tool in past decades to study microbial diversity in a wide range of environments (23, 63, 73). However, PCR is not bias free, and due to the large amount of new microbe sequences deposited in public data banks every year, PCR primer systems targeting microbial groups need to be constantly updated. In this study, we successfully developed a nested primer system suitable for combined DGGE and pyrosequencing analyses of TC-DNA samples. This system covered, with high specificity, the V4 and V5 regions of the 16S rRNA gene of the Archaea domain. The in silico analyses of the primer pairs used in the nested PCR showed that PCR system specificity was highly dependent on only one primer, ARCH958R-mod. This is not surprising, as primer pair specificity is determined by the combined target affinity of both reverse and forward primers; sequence types not recognized by both primers in the PCR system are subsequently excluded. Curiously, due to the high number of unknown archaeal sequences observed in this study, it is possible that the conserved nature of primers ARC344f-mod and 524F-10 in combination with the Archaea-specific primer ARCH958R-mod may have enabled the recovery of several unknown Archaea taxon groups; this has been shown previously for the primer 1380F in a study published by Amaral-Zettler et al. (2).
Unlike terrestrial plants and their associated microorganisms, the influence of mangrove roots on sediment microbial communities is relatively unknown (31, 32) and generally limited to bacterial communities. Here, there were significant differences in archaeal composition among microhabitats, indicating that mangrove roots influence archaeal sediment communities and that this is plant species specific. This is the first study to show such an effect on mangrove sediment archaeal communities. Interestingly, the complexity of the archaeal fingerprint profile detected here is substantially higher than that detected in other studies based on community fingerprinting analyses of Archaea (54, 64, 68, 72, 73). In addition to this, pyrosequencing has allowed us to classify the most dominant archaeal taxa in all mangrove microhabitats investigated in this study.
Taxonomic analyses of pyrosequencing data using the RDP classifier revealed that class distribution was similar for both rhizosphere microhabitats, but the distribution for both of these differed from that for the bulk sediment. Two of the more abundant classes represented here belong to the methanogenic Archaea. Members of the euryarchaeotal classes Methanomicrobia and Methanobacteria are known for their putative importance in sulfate reduction and methanogenesis in anoxic marine sediments, such as mangroves (51). Methanobacteria were represented only in rhizosphere microhabitats, indicating that plant roots influence microorganisms belonging to this class. High methanogenic archaeal abundance in mangrove environments may influence atmospheric warming (56). Crenarchaeotal microorganisms have, furthermore, been related to ammonia oxidation (17, 46, 71) and, in line with other studies, represent the dominant archaeal lineages in most soils (54). In agreement with other studies, archaeal richness is substantially lower than bacterial richness in the same environment (31, 41), although true richness for both groups is much larger than that observed, as indicated by a lack of asymptote using rarefaction and nonparametric richness estimators. At the level of the in silico and pyrosequencing analyses, the nested PCR system developed in this study was highly specific and revealed important archaeal diversity in mangrove sediments, probably with the highest values found so far for marine and freshwater sediments.
According to Yan et al. (72), archaeal communities in mangrove sediments had a higher percentage of Crenarchaeota than Euryarchaeota. The results obtained here agree with this study, since the crenarchaeotal class Thermoprotei was the most abundant class in all microhabitats (45 to 49%). Euryarchaeota represented 29 to 42% of the retrieved archaeal OTUs. However, the great majority of the OTUs assigned to Euryarchaeota could not be assigned to lower taxonomic levels. Nevertheless, our results show that most OTUs related to Euryarchaeota were specifically enhanced in the rhizosphere of L. racemosa and R. mangle. The Euryarchaeota phylum is divided into eight classes (5), of which four, Halobacteria, Methanomicrobia, Methanobacteria, and Thermoplasmata, were detected here. Among the Euryarchaeota which were assigned to the class level, the Methanomicrobia were the most abundant (0.94 to 1.82%) in all microhabitats and showed a higher abundance in the rhizosphere of R. mangle. Both Methanomicrobia and Methanobacteria are methanogens (25) involved in the carbon cycle through methanogenesis (73). OTUs belonging to Halobacteria, one of the less abundant classes (0.06 to 0.48%), are characterized by living in high-salinity environments and their distinct metabolic pathways (26, 59). The last euryarchaeotal class represented here (found only in bulk sediment) is Thermoplasmata, which is known to be acidophilic and contains species important in iron and sulfur cycling (29). In a previous study (31), we showed that R. mangle rhizospheres host abundant methanotrophic bacterial guilds. A possible effect of mangrove roots on microbes involved in the methane cycle is an important finding, given the rate of mangrove deforestation, and merits more-in-depth studies.
