

Abstract
Objectives
Genome-wide association studies (GWAS) in complex phenotypes, including psychiatric disorders, have yielded many replicated findings, yet individual markers account for only a small fraction of the inherited differences in risk. We tested the performance of polygenic models in discriminating between cases and healthy controls and among cases with distinct psychiatric diagnoses.
Methods
GWAS results in bipolar disorder (BD), major depressive disorder (MDD), schizophrenia (SZ), and Parkinson’s disease (PD) were used to assign weights to individual alleles, based on odds ratios. These weights were used to calculate allele scores for individual cases and controls in independent samples, summing across many single nucleotide polymorphisms (SNPs). How well allele scores discriminated between cases and controls and between cases with different disorders was tested by logistic regression.
Results
Large sets of SNPs were needed to achieve even modest discrimination between cases and controls. The most informative SNPs were overlapping in BD, SZ, and MDD, with correlated effect sizes. Little or no overlap was seen between allele scores for psychiatric disorders and those for PD.
Conclusions
BD, SZ, and MDD all share a similar polygenic component, but the polygenic models tested lack discriminative accuracy and are unlikely to be useful for clinical diagnosis.
Keywords: genome-wide association, polygenic model, allele burden, psychosis, prediction
Introduction
Genome-wide association studies (GWAS) have changed our thinking about the genetic architecture of the major psychiatric disorders. We now have molecular genetic evidence that their high heritability cannot solely be attributed to a few common polymorphic variants of major effect. Rather, many different alleles at various loci, each with a relatively small effect, appear to additively influence risk. Such loci are commonly referred to as “polygenes” (Mather, 1943); indeed, Gottesman and Shields pioneered the concept of polygenic etiology in psychiatric disorders as early as 1967 (Gottesman and Shields, 1967). GWAS have been able to identify some of these alleles, but together they account for only a small fraction of known heritability (Schulze, 2010). While complementary approaches, such as large-scale sequencing and sophisticated phenomics might yet uncover alleles of larger effect (Schulze, 2010), it presently seems likely that much of the risk for major psychiatric disorders is polygenic.
Polygenic effects underlying disease are difficult to study, but new methods based on GWAS data are being developed. Lee and colleagues (Lee et al., 2008) showed that genome-wide SNP-trait association information obtained in one group of heterogeneous stock mice could be used to predict many of the same traits in other, independent samples of mice. As expected, more heritable traits were more predictable. The International Schizophrenia Consortium (ISC) (International Schizophrenia Consortium et al., 2009) extended this concept to human case-control samples. GWAS information obtained in one schizophrenia sample could modestly but significantly discriminate between cases and controls in another sample. Interestingly, the schizophrenia GWAS information was also modestly informative for bipolar disorder (BD), but had no discriminative value for a variety of non-psychiatric disorders. In each case, only about 3% of the variance in case-control status was explained by the observed GWAS information but, using simulation, the authors argued that these results were consistent with common SNPs that tagged approximately 30% of the variance in liability.
The ISC study further suggested that BD and SZ share genetic risk factors. This would be consistent with some family and candidate gene studies (Berrettini, 2003; Lichtenstein et al., 2009; Schulze et al., 2005; Williams et al., 2006). However, the large number of SNPs needed to achieve even modest case prediction—in the tens of thousands—makes it difficult to rule out other interpretations. Different subsets of SNPs within the larger group could carry specific predictive value for SZ or BD, but not both. Even if many of the same SNPs are involved in both disorders, the risk alleles could differ. And even if some of the same risk alleles are involved in both disorders, the effect sizes could still differ.
Here, we consider several case-control samples with a total sample size over 16,000, including BD, major depressive disorder (MDD), SZ, and Parkinson’s Disease (PD). GWAS data from all of these samples were used to address several questions:
How well does GWAS information obtained in one BD case-control sample discriminate cases from controls in other, independent samples?
Do SNPs that show a consistent association in at least two BD samples have greater discriminative value in a third sample?
Is GWAS information drawn from a BD sample also informative for other psychiatric phenotypes such as SZ or MDD?
Does any apparent overlap between two disorders represent the same SNPs or risk alleles and, if so, are the effect sizes similar?
Does any discriminative value extend beyond psychiatric disorders?
Methods
Samples (Table 1)
Table 1.
