

Abstract
Wax esters are produced in certain bacteria as a potential carbon and energy storage compound. The final enzyme in the biosynthetic pathway responsible for wax ester production is the bifunctional wax ester synthase/acyl-coenzyme A (acyl-CoA):diacylglycerol acyltransferase (WS/DGAT), which utilizes a range of fatty alcohols and fatty acyl-CoAs to synthesize the corresponding wax ester. We report here the isolation and substrate range characterization for five WS/DGAT enzymes from four different bacteria: Marinobacter aquaeolei VT8, Acinetobacter baylyi, Rhodococcus jostii RHA1, and Psychrobacter cryohalolentis K5. The results from kinetic studies of isolated enzymes reveal a differential activity based on the order of substrate addition and reveal subtle differences between the substrate selectivity of the different enzymes. These in vitro results are compared to the wax ester and triacylglyceride product profiles obtained from each organism grown under neutral lipid accumulating conditions, providing potential insights into the role that the WS/DGAT enzyme plays in determining the final wax ester products that are produced under conditions of nutrient stress in each of these bacteria. Further, the analysis revealed that one enzyme in particular from M. aquaeolei VT8 showed the greatest potential for future study based on rapid purification and significantly higher activity than was found for the other isolated WS/DGAT enzymes. The results provide a framework to test prospective differences between these enzymes for potential biotechnological applications such as high-value petrochemicals and biofuel production.
INTRODUCTION
Wax esters are common in higher plants and animals, where they are known to serve a variety of functions, including protecting the organism from dehydration, UV light, and pathogens (9, 25). In certain plants, such as jojoba (Simmondsia chinensis), they also serve as a neutral lipid storage compound (9, 15), similar to triacylglyceride (TAG) storage in seed oil crops (canola, safflower, sunflower, and soybean) (23). While polyhydroxyalkanoic acids, starch, and glycogen are the primary forms of reduced carbon storage compounds in most bacteria, examples of bacteria that accumulate neutral lipids, such as TAGs and wax esters, have also been described (25, 26).
The enzyme responsible for catalyzing the esterification of a fatty acyl coenzyme A (acyl-CoA) and a fatty alcohol is referred to as the wax ester synthase/acyl-CoA:diacylglycerol acyltransferase (WS/DGAT; EC 2.3.1.75) (Fig. 1). The first bacterial WS/DGAT was initially identified from Acinetobacter calcoaceticus strain ADP1 (10). Homologs to the gene for this protein have been identified in a number of other bacteria, and the genes for several WS/DGAT enzymes have been cloned and overexpressed for enzyme characterization (1, 8). Although there are obvious regions of conservation in the primary amino acid sequence of WS/DGAT proteins, the overall similarity is quite low between homologs from different species, as well as homologs within the same species. The low overall similarity raises questions regarding the differences that can occur as a result, potentially affecting such properties as substrate specificity.
Fig 1.
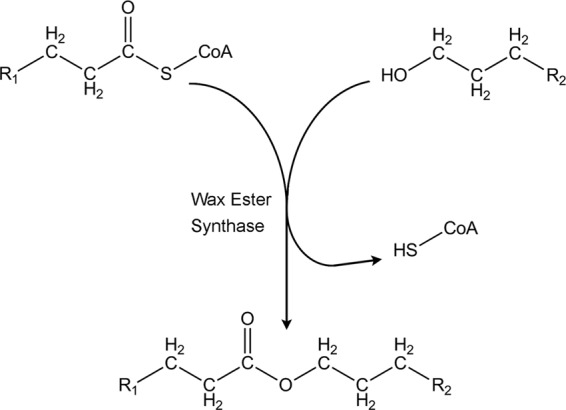
Reaction scheme of the wax ester synthase reaction. The wax ester synthase/acyl-CoA:diacylglycerol acyltransferase enzyme (EC 2.3.1.75) catalyzes the esterification of a fatty acyl-CoA and a fatty alcohol, yielding free coenzyme A and the corresponding wax ester.
In addition to the WS/DGAT enzymes, our laboratories and others have also sought to characterize additional enzymes that reduce fatty acyl-CoA (EC 1.2.1.50) and fatty aldehyde substrates (EC 1.1.1.2 or EC 1.1.1.21) to fatty alcohols to serve as the second substrate along with a fatty acyl-CoA that together compose the two required substrates for WS/DGAT when producing wax esters (16, 24, 27). Each substrate utilized by these enzymes is believed to be derived from the cellular fatty acyl-CoA pool (22, 25). An underlying property of the various enzymes involved in the production of wax esters in bacteria is their broad substrate range. This broad substrate range for WS/DGAT has led to interest in using WS/DGAT enzymes to produce a range of compounds, including the production of an ethyl ester which has been termed “microdiesel” as the fatty acid ethyl ester is produced directly by the microorganism (11). Several bacterial WS/DGAT enzymes have been reported to have a broad substrate range (20, 22), even though the natural products of these enzymes are generally limited to only a small range of biological wax esters (12).
In this report, we have cloned five different WS/DGAT genes so that when heterologously expressed in Escherichia coli, they produce a maltose-binding protein (MBP)–WS/DGAT–polyhistidine-tagged fusion protein. The products of the five different WS/DGAT genes selected share little overall primary protein sequence similarity and identity and were cloned from four different bacteria that are known or were shown here to accumulate wax esters, TAGs, or both. These enzymes were isolated and characterized to initiate a detailed study to directly compare and contrast the stability, substrate profiles, and differences in activity among these various homologs when treated under similar assay conditions. The results obtained provide insight into the differences between these proteins and the potential for substrate inhibition or activation and provide a blueprint to utilize for further studies to probe the importance of specific amino acid residues within these proteins that are responsible for the enzyme substrate selectivity.
We have also confirmed the accumulation of neutral lipids in each of the host bacteria from which the enzymes were cloned and isolated to characterize the chemical composition of the wax ester precursors from these bacteria. Utilizing endogenous waxes produced by the bacteria and the activities of the isolated enzymes, we can compare and contrast these results to infer potential control elements within the cell that affect the classes of neutral lipids produced in these bacteria.
MATERIALS AND METHODS
Materials.
Bacteria were obtained from the American Type Culture Collection (ATCC) or from Lindsay Eltis and James Tiedje (for Rhodococcus jostii RHA1 and Psychrobacter cryohalolentis K5, respectively). Fatty alcohols were obtained from Nu-Chek Prep (Elysian, MN) and Sigma Chemical Company (St. Louis, MO). DTNB [5,5′-dithiobis(2-nitrobenzoic acid)] and fatty acyl-CoAs were obtained from Sigma Chemical Company. Miller's Luria broth (LB) was obtained from Fisher Scientific (Pittsburgh, PA).
Sequence comparisons.
Protein primary sequence alignments were generated using the programs MultAlin (3) and ESPript (6).
Plasmid constructions.
All constructs were initially cloned into Escherichia coli JM109 for manipulation and sequencing. Final confirmed plasmids were then transferred to E. coli TB1 for protein expression (both strains obtained from New England Biolabs, Ipswich, MA). All genes were cloned from specific bacteria following standard molecular biology techniques. The gene for Ma1 (YP_957462) was cloned from Marinobacter aquaeolei VT8 (ATCC 700491) using the primers 5′-GACAGAATTCCACCATGGAACGCCCCTGAATCCCACTGACCAGCTC-3′ and 5′-GACATCTAGACCGGCGTTGAGCTCCAGCTCTGCCAGAC-3′, digested with EcoRI and XbaI, and ligated into a pUC19-derived vector to confirm the sequence (restriction enzyme sites are shown in bold). A single NcoI site was removed from the gene via a silent mutation by site-specific mutagenesis (SSM) according to the Stratagene protocol (Agilent Technologies, Santa Clara, CA). The gene encoding Ma2 (YP_960328) was cloned from M. aquaeolei VT8 using the primers 5′-GACAGAATTCAAACGTCTCGGAACCCTGGATGCCTCCTG-3′ and 5′-GACATCTAGACTCTTGCGGGTTCGGGCGCGCTTCTTCG-3′, digested with EcoRI and XbaI, and ligated into a pUC19-derived vector to confirm the sequence. The gene for Ac1 (YP_045555) was cloned from Acinetobacter baylyi (ATCC 33305) using the primers 5′-GACTAGGATCCATGGTCGCCCATTACATCCGATTGATTTTATATTC-3′ and 5′-GACTAGGTACCTCTAGACCATTGGCTGTTTTAATATCTTCCTGCTTTG-3′, digested with BamHI and KpnI, and ligated into a pUC19-derived vector to confirm the sequence. A single NcoI site was removed from the gene via a silent mutation by SSM. The gene for Ps1 (YP_579515) was cloned from Psychrobacter cryohalolentis K5 using the primers 5′-GACACCATGGACGACTTCTTACAGCAGTCGACCAG-3′ and 5′-GACATCTAGACTCGGAGCTAGCTTTTTTGTTTTAC-3′, digested with NcoI and XbaI, and ligated into a pUC19-derived vector to confirm the sequence. A single NdeI site was removed from the gene via a silent mutation by SSM. The gene for Rh1 (YP_701572) was cloned from Rhodococcus jostii RHA1 using the primers (5′-GACACGAATTCCCGGTTACCGATTCGATATTC-3′ and 5′-GATCAGTCTAGACCGAGCAATGCCGCCTCGAGCTC-3′, digested with EcoRI and XbaI, and ligated into a pUC19-derived vector to confirm the sequence. For each construct, the entire gene was transferred to a pMAL-derived vector that includes an insert with an in-frame XbaI site that results in a C-terminal His8 tag (27) when inserted into in-frame EcoRI or NcoI and XbaI sites already in the plasmid. Prior to introduction into the expression strain, all plasmids were confirmed by sequencing the entire inserted region.
