

Abstract
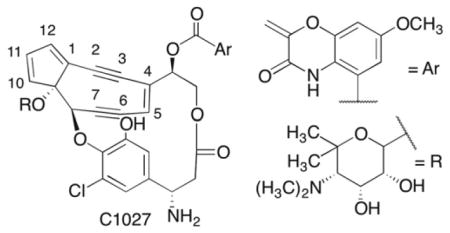
C1027 is a potent antitumor agent that damages DNA. It has the unusual ability to produce double strand breaks and interstrand cross-links (ICLs) intracellularly, which enable it to initiate concurrent ataxia-telangiestasia mutated (ATM) and Rad-3 related (ATR) independent damage responses. The latter form of damage is not well characterized. We have examined the effect of DNA sequence on C1027 reactivity and found it to be more diverse than previously thought. In addition, analysis of the chemical stability of ICLs suggests that they result from reaction with the deoxyribose ring on one strand but direct addition to a nucleobase on the opposite strand. 2012 Elsevier Ltd. All rights reserved
Keywords: DNA damage, enediynes, interstrand cross-link, oxidized abasic sites
1. Introduction
C1027 is a member of the general class of enediyne antitumor antibiotics that induce cell death by damaging DNA.1 Enediynes undergo a Bergman rearrangement to biradicals, which have been shown to abstract hydrogen atoms from the deoxyribose backbone of DNA (Scheme 1).2 Many of the enediynes (and related molecules, such as the neocarzinostatin chromophore (NCS)) are stable until they undergo a chemical reaction (e.g. reduction), which activates the molecule to undergo rearrangement to the biradical.3 C1027 is a member of a subset of enediynes that forms the requisite biradical reversibly without any prior activation.4–7 The enediyne pharmacophore is protected (from itself) in the absence of DNA by an apoprotein. The enediynes are some of the most cytotoxic antitumor agents known. For instance, C1027 is several thousand times more potent than adriamycin and mitomycin C.8–10 Their propitious cell killing and unique chemical reactivity generated a great deal of research activity. To some degree, the enediynes were a victim of their own success, as their high cytotoxicity and limited tumor specificity proved difficult to manage in the clinic.
Scheme 1.

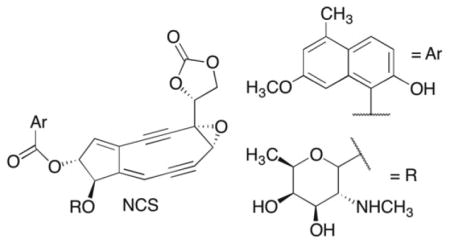
Calicheamicin's general toxicity was overcome by preparing an antibody conjugate (Mylotarg) that is used to treat acute myeloid leukemia.11 C1027 is unique in that it initiates ataxia-telangiestasia mutated (ATM) and Rad-3 related (ATR) checkpoint responses to DNA damage in cells due to its unique ability to form double strand breaks, as well as interstrand cross-links. Under hypoxic conditions such as those associated with solid tumors, C1027 induced strand breaks are repressed but formation of interstrand cross-links are stimulated (12). Perhaps of potential therapeutic relevance, it is the only known enediyne as well as ionizing radiation, that shows not decreased but enhanced cytotoxicity towards hypoxic cells (12). The novelty of C1027 mode of action provided the motivation for this study.12–14
The chemical basis for the cytotoxicity of the enediynes is often attributed to their ability to produce double strand breaks, although not all of these molecules generate high yields of double strand breaks in free (nonnucleosomal) DNA.15–18 These antitumor agents also produce bistranded DNA damage, and single strand breaks that are comprised of lesions that irreversibly inhibit repair enzymes and/or react further to produce interstrand cross-links that are converted to double strand breaks by DNA misrepair.19–26 C1027 is unusual in that it yields double strand breaks, interstrand cross-links in DNA, and DNA-drug adducts.5,6,27–29 Moreover, the formation of the former two types of damage account for initiation of the ATM and ATR pathways.12–14 Product analyses and isotopic labeling show that C1027 abstracts hydrogen atoms from the C1’, C4’, and C5’ positions of nucleotides on opposite strands in a 3 base pair region.6,28,29 Although the nucleotides at which adducts and cross-links form were identified, structural information on these products is lacking. However, it is believed that C1027 and the DNA are bonded to one another in adducts and cross-links via the deoxyribose backbone.29 This was proposed to result from hydrogen atom abstraction by the biradical, followed by reaction of the DNA radical with the reduced pharmacophore (Scheme 1).
2. Results and Discussion
2.1 Direct strand scission and alkali-labile lesion formation
2.1.1 Local sequence effects on strand damage specificity and yield
The antibiotic C1027 was reacted with the 3’-32P-labeled 158 bp HindIII/EcoRV restriction fragment obtained from pBR322. This fragment contains the same binding site identified by Goldberg as a strong cleavage site.6,28 Indeed, the 5’-d(GTTA1T)/d(ATA3A2C) sequence was the most intensely damaged in our hands as well. Strand damage formed predominantly on opposite strands at A1, A2, and A3. Using this as a lead we prepared a series of 19 bp duplex substrates (1a–1h, Table 1) from synthetic oligonucleotides to examine the flanking sequence effects and (in one instance) internal sequence (1e) on strand damage.