Most dominant OTUs (≥20 sequences) were distributed more or less equally in all microhabitats. Several OTUs (OTUs 30, 38, 41, 55, 60, 63, 64, 70, 78, and 182) were, however, more abundant in rhizosphere microhabitats than in the bulk sediment microhabitat. Some of these (OTUs 60, 64, and 182) showed strong associations with R. mangle and were closely related to sequences found in the BLAST data bank that have been recovered from salt marsh sediments, tropical estuarine sediments, and coastal waters. OTUs 60, 64, and 182 were related to Thermoprotei, an uncultured archaeon, and a Nitrosopumilus sp., respectively. The Thermoprotei archaeon also showed high relative abundance in mangrove sediment and a pronounced association with mangrove rhizospheres. The high prevalence of this taxon in mangrove environments may be related to its involvement in nitrogen, carbon, iron, or sulfur cycling (17, 46, 71), which are in turn essential nutrients for mangrove plants. Nitrosopumilus sp. is a novel marine ammonia-oxidizing archaeon (AOA) isolated for the first time from gravel collected from a tropical marine tank (46). The study of Herrmann et al. (36) suggests that the increased abundance of AOA was responsible for the enhanced nitrification activity observed in the rhizosphere of the freshwater macrophyle Littorella uniflora. In line with our results, Li et al. (49) showed that there was also an increase in the diversity and abundance of AOA in the surface layer of sediments near mangrove trees, indicating that mangrove trees might affect AOA and ammonia-oxidizing bacteria (AOB) community structure. The AOA Nitrosopumilus sp. detected in this study showed a strong association with R. mangle roots and may play an as yet undetermined role in the process of nitrification in the rhizosphere.
Concluding remarks.
Here we present a highly specific nested primer system for Archaea suitable for PCR-DGGE and barcoded pyrosequencing analyses. The advantage of a nested approach is that it amplifies 16S rRNA gene fragments that can subsequently be used for both DGGE and pyrosequencing analyses. In a comparison of DGGE and pyrosequencing data (13), we showed that DGGE fingerprinting data yield results significantly congruent with those obtained with barcoded pyrosequencing data using the standard 97% cutoff level. This enabled us to compare rapidly and in a cost-effective manner archaeal communities by assessing variation in composition with DGGE analysis and identifying the taxonomic classification of community members with pyrosequencing using a limited number of samples. Our results showed that the rhizosphere microhabitats of R. mangle and L. racemosa hosted distinct archaeal assemblages. This study also shows that various groups of Archaea with possibly important roles in sediment nutrient cycling have strong associations with mangrove rhizospheres. In summary, we have provided unprecedented information on Archaea diversity and composition in a mangrove environment. Future studies based on isolation, cultivation, and biochemical characterization of rhizocompetent archaeal populations will be of fundamental importance for a more complete understanding of Archaea-sediment-plant interactions.
ACKNOWLEDGMENTS
This study was funded by the Deutsche Forschungsgemeinschaft (SM59/4-1 and 4-2; www.dfg.de/en/index.jsp), FAPERJ-Brazil (www.faperj.br), the Foundation for Science and Technology (FCT; Portugal; PTDC/AAC-CLI/107916/2008; http://alfa.fct.mctes.pt), and the European Regional Development Fund (ERDF) through COMPETE-(FCOMP-01-0124-FEDER-008657).