Samples studied
| Sample | Cases | Case diagnosis | Controls | Platform |
|---|---|---|---|---|
| GAIN BD | 1001 | Bipolar I, schizoaffective disorder | 1033 | Affymetrix 6.0 |
| WTCCC BD | 1856 | Bipolar I, bipolar II, schizoaffective disorder | 2945 | Affymetrix 500K |
| German BD | 645 | Bipolar I | 1310 | Illumina HumanHap 550 |
| GAIN MDD | 1722 | Major depressive disorder | 1774 | Perlegen |
| GAIN SZ | 1343 | Schizophrenia | 1378 | Affymetrix 6.0 |
| NIA/NINDS PD | 984 | Parkinson’s disease | 809 | Illumina Infinium Human-I/HumanHap300 |
We studied three GWAS samples of BD (GAIN BD, WTCCC BD, German BD), and one each of MDD (GAIN MDD), and SZ (GAIN SZ). A GWAS sample of PD (NIA/NINDS PD) served as a non-psychiatric comparison sample (Simon-Sanchez et al., 2008).
GAIN BD
Ascertainment, diagnosis, and genotyping are detailed elsewhere (Smith et al., 2009). Unrelated subjects with a final diagnosis of bipolar 1 disorder (BD1) or schizoaffective bipolar disorder (SABP) were submitted to GAIN for genotyping. Genotyping was performed using the Affymetrix 6.0 array. After application for access was granted, genotype data were downloaded from the dbGaP website (http://dbgap.ncbi.nlm.nih.gov) on March 28, 2008 and April 18, 2008. There were 729,304 markers in 2099 individuals present in the original download. After several quality control measures and data cleaning, the final file included 2034 subjects (1001 cases, 1033 controls). For details, see Supplemental Data.
WTCCC BD
Ascertainment, diagnosis, and genotyping of the WTCCC data are detailed elsewhere (Wellcome Trust Case Control Consortium, 2007). After application for access was granted, data were downloaded from the WTCCC website (https://www.wtccc.org.uk) on December 20, 2007. A total of 500,568 Affymetrix 500K markers (called by CHIAMO) from 5002 individuals were present in the original download, including both the 1958 Birth Cohort and the UK Blood Service control groups. We used PLINK (vers. 1.04) (Purcell et al., 2007) for most quality control and data cleaning steps and the GRR software (Abecasis et al., 2001) to identify apparently related individuals. The final file included 4801 subjects (1856 cases, 2945 controls). For details, see Supplemental Data.
German BD
The sample comprised 702 cases with BD1 disorder diagnosed according to DSM-IV criteria and 1364 controls. Cases were recruited from consecutive hospital admissions and underwent multi-tiered phenotype characterization, as detailed elsewhere (Fangerau et al., 2004). The population-based control samples stemmed from three epidemiological cohorts from different parts of Germany, reflecting the population composition of the case sample: 493 from the PopGen cohort (Northern Germany; www.popgen.de); 488 from the KORA cohort (Southern Germany; www.gsf.de/KORA); and 383 from the Heinz Nixdorf Recall Study cohort (Western Germany; www.recall-studie.uni-essen.de). A total of 561,629 Illumina HumanHap550 SNP markers were present in the original study sample. After quality control and data cleaning, the final file included 1955 subjects (645 cases, 1310 controls). For details, see Supplemental Data.
GAIN MDD
Ascertainment, diagnosis, and genotyping of the GAIN MDD sample are detailed elsewhere (Sullivan et al., 2009). After application for access was granted, genotype and phenotype files were downloaded from the dbGaP website (http://dbgap.ncbi.nlm.nih.gov) on November 8, 2007. There were 459,248 Perlegen markers in 3,761 individuals present in the original download. After quality control and data cleaning, the final file included 3496 subjects (1722 cases, 1774 controls). For details, see Supplemental Data.
GAIN SZ
Ascertainment, diagnosis, and genotyping of the GAIN SZ sample are detailed elsewhere (Shi et al., 2009). After application for access was granted, genotype and phenotype files were downloaded from the dbGaP website (http://dbgap.ncbi.nlm.nih.gov) on February 21, 2009. There were 729,454 markers in 2,820 individuals present in the original download. The downloaded files included 29 trios and two parent-child pedigrees. After quality control and data cleaning, the final file included 2721 subjects (1343 cases, 1378 controls). For details, see Supplemental Data.