Table 1.
Duplex substrates used to study C1027 reactivity.

|
C1027 was reacted under aerobic conditions with the sequence taken directly from the 158 bp HindIII/EcoRV restriction fragment. The total strand damage detected by gel electrophoresis was more than 90% in each strand after piperidine treatment (1 M, 90 °C, 30 min) (1a, Tables 2 and 3). A slower moving product, identified as a DNA-drug adduct was also detected. The adduct yield was significantly greater in the bottom strand of the duplex, consistent with that previously reported.27,29 Consistent with the previous report, strand damage was detected at A11 (Table 2), A29 (Table 3), and A30 (Table 3).
Table 2.
| Duplex | Tot. Strand Damage (%) | Damage Site(s) | Yield C1027- DNA adduct (%) |
|---|---|---|---|
| 1a | 97.1 ± 0.1 | A11, G8 (4.6:1) | ~1 |
| 1b | 58.3 ± 2.3 | G8, T9, A11, A12, G13 | 4.6 ± 0.5 |
| 1c | 42.8 ± 2.5 | G8, T9, A11, G13 | 0 |
| 1d | 54.4 ± 1.2 | G8, G13 | 2.6 ± 0.2 |
| 1e | 81.3 ± 1.0 | T11 | 0 |
| 1f | 96.7 ± 0.7 | T10, A11 (1:3.0) | ~1 |
| 1g | 97.2 ± 0.7 | T10, A11 (2.5:1) | 0 |
| 1h | 95.2 ± 0.9 | A8, T10, A11 (1.3:1:8.1) | 0 |
Reactions carried out on 5'-32P-labeled DNA.
Yields are the average ± std. dev. of 3 individual reactions.
Table 3.
| Duplex | Tot. Strand Damage (%) | Damage Site(s) | Yield C1027- DNA adduct (%) |
|---|---|---|---|
| 1a | 95.0 ± 0.3 | A29, A30 (1.4:1) | 5.2 ± 0.1 |
| 1b | 46.4 ± 2.5 | T28, A29, A30, A33 | 4.3 ± 0.8 |
| 1c | 47.2 ± 2.4 | G27, T28, A29, A30, A33 | 7.0 ± 0.7 |
| 1d | 54.6 ± 3.0 | T28, A29, A30, C32, A33 | 4.7 ± 0.4 |
| 1e | 63.8 ± 3.8 | A29, A30 | 5.4 ± 0.2 |
| 1f | 92.4 ± 0.5 | A29, A30, G31 (4.0:5.9:1) | 11.1 ± 0.8 |
| 1g | 77.6 ± 0.3 | A29, A30, A31 (1:2.4:4.7) | 8.1 ± 0.8 |
| 1h | 96.5 ± 0.8 | A29, A30, T31 (3.5:6.8:1) | 4.7 ± 0.1 |
Reactions carried out on 5'-32P-labeled DNA.
Yields are the average ± std. dev. of 3 individual reactions.
Varying the N8:N31 base pair (1f – 1h) had little effect on the overall amount of strand damage. However, strand damage was distributed over a slightly greater number of nucleotides within the core pentanucleotide sequence.
In contrast, varying the N12:N27 base pair (1b – 1d) had a more severe effect on strand damage, reducing it by more than 40% compared to that in 1a (Tables 2 and 3). In addition, the damage pattern was much more diffuse, suggestive of weaker, less selective binding.
In general, the effects of O2 deprivation on strand damage were also consistent with previous reports.27,29 The amount of direct strand scission and alkali-labile lesions decreased 2–3 fold under anaerobic conditions. In the strong cleavage sequences (1a, 1e– h), strand damage was only observed at positions A29 and A30 (A30, A31 in 1g) in the bottom strand and at A11 (or T11) in the top strand. Strand damage in the weak cleavage duplexes (1b–1d) was dispersed over several nucleotides. The effect of O2 on DNA-drug adduct yield was strand dependent. Adduct formation was not observed in the top strand (nucleotides 1–19) of any duplex under anaerobic conditions. DNA-drug adduct yield in the bottom strand (nucleotides 20–38) was reduced under anaerobic conditions, but the magnitude of the effect was far more variable. For instance, while no adduct was detected from 1d, its yield was 4.7% in 1f under anaerobic conditions. The latter still represented more than a 50% reduction in yield under anaerobic conditions. In contrast, the reduction in adduct yield under anaerobic conditions was modest in 1a and 1h.
