

Abstract
Background
The N-methyl-D-receptor antagonist ketamine is metabolized in the liver into its active metabolite norketamine. No human data are available on the relative contribution of norketamine to ketamine-induced analgesia and side effects. One approach to assess the ketamine and norketamine contributions is by measuring ketamine-effect at varying ketamine and norketamine plasma concentrations using the CYP450 inducer rifampicin.
Methods
In 12 healthy male volunteers the effect of rifampicin versus placebo pretreatment on S-ketamine (a 2-h infusion of 20 mg/h)-induced analgesia and cognition was quantified. The relative ketamine and norketamine contribution to effect was estimated using a linear additive population pharmacokinetic-pharmacodynamic model.
Results
S-ketamine produced significant analgesia, psychotropic effects (drug high), and cognitive impairment (including memory impairment, reduced psychomotor speed, reduced reaction time, reduced cognitive flexibility). Modeling revealed a negative contribution of S-norketamine to S-ketamine-induced analgesia and absence of contribution to cognitive impairment. At ketamine and norketamine effect concentrations of 100 ng/ml and 50 ng/ml, respectievly, the ketamine contribution to analgesia is −3.8 cm (visual analogue pain score) versus a contribution of norketamine of +1.5 cm, causing an overall effect −2.3 cm. The blood-effect-site equilibration half-life ranged from 0 (cognitive flexibility) to 11.8 (pain intensity) min, and averaged across all end-points was 6.1 min.
Conclusions
This first observation that norketamine produces effects in the opposite direction of ketamine requires further proof. It can explain the observation of ketamine-related excitatory phenomena (such as hyperalgesia and allodynia) upon the termination of ketamine infusions.
Introduction
Many drugs used in clinical anesthesia and pain medicine are metabolized into active compounds. Often it is unknown how parent and metabolite contribute to the observed effects. One way to determine their relative contributions is to administer the metabolite and assess its potency. Next, pharmacokinetic-pharmacodynamic (PK-PD) modeling is required to obtain a precise estimates of the relative contributions as steady-state conditions are seldom reached after infusion of the parent drug. An illustrious example of a drug and its active metabolite is morphine and morphine-6-glucuronide (M6G). While early (descriptive) human and animal studies suggest a relative large contribution of M6G to morphine’s effects, later studies performed in humans that combined data on the separate infusions of morphine and M6G, showed just a minor contribution of M6G to effect.1,2
Another drug with an active metabolite is ketamine. Ketamine, an N-methyl-d-asparate receptor antagonist, is used as anesthetic and at low-dose (up to 30 mg/h) as analgesic.3-5 Upon administration, ketamine is rapidly metabolized into norketamine via cytochrome P450 enzymes in the liver, and norketamine is further metabolized into hydroxynorketamine; ketamine and norketamine are centrally acting N-methyl-d-asparate receptor antagonists, hydroxynorketamine is without pharmacological activity.6-11 Animal data indicate that norketamine has about 20-60% the potency of ketamine and is thought to contribute up to 30% to ketamine-induced analgesia and, to a lesser extent, to the development of psychotropic side effects.7-10,12 No human data are available on norketamine’s contribution to ketamine effect as norketamine is not available for human use. We previously showed that pretreating humans with rifampicin (an antibiotic that induces multiple hepatic P450s, including CYP 2B6 and 3A4, involved in the ketamine N-demethylation into norketamine) caused a 10% reduction in ketamine and a 50% reduction of S-norketamine concentrations.11 To get an indication of the contribution of S-norketamine to S-ketamine effect in that study, simulation studies were performed and we predicted a 20 % contribution of norketamine to ketamine effect.11
In the current placebo-controlled randomized trial, we assessed the contribution of S-norketamine to S-ketamine effect by measuring S-ketamine’s analgesia and cognitive impairment under two specific pharmacokinetic conditions: (1) a condition in which the metabolism of S-ketamine and S-norketamine was not influenced, and (2) a condition in which the metabolism of both compounds was induced by rifampicin. These conditions lead to variations in plasma concentration of S-ketamine and S-norketamine and allow determination of their relative contributions to effect. This design and the application of an additive ketamine-norketamine PK-PD model allows the estimation of the norketamine versus ketamine contribution to changes in effect observed after infusion of just ketamine.