Protein purification.
E. coli strain TB1 was transformed with plasmids based on the MBP- and polyhistidine-tagged dual fusion construct and selected on LB plates containing ampicillin (100 μg/ml). Cells were grown by transfer of an overnight seed stock culture (10 ml) to 1 liter of LB medium with the same concentration of antibiotic to an optical density at 600 nm of ∼0.6 and then induced with IPTG (isopropyl-β-d-thiogalactopyranoside) for 2 h or overnight, depending on the optimal conditions for each protein. Cells were collected by centrifugation at 7,000 × g for 8 min and stored at −20°C.
Frozen cells were thawed on ice in ∼30 ml of extraction buffer (50 mM phosphate buffer [pH 7.0], 300 mM NaCl) containing 30 mg of CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} detergent and 10 mg of lysozyme and several flakes (∼1 mg) of DNase. The cells were lysed by sonication (Misonix XL-2000) using three 20-s bursts to break the cells. Cell debris was pelleted at ∼17,500 × g for 8 min, and the supernatant was loaded onto a nickel-charged metal affinity column (Chelating Sepharose Fast Flow; GE Healthcare, Piscataway, NJ) and washed with the extraction buffer containing 50 mM imidazole to remove nonspecific proteins. The desired protein was eluted with 500 mM imidazole in extraction buffer adjusted to pH 7.8. Fractions were analyzed for protein content using the Coomassie Plus protein assay reagent (Thermo Scientific, Rockford, IL) and pooled and added to an amylose column (E8021L; New England Biolabs). Bound protein was washed with ∼20 ml of extraction buffer and eluted from the column using extraction buffer containing 10 mM maltose. Fractions containing protein were pooled and flash frozen in liquid nitrogen for later analysis and storage. All proteins described here have been found to be stable for many months when stored in this manner.
Protein purity was assessed on SDS-PAGE gels. Bands isolated from the gel were further analyzed for identification using an in-gel trypsin digest kit (P/N PP0100-1KT; Sigma), and samples were analyzed at the Center for Mass Spectrometry and Proteomics at the University of Minnesota.
Spectrophotometric protein assays.
A protein assay was developed for the present study using an approach that indirectly measures fatty alcohol and fatty acyl-CoA esterification catalyzed by the WS/DGAT enzyme using the coupled reaction of DTNB with free CoA liberated during the esterification reaction. A similar approach was reported by others (4, 8, 17) and by ourselves for other reactions that follow the hydrolysis of fatty acyl-CoA substrates (27) and is based on the protocol for assaying citrate synthase as provided by Sigma Chemical (C3260; Sigma Chemical). Colorimetric assays were run in real-time in quartz cuvettes (1-cm path length) containing 1 ml of 50 mM potassium phosphate (pH 7.0) and 300 mM NaCl buffer. To this was added 10 μl of 18 mg of DTNB/ml dissolved in dimethyl sulfoxide (DMSO). Unless otherwise stated, fatty alcohols were dissolved in DMSO, added as 50-μl quantities, and distributed thoroughly by mixing in the cuvette with a 200-μl pipettor. After mixing, 50 μl of a prediluted quantity of protein in the same buffer was added, mixed thoroughly, and followed on a spectrophotometer (Model Cary 50 Bio; Varian, Palo Alto, CA). After 1 min, 15 μl of palmitoyl-CoA (1 mM stock in buffer) was added to initiate the reaction, and the solution was again mixed thoroughly for 10 to 15 s with the pipettor, and then the reaction was monitored (generally to completion) for an additional 7 min. Quantities of enzyme used in these experiments were determined by analyzing a range of concentrations until a quantity was found that would result in the complete hydrolysis of the fatty acyl-CoA within approximately 2 to 4 min, since the level of activity for each protein was found to vary (see Table 2). Initial reaction rates were calculated in Excel (Microsoft, Redmond, WA) using the initial linear rates of reaction obtained from the spectrophotometric assays by obtaining the slope from the best-fit line and calculating nmol of NTB2− dianion formed per s using the extinction coefficient of the NTB2− dianion at 412 nm of 14,150 M−1 cm−1. For the order of addition and control experiments, assays were run in the same manner, but the final component used to initiate the reaction (fatty acyl-CoA) was exchanged with either the fatty alcohol or the enzyme. For fatty acyl-CoA comparisons, palmitoyl-CoA was substituted with smaller fatty acyl-CoA substrates, and for the determination of approximate values of kinetic parameters, lower amounts of palmitoyl-CoA were added while maintaining the fatty alcohol at a set concentration. All assays were run at room temperature (∼22°C).
Table 2.
Specific wax synthase activity of WS/DGAT enzymes with dodecanol or hexadecanol substrates and palmitoyl-CoA for isolated WS/DGAT proteins
| Proteina | Sp act [nmol of product min−1 (mg of protein)−1]b |
C12/C16 ratioc | Ratio (%) of activity (based on order of addition)d | |
|---|---|---|---|---|
| Dodecanol (C12) | Hexadecanol (C16) | |||
| Ma1 | 37,300 ± 3,900 | 680 ± 20 | 55 | 12 |
| Ma2 | 4,960 ± 510 | 240 ± 20 | 20 | 79 |
| Rh1 | 3,370 ± 170 | 120 ± 5 | 27 | 34 |
| Ac1 | 840 ± 140 | 80 ± 5 | 11 | 5 |
| Ps1 | 830 ± 30 | 80 ± 2 | 10 | 78 |
The acronyms for the enzymes listed here are defined in Materials and Methods.
It should be noted that the protein concentration is the mass obtained for the fusion construct, which contains >50% mass as the MBP fusion. The results are the means of at least three independent measurements.
Calculated as the ratio of the activity of dodecanol and palmitoyl-CoA over the activity of hexadecanol and palmitoyl-CoA using the same quantity of each enzyme.
Calculated as the rate of activity when dodecanol was added as the last component in order of addition assays divided by the rate of activity when palmitoyl-CoA was added as the last component in order of addition assays, as illustrated in Fig. S1 in the supplemental material.
Lipid analysis by GC and MS.
Gas chromatography (GC) was utilized to track and quantify neutral lipids and derivatized lipids described below as part of these studies. For routine analysis, a flame ionization detector (FID) was utilized due to the lower limits of detection possible by this method versus mass spectrometry (MS). The protocol for analysis utilized a high-temperature column (RTX-Biodiesel TG; 15 M, 0.32 mm [inner diameter], 0.10-μm df; Restek, Bellefonte, PA) and a GC-2010 gas chromatograph (Shimadzu Scientific Instruments, Columbia, MD) equipped with a PTV injector. The program utilized a temperature profile of 60°C for 1 min, with a 10°C/min temperature ramp to 360°C for 15 min and a carrier gas of helium running at a linear velocity of 20 cm/s. Further analysis for identification of specific compounds was done using a QP2010 GC/MS (Shimadzu Scientific Instruments, Columbia, MD) equipped with a split/splitless injector and a column designed for better separation of fatty acid methyl esters (FAMEs; Stabilwax DA; 30 M, 0.25 mm [inner diameter], 0.25-μm df; Restek). For this analysis, we utilized a temperature profile of 60°C for 4 min, with a 10°C/min temperature ramp to 240°C for 5 min and a carrier gas of helium running at a linear velocity of 35 cm/s. Peaks were quantified and identified using the software packages GCsolution Analysis version 2.32 and GCMS Solutions version 2.53 (Shimadzu Scientific Instruments, Columbia, MD) and the NIST08 Library.