2.1.2 Direct strand breaks and alkali-labile lesion product distribution
Previously, the products at specific nucleotides formed by C1027 were identified to result from C1’, C4’, and C5’ oxidation.6,28,29 In DNA containing the 5’-GTTAT/ATAAC sequence found in 1a the nucleotides identified as A11, A29, and A30 accounted for the C4’, C1’, and C5’ oxidation products respectively.28 Minor amounts of C4’-oxidation were also detected at the position equivalent to A30.29 We employed direct strand scission and a variety of chemical reactions to identify the C4’-, C5’-oxidation, and alkali-labile products in the strong cleavage sequences (1a, 1f–1h). Direct strand breaks of 5’-32P-oligonucleotides containing 3’-phosphate and 3’-phosphoglycolate termini were attributed to C5’-and C4’-oxidation, respectively. Evidence for C5’-oxidation was qualitatively confirmed using 3’-32P-1a, which enabled the detection of the characteristic 5’-aldehyde (2). Basing the yield of 5’-oxidation on 3’-phosphate overstates the amount of this pathway due to adventitious cleavage of alkali-labile abasic lesions. However, the additional cleavage is small because only a few percent of independently generated alkali-labile lesions are cleaved in our hands.
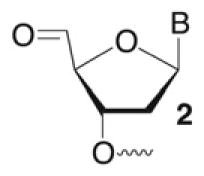
Quantitative analysis of direct strand scission (Table 4) indicated that C4’-oxidation was the dominant reactive pathway in the top strand of the strongly cleaved sequences (1a, 1f–h).28 No more than ~1.5% direct strand scission from C5’-oxidation was observed (data not shown). However, C1027 was not as selective with respect to direct strand break formation in the bottom strand (Table 5). Although C5’-oxidation was the major path to direct strand scission in the bottom strand of 1a and 1f–h, 3’-phosphoglycolates were formed in varying amounts, indicating that hydrogen atom abstraction from the C4’-position also occurred.
Table 4.
C1027 strand damage distribution (% yield) under aerobic conditions in strands containing nucleotides 1–19.a,b
| Duplex (position) | Direct Strand Break | C4-AP | L |
|---|---|---|---|
| 1a (A11) | 37.3 ± 1.5 | 12.5 ± 1.7 | 10.8 ± 2.6 |
| 1f (T10) | 2.9 ± 0.4 | 6.3 ± 0.6 | 4.2 ± 0.6 |
| 1f (A11) | 20.9 ± 1.6 | 9.4 ± 2.6 | 10.5 ± 0.8 |
| 1g (T10) | 16.2 ± 0.6 | 13.1 ± 0.7 | 15.2 ± 6.2 |
| 1g (A11) | 17.0 ± 0.2 | 7.4 ± 0.8 | 6.2 ± 1.4 |
| 1h (A11) | 44.1 ± 2.0 | 11.2 ± 2.5 | 6.7 ± 1.4 |
Reactions carried out on 3'-32P-labeled DNA.
Yields are the average ± std. dev. of 3 individual reactions.
Table 5.
C1027 strand damage distribution (% yield) under aerobic conditions in strands containing nucleotides 20–38.a,b
| Duplex (position) | Direct Strand Break | L | |
|---|---|---|---|
| 3'-Phosphate | 3'-Phosphoglycolate | ||
| 1a (A29) | - | - | 38.7 ± 1 |
| 1a (A30) | 46.0 ± 0.9 | 5.5 ± 0.6 | - |
| 1f (A29) | - | - | 30.2 ± 4.6 |
| 1f (A30) | 33.8 ± 1.5 | 8.2 ± 0.3 | - |
| 1g (A29) | - | - | 9.2 ± 0.4 |
| 1g (A30) | 18.0 ± 1.0 | 5.8 ± 1.0 | 4.6 ± 2.0 |
| 1g (A31) | 51.7 ± 0.9 | 3.3 ± 0.1 | - |
| 1h (A29) | - | - | 38.2 ± 2.6 |
| 1h (A30) | 37.8 ± 0.8 | 16.8 ± 1.3 | - |
Reactions carried out on 5'-32P-labeled DNA.
Yields are the average ± std. dev. of 3 individual reactions.
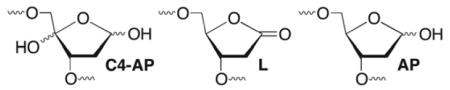
The distribution of alkali-labile lesions demonstrated that the promiscuity of C1027 mediated damage was greater than previously thought.28,29 Established chemical reagents were utilized to determine the distribution of alkali-labile lesions (Table 6). Qualitative evidence for 2-deoxyribonolactone (L) at A29 in 1a was obtained using a series of fingerprint reactions, but was inconvenient for quantitative purposes.28,30 Instead, L and C4-AP were analyzed for quantitatively by taking advantage of their complimentary reactivity with methoxyamine and hydrazine (Table 6). C4-AP is cleaved by treatment with hydrazine.31 Unlike C4-AP, which contains an aldehyde, 2-deoxyribonolactone (L) is not protected from NaOH induced cleavage by prior reaction with methoxyamine. L was previously shown to not react with such reagents.32
Table 6.