NIA/NINDS PD
This dataset was provided by A. Singleton and M. Nalls on January 7, 2009. Ascertainment, diagnosis, and genotyping of the NIA/NINDS PD sample are detailed elsewhere (Simon-Sanchez et al., 2008). The sample comprised 984 PD cases and 809 controls. Genotyping was performed using a combination of the Illumina Infinium Human-550kv1 and 550kv3 BeadChips, assaying a total of unique 545,066 SNPs. After quality control and data cleaning, the final file included 1793 subjects (984 cases, 809 controls). For details, see Supplemental Data.
Whole-genome imputation
To facilitate comparison across different genotyping platforms, we performed whole-genome imputation for the following test samples: WTCCC BD, German BD, GAIN MDD, and NIA/NINDS PD. For imputation, we used the MArkov Chain Haplotyping (MACH) program, version 1.0 (Li et al., 2010). MACH uses Markov chain haplotyping to resolve haplotypes—and therefore missing genotypes—from observed genotypes in unrelated individuals. For details, see Supplemental Data.
Polygenic modeling
We performed several polygenic analyses using predefined discovery and test samples. Although control samples overlapped between GAIN BD and GAIN SZ, all comparisons were performed between independent, non-overlapping samples. Whole-genome prediction, as previously suggested (Lee et al., 2008), was pursued; that is, all SNPs genotyped in the discovery sample were used to discriminate affection status in the test samples. The following discovery-test-sample pairings were studied:
Whole genome-data from GAIN BD were used to discriminate affection status in WTCCC BD
Whole genome-data from GAIN BD were used to discriminate affection status in German BD
Whole genome-data from GAIN BD were used to discriminate affection status in GAIN MDD
Whole genome-data from WTCCC BD were used to discriminate affection status in GAIN SZ
Whole genome-data from GAIN BD were used to discriminate affection status in NIA/NINDS PD
Whole genome-data from GAIN SZ were used to discriminate affection status in NIA/NINDS PD
Whole genome-data from GAIN MDD were used to discriminate affection status in NIA/NINDS PD
Polygenic modeling was performed by weighting each SNP in the specified discovery sample, and then assigning a score to each individual in the test sample on the basis of the weighted sum of alleles across all SNPs. The weighting was performed as follows: in the designated discovery sample, one allele of each SNP was assigned a weight based on the log10 of the estimated odds ratio (OR). Thus, alleles with OR<1, which are effectively protective, received a negative weighting; alleles with OR>1, which increases risk, received a positive weighting, proportional to their ORs. Alleles with OR=1 were assigned a weight of zero. Using these weightings, the PLINK --score function was used to assign a score to each individual in the independent test sample. Thus each person was scored with a weighted sum of “risk” and “protective” alleles.
This allele score was then used as a predictor in a standard logistic regression analysis with case status as the dependent variable. The logistic model was also used to estimate a receiver operator characteristic (ROC) curve and an area under the curve (AUC) as an estimate of discriminant accuracy. An AUC=0.5 is equivalent to chance discrimination between cases and controls, while values over 0.5 indicate a better than chance probability that any random case-control pair would be assigned correct diagnoses.
AUC was translated into individual risk on the liability scale by use of the QIMR GENROC Calculator (http://gump.qimr.edu.au/genroc/; (Wray et al., 2007)).
Correlation of effect sizes
We investigated the potential correlation of effect sizes across disorders in the following four independent samples: WTCCC BD, GAIN SZ, GAIN MDD, and NIA/NINDS PD. A Pearson correlation analysis of the log ORs was performed for all six pairings of these four samples. To limit potential inflation of test statistics due to high levels of linkage disequilibrium (LD) in the imputed data sets, we applied LD-based SNP pruning using the PLINK --indep-pairwise routine (window size of 2, shift by 1 SNP, r2 =0.2).
Results
Using the information from the GAIN BD sample, we discriminated cases from controls in the WTCCC BD sample with high statistical significance (p = 2.5 × 10−10) but modest diagnostic accuracy (AUC = 0.55) (Fig. 1a). A similar level of discrimination was achieved in the German BD (p=4.6 × 10−8; AUC=0.57; Fig. 1b) and the GAIN MDD samples (p=7.32 × 10−7; AUC=0.55; Fig. 1c). The weighted burden of risk alleles derived from the WTCCC BD sample could also discriminate cases from controls in the GAIN SZ sample with high statistical significance (p=2.9 × 10−9) and similar accuracy to that seen for BD (AUC=0.56; Fig. 1d). These AUC values mean that common genetic markers account for about 1% of the variance in individual risk for major mental illness and are thus not clinically useful.