GC substrate specificity assays.
In addition to the real-time assay described above, an approach was developed to test the substrate preference of different enzymes by subjecting a specific substrate (dodecanol or palmitoyl-CoA) to a range of complementary substrates to determine whether the different enzymes shared similar substrate profiles. The assay was run by placing 1.5 ml of 50 mM potassium phosphate and 300 mM NaCl buffer at pH 7.0 in a clean test tube, followed by the addition of 20 μl of DTNB (18 mg/ml in DMSO), 20 to 50 μl of stock enzyme solution, and 50 μl of an alcohol stock solution containing 50 nmol each of several alcohols (hexanol, octanol, nonanol, decanol, undecanol, dodecanol, tetradecanol, hexadecanol, heptadecanol, and octadecanol) dissolved in DMSO. The assay was initiated by adding 200 μl of a stock solution containing 200 nmol of palmitoyl-CoA. The test tube was mixed thoroughly after the addition of each component by vortexing, and reactants were added rapidly (within 1 min) to minimize precipitation of insoluble substrates. The reaction was allowed to occur for 15 min, during which a yellow color developed (usually within the first 2 min) related to the release of free CoA by the reaction, indicating completion of the reaction. A 1.2-ml portion of hexane was then added to the test tube, and the solution was vigorously mixed by vortexing three times for 30 s. The test tube was then centrifuged at 3,000 rpm on a tabletop centrifuge, and the hexane layer was removed to analyze the generated wax esters. Quantities and compositions were determined by a combination of the GC/FID and GC/MS methods described above. Separate experiments were run substituting the alcohols described above with 100 μl of a stock containing 0.5 mg of hexanol/ml, 11 mg of isoamyl alcohol/ml, 16 mg of phenyl ethanol/ml, and 100 nmol of palmitoyl-CoA. These concentrations were selected by testing a range of concentrations with Ma1 to find a concentration of each that resulted in approximately equivalent amounts of each wax ester produced with this protein.
Neutral lipid accumulation in bacteria.
For neutral lipid production, strains were grown in the following lipid-inducing media (all defined concentrations per liter) as specified for each strain: R. jostii RHA1 (5 g of NaCl, 6 g of sodium acetate, 100 mg of Casamino Acids, 50 mg of CaCl2·2H2O, 200 mg of MgSO4·7H2O, 500 mg of K2HPO4, 10 mg of FeSO4·7H2O, 1.5 g of Na2SO4, and 180 mg of urea, adjusted to pH 7.2), P. cryohalolentis K5 (5 g of NaCl, 6 g of sodium acetate, 100 mg of Casamino Acids, 50 mg of CaCl2·2H2O, 200 mg of MgSO4·7H2O, 500 mg of K2HPO4, 10 mg of FeSO4·7H2O, 1.5 g of Na2SO4, and 270 mg of NH4Cl, adjusted to pH 7.2), A. baylyi (2 g of NaCl, 500 mg of tryptone, 150 mg of yeast extract, 3 g of glycerol, 2 g of sodium gluconate, 50 mg of CaCl2·2H2O, 200 mg of MgSO4·7H2O, 500 mg of K2HPO4, 10 mg of FeSO4·7H2O, and 1.5 g of Na2SO4, adjusted to pH 7.2), and M. aquaeolei VT8 (18 g of NaCl, 7 g of sodium succinate, 100 mg of Casamino Acids, 50 mg of CaCl2·2H2O, 5 g of MgSO4·7H2O, 500 mg of K2HPO4, 10 mg of FeSO4·7H2O, 1.5 g of Na2SO4, and 250 mg of NaNO3, adjusted to pH 7.2). All cultures were grown in 2-liter Erlenmeyer flasks on a shaker table at ∼180 rpm at 30°C, except for P. cryohalolentis K5, which was grown at 25°C. Cultures were grown for at least 12 h after depletion of the nitrogen source to induce lipids. Cultures for each species were grown multiple (minimum of three) times to compare the consistency of the results. For experiments with extraneously added alcohols, M. aquaeolei VT8 was grown in the same media as listed above, except that 50 mg of pentadecanol or tridecanol was added after 12 h of growth.
Analysis of natural neutral lipid profiles for target bacteria.
Cells of each organism grown under the lipid accumulating conditions described above were used to determine the composition of native neutral lipids. The approach utilized to optimize the medium composition is similar to what has been reported by others (10, 13). Cells from each organism were harvested by centrifugation, frozen, and dried by lyophilization. A known quantity of dried cells (usually 200 mg) was extracted using sonication and a three-component solvent mixture consisting of equal volumes of methylene chloride, tetrahydrofuran, and hexane. The neutral lipids were analyzed by GC/FID for preliminary identification of the primary component fraction (wax esters or TAGs) and the level of heterogeneity (distribution of unique peaks) of each fraction. To further analyze the composition of each component (fatty alcohol and fatty acid) within the neutral lipid fraction, TAGs were converted to FAMEs, and wax esters were converted to FAMEs and free alcohols. Briefly, 10 to 20 mg of isolated neutral lipid extracted from the cells were dried in a COD digestion vial (Hach, Loveland, CO), and 10 μl of concentrated sulfuric acid was added along with 1.5 ml of methanol. The vial was then sealed and heated to 115°C for 30 min on a digital reactor heating block (DRB200; Hach, Loveland, CO), while removing the bottles every 5 min to gently mix by swirling to further dissolve the lipid. Heavy leather gloves and goggles were used during this procedure based on the inherent danger from superheating the solution. After the reaction, the sample was cooled, 1 ml of saturated sodium bicarbonate in distilled water was added to neutralize the remaining acid, and then 3 ml of methylene chloride and 2 ml of distilled water were added and mixed by vortexing. The water layer was removed, and the remaining methylene chloride was washed two more times with an additional 3 ml of distilled water each time. The resulting neutral lipid components were then analyzed again by GC/FID to confirm complete conversion to fatty alcohols and FAMEs. Samples were then analyzed by GC/MS to identify individual component compounds as described above.
RESULTS
Natural neutral lipid distributions.
Initially, we analyzed the neutral lipids obtained from each of the oleaginous microorganisms used in the present study to determine the fatty acid and fatty alcohol composition. The selected approach utilizes a solvent mixture that has been found in our laboratory to successfully isolate neutral lipids from a range of biological samples (5). Figure 2 shows the GC chromatograms for the lipid fraction obtained from each of these lipid-accumulating bacteria. For these studies, we selected high-temperature GC separation versus thin-layer chromatography to provide a greater level of separation to detail the diversity of compounds present within each class of neutral lipids (i.e., TAGs, wax esters, etc.) and to allow quantification of the various products extracted. Marinobacter aquaeolei VT8 and Psychrobacter cryohalolentis K5 were found to accumulate only wax esters under the conditions tested in these studies, whereas both Rhodococcus jostii RHA1 and Acinetobacter baylyi were found to produce a mixture of wax esters and TAGs (Fig. 2), similar to the findings of previous reports (7, 10), with the proportions of these classes of lipids varying from one growth to another, regardless of medium composition, agitation speed, and temperature. Particularly for R. jostii RHA1, substantial differences were found in the dominance of different lipid classes (wax esters and TAGs) among the multiple growths performed with this bacterium, as illustrated by two separate samples representing different extremes that are presented in Fig. 2. Although a detailed analysis of the factors responsible for the differences between types and quantities of the neutral lipids obtained in separate R. jostii RHA1 growths is beyond the scope of our study, a general trend can be established to detail the diversity present and identify the primary components within each class of neutral lipid (TAGs and wax esters) based on these results and the conversion of the isolated lipids to FAMEs and the fatty alcohol components (specifically for wax esters).
Fig 2.

Representative gas chromatograms of the neutral lipids obtained from various bacteria utilized to clone enzymes as part of the present study. Shown are the results obtained using GC with a flame ionization detector analysis of the neutral lipids from four different bacteria. An internal standard (octacosane) elutes at ∼21 min, with the wax esters eluting between 22 and 28 min and the TAGs eluting between 30 and 40 min. The samples shown were utilized to isolate and convert the neutral lipids to fatty alcohols and FAMEs as described in Materials and Methods, and the concentrations of individual fatty alcohols and FAMEs are presented in Table 2. Two samples are presented for R. jostii RHA1, since this strain produced a greater degree of variability in percentages of wax esters or TAGs in different growths, independent of the media and growth conditions utilized as part of the present study, as detailed in Table 1.
