

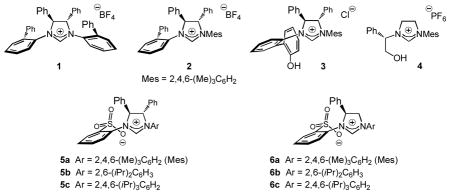
Catalytic enantioselective allylic substitution (EAS) reactions are of prominent utility in chemical synthesis.[i] Such transformations generate a stereogenic center adjacent to an alkene, which can be further functionalized to afford a range of other enantiomerically enriched molecules. Despite noteworthy advances, several issues remain unresolved in this area. Nearly all the existing protocols require nucleophilic metal-based reagents, which, while often the source of high reactivity, are not well suited when certain commonly occurring functional groups are present. Moreover, whereas significant focus has been placed on additions of alkyl groups,[i] reported examples of catalytic processes that lead to incorporation of aryl or heteroaryl,[ii] allyl,[iii] alkynyl[iv] or allenyl[v] moieties are markedly small in number.
The corresponding reactions resulting in additions of vinyl units have received attention only recently. Efforts in these laboratories have led to the development of catalytic EAS with vinylmetals, generated through hydroaluminations of alkynes[vi] (with di-iso-butylaluminum hydride) and used in situ for site-and enantioselective C–C bond formation.[vii] Nonetheless, vinylaluminums with a heteroatom substituent at their allylic position cannot be efficiently prepared by hydrometallation.[viii] The above shortcoming is underlined by the sequence in Scheme 1, regarding a projected enantioselective synthesis of Pummerer ketone,[ix] an intermediate in morphine biosynthesis.[x] The key step would ideally involve EAS with a carbonyl-substituted reagent, leading to the formation of an α,β-unsaturated ester that could then be subjected to a diastereoselective intramolecular conjugate addition. The analogous vinylaluminums are inaccessible. In contrast, the corresponding vinylborons are prepared readily; however, an efficient method for catalytic EAS involving such entities does not exist. Such protocols would notably enhance the general utility of this important class of transformations.
Scheme 1.

A route for enantioselective synthesis of Pummerer ketone might involve catalytic allylic substitution with a carbonyl-containing vinyl group followed by a catalytic diastereoselective intramolecular conjugate addition and catalytic ring-closing metathesis. pg = protecting group.
Herein, we outline a method for catalytic EAS reactions that proceed with vinylborons,[xi] including those that contain a carboxylic ester or an acetal unit; reagents are either purchased or can be prepared by Cu-catalyzed processes.[xii] The couplings are promoted by sulfonate-bridged bidentate N-heterocyclic carbene (NHC) complexes of copper and generate all-carbon quaternary stereogenic centers.[xiii] The desired products, including those with an ester, an aldehyde or acetal group, are formed in up to 98% yield, >98% SN2′ selectivity and >98:2 enantiomeric ratio (e.r.). Utility is demonstrated by concise enantioselective syntheses of Pummerer ketone as well as its hitherto undisclosed anti isomer, where diastereoselective intramolecular conjugate additions, controlled by cinchona alkaloid catalysts, and Ru-catalyzed ring-closing metathesis complement the NHC–Cu-catalyzed EAS process.
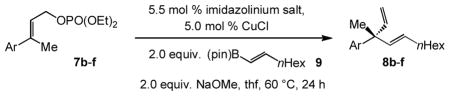
We initiated our investigations by probing the ability of different NHC–Cu complexes to promote reaction between commercially available n-hexyl(pinacolato)vinylboron 9 and o-methoxyphenyl- containing allylic phosphate 7a; the expected product (8a) contains an ortho-alkoxy-substituted aryl unit, as required in the proposed approach to enantioselective synthesis of Pummerer ketones (cf. Scheme 1). The presence of NaOMe is to assist the formation of the NHC–Cu-vinyl intermediate (via the methoxy-boronate).[xiv] Preliminary studies indicated that the transformation is sluggish at ambient temperature (≤ 30% conv. with various catalysts). Therefore, in contrast to Cu-catalyzed reactions with the related allenylboron,[v] which proceed to completion at 22 °C, it appeared that EAS with the relatively more hindered vinylboron demands more forcing conditions. Whether high site- and enantioselectivity would be achievable under the latter circumstances remained to be established.