Complimentary reactivity of abasic lesions.
| Cleavage conditions | |||
|---|---|---|---|
| CH3ONH2/NaOH | Hydrazine | NaOH | |
| AP | − | − | + |
| C4-AP | − | + | + |
| L | + | − | + |
The selectivity for 2-deoxyribonolactone at A29 in 1a, 1f, and 1h was consistent with previous data from similar sequences (Table 5).28 However, C1’-oxidation also occurred at A30 in 1g, where C4’- and C5’-damage were detected. A more diverse set of damage products was detected in the top strand of 1a and 1f–h (Table 4). The formation of C4-AP was not surprising, as analysis of the direct strand scission products had already established that C4’-oxidation was a major contributor to damage at the N10 and N11 positions. However, the formation of L resulting from C1’-hydrogen atom abstraction was unexpected and it was observed in as high as 15.2% yield.
The sum of the individual yields of L and C4-AP determined as described above were consistently less than the total amount of NaOH labile lesions. The shortfall ranged between 3.9 and 7.6%. Although this could be due to errors in the individual experiments, an alternative explanation is that a third lesion is formed. AP sites would have the appropriate reactivity. Unlike L and C4-AP formation, the mechanism of AP formation would not involve hydrogen atom abstraction from the deoxyribose backbone. A more likely source of AP would be reaction of the drug with the corresponding nucleobase, followed by deglycosylation. Direct evidence for this process is lacking. However, it is consistent with the postulated structure of interstrand cross-links (see below). Overall, with or without AP, we find that C1027 reacts more promiscuously in some sequences than previously believed (Figure 1).
Figure 1.

C1027 reactivity distribution with respect to duplex and nucleotide (A) Strands containing nucleotides 1–19 (B) Strands containing nucleotides 20–38.
2.2 C1027-DNA interstrand cross-link formation
2.2.1 Interstrand cross-link yield and location
Interstrand cross-links (ICLs) were detected only under anaerobic conditions. Their quantities generally correlated with overall damage yields under aerobic conditions. Cross-link yields in the weakly damaged sequences (1b–d) ranged from 10.7 – 20.8% and were not characterized further. More strongly damaged sequences (1a, 1e–h) yielded cross-links in as high as 57.6% (Table 7). As mentioned earlier (Section 2.1.1), ICLs form at the expense of single strand lesions, including C1027-DNA adducts.
Table 7.
C1027-DNA interstrand cross-link formation
| Duplex (local sequence) | ICL Yield (%)a | Nucleotides Cross-linked | |
|---|---|---|---|
| 1a | 5’-G8 T9 T10 A11-T12
3’-C31 A30 A29 T28 A27 |
36.0 ±16.1 | A11, ,A30; A11, A29(~4:1) |
| 1e | 5’-G8 T9 T10 T11 T12 3’-C31 A30 A29 A18 A27 |
19.1b | T11. A30 |
| 1f | 5’- C8 T9 T10 A11 T12 3’-G31 A30 A29 T28 A27 |
57.6 ±0.1 | A11, A30 |
| 1g | 5’-T8 T9 T10 A11 T12 3’-A31 A30 A29 T28 A27 |
51.9 ±13.0 | T10, A31 |
| 1h | 5’-A8 T9 T10 A11 T12 3’-T31 A30 A29 T29 A27 |
38.9 ± 7.9 | A11 ,A30 |
Yields are the average of 3 measurements.
Single measurement.
Although we were unable to characterize the ICLs using mass spectrometry, the location of cross-linking on each strand was determined using hydroxyl radical cleavage.19,33,34 With one exception, 1a, a single pair of nucleotides was cross-linked in each duplex, and the nucleotides involved spanned 3 base pairs.27,29 In the strand containing two or more consecutive dA’s, cross-linking occurred at the 3’-terminal dA in this sequence, with the exception of the aforementioned minor product from 1a. However, the 3’-terminal dA was cross-linked in the major ICL of 1a.
2.2.2 Interstrand cross-link stability
C1027-DNA ICL formation has been proposed to involve a rebound mechanism in which the (bi)radical drug abstracts two hydrogen atoms sequentially from the deoxyribose backbone. Covalent bond formation results from reaction between the respective sugar radical and the reduced drug, presumably via addition into one of the π-bonds in C1027. Despite the importance of the ICL product(s), other than mass data confirming its molecular weight, structural information is unavailable. We attempted to gain more information by analyzing enzyme-digested material using MS/MS but were unsuccessful. Hence, we subjected the ICL from 1a and 1g to a simple set of chemical reactions. The reactivity of the ICL with acid (citrate) or base (piperidine) varied depending upon whether we utilized product that was isolated by denaturing polyacrylamide gel electrophoresis or carried out reactions on crude product mixtures.