Table 1 shows the breakdown of the different fatty acid and fatty alcohol components derived from the wax esters and TAGs (fatty acid component only) after an acid treatment as described in the Materials and Methods. Each microorganism produced a unique range of lipids under the nutrient limited conditions utilized here. The neutral lipid fraction obtained from P. cryohalolentis K5 did not contain detectable amounts of TAGs, and the composition of fatty acids and fatty alcohols obtained from the wax esters were similar in composition, with a strong tendency for C18 derived components (more than 90% for both fatty alcohol and fatty acid components). The neutral lipids isolated from M. aquaeolei VT8 were also predominantly composed of wax ester without detectable amounts of TAGs. In contrast to P. cryohalolentis, the composition of fatty acids and fatty alcohols differed slightly from one another. The wax composition contained very little unsaturated C16 fatty alcohol, while containing a significant amount of unsaturated C16 fatty acid (1.4% of fatty alcohols and 26.1% of fatty acids, respectively). In addition, a higher quantity of unsaturated C18 fatty alcohol was found relative to unsaturated C18 fatty acid (45.3% of fatty alcohols and 26.8% of fatty acids, respectively). The results from A. baylyi are a bit more convoluted due to the presence of TAGs but yielded primarily unsaturated fatty alcohols (46.3% of fatty alcohols as C16:1 and 53.7% of fatty alcohols as C18:1), even though the fatty acid component had significant percentages (34.2% of fatty acids) of saturated C16 fatty acid. Perhaps the most interesting result found in the natural neutral lipid analysis was for R. jostii RHA1, which fluctuated substantially in various growths, and either produced primarily wax esters or TAG, but generally contained at least some of both classes of neutral lipids (Fig. 2). In the first sample presented from R. jostii RHA1, distribution of the fatty alcohol component was similar to that of the fatty acids and were found to be primarily C16 and C18, while in the second sample, the TAG components were a combination of C14, C15, C16, C17, and C18 saturated and unsaturated fatty acids when TAGs were the predominant neutral lipid found (Table 1, sample 2). The lack of odd numbered fatty alcohols (and a lower yield of odd fatty acids) from the predominantly wax ester containing sample (sample 1) versus the TAG dominated sample (sample 2), is an intriguing feature, and based on the selectivity assays described (see below), would not be related to enzyme specificity for the enzyme product of the gene cloned from R. jostii RHA1. Further, since the GC method utilized here can differentiate between odd and even (carbon number) wax esters (see below), it is also interesting that none of the R. jostii RHA1 growths analyzed as part of this study showed significant percentages of odd wax esters, whereas odd fatty acids were found in similar concentrations to the even fatty acids whenever TAGs were present. Others have also reported differences between odd and even fatty acid components obtained from different Rhodococcus strains using different medium compositions (2, 7).
Table 1.
Percent fatty acid and fatty alcohol composition of isolated natural neutral lipid fractions (wax ester and triacylglyceride) from different bacteria
| Product | Neutral lipid component obtained from specific bacteria (%)a |
||||
|---|---|---|---|---|---|
| M. aquaeolei | P. cryohalolentis | A. baylyi | R. jostii, sample 1 | R. jostii, sample 2 | |
| Acids | |||||
| C14 | 3.2 | 0.5 | 3.2 | 2.9 | |
| C15 | 2.0 | 12.4 | |||
| C15:1 | 1.0 | ||||
| C16 | 43.9 | 8.6 | 34.2 | 34.0 | 24.5 |
| C16:1b | 26.1 | 10.8 | 9.0 | 6.6 | |
| C17 | 2.4 | 19.4 | |||
| C17:1 | 3.4 | 20.0 | |||
| C18 | 70.5 | 11.7 | 26.4 | 3.7 | |
| C18:1 | 26.8 | 20.9 | 42.7 | 19.6 | 9.6 |
| Alcohols | |||||
| C16 | 47.6 | 8.7 | 29.2 | ||
| C16:1 | 1.4 | 46.3 | 6.8 | ||
| C18 | 5.6 | 79.7 | 24.6 | ||
| C18:1 | 45.3 | 11.6 | 53.7 | 29.4 | |
The results presented are area percent values for all identified fatty acids or all identified fatty alcohols from the neutral lipids isolated and shown in Fig. 2. R. jostii was grown multiple times under the conditions described in Materials and Methods but partitioned into different percentages of wax esters and TAGs in individual growths, independent of the growth conditions and medium composition described. The two isolations presented here (R. jostii 1 and R. jostii 2) represent growths that yielded predominantly one or the other class (as shown in Fig. 2) and are used here to illustrate the differences in the fatty acid profiles for these two growths.
Two peaks were identified to be C16:1 by GC/MS and are assumed to be differing isomers of this acid; for these results, the areas of both peaks were combined and are reported as one value here.
As a further test of in vivo selectivity of the WS/DGAT enzymes from M. aquaeolei VT8, an experiment was also performed in which pentadecanol was added to the growth media during lipid accumulation for this strain. Figure 3 shows the differences in the native wax esters, which were confirmed by GC/MS and are labeled as to total carbon size, and the change in the wax ester profile when the odd pentadecanol was added to the medium. This result shows a substantial increase in the presence of C31 and C33 wax esters (which are only trace components in the native waxes, and are proposed to be derived from reactivity with palmitoyl-CoA and oleyl-CoA, respectively), but no presence of a C35 wax ester. Similar experiments (not shown) have been performed with tridecanol, yielding further support for the potential of the native enzymes to utilize these extraneously provided fatty alcohols.
Fig 3.

Shift in wax ester profile in Marinobacter aquaeolei VT8 when grown with extraneously added pentadecanol. Shown are GC/FID chromatograms of a representative wax ester profile from M. aquaeolei VT8 grown under normal wax ester inducing conditions (below) and a similar culture of M. aquaeolei VT8 grown in a similar media with extraneously added pentadecanol. The primary wax ester peaks found in the native waxes are composed of C16 and C18 fatty acid and fatty alcohol components, yielding primarily C32, C34, and C36 waxes, whereas the addition of the C15 fatty alcohol results in the production of C31 and C33 wax esters and an increase in C30 wax esters.
Isolation of WS/DGAT proteins.
Five genes encoding putative WS/DGAT enzymes from four different bacteria were cloned, and the proteins heterologously expressed from these genes were isolated in order to conduct comparisons of the differences in the in vitro stability, activity, and selectivity for each enzyme. Rapid isolation was enabled by cloning each gene into a vector that incorporated an MBP fusion on the N terminus and an His8 tag on the C terminus of the protein, similar to the approach utilized to purify other proteins involved in the production of wax esters from M. aquaeolei VT8 (24, 27). M. aquaeolei VT8 contains four genes coding for potential WS/DGAT homologs. Two of these genes (Ma1 and Ma2) were cloned and the expressed enzyme product isolated. From the SDS-PAGE gel of the isolated protein, it can be seen that Ma1 was highly purified by this approach, as indicated by the presence of only a single band on the gel, demonstrating little degradation during expression and isolation of the protein. Ma2 and the protein fusion from A. baylyi (Ac1) showed a prominent band with only minor smaller fragments, while the protein fusions from P. cryohalolentis K5 (Ps1) and R. sp. strain RHA1 (Rh1) both showed some degree of degradation or contamination from other proteins (Fig. 4). Despite the potential degradation found in certain fusions, all of the preparations showed a dominant band corresponding to the complete fusion protein, and the inclusion of both MBP on the N terminus and a polyhistidine tag on the C terminus of the protein resulted in reasonable yields of a protein that was sufficiently soluble in aqueous solutions to perform enzyme assays, although only the Ma1 protein appears to be homogeneous based on this rapid isolation approach. Similar to other experiences with enzymes involved in wax ester production from M. aquaeolei VT8 (24, 27), Ma1 was found to be practically insoluble when heterologously expressed in E. coli without the MBP fusion. Conventional column purifications of the soluble fraction of lysed cells resulted in poor protein recovery despite Ma1 being highly expressed, as determined by the appearance of a strong band of expected size by SDS-PAGE analysis of whole cells and membrane fractions (not shown). Isolation of MBP fusion protein and subsequent digestion with Factor Xa resulted in the two expected cleaved bands when analyzed by SDS-PAGE but also resulted in a lower activity (potentially due to protein instability during the enzymatic cleavage or possibly from protein insolubility). Based on this finding, the MBP fusion product from each purification was utilized for the purposes of the activity assays described below.
Fig 4.