Figure 1.
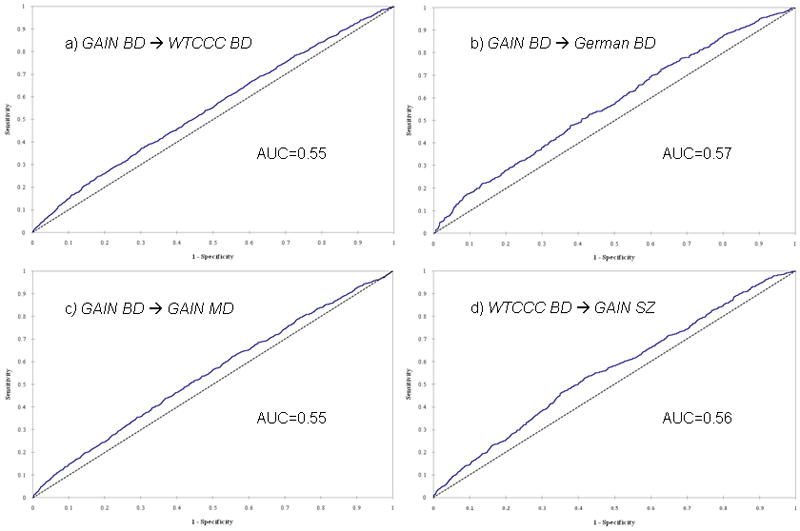
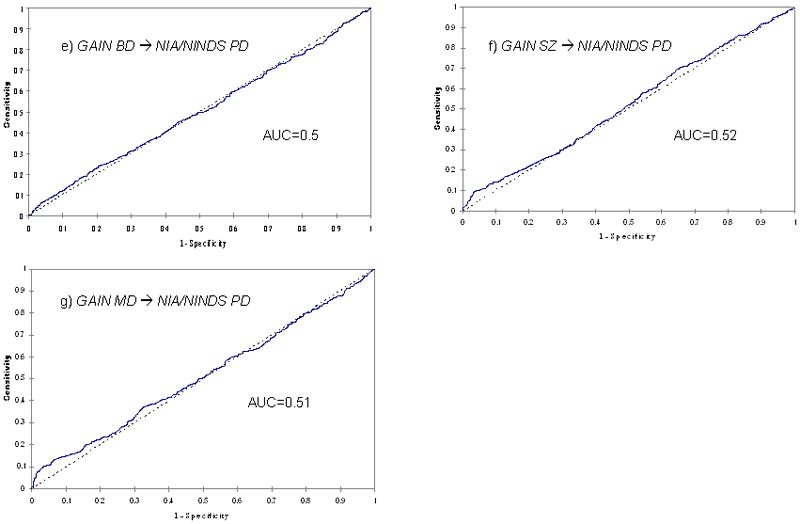
Whole-genome prediction: Areas under the curve (AUC) for various prediction pairings (psychiatric disorders, a. to d., and between psychiatric disorders and Parkinson disease [PD], e. to g.)
In contrast, the weighted burden of risk alleles from the GAIN BD sample had no predictive value in the NIA/NINDS PD dataset (AUC=0.50, p=ns; Fig. 1e). Given that the GAIN BD sample is one of the smaller ones in this study, one may argue to combine the three BD data sets for the discovery as this may lead to a more robust prediction. We tested this possibility: while the AUC improved slightly (0.51), significance could not be established.
The weighted burden of risk alleles from both the GAIN SZ and GAIN MDD samples had some predictive value in the NIA/NINDS PD data (AUC=0.52, p=0.01, Fig. 1f; and AUC = 0.51, p=0.001, Fig. 1g, respectively). The prediction based on the SZ sample, however, was not significant after correction for multiple comparison.