Isolated cross-link from 1a and 1g was partially decomposed by piperidine treatment (1 M, 90 °C, 30 min). Approximately 20% of the ICL was decomposed by this treatment. When the bottom strand of 1a (containing nucleotides 20–38) was 5’-32P-labeled, the major decomposition product observed comigrated with the C1027-DNA adduct (Table 8), suggesting that the labeled oligonucleotide was uncleaved and at least a portion of the drug remained covalently bound. A minor amount of strand scission at the cross-linked nucleotide (A30) was observed. In contrast, strand scission was the major result in the top strand of 1a following piperidine treatment. Cross-link isolated from 1g showed the opposite preferences. Adduct was the major product formed in the strand containing nucleotides 1–19, but strand scission was preferred in the complementary strand. A significant difference between 1a and 1g is that the strand in the latter which retains the drug adduct contains a thymidine at the modified position. The reactivity of the isolated cross-link was unaffected by incubation with citrate buffer (pH 4.6, 10 mM, 70 °C, 3 h) prior to reacting it with hot piperidine. In contrast, crude ICLs from either duplex were completely destroyed by incubation in acidic citrate buffer. The crude interstrand cross-link was also more labile than isolated material to direct piperidine treatment.
Table 8.
Decomposition product distribution from piperidine treated isolated interstrand cross-link.a
| Duplex | Nucleotide | % Strand Scission | % Adduct |
|---|---|---|---|
| 1a | A11 | 17.0 ± 2.4 | 5.2 ± 0.5 |
| 1a | A30 | 6.6 ± 0.3 | 11.3 ± 0.5 |
| 1g | T10 | 2.9 ± 0.5 | 11.3 ± 0.5 |
| 1g | A31 | 7.4 ± 0.5 | 3.1 ± 0.1 |
Yields are the average ± std. dev. of 3 individual reactions.
2.2.3 A proposal for the general structure of C1027-DNA cross-links
The reactivity of isolated and crude ICLs with piperidine and citrate provides inferential information as to how the enediyne chromophore is bonded to the DNA. The increased stability of gel purified ICL compared to crude material suggests that the material undergoes a chemical transformation during isolation. We suggest that at least some of the cross-links formed from 1a the top strand is bonded to the C1027 chromophore via its nucleobase and that the initially formed product undergoes further reaction during cross-link purification. The transformation stabilizes the glycosidic bond, explaining why the isolated ICL is more stable to citrate than unpurified product. Piperidine treatment of nucleobase modified DNA would be expected to result in strand scission at that site (e.g. A11). Since C1027 binds in the minor groove, the drug likely reacts with N1 or N3. There is precedent for addition products at these positions to be alkaline labile and to react with water to produce molecules that are more stable to acidic conditions (e.g. citrate buffer) but that are still somewhat labile to piperidine.34,36–40 We suggest that the drug (bi)radical adds directly to the nucleobase. This reactivity pattern helps explain why the strands of the cross-linked product from 1g behave differently. Nucleotide T10 in the top strand of 1g yields mostly adduct upon piperidine treatment and strand scission is preferred on the bottom strand (Table 8). The thymine ring does not offer C1027 a site to react in the minor groove. Consequently, the drug adds to A31 in the bottom strand.
Nucleotides alkylated (or arylated) on their sugar are not usually alkali-labile. Hence, we propose that the major pathway for cross-linking to the bottom strand of 1a (A30) or top strand of 1g (T10) involves carbon-carbon bond formation between the sugar and the drug chromophore. Decomposition of the ICL yields an intact bottom strand containing at least a part of C1027. Bond formation could occur via a rebound mechanism in which the activated C1027 abstracts a hydrogen atom from the deoxyribose, and this radical adds to a π-bond of the drug.
The π-bond that reacts with the deoxyribose radical need not be from the six membered ring formed upon Bergman cyclization (C2-C7). Inspection of molecular models indicates that formation of two covalent bonds to this ring with opposing DNA strands would require severe distortion of the duplex and is unlikely. The cross-linked products do not migrate anomalously in denaturing gels compared to size markers, which is consistent with a nondistorted structure. The C1027 chromophore could more readily span the duplex minor groove if it utilized other π-bonds, such as those in the cyclopentadiene ring to react with the DNA sugar radical. Alternatively, the chlorinated phenol ring could also undergo radical addition. NMR analysis of the reduced C1027 chromophore bound to DNA indicates that the macrocyclic domain containing the chlorinated phenol ring does not interact with DNA.35 However, that does not preclude the phenol ring being involved in ICL formation. Invoking the phenol ring in cross-linking is attractive because the deshydroxy and deschloro analogues of C1027 do not yield cross-linked DNA under oxygen conditions where they are readily observed for C-1027.13 One possibility is that these analogues are less reactive with the deoxyribose radical. Another explanation is that the deshydroxy and deschloro analogues adopt conformations that are not conducive to cross-linking.