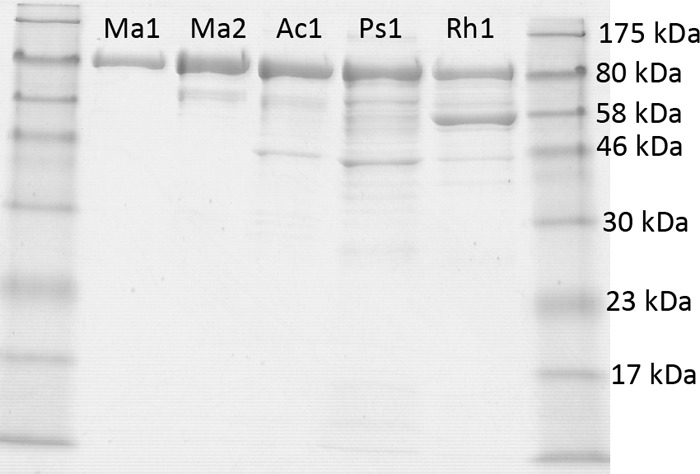
SDS-PAGE gel of various MBP–WS/DGAT–His-tagged fusion proteins isolated using the rapid two-step purification procedure described in Materials and Methods. Samples are flanked by a protein standard (New England Biolabs) on either side. A prominent band is seen for each sample corresponding to the complete fusion protein, and various degrees of degradation or premature translational termination are found for several of the proteins shown. Acronyms for the enzymes reported are defined in Materials and Methods.
Although several of the proteins shown in Fig. 4 could be further purified with conventional size exclusion or other chromatography steps, the purpose of the figure is to compare protein stability under similar conditions utilized to express and rapidly isolate each protein. Further chromatography steps did result in removal of degraded or contaminating proteins in some cases but were also accompanied by a further loss in specific activity and yield. Analysis of several smaller bands from the gel shown in Fig. 4 by in-gel trypsin digestion and matrix-assisted laser desorption ionization MS revealed that degradation was likely occurring in the WS/DGAT portion of the MBP-WS/DGAT fusion, since multiple fragments related to the MBP were identified from these degradation products (smaller bands on Fig. 4, lanes Ps1 and Rh1), indicating that the MBP portion was still intact. Further analysis with a polyhistidine-tag indicator dye (Pierce His6 protein tag staining kit; Thermo Scientific) also indicated that the majority of these bands are lacking the C-terminal polyhistidine sequence. The presence of these contaminating bands also increased if the cells were induced for longer periods of time in specific preparations but decreased if the induction was halted within 2 h of the IPTG addition.
Enzyme activity assays.
Spectrophotometric assays were used to monitor the activity of the isolated enzymes in real time as described in Materials and Methods. The robustness and reproducibility of the assay was tested under various conditions, and the resulting products were confirmed by direct approaches using GC/MS analysis of isolated products in addition to direct quantification of specific substrates by GC with a flame ionization detector (GC/FID), taking advantage of the lower limit of detection for this approach versus GC/MS. The identities of products found here agree with previous reports related to the activity of these enzymes and the broad range of products that these enzymes can produce (18, 20, 22). DMSO concentrations used here (ca. 5% by volume) were well below the levels found to cause a minor inhibition of the enzyme assay (ca. 10% DMSO by volume). A pH of 7 was selected for this analysis since it was found to be the optimal pH value for Ma1 and is closer to the pKa of the buffer system, although the enzyme was active over a broad pH range, with only minor losses at pH 8, which is the optimal pH reported by others for Ac1 (20). Although others have reported higher activities for WS/DGAT enzymes at higher temperatures (20), 22°C was selected for simplicity and because the growth conditions of the different organisms under native wax production detailed above was done at 25 or 30°C. All enzymes were thawed and stored on ice until addition to the assay, since storage for several hours at room temperature was found to decrease enzyme activity, even though no proteolytic degradation was found by SDS-PAGE analysis (data not shown).
From the activities shown in Table 2, it was found that the Ma1 protein had the highest specific activity of each of the proteins purified, with nearly 10-fold-higher activity than Rh1 and Ma2, which were both ∼4-fold higher than Ac1 and Ps1. These results compare well with previous reports for WS/DGAT activity from Acinetobacter sp. strain ADP1 when purified without the inclusion of any protein fusions or affinity tags (20), although different techniques were applied in those reports to purify the enzyme. It should also be noted that the activity for the proteins in this present study are reported per mg of protein, as was done in other studies (20), but includes the mass of the MBP, which accounts for nearly 50% of the mass of the fusion protein construct and, as such, the reported activity is lower than would be reported if it were calculated only on the percentage of the protein consisting of the WS/DGAT portion. Each enzyme isolated as part of the present study was found to have much higher activity with dodecanol as the alcohol substrate than was found with hexadecanol (Table 2), which is expected to be a natural substrate in vivo based on analysis of wax ester fractions obtained from each organism (Table 1), and for this reason, dodecanol was selected as the primary fatty alcohol substrate in assays for activity for the remainder of the comparisons. In addition, each of the fatty alcohols was first dissolved in DMSO to facilitate rapid addition and dispersion of the substrate to the assay buffer just prior to initiation of the reaction. Hexadecanol had a tendency to precipitate or separate from the DMSO solution if not warmed occasionally, while dodecanol remained dissolved in the DMSO with no adverse affects to measured activity rates even after several days. While several of the protein isolates showed some degree of protein degradation, the degree of degradation does not correlate with the differences in activities reported here (Fig. 4 and Table 2).
Effect of order of addition of assay components on in vitro specific WS activity.
As part of efforts to characterize activity for the proteins tested here, experiments were undertaken to confirm that the activity measured was the result of the added enzyme by leaving out a specific component in the assay. These experiments confirmed that activity (measured as the change in absorbance at 412 nm during the first 1 to 4 min of the assay) was only found if all components were included in the assay. There are three primary components in each assay, including each substrate (fatty alcohol and fatty acyl-CoA) and the protein, in addition to the coupled color indicator component DTNB and the selected buffer solution. Assays were run by adding all components except for one to first establish a stable baseline on the spectrophotometer prior to initiation of the reaction by adding the final component. In the case of several of these enzymes, it was found that the order of addition (or more specifically the substrate that the protein was incubated with during the first minute while the background absorbance was measured) had a significant impact on the rate of reaction. With certain WS/DGAT enzymes, this order of addition effect was quite striking (see Fig. S1 in the supplemental material for an example of the time course assays), indicating that as much as ∼95% of the enzyme activity could be lost (in the case of Ac1) by incubating the enzyme with palmitoyl-CoA during the initial 1 min (prior to adding the fatty alcohol to initiate the reaction) versus the result that was obtained if Ac1 were incubated with fatty alcohol first (with addition of fatty acyl-CoA to initiate the reaction). When the enzyme was added as the final component, an activity was observed that was between the higher rate found when incubating with the fatty alcohol first (see Fig. S1 in the supplemental material). While two of the enzymes showed only a minimal loss of activity when first incubated with fatty acyl-CoA (Ma2 and Ps1, Table 2), the other two (Rh1 and Ma1) showed more substantial losses in activity based on the order of addition of assay components, in a manner similar to Ac1. In none of these five enzymes was a higher activity found by incubating with the fatty acyl-CoA substrate first.
Kinetic characterization of purified recombinant WS/DGAT proteins.
Several attempts were made to determine kinetic parameters for the enzymes in these studies as have been reported by others (20). However, as described above, the order of addition of specific substrates and the required substrate ratios made it difficult to determine whether changes in rate for fatty alcohols at lower concentration were the result of the actual rate of reaction or the inclusion of higher levels of fatty acyl-CoA in the assay (which could inhibit the reaction). Although the rates of palmitoyl-CoA could be followed based on the higher activity when first incubated with dodecanol (or other fatty alcohols), we found that the rate was constant (see Fig. S2 in the supplemental material) even at very low concentrations of palmitoyl-CoA (2.5 nmol in 1.2 ml of total volume or 2.1 μM) and remained constant until just before the substrate was completely consumed, indicating a Km for palmitoyl-CoA that was likely below 1 μM. These results contrast with other reports of Km values for acyl-CoAs that were much higher (8, 20), although the authors in the previous studies may not have focused on the potential importance of the order of substrate addition to the measured activities, and utilized other techniques to keep the insoluble substrates in solution (e.g., inclusion of bovine serum albumin [BSA] [8]). Similar absorbance versus time responses for the specific activity measurements seem to support a low Km value for the fatty acyl-CoA components for all five of the proteins tested in these studies, and similar changes in the sharp drop in activity just before substrate was completely consumed was observed when using either dodecanol or hexadecanol (even though the specific activity for each enzyme was different, Table 2). A precise measurement of Km values below 1 μM are below the dynamic range of the spectrophotometric assay utilized here. The change in absorbance based on the quantity of palmitoyl-CoA utilized (10 nmol in 1.2 ml, or 8.3 μM multiplied by the extinction coefficient of 14,150 M−1 cm−1 when using a 1-cm path length) is calculated to be 0.117, which is in reasonable agreement with the obtained difference of ∼0.09 absorbance units, and appears to follow a linear relationship related to increasing amounts of palmitoyl-CoA (see Fig. S2 in the supplemental material), illustrating the further utility of this approach for monitoring WS/DGAT activity and assessing potential Km values from a single real-time assay.