SNPs that were consistently associated with the same phenotype in other samples were more informative. The 107,074 SNPs that were consistently associated with BD in both the GAIN BD and WTCCC BD samples at the p<0.05 level had greater discriminant accuracy in the German BD sample (AUC = 0.63), although the improvement was modest. This AUC value means that common genetic markers account for about 3% of the variance in individual risk for BD. The application of a more stringent p-level threshold actually led to a drop in discriminant accuracy, suggesting that many informative SNPs are not statistically significant even in large samples. In agreement with a polygenic model, an increase in allele score led to an increase in the ratio of case to control status. In the fourth quartile of allele scores, the discriminant accuracy was highest, with a case/control ratio of 3.6 (Fig. 2).
Figure 2. Relationship between case status and allele score (in German sample), when SNPs consistently associated with bipolar disorder at p<0.05 in two other samples (GAIN and WTCCC) are used.
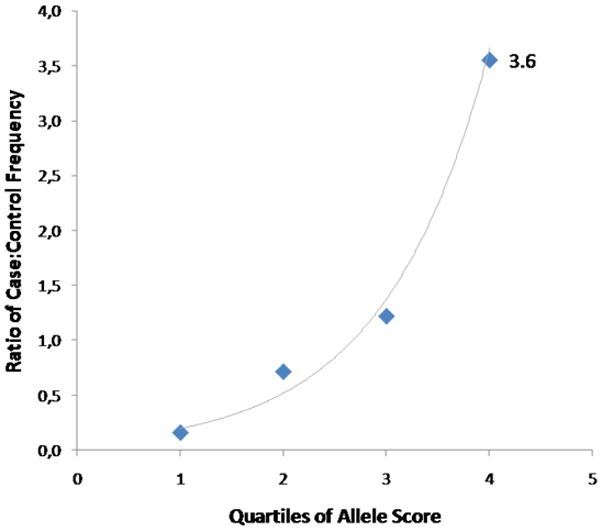
The allele score as obtained through the --score function in PLINK was divided into quartiles (x-axis) and plotted against the ratio of cases to controls (relative frequency) in each quartile range. An exponential line (gray) gives a good fit to the data (r2=0.98; lowess goodness of fit). To give an example for the sensitivity and specificity of BD, at a threshold allele score of 0.003 (97th percentile) the test has 47% specificity and 70% sensitivity.
We also assessed the level of correlation between log ORs of SNPs in all pairs of samples. The results (Table 2) show a weak positive correlation when psychiatric samples were compared, with r2-values ranging from 0.014 (BD vs. MDD) to 0.039 (BD vs. SZ), attributable to a significant excess of SNPs that show the same direction of association in pairs of samples. Virtually no correlation was observed in log ORs between BD and PD, but log ORs were weakly correlated between PD and MDD (r2=0.036) and between PD and SZ (r2=0.024). These results did not change when we applied more stringent LD-based pruning (data not shown).
Table 2.
Correlation of effect sizes
| Pearson correlation of log ORs | ||
|---|---|---|
| r | p | |
| WTCCC BD and GAIN SZ | 0.039 | <2.2e-16 |
| WTCCC BD and GAIN MDD | 0.014 | <2.2e-16 |
| WTCCC BD and NIA/NINDS PD | 0.002 | 0.01629 |
| GAIN SZ and GAIN MDD | 0.029 | <2.2e-16 |
| GAIN SZ and NIA/NINDS PD | 0.024 | <2.2e-16 |
| GAIN MDD and NIA/NINDS PD | 0.036 | <2.2e-16 |
Discussion
The term “polygenic” was first used to describe a “…heritable difference [that is] dependent on the joint action of many genes, each having an effect small…” (Mather, 1943). This is the sense in which the word is still used today, although polygenic effects have typically not been measured directly, but rather have been inferred when a significant fraction of the known heritability of a trait or disease could not be explained by individually identified genes. Polygenic effects based on such evidence of absence can only be judged heuristically, by how well they fill the gap that individual genes have left unfilled.
The advent of genome-wide SNP arrays has made it possible, for the first time, to measure polygenic effects at the molecular level in humans, based on series of hundreds of thousands to millions of common polymorphisms. This measurement is inherently imprecise, since it is not possible to differentiate well between SNPs that truly make a small contribution to disease risk and SNPs that do not. These data are statistical, and do not pinpoint specific genes. SNP arrays are also biased toward common alleles, and do not represent whatever contribution to risk might come from rarer alleles. Nevertheless, SNP arrays allow the best available estimate of polygenic effects and comparisons between samples ascertained from different disorders.