3. Conclusions
Examining DNA cleavage by C1027 largely reaffirms the distribution of oxidized sugar damage that leads to direct strand scission and alkali labile lesions. However, strand damage is more chemically diverse than previously thought, in particular with respect to the formation of 2-deoxyribonolactone. In addition, small amounts of AP sites may also form. Subtle changes in DNA structure induced in and around the binding region may cause changes in drug binding, which in turns alters accessibility of individual hydrogen atoms. Examination of the reactivity of interstrand cross-links, a unique product produced by C1027 amongst the enediyne family of products, indicates that at least some of these products involve covalent modification of one strand via its deoxyribose backbone and the other via a nucleobase. Detailed structural characterization of the cross-linked products is desirable and could shed light on the effects of substituents on the ability of C1027 analogues to form such products.
4. Experimental
4.1 General Methods
Oligonucleotides were synthesized on an Applied Biosystems Incorporated 394 oligonucleotide synthesizer. Commercially available oligonucleotide synthesis reagents were purchased from Glen Research (Sterling, VA). T4 polynucleotide kinase and terminal deoxynucleotide transferase were obtained from New England Biolabs. 32P-labeled nucleotide triphosphates were purchased from Perkin Elmer. C18-Sep-Pak cartridges were obtained from Waters. Quantification of radiolabeled oligonucleotides was carried out using a Molecular Dynamics Phosphorimager 840 equipped with ImageQuant TL software. DNA was radiolabeled at the 5’-terminus with γ-32P-ATP (45 min, 37 °C) using T4 polynucleotide kinase in the supplied buffer (70 mM Tris-HCl, 10 mM MgCl2, 5 mM dithiothreitol, pH 7.6). Oligonucleotides were radiolabeled at their 3’-termini with α-32P-dATP (cordycepin) (45 min, 37 °C) using terminal transferase in the reaction buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM dithiothreitol, 0.25 mM CoCl2 pH 7.9).
4.2 Preparation of 3'-labeled restriction fragments
A 100 μL reaction containing pBR322 plasmid DNA (~10–15 μg), NEB buffer 2 (10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, pH 7.9), HindIII (3 μL, 20,000 U/mL, 60 units) and EcoRV (1 μL, 20,000 U/mL, 20 units) restriction enzymes were incubated at 37 °C for 1 h. Xylene cyanol dye was added to the reaction and ran down on a 1.8% agarose gel to separate fragments. The band corresponding to the 158 bp fragment was excised and purified using a QIAquick Gel Extraction Kit (Qiagen) and eluted with 30 μL of the provided elution buffer. The 3’-labeling was carried out in 100 μL volume in NEB buffer 2; 0.5 mM of dTTP, dATP and dGTP; 1 μL large Klenow fragment and 32P-α-dCTP. This reaction was incubated at room temperature for 20 min, then 5 μL of 100 μM dNTP mixture was added and the solution further incubated at room temperature for 10 min. The solution was desalted using a Sephadex G-50 Spin column, and the enzyme removed by phenol extraction (2x), followed by precipitation (0.3 M NaOAc, pH 5.2). A 1 μL aliquot was analyzed by 5% nondenaturing PAGE (with an appropriate DNA ladder) to check the purity of the radiolabeled restriction fragment.
4.3 Aerobic reactions of DNA with C1027
A typical drug reaction consisted of 0.4 μM of DNA and 10 μM C1027 (10 mM sodium phosphate buffer, pH 7.2, 3 h, 37 °C) for aerobic experiments.
4.4 Anaerobic reactions of DNA with C1027
Experiments carried out under anaerobic conditions were incubated with the drug overnight, at 37 °C in the buffer described above. The anaerobic reactions were prepared using a glass vessel with 2 separate chambers for the drug and the DNA solution (10 mM sodium phosphate buffer, pH 7.2) (Supporting Information Figure 1) Solutions were degassed five times by the freeze-pump-thaw-method using a high vacuum line. After degassing, the tubes were inverted, centrifuged to force liquids down and vortexed. The yields of the ICLs were quantified by adding formamide loading buffer to an aliquot, separating products by 20% denaturing PAGE, and detected via phosphor imaging analysis.
4.5 Identification of lesions at individual sites
Aliquots from the DNA-drug reaction were taken and treated with 0.1 M NaOH (30 min, 37 °C), 100 mM hydrazine hydrochloride (pH 8, 1 h, 25 °C), or 5 mM methoxyamine (pH of aliquot is adjusted with 10 mM sodium phosphate buffer (pH 7.2), 30 min, room temp.). One half of the solutions reacted with methoxyamine and hydrazine were subsequently treated to NaOH (0.1 M, 30 min, 37 °C; then quench with 0.1 M HCl). An equal volume of formamide loading buffer was added to the samples and products were separated by 20% denaturing PAGE, and quantified by phosphor imaging analysis.