Substrate specificity.
Because of the low global conservation in the protein primary sequence of the enzymes studies here, it was of interest to develop a protocol to test the wax ester profiles when the enzymes were exposed to a range of substrates. A GC assay was developed based on the ability to screen a broad array of substrates quickly by exposing the enzyme to a mixture containing equimolar quantities of a variety of one type of substrate (such as the fatty alcohols) and an excess of a single component of the complementary substrate (such as palmitoyl CoA). An example of the assay controls and results obtained for testing specificity for a range of straight-chain fatty alcohols is shown in the supplemental material. These experiments were run with palmitoyl-CoA at a 4-fold molar excess to each of the 10 fatty alcohols, which were added as a mixture containing equimolar quantities. The resulting wax esters were identified, and the percentages of those obtained are shown in Fig. 5. Although the overall profile of the resulting wax esters were globally similar, differences were seen between specific proteins, such as Rh1, which produced significantly higher amounts of nonanol- and decanol-containing wax esters and significantly less of tetradecanol- and higher alcohol-containing wax esters than the other enzymes tested. None of the enzymes produced measurable amounts of hexanol containing wax esters above trace levels, even though hexanol could serve as a substrate in the absence of higher alcohols (see below). Similar sharp differences in activity for lower alcohols have been reported by others (20). The results also reveal that nearly all of the C11-OH and C12-OH (dodecanol) substrates were consumed (see Fig. S4 in the supplemental material), while significant amounts of the other alcohols still remained, indicating a strong specificity for certain alcohols over others. The assays yielded consistent results, providing a snapshot of the substrate profiles for each enzyme. This assay approach should aid in future screening to better study these enzymes, and the potential contributions of specific amino acids in the primary sequence to substrate specificity. The selectivity of this assay for dodecanol versus hexadecanol for each of these GC results also agrees well with the real-time spectrophotometric results obtained (Table 2), which showed significantly higher specific activity rates for dodecanol versus hexadecanol, and the nearly complete consumption of these substrates agrees well with a likely low Km value that is proposed for dodecanol.
Fig 5.

Wax ester product distribution indicating fatty alcohol selectivity of different WS/DGAT enzymes in the presence of palmitoyl-CoA. Shown are the percentages of fatty alcohol derived wax esters formed when 50 nmol of each of 10 different fatty alcohols were combined with 200 nmol of palmitoyl-CoA (C16-CoA). Assays were run for 15 min in the presence of each enzyme, and then the lipids were extracted from the assay as described in Materials and Methods. The area of each peak was converted to a percentage of the total of all wax esters obtained. The x axis is labeled as to the alcohol component of the wax ester (i.e., octyl hexadecanoate derived from octanol and palmitoyl-CoA [hexadecanoyl-CoA]).
A GC assay similar to that described above was conducted using a single fatty alcohol (dodecanol) at 3 molar equivalents combined with a single molar equivalent of each of five different fatty acyl-CoA substrates. To effectively extract the products from the assay mixture, a second alcohol (in this case an odd alcohol that could be differentiated from the dodecanol) was added in slight excess to react with the remaining two equivalents of fatty acyl-CoA substrates following the initial assay, which acted as potential detergents if not consumed completely and made product isolation difficult. This approach revealed a high selectivity of the enzymes for palmitoyl-CoA (C16), myristoyl-CoA (C14), and lauroyl-CoA (C12) but showed little production of the smaller wax esters (results not shown). In this instance, an additional measure of specific activity was done using the real-time spectrophotometric assay with each of the different fatty acyl-CoA substrates to obtain a clearer understanding of the specificity of each enzyme for different fatty acyl-CoA substrates. To illustrate these results, the specific activity for each acyl-CoA substrate is plotted as a percentage of the specific activity obtained for palmitoyl-CoA (Fig. 6). The activity of each individual WS/DGAT enzyme varied with fatty acyl-CoA molecules of decreasing chain length. For example, Ac1 showed similar activities for C16-CoA and C14-CoA, but the activity dropped substantially for the C12-CoA, C10-CoA, and C8-CoA substrates. In contrast, Ps1 and Ma2 both showed higher activity for C14-CoA versus C16-CoA and retained a significant portion of activity for the smaller acyl-CoA substrates, albeit at steadily lower rates as the size of the substrates decreased below C14-CoA. Ma1 and Rh1 showed similar specific activity percentages to one another for each of the acyl-CoA substrates, but the percentage of remaining activity was lower for substrates below C14-CoA than was found for Ma2 and Ps1.
Fig 6.

Rates of specific activity plotted as a percentage of the specific activity found using palmitoyl-CoA (C16-CoA). Assays were run with 20 nmol of dodecanol and 15 nmol of various fatty acyl-CoA substrates in a final volume of ∼1.2 ml of buffer, and the specific activity was monitored spectrophotometrically at 412 nm. Quantities of protein added ranged from 0.3 to 4.5 μg based on the specific activity results obtained with dodecanol and palmitoyl-CoA for each protein, as listed in Table 1.
As an example of the feasibility to use these approaches to test other potential substrates for these promiscuous enzymes, an additional GC assay was conducted using palmitoyl-CoA and each of three alternative alcohols, including hexanol, isoamyl alcohol, and phenyl ethanol. Each substrate was chosen based on potential differences in the size and degree of branching or aromaticity of the compounds, since it was presumed that these would be good substrates to probe potential differences in the active site environment of the different enzymes. Substrates were added at concentrations that resulted in similar product percentage yields of each with the Ma1 enzyme (as equimolar quantities of these substrates to palmitoyl-CoA were too low to produce sufficient product yield for the analysis) and were then utilized with the other four enzymes at the same concentrations for comparison. Figure 7 shows the results obtained for the isolated products, with the starkest differences found for phenyl ethanol, which showed the highest activity with Ma2, and the lowest with Ac1. Again, this approach illustrates the potential of the assay as a screening tool to probe the differences in various WS/DGAT enzymes.
Fig 7.

Medium-chain, branched and aromatic alcohol selectivity of different WS/DGAT enzymes. Shown are the percentages of different wax esters formed when specific concentrations of three different branched (isoamyl alcohol), medium (hexanol), and aromatic alcohols were combined with 100 nmol of palmitoyl-CoA (C16-CoA). Assays were run for 20 min after the addition of each enzyme, and then the wax ester products were analyzed as described in Materials and Methods. The area of each peak was quantified as a percentage of the total of all wax esters produced.
DISCUSSION
Homologs of the genes for wax ester synthase/acyl-CoA:diacylglycerol acyltransferase (WS/DGAT) enzymes have been found in a range of different bacteria through sequencing efforts and in some cases by direct analysis of the gene products (12, 22, 25). The wax ester accumulation pathway, which represents a potential mechanism for reduced carbon storage, is limited to only a small percentage of bacteria overall, and the presence of neutral lipids such as wax esters or TAGs has been confirmed in only a small number of bacterial species (1, 7, 10, 13). In contrast, alternative methods for carbon storage such as the synthesis of polyhydroxybutyrate, starch, or glycogen seem to be a predominant reduced carbon storage mechanism for most bacteria (22, 25). Studies to date on lipid accumulating bacteria have focused primarily on the WS/DGAT enzyme isolated from Acinetobacter species and have not made direct comparisons between WS/DGAT enzymes isolated from multiple bacteria, although a very recent report demonstrated expression of a variety of WS/DGAT enzymes in yeast for a comparison of the lipids produced in vivo and further analyzed these enzymes using crude cell extracts (19). In this report, we cloned five genes for the WS/DGAT protein products from four different bacteria and compared the activities and selectivity of the isolated proteins to the composition of the natural wax esters or TAGs produced under nutrient-stressed conditions for each bacterium. Genes were selected for expression based on either the presence of only one gene (for A. baylyi and P. cryohalolentis K5) or based on high primary amino acid sequence similarity to the A. baylyi gene product (for M. aquaeolei VT8 and R. jostii RHA1). The natural lipid profiles obtained showed significant differences between the types of neutral lipids they accumulated (wax esters or TAGs, Fig. 2). In contrast, the individual components that comprise the neutral lipids (fatty acids and fatty alcohols, Table 1) vary only slightly between species. Saturated and unsaturated C16 and C18 fatty acids and fatty alcohols were the dominant components of the neutral lipids with exception to the lipids produced by R. jostii RHA1, which additionally produced fatty acids with odd numbered chain lengths (Table 1), a finding consistent with previous reports (2, 7).