In a groundbreaking study, the ISC (International Schizophrenia Consortium et al., 2009) previously published a similar analysis focused on SZ. They selected SNPs at various association p-value thresholds and showed that these SNPs explained between 2–3% of the total variance of the SZ phenotype in independent SZ datasets. The same SNPs explained about 1–2% of the variance in each of two large BD GWAS samples, but could not predict disease status in any of six non-psychiatric phenotypes. The authors concluded that their data supported a polygenic basis of SZ that is substantially shared with BD.
In the present study we pursued a distinct but related approach. Instead of selecting SNPs based on p-value thresholds, we used complete genome-wide SNP data, which should carry more information (Lee et al., 2008). We did not limit our analyses to SZ and BD, but also included MDD. Like the ISC study, we used a non-psychiatric dataset as a negative control to test the psychiatric specificity of the genome-wide set of SNPs. As we were very much interested in the issue of genetic overlap across psychiatric phenotypes, we chose PD as -in contrast to BD, SZ, and MDD- it presents with distinct, predominantly tangible somatic symptoms but also shares some clinical features with them. For these, it would be interesting to see whether they can be attributed to the same genetic underpinnings or rather be considered phenocopies. Unlike the ISC study, we also showed that the apparent overlap in genetic markers among the psychiatric disorders we studied corresponds to a correlation in effect size and direction of association, offering further support for overlapping genetic risk factors.
Our results demonstrate that a weighted, whole-genome set of SNPs derived from one BD dataset discriminated between BD cases and healthy controls in each of two independent datasets at high levels of statistical significance (p<10−8). The p-values obtained in such comparisons are based on a single test and thus are not subject to genome-wide correction for multiple testing. However, the discriminant accuracy was modest, with AUC values in the range of 55 to 57%.
SNPs consistently associated with BD in two samples were better at discriminating cases from controls in a third sample (AUC 63%). This level of accuracy is comparable to that reported by the ISC (International Schizophrenia Consortium et al., 2009) for SZ, and similar to that observed by Evans and colleagues (Evans et al., 2009) in the WTCCC BD dataset, where the dataset was split into training and prediction subsets. Similar AUC values were also reported for highly-selected SNPs used to predict Type 2 diabetes (Lyssenko et al., 2008) and heart disease (van der Net et al., 2009). The convergence of results suggests that these AUC values may be close to the maximum that could be obtained from common genetic markers applied to common disorders. In a subsequent analysis, we also tested the hypothesis whether an increase in discriminative power through the use of consistently associated SNPs could also be achieved by simply increasing the discovery sample, here by combining the three BD samples to predict affection status in MDD. However, this was not the case (AUC=0.51; p=n.s.).
Such models are far from useful in clinical diagnosis. Even for complex phenotypes with a better GWAS yield than typically seen in psychiatric disorders, diagnosis based on genetic markers is often less accurate than family history or other long-known clinical or epidemiological risk factors (Janssens and van Duijn, 2009). While it would be desirable to be able to truly identify causal genetic variants that would be useful in diagnosis (Wray et al., 2007), this may not be realistic for common diseases with many genetic risk factors.
On the other hand, polygenic models do provide some information about shared genetic risk factors. We found that GWAS information from one BD sample could discriminate between cases and controls with SZ or MDD, at similar levels of accuracy and statistical significance. Informative SNPs were largely overlapping, with correlated effect sizes for most of the comparisons performed. Clearly, the clinical differences between BD, SZ, and MDD cannot be explained by common genetic markers. In contrast, no correlation was noted between SNPs informative for BD and SNPs informative for PD, and very little correlation between SNPs informative for SZ or MDD and SNPs informative for PD. We conclude that polygenic effects shared among psychiatric illnesses are largely distinct from those we can measure in a neurological disorder such as PD.
These data contribute to an emerging picture that supports the ample body of knowledge about shared genetic risk factors in SZ, BD, and MDD (Berrettini, 2000; Gershon et al., 1988; Lichtenstein et al., 2009; Maier et al., 1993). Does this mean that SZ, BD, and MDD are somehow the same disorders? We think not. While shared genetic pathways cannot be denied or discounted at this point, weak genetic risk factors do not outweigh the largely distinct course of illness, symptomatology, and treatment response long observed in SZ, BD, and MDD. Notably, an example from outside psychiatry may help illustrate this point; while strong genetic overlap exists between Crohn’s disease and ankylosing spondylitis, each of these disorders has a distinct target organ, pathophysiology, and preferred treatment indication (Thomas and Brown, 2010).