As illustrated in entries 1–4 of Table 1, there is reasonable efficiency (58–92% conv., ≤ 60% yield) and low to appreciable site and enantioselectivity with monodentate and aryloxy-or alkoxy-bridged NHC–Cu complexes (80–93% SN2′ and up to 80:20 e.r.). Conversely, as depicted in entries 5–10 of Table 1, with sulfonate-bridged imidazolinium salts[xv] 5a-c and 6a-c as catalyst precursors, transformations are efficient, generating divinyl-substituted quaternary carbon stereogenic centers with nearly complete site and enantioselectivity (up to 95% yield, >98% SN2′ and >98:2 e.r.).[xvi]
Table 1.
Initial examination of various chiral NHC–Cu complexes.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Imidazolinium salt | Conv. [%][b] | Yield [%][c] | SN2′:SN2[b] | e.r.[d] |
| 1 | 1 | 87 | 51 | 86:14 | 80:20 |
| 2 | 2 | 92 | 59 | 87:13 | 73:27 |
| 3 | 3 | 58 | 12 | 80:20 | n.d.[e] |
| 4 | 4 | 89 | 60 | 93:7 | 52:48 |
| 5 | 5a | >98 | 95 | 98:2 | 90:10 |
| 6 | 5b | >98 | 90 | 98:2 | 97:3 |
| 7 | 5c | >98 | 91 | >98:2 | >98:2 |
| 8 | 6a | 95 | 87 | >98:2 | 73:27 |
| 9 | 6b | >98 | 94 | 98:2 | >98:2 |
| 10 | 6c | >98 | 86 | 98:2 | >98:2 |
Reactions were performed under N2 atmosphere.
Determined through analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yield of isolated and purified products.
Determined by HPLC analysis (±2%); see the Supporting Information for details.
Enantioselectivity not determined due to low yield of isolated product (12%). n.d. = not determined.
Catalytic EAS with vinylboron 9 can be performed with various aryl-substituted allylic phosphates; representative cases are shown in Table 2. Reactions with substrates that contain an ortho aryl group, although somewhat less facile (82–87% conv. in entries 1–3 vs. >98% conv. in entries 4–5), are generally more enantioselective (87:13 to >98:2 e.r. vs. 84:16–86:14 e.r.). There is high site selectivity in favor of the branched 1,4-diene isomer in every case (96–98% SN2′).[xvii]
Table 2.
Site- and enantioselective NHCŠCu-catalyzed EAS with Ar-substituted substrates and alkenylboron 9.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate [Ar] | Imidazolinium salt | Conv. [%][b] | Yield [%][c] | SN2′:SN2[b] | e.r.[d] |
| 1 | 7b [oMeC6H4] | 6b | 87 | 82 | 96:4 | 92:8 |
| 2 | 7c [oBrC6H4] | 6b | 87 | 85 | 98:2 | >98:2 |
| 3 | 7d [oNO2C6H4] | 5a | 82 | 50 | 98:2 | 87:13 |
| 4 | 7e [C6H5] | 6c | >98 | 90 | 98:2 | 86:14 |
| 5 | 7f [pClC6H4] | 6b | >98 | >98 | 98:2 | 84:16 |
Reactions were performed under N2 atmosphere.
Determined through analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yields of isolated purified products (±5%).
Determined by HPLC analysis (±2%); see the Supporting Information for details.
The examples provided in Scheme 2 illustrate that the NHC–Cu-catalyzed EAS can be performed with different robust vinylborons and allylic phosphates, including those that contain an alkyl or a carboxylic ester unit. Thus, (pinacolato)vinylborons that carry a halogen atom (cf. 8g) or an aryl group (cf. 8h, 10 and 11) can be utilized. As before, high efficiency (75–98% yield) and exceptional site selectivity (>98% SN2′) is observed, and allylic phosphates with ortho-substituted aryls are converted to products of higher enantiomeric purity (>98:2 vs. 85:15–87:13 e.r.).
Scheme 2.

Products of NHC–Cu-catalyzed EAS of various trisubstituted allylic phosphates and different readily accessible vinylborons. All reactions were performed with 5.5 mol % 6b; see Table 2 for conditions and the Supporting Information for details).
The transformations discussed up to this point establish the feasibility of using a readily accessible and robust class of boron-based reagents. Next, we turned our attention to examining C–C bond forming processes where use of a vinylmetal is not possible. With ester-substituted vinylboron 13, as presented in Scheme 3, NHC–Cu-catalyzed EAS furnishes the desired product with high selectivity. Allylic substitutions proceed in >98% SN2′ selectivity and 96:4 to >98:2 e.r. (Scheme 3)[xviii] and the desired products are obtained in 51–69% yield. It is possible that slower rate of reaction (80 °C required vs. 60 °C for alkyl-substituted vinylborons) and moderate yields are because the electron withdrawing ester substituent diminishes the facility of the addition of the vinylcuprate to the allylic phosphate, which is accompanied by the conversion of the Cu(I) complex to a Cu(III) intermediate; subsequent reductive elimination generates the C–C bond.[v,viic]
Scheme 3.