Footnotes
Published ahead of print 1 June 2012
REFERENCES
- 1. Aburto-Oropeza O, et al. 2008. Mangroves in the Gulf of California increase fishery yields. Proc. Natl. Acad. Sci. U. S. A. 105:10456–10459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amaral-Zettler LA, McCliment EA, Ducklow HW, Huse SM. 2009. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS One 4:e6372 doi:10.1371/journal.pone.0006372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bano N, Ruffin S, Ransom B, Hollibaugh JT. 2004. Phylogenetic composition of Arctic Ocean archaeal assemblages and comparison with Antarctic assemblages. Appl. Environ. Microbiol. 70:781–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bent SJ, Forney LJ. 2008. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J. 2:689–695 [DOI] [PubMed] [Google Scholar]
- 5. Boone D, Castenholz R. 2001. The Archaea and the deeply branching and phototrophic Bacteria, p 721 In Chief G, Garrity ED. (ed), Bergey's manual of systematic bacteriology, 2nd ed, vol 1 Springer-Verlag, New York, NY [Google Scholar]
- 6. Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P. 2008. Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6:245–252 [DOI] [PubMed] [Google Scholar]
- 6a. Casamayor EO, et al. 2002. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4:338–348 [DOI] [PubMed] [Google Scholar]
- 7. Chaban B, Ng SYM, Jarrell KF. 2006. Archaeal habitats—from the extreme to the ordinary. Can. J. Microbiol. 52:73–116 [DOI] [PubMed] [Google Scholar]
- 8. Chao A, Chazdon RL, Colwell RK, Tsung-Jen S. 2005. A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol. Lett. 8:148–159 [Google Scholar]
- 9. Claesson MJ, et al. 2009. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4:e6669 doi:10.1371/journal.pone.0006669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cleary DF. 2004. Community composition and species richness of parasitoids infesting Yponomeuta species in the Netherlands. Contrib. Zool. 73:255–261 [Google Scholar]
- 11. Cleary DF. 2003. An examination of scale of assessment, logging and ENSO-induced fires on butterfly diversity in Borneo. Oecologia 135:313–321 [DOI] [PubMed] [Google Scholar]
- 12. Cleary DFR, Genner MJ. 2004. Changes in rain forest butterfly diversity following major ENSO-induced fires in Borneo. Global Ecol. Biogeogr. 13:129–140 [Google Scholar]
- 13. Cleary DFR, Smalla K, Mendonça-Hagler L, Gomes NCM. 2012. Assessment of variation in bacterial composition among microhabitats in a mangrove environment using DGGE and barcoded pyrosequencing. PLoS One 7:e29380 doi:10.1371/journal.pone.0029380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colwell RK, Coddington JA. 1994. Estimating terrestrial biodiversity through extrapolation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 345:101–118 [DOI] [PubMed] [Google Scholar]
- 15. Curtis TP, et al. 2006. What is the extent of prokaryotic diversity? Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:2023–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Lacerda LD. 2002. Mangrove ecosystems: function and management. Springer-Verlag, Berlin, Germany [Google Scholar]
- 17. de la Torre JR, Walker CB, Ingalls AE, Koenneke M, Stahl DA. 2008. Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol. 10:810–818 [DOI] [PubMed] [Google Scholar]
- 18. Reference deleted.