Our results may be compatible with a small but significant polygenic overlap between PD and SZ or MDD. One could speculate that this shared polygenic component may contribute to the cognitive, depressive, and psychotic symptomatology common to all three disorders. Although PD is primarily a neurological disorder, its clinical picture often includes symptoms of dementia, depression, and psychosis (Cummings, 1992; Schneider et al., 2008). However, the polygenic models based on the GAIN SZ or the GAIN MDD datasets yielded small AUC values for PD in our analysis — slightly better than chance — and the p-values associated with these AUC values did not survive correction for multiple testing across the seven disorder pairings we explored.
We have demonstrated that 1) there is molecular genetic overlap among MDD, BD, and SZ in whole genome SNP data derived from large case-control samples; 2) among these disorders, the most informative SNPs show correlated effect sizes and direction of association; and 3) common genetic markers have modest ability to distinguish psychiatric cases from controls. Larger samples, better clinical diagnosis, and a more complete sampling of all of the relevant genetic variation — including rare alleles of potentially larger effect — are needed to complete the picture (Cirulli and Goldstein, 2010; Lango Allen et al., 2010). Nevertheless, clinical utility may be out of reach for many complex diseases. Methods that can account for the imperfect correlation between causal variants and genotyped SNPs may help increase the predictive value of GWAS data (Yang et al., 2010), but it seems clear that common alleles cannot explain the clinical differences between MDD, BD, and SZ. Future studies aimed at explaining these differences will need to consider environmental as well as genetic risk factors.
Supplementary Material
Acknowledgments
The authors are greatly indebted to Naomi Wray for highly inspirational discussions. Ioline Henter provided outstanding editorial assistance.
Funded by the Intramural Research Program of the National Institute of Mental Health (NIMH), National Institutes of Health, Department of Health and Human Services (IRP-NIMH-NIH-DHHS), Deutsche Forschungsgemeinschaft (DFG), the National German Genome Research Network (NGFN), NARSAD (Independent/Junior Investigator Awards to FJM/TGS), and the Alfried Krupp von Bohlen und Halbach-Stiftung.
Genotyping of the GAIN BD, GAIN MDD, and GAIN SZ samples was provided through the Genetic Association Information Network (GAIN). The datasets used for the analyses described in this manuscript were obtained from the database of Genotypes and Phenotypes (dbGaP). Samples and associated phenotype data were provided by the contributing studies. We thank the Wellcome Trust Case Control Consortium, the Netherlands Study of Depression and Anxiety, and the Netherlands Twin Registry for making data/results available for analysis.
The contributions of ABS and MAN were supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services (project Z01 AG000932-02, human subjects protocols 2004-147 and 2003-081).
The Bipolar Genome Study contributors
University California, San Diego: John R. Kelsoe, Tiffany A. Greenwood, Caroline M. Nievergelt, Rebecca McKinney, Paul D. Shilling
Scripps Translational Science Institute: Nicholas J. Schork, Erin N. Smith, Cinnamon S. Bloss
Indiana University: John I. Nurnberger, Jr., Howard J. Edenberg, Tatiana Foroud, Daniel L. Koller
University of Chicago: Elliot S. Gershon, Chunyu Liu, Judith A. Badner
Rush University Medical Center: William A. Scheftner
Howard University: William B. Lawson, Evaristus A. Nwulia, Maria Hipolito
University of Iowa: William Coryell
Washington University: John Rice
University California, San Francisco: William Byerley
National Institute of Mental Health: Francis J. McMahon, David T.W. Chen, Thomas G. Schulze
University of Pennsylvania: Wade H. Berrettini
Johns Hopkins University: James B. Potash, Peter P. Zandi, Pamela B. Mahon
University of Michigan: Melvin G. McInnis, Sebastian Zöllner, Peng Zhang
The Translational Genomics Research Institute: David W. Craig, Szabolcs Szelinger
Portland Veterans Affairs Medical Center: Thomas B. Barrett
Georg-August-University Göttingen: Thomas G. Schulze
Footnotes
Conflict of Interest
None to declare