Alignments of the protein primary sequences of the gene products cloned and heterologously expressed in these studies are provided in the supplemental material, and reveal that only ca. 20% of the sequence is identical in each of these proteins, while the conservation is somewhat higher if common substitutions are included (phenylalanine and tyrosine or isoleucine and leucine, for example) at ca. 25%. Each enzyme showed identity of the residues believed to comprise the active-site motif (HHXXXDG) (21), but aside from a few regions of strong conservation, the amino acid primary sequences show a high degree of variation from one another, while the activity toward either fatty alcohols or fatty acyl-CoA substrates is well conserved based on our results for each enzyme (Fig. 5 and 6).
The characterization of membrane bound or membrane associated enzymes such as those involved in lipid metabolism is difficult by comparison to cytosolic enzymes which commonly act on substrates dissolved in aqueous solution. Generally, membrane associated enzymes are difficult to purify using techniques that work well for soluble proteins. Although detergents can improve the solubility of membrane-associated enzymes in certain cases, the addition of the detergent may also result in losses of enzyme activity. We have utilized a gene fusion approach to isolate various proteins believed to be involved in the wax ester synthesis pathways in bacteria (27). The approach utilizes two different affinity tags: (i) an MBP that improves the solubility of the protein and also allows the protein to be isolated by using an amylose resin and (ii) a polyhistidine tag that allows the protein to be further purified using metal affinity chromatography. To assist in the removal of degradation products resulting from incomplete translation or potential proteolytic processes in the cell, these tags are placed on opposite terminal ends of the proteins. This approach has been successful in isolating proteins that reduce fatty acyl-CoAs and also fatty aldehydes (24, 27) and was utilized here to overcome solubility issues with the WS/DGAT enzymes from the selected species. The use of the dual affinity tag constructs of the WS/DGAT enzymes resulted in enzymes with activity comparable to that reported by others (1, 20), indicating that the approach offers an alternative for the rapid isolation of this membrane-associated protein without adversely affecting activity, which should assist in future studies of these enzymes.
A key aim of the present study was to establish which of the enzymes included here would be the best candidate for future structural and mechanistic studies, and a key component of this analysis was protein stability and purity using the rapid two-step affinity isolation protocol. Although several enzymes in these studies could be further purified with additional chromatography steps, the proteins from M. aquaeolei VT8 (Ma1 and Ma2) and A. baylyi (Ac1) produced the highest quantities of protein with the least signs of degradation or impurities using the rapid purification steps. SDS-PAGE analysis of the other purified WS/DGAT proteins (Rh1 and Ps1) revealed a prominent band as well as several less prominent bands with decreasing molecular weight. Further analysis of the less-prominent bands revealed that they were likely degradation products of the fusion protein. The degradation could be the result of proteolysis or premature termination during translation, since all of the genes were cloned directly from the host organisms with no attempt to optimize the DNA sequences for heterologous expression in E. coli. The limited degradation found in these purifications did not appear to significantly affect the activity since the rates of wax ester formation from these isolated protein preparations were comparable to the rates observed in preparations with higher purity. Others have reported requiring high concentrations of EDTA and the addition of protease inhibitor cocktails to lessen degradation during purification (20). Based on the assessment made here, Ma1 seems to be the best candidate enzyme for future study, based on overall stability under simple growth conditions, rapid purification to homogeneity and significantly higher activities compared to the other enzymes tested (Table 2 and Fig. 4).
The results presented here reveal a stark difference between the natural products of neutral lipid synthesis found in each specific bacteria and the potential substrate range for each of the enzymes. For example, P. cryohalolentis K5 was found to produce predominantly wax esters with a fatty alcohol composition that was >90% derived from C18-based alcohols, yet the activity for the isolated enzyme showed the highest specificity for C14 alcohols. The general selectivity of the WS/DGAT enzymes studied here for fatty alcohol was substantially different from the selectivity found for different fatty acyl-CoA substrates, with a general selectivity for longer fatty acyl-CoA substrates (C16 and C14 chain lengths), while the enzymes appeared to prefer medium range fatty alcohols (approximately C12 for most enzymes). Although it may be argued that solubility effects could explain some of these differences, the variation in the selectivity for fatty alcohols found for different enzymes and the reproducibility of the results overall support a likelihood that these differences are enzyme specific. Further, based on the low level of conservation in the amino acid sequence between the WS/DGAT enzymes studied, one would expect some differences in the substrate selectivity profiles of these different enzymes.
Taken as a whole, the differences between the enzyme substrate profiles and the profile of the neutral lipids obtained from each bacterium supports the hypothesis that the primary determinant of natural wax ester length and composition lies upstream in the metabolic pathways that produce the fatty acyl-CoA and fatty alcohol substrates for the downstream WS/DGAT protein. The substrate promiscuity of the WS/DGAT proteins serves to accept any number of alcohols that the cell might provide for the acyl transferase reaction. Indeed, this hypothesis is supported using M. aquaeolei VT8 cells supplemented with C15 fatty alcohol (Fig. 3) during growth, which was found to substantially shift the product profiles, supporting this hypothesis that the native enzymes are capable of producing a broader range of wax esters in vivo if provided different precursors, including odd numbered carbon alcohols as was found in our in vitro experiments. This feature has been utilize by others to produce potential commodity bioproducts such as ethyl esters to make microdiesel (11). By cloning and heterologously expressing the genes for a range of WS/DGAT enzymes in a similar protein fusion construct and then testing the activities of each enzyme under similar conditions, we provide here a set of data that allow the direct comparison of results to contrast these enzymes. These data will be valuable in the development of future experiments to probe the relationships between sequence and function within this class of enzymes.
It should be noted that the exact cellular environment in which the WS/DGAT enzymes function could not be replicated in vitro. As a consequence, the results of this or any other current in vitro experiments with WS/DGAT may not reflect the true kinetic parameters of these enzymes in vivo. The enzyme is associated with the membrane and utilizes a soluble (fatty acyl-CoA) substrate and an insoluble (fatty alcohol) substrate. We have attempted to mitigate these two issues by using an MBP fusion construct of each WS/DGAT protein and by adding the fatty alcohol substrates to the enzyme assays under conditions where the fatty alcohol is already dispersed within a volume of DMSO. This approach offers alternatives to the inclusion of additional proteins (such as BSA) or detergents to aid in solubilization of substrates and protein (8), since the assays utilized here relied only on DMSO to initially aid in dissolving and disbursing the fatty alcohol component during the assay. Despite our best efforts, the exact cellular conditions could not be replicated. However, the methods used here provide an opportunity to compare the activities from several WS/DGAT homologs toward a variety of substrates. Because each enzyme is treated in the same manner, our results and conclusions should correlate to real differences between each enzyme functioning in vivo. This approach is not meant to replace techniques that utilize detergents or membrane emulsions that should provide additional information but instead provides an alternative method for testing complex systems involving both soluble and membrane associated substrates and products.
A key finding in these experiments relates to the importance of substrate addition order and the effects that incubation of specific substrates have on enzyme activity for certain WS/DGAT enzymes (see Fig. S1 in the supplemental material and Table 2). The effect of substrate addition order seemed to be more prominent for certain proteins, was reproducible, and may indicate a mechanism of allosteric control of protein activity based on substrate levels within the cell. Since the effect was minimal in certain proteins (Table 2), these findings provide a potential blueprint for future studies to probe the role specific residues have in gating the activity and selectivity of the enzyme based on substrate concentrations. This also may signify an important cellular mechanism to lower enzyme activity when fatty alcohol concentrations within the cell are dropping or increase the activity if fatty alcohol concentrations within the cell begin to rise, which may represent a scenario that is detrimental to the cell. Most importantly, the findings illustrate the importance of the order of addition of substrates when attempting to calculate maximum specific activity with WS/DGAT enzyme. Since this difference in the order of addition results was only observed with certain enzymes, it is not believed that this effect is the result of a physical phenomenon such as the insolubility of an assay component. Utilization of this optimized assay seems to support Km values for these enzymes that are significantly lower than previous reports (8, 20).