- 19. DeLong EF. 1992. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U. S. A. 89:5685–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeLong EF, Pace NR. 2001. Environmental diversity of Bacteria and Archaea. Syst. Biol. 50:470–478 [PubMed] [Google Scholar]
- 21. DeSantis TZ, et al. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dowd SE, et al. 2008. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 8:43 doi:10.1186/1471-2180-8-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edwards RA, et al. 2006. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7:57 doi:10.1186/1471-2164-7-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellingsen KE. 2002. Soft-sediment benthic biodiversity on the continental shelf in relation to environmental variability. Marine Ecol. Prog. Ser. 232:15–27 [Google Scholar]
- 25. Embley TM, Finlay B. 1994. The use of small subunit rRNA sequences to unravel the relationships between anaerobic ciliates and their methanogen endosymbionts. Microbiology 140:225–235 [DOI] [PubMed] [Google Scholar]
- 26. Falb M, et al. 2008. Metabolism of halophilic archaea. Extremophiles 12:177–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Food and Agriculture Organization of the United Nations 2007. The world's mangroves 1980–2005. A thematic study prepared in the framework of the global forest resources assessment 2005. FAO forestry paper. Food and Agriculture Organization of the United Nations, Rome, Italy [Google Scholar]
- 28. Francis CA, Beman JM, Kuypers MM. 2007. New processes and players in the nitrogen cycle: the microbial ecology of anaerobic and archaeal ammonia oxidation. ISME J. 1:19–27 [DOI] [PubMed] [Google Scholar]
- 29. Golyshina OV, Timmis KN. 2005. Ferroplasma and relatives, recently discovered cell wall-lacking archaea making a living in extremely acid, heavy metal-rich environments. Environ. Microbiol. 7:1277–1288 [DOI] [PubMed] [Google Scholar]
- 30. Gomes NCM, et al. 2007. Diversity of ndo genes in mangrove sediments exposed to different sources of polycyclic aromatic hydrocarbon pollution. Appl. Environ. Microbiol. 73:7392–7399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gomes NCM, et al. 2010. Taking root: enduring effect of rhizosphere bacterial colonization in mangroves. PLoS One 5:e14065 doi:10.1371/journal.pone.0014065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gomes NCM, et al. 2010. Mangrove microniches determine the structural and functional diversity of enriched petroleum hydrocarbon-degrading consortia. FEMS Microbiol. Ecol. 74:276–290 [DOI] [PubMed] [Google Scholar]
- 33. Gomes NCM, et al. 2001. Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil 232:167–180 [Google Scholar]
- 34. Gontcharova V, et al. 2010. Black Box Chimera Check (B2C2), a Windows-based software for batch depletion of chimeras from bacterial 16S rRNA gene datasets. Open Microbiol. J. 4:47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reference deleted.
- 36. Herrmann M, Saunders AM, Schramm A. 2008. Archaea dominate the ammonia-oxidizing community in the rhizosphere of the freshwater macrophyte Littorella uniflora. Appl. Environ. Microbiol. 74:3279–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reference deleted.
- 38. Heuer H, et al. 2001. Bacterial community profiling using DGGE or TGGE analysis, p 177–190 In Environmental molecular microbiology: protocols and applications. Horizon Press, Norwich, United Kingdom [Google Scholar]
- 39. Höflich G, Wiehe W, Kuhn G. 1994. Plant-growth stimulation by inoculation with symbiotic and associative rhizosphere microorganisms. Experientia 50:897–905 [Google Scholar]
- 40. Holguin G, Vazquez P, Bashan Y. 2001. The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: an overview. Biol. Fertil. Soils 33:265–278 [Google Scholar]
- 41. Huber JA, et al. 2007. Microbial population structures in the deep marine biosphere. Science 318:97–100 [DOI] [PubMed] [Google Scholar]
- 42. Reference deleted.