The main aims of this study were: (i) to assess the effect of low-dose ketamine on pain responses and cognition during and following a 2-h infusion; and (ii) to get an estimate of the contribution of norketamine to ketamine effect. We hypothesize that in agreement with our previous simulation study, norketamine contribute to 20 % to ketamine-induced effect. In order to assess the contribution of norketamine we performed a population PK-PD analysis using the pharmacokinetic data from our previous study.11
Materials and Methods
After the protocol was approved by the local Human Ethics Committee (Commissie Medische Ethiek, Leiden, The Netherlands) and the Central Committee on Research involving Human Subjects (Centrale Commissie Mensgebonden Onderzoek, The Hague, The Netherlands) participants were recruited and informed consent was obtained according to the Declaration of Helsinki. The study was registered under number NTR1328.*
Participants
Twelve healthy male volunteers aged 18-37 were enrolled in the study. Participants were excluded from participation in the presence of one or more of the following criteria: body mass index > 30 kg/m2; presence or history of major heart, lung, liver, kidney, neurological or psychiatric disease; history of chronic alcohol or illicit drug use; medication use, allergy to study medication; use of contact lenses during the study (to prevent damage by rifampicin) and color-blindness. All participants were subjected to a medical history and physical examination before participation. Participants had to refrain from food and drinks 8 hours prior to the start the study day. Alcohol, coffee and chocolate were not allowed for 24 hours and grapefruit or grapefruit juice was not allowed for 6 days prior to the study day.
Study Design
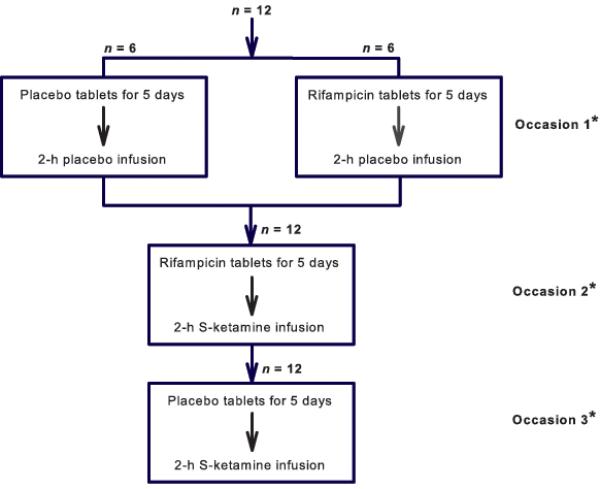
This study had as a randomized single-blind, placebo-controlled, crossover design. Participants were studied on three occasions, with at least three weeks between sessions (Fig. 1). In the five days before study occasion 1, six subjects took rifampicin 600 mg tablets (Sandoz BV, Almere, The Netherlands) (1 tablet/day taken just before going to sleep), six others took placebo tablets (cellulose tablets produced by the local pharmacy). On the study day all 12 subjects received a 2-h treatment with normal saline (NaCl 0.9%) (Study Rifampicin/Placebo-Placebo). In the five days before study occasion 2, all 12 subjects took rifampicin 600 mg tablets (1 tablet/day, taken before going to sleep). On the study day all subjects received a 2-h treatment with S(+)-ketamine (S-ketamine, Pfizer BV, Capelle aan de IJssel, The Netherlands) (Study Rifampicin-Ketamine). Finally, in the five days before study occasion 3, all 12 subjects took placebo tablets (1 tablet/day, taken before going to sleep). On the study day all subjects received a 2-h treatment with S-ketamine (Study Placebo - Ketamine). The S-ketamine intravenous infusion dose was 0.29 mg/kg per h (= 20 mg/h for a volunteer of 70 kg). The order of the three occasions was random. Randomization was performed upon inclusion of the subject by the local pharmacy that provided the blinded study material (rifampicin/placebo tablets and S-ketamine/saline infusion).