NHC–Cu-catalyzed EAS with allylic phosphates and carboxylic ester containing vinylboron 13.
In spite of the appreciable reactivity and exceptional site- and enantioselectivity in the latter processes, we decided to search for a more efficient protocol for obtaining products with carbonyl-containing vinyl units. We were especially interested in identifying a higher yielding approach to the enantioselective preparation of α,β-unsaturated ester 14, to be incorporated in the projected Pummerer ketones synthesis (cf. Scheme 1). We accordingly explored the possibility of EAS with another commercially available vinylboron: the acetal-containing 16 (Scheme 4). Based on the abovementioned electronic effects regarding the slower rate of transformations with vinylboron 13, we envisioned that catalytic EAS with 16, which carries a less electron-withdrawing substituent, would be more facile and efficient. Reaction of allylic phosphate 7a with 16 in combination with 5.5 mol % imidazolinium salt 6b, under otherwise identical conditions (see above), followed by treatment of the mixture with a suspension of silica gel for one hour (22 °C) delivers enal 15a in 88% yield and >98:2 e.r. (<2% SN2 addition). Other examples in Scheme 4 (15b-c and 17–18) demonstrate the generality of the approach. Use of organoboron 16 offers the attractive combination of furnishing the desired products in higher yields (vs. 13), while allowing access to the more reactive α,β-unsaturated aldehydes by a simple hydrolytic procedure. The enantiomerically enriched acetals can be accessed through chromatography with silica gel neutralized with Et3N (3% by volume);[xix] for example, the precursor to 15a is isolated in 86% yield and >98:2 e.r. The vinylaluminums corresponding to 16 or its derived aldehyde cannot be prepared by hydroalumination procedures.
Scheme 4.

NHC–Cu-catalyzed EAS of allylic phosphates with acetal-containing alkenylboron 16.
In connection with the synthesis of Pummerer ketones, treatment of allylic phosphate 19 with the aforementioned conditions delivers unsaturated aldehyde 20 in 77% yield, >98% SN2′ selectivity and 98:2 e.r. Subjection of the enal to oxone in MeOH (22 °C, 18 h) delivers the methyl ester concomitant with the removal of the methoxymethyl (MOM) group, affording 21 in 83% yield.[xx] The two-step sequence that commences with allylic phosphate 19 and culminates in unprotected phenol 21 in high enantiomeric purity proceeds in 64% overall yield [vs. 51% yield of methoxymethyl (MOM) ether 14 in Scheme 3].
With an efficient route for enantioselective synthesis of α,β-unsaturated ester 21 outlined, we explored the possibility of converting the EAS-derived product to the desired benzofuran in a diastereoselective manner (cf. Scheme 1). Due to the similar size of methyl and vinyl substituents at the quaternary carbon center, high diastereoselectivity can only be achieved if an effective chiral catalyst promotes the cyclization. We thus considered the possibility that cinchona alkaloids might catalyze the desired intramolecular phenol conjugate addition[xxi] efficiently and stereoselectively. We pursued the latter strategy with the knowledge that every extant example thus far is to obtain a benzopyran.[xxi] As illustrated in Scheme 5, treatment of 21 with 10 mol % of commercially available and inexpensive cinchonine (22) leads to the formation of anti-23 in 87% yield and 89:11 diastereomeric ratio (d.r.; 0 °C, 2.0 h). Conversion to Weinreb amide anti-25 proceeds efficiently (76% yield). The latter reaction generates ~20% of the α,β-unsaturated amide 26 as a byproduct; however, as depicted in Scheme 5, the catalytic diastereoselective benzofuran synthesis can be performed with 26 to afford additional amounts of anti-25 with similar efficiency and stereoselectivity (92% yield, 88:12 d.r.).[xxii] Subsequent treatment with vinylmagnesium bromide and ring-closing metathesis catalyzed by Ru-based carbene 27[xxiii] delivers the anti isomer of Pummerer ketone in 80% yield (>98:2 e.r.). Since the stereochemical outcome of the C–O bond generation by the intramolecular conjugate addition can be controlled by a chiral catalyst, the alternative diastereomer can be obtained simply through the use of 10 mol % cinchonidine (24, Scheme 5). Benzofuran syn-23 is therefore generated in 89% yield and 90:10 d.r. and can be carried on to complete the enantioselective synthesis of Pummerer ketone.[xxiv]
Scheme 5.