Although the production of neutral lipids such as wax esters as a potential storage compound is rare among bacteria, WS/DGAT homolog enzymes are found in a broad range of bacteria (12, 13, 22, 25). Here, we confirm the production of wax esters in the psychrophilic bacterium P. cryohalolentis K5 (and have additionally confirmed their presence in Psychrobacter arcticus 273-4 [results not shown]), each of which are strains of bacteria isolated from polar environments. In P. arcticus 273-4 and P. cryohalolentis K5, only a single copy of the WS/DGAT gene is found, similar to what is found in Acinetobacter species, whereas M. aquaeolei VT8 and R. jostii RHA1 each contain multiple homologs for the WS/DGAT (4 and 15 or more genes in each, respectively [1, 8, 14]). The precise reason certain species contain multiple copies of these genes is not yet clear. We found, as others have reported (10), that A. baylyi contains a mixture of wax esters and TAGs even though it contains only one WS/DGAT coding gene, while it is possible that certain strains such as M. aquaeolei VT8 and R. jostii RHA1 may have multiple homologs expressed at any point in time during lipid accumulation. As such, we cannot discount that some aspect of the selectivity for natural products may be related to the presence of different enzymes. Although the WS/DGAT enzyme found in M. aquaeolei VT8 and related species has been implicated in the production of wax esters derived from phytols (8), our analysis of waxes isolated under nutrient limitation show a lipid profile composed primarily of C16 and C18 derived fatty acids, although our media did not include any extraneous phytol.
Interest in the utilization of enzymes such as WS/DGAT and other enzymes involved in the production of storage lipids requires an understanding of not only the substrate ranges of these proteins but also the potential for expanding or altering the profiles of the specific enzyme and selection of the optimal enzyme for biotechnological applications. By isolating a range of these enzymes and characterizing each under the same conditions, we are able to make direct comparisons, which, along with the primary sequences of each, should assist in future studies to test whether the specificity of the enzyme can be altered without dramatically decreasing activity overall. Although a published structure of a functional WS/DGAT enzyme would also assist in such studies, crystallization studies to date have been unsuccessful (21). In the absence of structural information, direct comparison studies such as were pursued here offer an additional avenue to probe primary structure as it relates to enzyme function.
Supplementary Material
ACKNOWLEDGMENTS
This study is supported by a grant from the National Science Foundation (award 0968781) to B.M.B. and L.C.S.
We thank Lindsay Eltis and James Tiedje for providing several of the strains used in this study. We are grateful to the anonymous reviewers, who provided many detailed and helpful suggestions related to the manuscript.
Footnotes
Published ahead of print 8 June 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Alvarez AF, Alvarez HM, Kalscheuer R, Wältermann M, Steinbüchel A. 2008. Cloning and characterization of a gene involved in triacylglycerol biosynthesis and identification of additional homologous genes in the oleaginous bacterium Rhodococcus opacus PD630. Microbiology 154:2327–2335 [DOI] [PubMed] [Google Scholar]
- 2. Alvarez HM, Steinbüchel A. 2002. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 60:367–376 [DOI] [PubMed] [Google Scholar]
- 3. Corpet F. 1988. Multiple sequence alignment with hierarchical-clustering. Nucleic Acids Res. 16:10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellman GL. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70–77 [DOI] [PubMed] [Google Scholar]
- 5. Gardner R, Peters P, Peyton B, Cooksey KE. 2011. Medium pH and nitrate concentration effects on accumulation of triacylglycerol in two members of the Chlorophyta. J. Appl. Phycol. 23:1005–1016 [Google Scholar]
- 6. Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
- 7. Hernández MA, et al. 2008. Biosynthesis of storage compounds by Rhodococcus jostii RHA1 and global identification of genes involved in their metabolism. BMC Genomics 9:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holtzapple E, Schmidt-Dannert C. 2007. Biosynthesis of isoprenoid wax ester in Marinobacter hydrocarbonoclasticus DSM 8798: identification and characterization of isoprenoid coenzyme A synthetase and wax ester synthases. J. Bacteriol. 189:3804–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jetter R, Kunst L. 2008. Plant surface lipid biosynthetic pathways and their utility for metabolic engineering of waxes and hydrocarbon biofuels. Plant J. 54:670–683 [DOI] [PubMed] [Google Scholar]
- 10. Kalscheuer R, Steinbüchel A. 2003. A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J. Biol. Chem. 278:8075–8082 [DOI] [PubMed] [Google Scholar]
- 11. Kalscheuer R, Stölting T, Steinbüchel A. 2006. Microdiesel: Escherichia coli engineered for fuel production. Microbiology 152:2529–2536 [DOI] [PubMed] [Google Scholar]
- 12. Manilla-Pérez E, Lange AB, Hetzler S, Steinbüchel A. 2010. Occurrence, production, and export of lipophilic compounds by hydrocarbonoclastic marine bacteria and their potential use to produce bulk chemicals from hydrocarbons. Appl. Microbiol. Biotechnol. 86:1693–1706 [DOI] [PubMed] [Google Scholar]
- 13. Manilla-Pérez E, Lange AB, Luftmann H, Robenek H, Steinbüchel A. 2011. Neutral lipid production in Alcanivorax borkumensis SK2 and other marine hydrocarbonoclastic bacteria. Eur. J. Lipid Sci. Technol. 113:8–17 [Google Scholar]
- 14. McLeod MP, et al. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U. S. A. 103:15582–15587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Metz JG, Pollard MR, Anderson L, Hayes TR, Lassner MW. 2000. Purification of a jojoba embryo fatty acyl-coenzyme A reductase and expression of its cDNA in high erucic acid rapeseed. Plant Physiol. 122:635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reiser S, Somerville C. 1997. Isolation of mutants of Acinetobacter calcoaceticus deficient in wax ester synthesis and complementation of one mutation with a gene encoding a fatty acyl coenzyme A reductase. J. Bacteriol. 179:2969–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol. 91:49–60 [DOI] [PubMed] [Google Scholar]
- 18. Röttig A, Wenning L, Bröker D, Steinbüchel A. 2010. Fatty acid alkyl esters: perspectives for production of alternative biofuels. Appl. Microbiol. Biotechnol. 85:1713–1733 [DOI] [PubMed] [Google Scholar]
- 19. Shi SB, Valle-Rodríguez JO, Khoomrung S, Siewers V, Nielsen J. 2012. Functional expression and characterization of five wax ester synthases in Saccharomyces cerevisiae and their utility for biodiesel production. Biotechnol. Biofuels 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stöveken T, Kalscheuer R, Malkus U, Reichelt R, Steinbüchel A. 2005. The wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J. Bacteriol. 187:1369–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stöveken T, Kalscheuer R, Steinbüchel A. 2009. Both histidine residues of the conserved HHXXXDG motif are essential for wax ester synthase/acyl-CoA:diacylglycerol acyltransferase catalysis. Eur. J. Lipid Sci. Technol. 111:112–119 [Google Scholar]
- 22. Stöveken T, Steinbüchel A. 2008. Bacterial acyltransferases as an alternative for lipase-catalyzed acylation for the production of oleochemicals and fuels. Angew. Chem. Int. Ed. Engl. 47:3688–3694 [DOI] [PubMed] [Google Scholar]
- 23. Voelker T, Kinney AT. 2001. Variations in the biosynthesis of seed-storage lipids. Annu. Rev. Plant Physiol. 52:335–361 [DOI] [PubMed] [Google Scholar]
- 24. Wahlen BD, Oswald WS, Seefeldt LC, Barney BM. 2009. Purification, characterization, and potential bacterial wax production role of an NADPH-dependent fatty aldehyde reductase from Marinobacter aquaeolei VT8. Appl. Environ. Microbiol. 75:2758–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wältermann M, et al. 2005. Mechanism of lipid-body formation in prokaryotes: how bacteria fatten up. Mol. Microbiol. 55:750–763 [DOI] [PubMed] [Google Scholar]
- 26. Wältermann M, Stöveken T, Steinbüchel A. 2007. Key enzymes for biosynthesis of neutral lipid storage compounds in prokaryotes: properties, function and occurrence of wax ester synthases/acyl-CoA/diacylglycerol acyltransferases. Biochimie 89:230–342 [DOI] [PubMed] [Google Scholar]
- 27. Willis RM, Wahlen BD, Seefeldt LC, Barney BM. 2011. Characterization of a fatty acyl-CoA reductase from Marinobacter aquaeolei VT8: a bacterial enzyme catalyzing the reduction of fatty acyl-CoA to fatty alcohol. Biochemistry 50:10550–10558 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.