Figure 1.
Flow chart of the study. * The occasion sequence was random.
Prior to the first study occasion all subjects participated in two training sessions to get accustomed to the cognitive function tests. On the study day, baseline parameters were obtained (cognitive function tests, pain tests) before treatment. Next, during the 2-h treatment and 3-h following infusion all tests and scores were performed at regular intervals.
Heat pain
Heat pain was induced with the TSA-II NeuroSensory Analyzer (Medoc, Ramat Yishai, Israel). A 3 × 3 cm thermode was placed on the skin of the volar side of the forearm. The temperature was increased from 32 °C by 0.5 °C/s to ‘peak temperature’, after which the temperature was rapidly returned to 32 °C. After each stimulus the Visual Analogue Score (VAS) for pain intensity and pain appreciation was obtained using a 10 cm scale ranging from 0 (= no pain) to 10 (= most severe pain). ‘Peak temperature’ was determined for each subject individually during a test phase. ‘Peak temperature’ was varied from 46 to 52 °C at 1°C intervals. The lowest temperature that caused a VAS of 6 or greater was used in the study. Pain tests were performed at t = 0 (baseline), 5, 10, 15 min following the start of drug infusion and subsequently at 30-min intervals. In order to prevent sensitization of the skin, the thermode was repositioned after each stimulus.13
Side effects: Drug high
Drug high was scored at the end of the S-ketamine infusion on a 10-point numerical rating scale from 0 = no effect to 10 = maximal effect. Only integers were allowed as scores.
Cognition
Cognition was measured with a neurocognitive test battery (CNS Vital Signs, Morrisville, NC) and performed on a laptop computer.14 The battery consisted of seven tests: 1. Symbol digit coding; 2. Stroop test; 3. Shifting attention test; 4. Finger tapping; 5. Continuous performance tests; 6. Verbal and visual test; 7. Verbal and visual memory delay. See for a further explanation of the tests Appendix 1. The full battery (i.e, all 7 tests) were performed prior to drug infusion (baseline) and at t = 120 and 300 min following the start of infusion (the duration of the battery was 30 min). At t = 30, 60, 90, 150, 180, 210, 240 and 270 min a short battery was performed that included symbol digit coding, Stroop test and shifting attention test. All tests were in the Dutch language. The full battery generates scores on 5 separate domains: memory, psychomotor speed, reaction time, complex attention and cognitive flexibility (see Appendix 1). The short battery generates scores on the domains: reaction time and cognitive flexibility. Data analysis was performed on the domain scores.
Domain scores are reported as standard scores (z-scores standardized to a mean of 100 and a standard deviation of 15).11 The average of the z-scores for the five domains generates a summary score, the NeuroCognition Index (NCI), which is reported as a standard score as well. The NCI is similar to an IQ score which is generated by averaging the z-scores of different sub-tests. (An NCI score of 100 is at the 50th percentile; 80% of the population scores between 80 and 120, 90% between 75 and 125). The NCI score gives an indication of the impact of treatment on the cognitive functions altogether.
Power Analysis and Statistical Analysis
Power Analysis
Taking into account our previous estimations,6,11 we assumed a difference in effects between rifampicine and placebo runs of 20%.4,9 Further assuming a SD of 20%, and α = 0.05 and β > 0.80, at least 11 subjects are needed per treatment (SigmaPlot v 12 for windows, Systat Software, Inc., San Jose, CA). In the current study we choose somewhat arbitrarily to test12 subjects (3 subjects were added to this number and served as reserve subjects in case some subjects did not complete all three visits; consequently 15 subjects were mentioned on trialregister.nl*).