Syntheses of Pummerer ketone and its anti isomer involving NHC–Cu-catalyzed EAS, cinchona alkaloid-catalyzed diastereoselective intramolecular conjugate addition and Ru-catalyzed ring-closing metathesis.
The possibility of utilizing commercially available and/or easily accessible (pinacolato)vinylboron reagents in Cu-catalyzed enantioselective allylic substitution reactions enhances the value of catalytic EAS reactions. Furthermore, the high site- and enantioselectivities obtained in the transformations detailed above provide further testimony to the unique ability of sulfonate-bridged bidented NHC–Cu complexes to serve as uniquely efficient catalysts for this important class of enantioselective C–C bond forming processes.
Footnotes
Financial support was provided by the NIH (GM-47480) and the NSF (CHE-1111074). F. G. is an AstraZeneca (2010–11) and was a Bristol-Myers Squibb graduate fellow (2011–12). We are grateful to Dr. B. Jung and Dr. F. Haeffner for helpful discussions, to Frontier Scientific, Inc. for gifts of vinylboronic acid pinacol esters and to Boston College Research Services for providing access to computational facilities.
References & Footnotes
- i.For reviews of catalytic enantioselective allylic substitution (EAS) reactions with “hard” organometallic nucleophiles, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem Commun. 2004:1779–1785. doi: 10.1039/b401123f.Yorimitsu H, Oshima K. Angew Chem Int Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653.Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem Rev. 2008;108:2824–2852. doi: 10.1021/cr068424k.Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515.
- ii.a) Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew Chem Int Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841. [DOI] [PubMed] [Google Scholar]; b) Selim KB, Yamada K-i, Tomioka K. Chem Commun. 2008:5140–5142. doi: 10.1039/b809140d. [DOI] [PubMed] [Google Scholar]; c) Falciola CA, Alexakis A. Chem Eur J. 2008;14:10615–10627. doi: 10.1002/chem.200801309. [DOI] [PubMed] [Google Scholar]; d) Selim KB, Matsumoto Y, Yamada K-i, Tomioka K. Angew Chem Int Ed. 2009;48:8733–8735. doi: 10.1002/anie.200904676. [DOI] [PubMed] [Google Scholar]; e) Polet D, Rathgeb X, Falciola CA, Langlois JB, Hajjaji SE, Alexakis A. Chem Eur J. 2009;15:1205–1216. doi: 10.1002/chem.200801879. [DOI] [PubMed] [Google Scholar]; f) Gao F, Lee Y, Mandai K, Hoveyda AH. Angew Chem Int Ed. 2010;49:8370–8374. doi: 10.1002/anie.201005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iii.a) Zhang P, Brozek LA, Morken JP. J Am Chem Soc. 2010;132:10686–10688. doi: 10.1021/ja105161f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang P, Le H, Kyne RE, Morken JP. J Am Chem Soc. 2011;133:9716–9719. doi: 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv.Dabrowski JA, Gao F, Hoveyda AH. J Am Chem Soc. 2011;133:4778–4781. doi: 10.1021/ja2010829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v.Jung B, Hoveyda AH. J Am Chem Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.For a review on hydroalumination of alkynes, see: Eisch JJ. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Schreiber SL, editors. Pergamon Press; Oxford: 1991. pp. 733–761.For site-selective Ni-catalyzed hydroalumination of terminal alkynes, see: Gao F, Hoveyda AH. J Am Chem Soc. 2010;132:10961–10963. doi: 10.1021/ja104896b.
- vii.a) Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:446–447. doi: 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]; b) Akiyama K, Gao F, Hoveyda AH. Angew Chem Int Ed. 2010;49:419–423. doi: 10.1002/anie.200905223. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao F, McGrath KP, Lee Y, Hoveyda AH. J Am Chem Soc. 2010;132:14315–14320. doi: 10.1021/ja106829k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.The exception is the hydroalumination of the t-butylether derivative of propargyl alcohol, which delivers the Z-vinylaluminum predominantly. There is <2% conversion with other derivatives of propargyl alcohol, such as the t-butyldimethylsilyl ether or the benzyl ether. See ref. [6b] and references cited therein.
- ix.For a previous synthesis of racemic Pummerer ketone, see: Vierfond J-M, Reynet A, Moskowitz H, Thal C. Syn Commun. 1992;22:1783–1792.