4.6 Determination of ICL connectivity using the hydroxyl radical cleavage reaction
Duplex DNA previously incubated with C1027 (anaerobic conditions, overnight, 37 °C) was concentrated to dryness, resuspended in formamide loading buffer, and purified by 20% (0.4 mm thickness) denaturing PAGE. The bands corresponding to the ICL products were carefully excised from the gel, crushed, and eluted in ~500 μL of elution buffer (1 mM EDTA, 100 mM NaCl) overnight at room temperature. The solution was filtered using a 10 mL Polyprep Column (Biorad) and desalted using a C18 column (100 mg Sep-Pak Vac-Pak, Waters). After concentration, a portion of the DNA was dissolved in 8 μL of water and mixed with 10 μL of the 2x oxidation buffer (20 mM NaCl, 20 mM sodium phosphate, pH 7.2, 2 mM sodium ascorbate, and 1 mM hydrogen peroxide). To initiate the reaction, 2 μL of the Fe•EDTA solution (1 mM EDTA and 0.5 mM Fe(NH4)2(SO4)2•6H2O) was added, and the reaction was allowed to proceed for no longer than 5 min. The reaction was quenched with 10 μL of 100 mM thiourea and concentrated in vacuo. The DNA was resuspended in formamide loading buffer, the products were separated by 20% denaturing PAGE, and analyzed by phosphor imaging.
4.7 Depurination of DNA
Crude or isolated ICLs were subjected to depurination conditions (100 mM sodium citrate, 100 mM NaH2PO4 and 100 mM NaCl, pH 4.6, 4 h, 70 °C). Acid treated DNA was then either treated with NaOH (0.1 M, 30 min, 37 °C) or piperidine (1 M, 30 min, 90 °C). Piperidine treated samples were concentrated to dryness in vacuo to remove piperidine, and then lyophilized to dryness 2x from water (50 μL). Control DNA (crude or isolated ICL) were also heated (4 h, 70 °C or 30 min, 90 °C) without prior treatment with base or acid.
4.8 Stability tests for crude and isolated ICLs
Crude ICLs (obtained without prior purification of anaerobic DNA-C1027 solutions) or isolated ICLs were subjected to heat or base treatments. Samples were heated (30 min, 90 °C), treated with NaOH (0.1 M, 30 min, 37 °C) or piperidine (1 M, 30 min, 90 °C). NaOH treated samples were neutralized with 0.1 M HCl. Piperidine treated samples were concentrated to dryness in vacuo to remove piperidine, and then lyophilized to dryness 2x from water (50 μL). The DNA was resuspended in formamide loading buffer, the products were separated by 20% denaturing PAGE, and analyzed by phosphor imaging.
Supplementary Material
Acknowledgments
We are grateful for support of this research from the National Institute of General Medical Sciences (GM-054996) and National Cancer Institute research grant CA106312 and National Cancer Institute Center Support grant CA16056 (T. Beerman). C1027 was a generous gift from Taiho Pharmaceutical CO.
Footnotes
Image of tube used for degassing samples. Autoradiograms showing effects of acid/base treatment on crude and isolated ICLs from 1a and 1b. Representative autoradiogram of hydroxyl radical cleavage of ICL
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xi Z, Goldberg IH. In: Comprehensive Natural Products Chemistry. Kool ET, editor. Vol. 7. Elsevier; Amsterdam: 1999. pp. 553–592. [Google Scholar]
- 2.Bergman RG. Acc Chem Res. 1973;6:25–31. [Google Scholar]
- 3.Goldberg IH. Acc Chem Res. 1991;24:191–198. [Google Scholar]
- 4.Usuki T, Mita T, Lear MJ, Das P, Yoshimura F, Inoue M, Hirama M, Akiyama K, Tero-Kubota S. Angew Chem Int Ed. 2004;43:5249–5253. doi: 10.1002/anie.200454133. [DOI] [PubMed] [Google Scholar]
- 5.Sugiura Y, Matsumoto T. Biochemistry. 1993;32:5548–53. doi: 10.1021/bi00072a008. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Zhen Y, Goldberg IH. Biochemistry. 1994;33:5947–5954. doi: 10.1021/bi00185a036. [DOI] [PubMed] [Google Scholar]
- 7.Hirama M, Akiyama K, Tanaka T, Noda T, Iida K-i, Sato I, Hanaishi R, Fukuda-Ishisaka S, Ishiguro M, Otani T, Leet JE. J Am Chem Soc. 2000;122:720–721. [Google Scholar]
- 8.Xu Y, Li D, Zhen Y. Cancer Chemother Pharmacol. 1990;27:41–46. doi: 10.1007/BF00689274. [DOI] [PubMed] [Google Scholar]
- 9.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739–758. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 10.Zhen Y, Ming X, Yu B, Otani T, Saito H, Yamada Y. J Antibiot. 1989;42:1294–1298. doi: 10.7164/antibiotics.42.1294. [DOI] [PubMed] [Google Scholar]