- 43. Huber T, Faulkner G, Hugenholtz P. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319 [DOI] [PubMed] [Google Scholar]
- 44. Huse SM, et al. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255 doi:10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Isenbarger TA, Finney M, Rios-Velazquez C, Handelsman J, Ruvkun G. 2008. Miniprimer PCR, a new lens for viewing the microbial world. Appl. Environ. Microbiol. 74:840–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Könneke M, et al. 2005. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543–546 [DOI] [PubMed] [Google Scholar]
- 47. Legendre P, Gallagher E. 2001. Ecologically meaningful transformations for ordination of species data. Oecologia 129:271–280 [DOI] [PubMed] [Google Scholar]
- 48. Leininger S, et al. 2006. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806–809 [DOI] [PubMed] [Google Scholar]
- 49. Li M, Cao H, Hong Y, Gu J-D. 2011. Spatial distribution and abundances of ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB) in mangrove sediments. Appl. Microbiol. Biotechnol. 89:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ludwig W, et al. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lyimo TJ, Pol A, Jetten MS, den Camp HJ. 2009. Diversity of methanogenic archaea in a mangrove sediment and isolation of a new Methanococcoides strain. FEMS Microbiol. Lett. 291:247–253 [DOI] [PubMed] [Google Scholar]
- 52. Mehta MP, Baross JA. 2006. Nitrogen fixation at 92 degrees C by a hydrothermal vent archaeon. Science 314:1783–1786 [DOI] [PubMed] [Google Scholar]
- 53. Muyzer G. 1999. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 2:317–322 [DOI] [PubMed] [Google Scholar]
- 54. Nakatsu CH, Torsvik V, Ovreas L. 2000. Soil community analysis using DGGE of 16S rDNA polymerase chain reaction products. Soil Sci. Soc. Am. J. 64:1382–1388 [Google Scholar]
- 55. Nawrocki EP, Eddy SR. 2007. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 3:540–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nelson DM, Cann IK, Mackie RI. 2010. Response of archaeal communities in the rhizosphere of maize and soybean to elevated atmospheric CO2 concentrations. PLoS One 5:e15897 doi:10.1371/journal.pone.0015897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nübel U, et al. 1996. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 178:5636–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oksanen J, et al. 2008, posting date vegan: community ecology package. http://vegan.r-forge.r-project.org/
- 59. Oren A. 2006. The order Halobacteriales, p 113–164 In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E. (ed), The prokaryotes. A handbook on the biology of bacteria: ecophysiology and biochemistry, vol 3 Springer, New York, NY [Google Scholar]
- 60. Perreault NN, Andersen DT, Pollard WH, Greer CW, Whyte LG. 2007. Characterization of the prokaryotic diversity in cold saline perennial springs of the Canadian high Arctic. Appl. Environ. Microbiol. 73:1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pruesse E, et al. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ranjard L, Poly F, Nazaret S. 2000. Monitoring complex bacterial communities using culture-independent molecular techniques: application to soil environment. Res. Microbiol. 151:167–177 [DOI] [PubMed] [Google Scholar]
- 63. Roesch LF, et al. 2007. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 1:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roh SW, et al. 2010. Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing. ISME J. 4:1–16 [DOI] [PubMed] [Google Scholar]
- 65. Smalla K, et al. 2001. Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67:4742–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stoeck T, et al. 2010. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19(Suppl 1):21–31 [DOI] [PubMed] [Google Scholar]
- 67. Takai K, Moser DP, DeFlaun M, Onstott TC, Fredrickson JK. 2001. Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 67:5750–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taketani RG, Yoshiura CA, Dias AC, Andreote FD, Tsai SM. 2010. Diversity and identification of methanogenic archaea and sulphate-reducing bacteria in sediments from a pristine tropical mangrove. Antonie Van Leeuwenhoek 97:401–411 [DOI] [PubMed] [Google Scholar]
- 69. Vetriani C, Jannasch HW, MacGregor BJ, Stahl DA, Reysenbach AL. 1999. Population structure and phylogenetic characterization of marine benthic archaea in deep-sea sediments. Appl. Environ. Microbiol. 65:4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Weinert N, et al. 2009. Rhizosphere communities of genetically modified zeaxanthin-accumulating potato plants and their parent cultivar differ less than those of different potato cultivars. Appl. Environ. Microbiol. 75:3859–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wuchter C, et al. 2006. Archaeal nitrification in the ocean. Proc. Natl. Acad. Sci. U. S. A. 103:12317–12322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yan B, Hong K, Yu Z. 2006. Archaeal communities in mangrove soil characterized by 16S rRNA gene clones. J. Microbiol. 44:566–571 [PubMed] [Google Scholar]
- 73. Yu Z, Garcia-Gonzalez R, Schanbacher FL, Morrison M. 2008. Evaluations of different hypervariable regions of archaeal 16S rRNA genes in profiling of methanogens by Archaea-specific PCR and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 74:889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]