- x.a) Pummerer R, Melamed D, Puttfarcken H. Ber Dtsch Chem Ges. 1922;55:3116–3132. [Google Scholar]; b) Casy AF, Parfitt RT, editors. Opioid Analgesics Chemistry and Receptors. Plenum Press; 1986. [Google Scholar]; c) Winternitz F, Autia NJ, Tumlirova M, Lacharette R. Bull Soc Chim Fr. 1956:1817. [Google Scholar]; d) Barton DHR, Deflorin AM, Edwards OE. J Chem Soc. 1956:530–534. [Google Scholar]
- xi.For non-enantioselective Cu-catalyzed allylic substitutions with arylboron reagents, see: Ohmiya H, Yokokawa N, Sawamura M. Org Lett. 2010;12:2438–2440. doi: 10.1021/ol100841y.Whittaker AM, Rucker RP, Lalic G. Org Lett. 2010;12:3216–3218. doi: 10.1021/ol101171v.For an enantioselective additions of aryl groups (cf. ref. [2f]) promoted by an NHC–Cu complex involving the more reactive and sensitive arylboronic acid neopentyl glycol esters (vs. pinacol esters), see: Shintani R, Takatsu K, Takeda M, Hayashi T. Angew Chem Int Ed. 2011;50:8656–8659. doi: 10.1002/anie.201103581.
- xii.a) Takahashi K, Ishiyama T, Miyaura N. J Organomet Chem. 2001;625:47–53. [Google Scholar]; b) Lee J-E, Kwon J, Yun J. Chem Commun. 2008:733–734. doi: 10.1039/b716697d. [DOI] [PubMed] [Google Scholar]; c) Lipshutz BH, Boskovic ZV, Aue DH. Angew Chem Int Ed. 2008;47:10183–10186. doi: 10.1002/anie.200804912. [DOI] [PubMed] [Google Scholar]; d) Kim HR, Jung IG, Yoo K, Jang K, Lee ES, Yun J, Son SU. Chem Commun. 2010;46:758–760. doi: 10.1039/b919515g. [DOI] [PubMed] [Google Scholar]; e) Kim HR, Yun J. Chem Commun. 2011;47:2943–2945. doi: 10.1039/c0cc04496b. [DOI] [PubMed] [Google Scholar]; f) Jang H, Zhugralin AR, Lee Y, Hoveyda AH. J Am Chem Soc. 2011;133:7859–7871. doi: 10.1021/ja2007643. [DOI] [PubMed] [Google Scholar]; g) Semba K, Fujihara T, Terao J, Tsuji Y. Chem Eur J. 2012;18:4179–4184. doi: 10.1002/chem.201103612. [DOI] [PubMed] [Google Scholar]
- xiii.For a review on catalytic enantioselective methods that generate quaternary carbon stereogenic centers, see: Das JP, Marek I. Chem Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a.
- xiv.Ohishi T, Nishiura M, Hou Z. Angew Chem Int Ed. 2008;47:5792–5795. doi: 10.1002/anie.200801857. b) Ref [5] [DOI] [PubMed] [Google Scholar]
- xv.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- xvi.For relevant stereochemical models, see ref. [5].
- xvii.Catalysts prepared from 5b-c and 6b-c generate high enantioselectivity for substrates that contain an ortho-substituted aryl substituent. However, only NHC–Cu complexes derived from 6b-c reliably deliver high e.r. values with other allylic phosphates.
- xviii.With the catalyst originating from 6b, 14 is isolated in 42% yield (>98% SN2′, 96:4 e.r.). The results obtained with 6b or 6c are often similar but the outcome can be, at times, slightly different. Currently, we cannot precisely predict the identity of the most optimal complex for a particular application.
- xix.See the Supporting Information for details.
- xx.Travis BR, Sivakumar M, Hollist GO, Borhan B. Org Lett. 2003;5:1031–1034. doi: 10.1021/ol0340078. [DOI] [PubMed] [Google Scholar]
- xxi.For related enantioselective intramolecular conjugate additions catalyzed by derivatives of cinchona alkaloids, see: Biddle MM, Lin M, Scheidt KA. J Am Chem Soc. 2007;129:3830–3831. doi: 10.1021/ja070394v.and references cited therein.
- xxii.Phenolic ester 21 undergoes intramolecular conjugate addition to afford an equal mixture of syn- and anti-23 upon standing for a few hours (22 °C), and is unstable under the conditions required for its conversion to a Weinreb amide.
- xxiii.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- xxiv.The catalytic RCM that affords Pummerer ketone must be carried out for 46 hours at 22 °C (vs. 50 °C and 20 h), since, in contrast to its anti isomer, it is prone to racemization. When RCM of enone derived from syn-25 is performed at 50 °C, under otherwise identical conditions, the tricyclic enone is generated with substantial loss of enantiomeric purity (~60:40 e.r.; >98% conv, 90% yield). See the Supporting Information for mechanistic rationale and additional details.