- 11.Sievers EL, Appelbaum FR, Spielberger RT, Forman SJ, Flowers D, Smith FO, Shannon-Dorcy K, Berger MS, Bernstein ID. Blood. 1999;93:3678–3684. [PubMed] [Google Scholar]
- 12.Beerman TA, Gawron LS, Shin S, Shen B, McHught MM. Cancer Research. 2009;69:593–598. doi: 10.1158/0008-5472.CAN-08-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy DR, Lu J, Shen B, Beerman TA. Proc Nat Acad Sci USA. 2007;104:17632–17637. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy DR, Gawron LS, Ju J, Liu W, Shen B, Beerman TA. Cancer Res. 2007;67:773–781. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]
- 15.Dedon PC, Salzberg AA, Xu J. Biochemistry. 1993;32:3617–22. doi: 10.1021/bi00065a013. [DOI] [PubMed] [Google Scholar]
- 16.Kishikawa H, Jiang YP, Goodisman J, Dabrowiak JC. J Am Chem Soc. 1991;113:5434–5440. [Google Scholar]
- 17.Epstein JL, Zhang X, Doss GA, Liesch JM, Krishnan B, Stubbe J, Kozarich JW. J Am Chem Soc. 1997;119:6731–6738. [Google Scholar]
- 18.Meschwitz SM, Schultz RG, Ashley GW, Goldberg IH. Biochemistry. 1992;31:9117–9121. doi: 10.1021/bi00153a001. [DOI] [PubMed] [Google Scholar]
- 19.Sczepanski JT, Jacobs AC, Greenberg MM. J Am Chem Soc. 2008;130:9646–9647. doi: 10.1021/ja8030642. [DOI] [PubMed] [Google Scholar]
- 20.Sczepanski JT, Jacobs AC, Majumdar A, Greenberg MM. J Am Chem Soc. 2009;131:11132–11139. doi: 10.1021/ja903404v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sczepanski JT, Jacobs AC, Van Houten B, Greenberg MM. Biochemistry. 2009;48:7565–7567. doi: 10.1021/bi901006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan L, Greenberg MM. J Am Chem Soc. 2009;131:15225–15231. doi: 10.1021/ja9061695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guan L, Greenberg MM. J Am Chem Soc. 2010;132:5004–5005. doi: 10.1021/ja101372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan LR, Bebenek K, Kunkel TA, Greenberg MM. Biochemistry. 2010;49:9904–9910. doi: 10.1021/bi101533a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Biochemistry. 2003;42:2449–2455. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto M, Greenberg MM, Kow YW, Hwang JT, Cunningham RP. J Am Chem Soc. 2001;123:3161–3162. doi: 10.1021/ja003354z. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Zhen Y, Goldberg IH. J Am Chem Soc. 1997;119:1133–1134. [Google Scholar]
- 28.Xu Y, Xi Z, Zhen Y, Goldberg IH. Biochemistry. 1995;34:12451–12460. doi: 10.1021/bi00038a044. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y-j, Xi Z, Zhen Y-s, Goldberg IH. Biochemistry. 1997;36:14975–14984. doi: 10.1021/bi972101o. [DOI] [PubMed] [Google Scholar]
- 30.Hwang JT, Tallman KA, Greenberg MM. Nucleic Acids Res. 1999;27:3805–3810. doi: 10.1093/nar/27.19.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugiyama H, Kawabata H, Fujiwara T, Dannoue Y, Saito I. J Am Chem Soc. 1990;112:5252–5257. [Google Scholar]
- 32.Sato K, Greenberg MM. J Am Chem Soc. 2005;127:2806–2807. doi: 10.1021/ja0426185. [DOI] [PubMed] [Google Scholar]
- 33.Millard JT, Weidner MF, Kirchner JJ, Ribeiro S, Hopkins PB. Nucleic Acids Res. 1991;19:1885–1892. doi: 10.1093/nar/19.8.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 35.Okuno Y, Iwashita T, Sugiura Y. J Am Chem Soc. 2000;122:6848–6854. [Google Scholar]
- 36.Veldhuyzen WF, Shallop AJ, Jones RA, Rokita SE. J Am Chem Soc. 2001;123:11126–11132. doi: 10.1021/ja011686d. [DOI] [PubMed] [Google Scholar]
- 37.Veldhuyzen WF, Parde P, Rokita SE. J Am Chem Soc. 2003;125:14005–14013. doi: 10.1021/ja036943o. [DOI] [PubMed] [Google Scholar]
- 38.Peng X, Hong IS, Li H, Seidman MM, Greenberg MM. J Am Chem Soc. 2008;130:10299–10306. doi: 10.1021/ja802177u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding H, Majumdar A, Tolman JR, Greenberg MM. J Am Chem Soc. 2008;130:17981–17987. doi: 10.1021/ja807845n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. J Am Chem Soc. 2002;124:3263–3269. doi: 10.1021/ja012135q. [DOI] [PubMed] [Google Scholar]
- 41.Regulus P, Duroux B, Bayle PA, Favier A, Cadet J, Ravanat JL. Proc Nat Acad Sci USA. 2007;104:14032–14037. doi: 10.1073/pnas.0706044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.