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- Berrettini W. Evidence for shared susceptibility in bipolar disorder and schizophrenia. Am J Med Genet C Semin Med Genet. 2003;123C:59–64. doi: 10.1002/ajmg.c.20014. [DOI] [PubMed] [Google Scholar]
- Berrettini WH. Are schizophrenic and bipolar disorders related? A review of family and molecular studies. Biol Psychiatry. 2000;48:531–538. doi: 10.1016/s0006-3223(00)00883-0. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Depression and Parkinson's disease: a review. Am J Psychiatry. 1992;149:443–454. doi: 10.1176/ajp.149.4.443. [DOI] [PubMed] [Google Scholar]
- Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Human Mol Genet. 2009;18:3525–3531. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- Fangerau H, Ohlraun S, Granath RO, Nothen MM, Rietschel M, Schulze TG. Computer-assisted phenotype characterization for genetic research in psychiatry. Hum Hered. 2004;58:122–130. doi: 10.1159/000083538. [DOI] [PubMed] [Google Scholar]
- Gershon ES, DeLisi LE, Hamovit J, Nurnberger JI, Jr, Maxwell ME, Schreiber J, et al. A controlled family study of chronic psychoses. Schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 1988;45:328–336. doi: 10.1001/archpsyc.1988.01800280038006. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci USA. 1967;58:199–205. doi: 10.1073/pnas.58.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens AC, van Duijn CM. Genome-based prediction of common diseases: methodological considerations for future research. Genome Med. 2009;1:20. doi: 10.1186/gm20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, van der Werf JH, Hayes BJ, Goddard ME, Visscher PM. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 2008;4:e1000231. doi: 10.1371/journal.pgen.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- Maier W, Lichtermann D, Minges J, Hallmayer J, Heun R, Benkert O, et al. Continuity and discontinuity of affective disorders and schizophrenia. Results of a controlled family study. Arch Gen Psychiatry. 1993;50:871–883. doi: 10.1001/archpsyc.1993.01820230041004. [DOI] [PubMed] [Google Scholar]
- Mather K. Polygenic inheritance and natural selection. Biol Revs. 1943;18:32–64. [Google Scholar]
- Purcell SM, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Althaus A, Backes V, Dodel R. Psychiatric symptoms in Parkinson's disease. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 5):55–59. doi: 10.1007/s00406-008-5012-4. [DOI] [PubMed] [Google Scholar]
- Schulze TG. Genetic research into bipolar disorder: the need for a research framework that integrates sophisticated molecular biology and clinically informed phenotype characterization. Psychiatr Clin North Am. 2010;33:67–82. doi: 10.1016/j.psc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, Ohlraun S, Czerski PM, Schumacher J, Kassem L, Deschner M, et al. Genotype-phenotype studies in bipolar disorder showing association between the DAOA/G30 locus and persecutory delusions: a first step toward a molecular genetic classification of psychiatric phenotypes. Am J Psychiatry. 2005;162:2101–2108. doi: 10.1176/appi.ajp.162.11.2101. [DOI] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Scholz S, Matarin Mdel M, Fung HC, Hernandez D, Gibbs JR, et al. Genomewide SNP assay reveals mutations underlying Parkinson disease. Hum Mutat. 2008;29:315–322. doi: 10.1002/humu.20626. [DOI] [PubMed] [Google Scholar]
- Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, de Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T, et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry. 2009;14:359–375. doi: 10.1038/mp.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GP, Brown MA. Genetics and genomics of ankylosing spondylitis. Immunol Rev. 2010;233:162–180. doi: 10.1111/j.0105-2896.2009.00852.x. [DOI] [PubMed] [Google Scholar]
- van der Net JB, Janssens AC, Sijbrands EJ, Steyerberg EW. Value of genetic profiling for the prediction of coronary heart disease. Am Heart J. 2009;158:105–110. doi: 10.1016/j.ahj.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NM, Green EK, MacGregor S, Dwyer S, Norton N, Williams H, et al. Variation at the DAOA/G30 locus influences susceptibility to major mood episodes but not psychosis in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2006;63:366–373. doi: 10.1001/archpsyc.63.4.366. [DOI] [PubMed] [Google Scholar]
- Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–1528. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nature. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.