Descriptive analysis
Prior to the group comparisons the placebo-placebo and rifampicin-placebo data were compared. Since no significant differences were present, these two groups were combined in the remainder of the analysis. The area-under-the-curve divided by the 300 min duration of the study (AUC/300) of pain intensity and appreciation were calculated. These area-under-the-curves of the three treatments were compared with an analysis of variance (and post-hoc Bonferroni’s test) or Kruskal-Wallis test (and post-hoc Dunnett’s test). Drug high scores at the end of infusion were compared with an analysis of variance (and post-hoc Bonferroni’s test). The NCI and the five cognition domains were analyzed with a repeated measures analysis of variance (factors: time and medication) with post-hoc Bonferroni test. Data analysis was performed with SPSS 16.0. P-values < 0.05 were considered significant. Data are presented as mean ± standard error of the mean (SEM) unless otherwise stated.
Pharmacokinetic-pharmacodynamic analysis
Since blood sampling has stimulatory effects that may interfere with the measurement of pain, side effects and cognition we decided to perform this study without the drawing of blood. Under these conditions, to be able to perform a PK-PD analysis, we assumed that S-ketamine and S-norketamine concentrations are well described by earlier established pharmacokinetic models. The pharmacokinetic model that we used has three compartments for S-ketamine and two for S-norketamine linked by three metabolism compartments.6,11
To eliminate a possible hysteresis between plasma concentration and effect, an effect compartment was postulated that equilibrates with the plasma compartment with a half-life t½ke0 (i.e., the blood-effect-site equilibration half-life). A similar value of t½ke0 was assumed for S-ketamine and S-norketamine.
To estimate the contribution of S-norketamine on S-ketamine-induced changes in pain responses, side effects (dizziness, drug high) and cognition (reaction time and cognitive flexibility) the following linear model was fitted to the data:
| (1) |
YE(t) = Y0 + FK.CE,K(t) + FN.CE,N(t) (1) where YE(t) = the effect at time t, Y0 = predrug baseline effect, FK the ketamine contribution to effect, CE,K = the ketamine effect-site concentration, FN the norketamine contribution to effect and CN,K = the norketamine effect-site concentration. FN is parameterized as fraction of FK, as follows: FN = FN*.FK. For example, when FK = 0.2 and CE,K = 100, the ketamine contribution to effect = 20%. When FN* = 1 the value of FN = 1×0.2 = 0.2 indicating that norketamine contributes as much to the effect as ketamine (both cause a 20% change in effect).
The sensitivity of the pharmacodynamic parameters on the pharmacokinetic parameters was assessed as follows. First, 95% confidence intervals of the pharmacokinetic parameters were constructed based on the inter-individual and inter-occasion variability available from an earlier study.9 Next, the pharmacodynamic analyses of the pain intensity data were rerun in turn for all pharmacokinetic parameters at both endpoints of those intervals.
The PK-PD data were analyzed with the statistical package NONMEM VII (ICON Development Solutions, Ellicott City, MD).15 Model parameters were assumed to be log-normally distributed. Residual error was assumed to be additive with variance σ2. Model selection was based on the χ2-test with P-values less than 0.01 considered significant (to select highly significant model components).
Results
All subjects completed the protocol without unexpected side effects. The subject’s age, weight, height and body mass index averaged to 23 ± 5 years, 184 ± 6 cm, 75 ± 12 kg and 22 ± 3 kg/m2, respectively (values are mean ± SD).
Descriptive Analysis – Comparison to Placebo (Tables 1 and 2)
Table 1.
Descriptive analysis of the ketamine-induced pain relief and side effects (drug high and dizziness
| Rifampicin/Placebo- Placebo |
Placebo- Ketamine |
Rifampicin- Ketamine |
|
|---|---|---|---|
| Pain intensity | |||
| AUC/300 (cm) | 6.8 ± 0.4 | 6.0 ± 0.4* | 5.7 ± 0.4* |
| Pain Appreciation | |||
| AUC/300 (cm) | 7.5 ± 0.6 | 6.4 ± 0.5* | 6.0 ± 0.4* |
| Drug high | |||
| Score at end of infusion |
0 ± 0 | 7.0 ± 0.4∥ | 5.2 ± 0.6∥§ |
Values are mean ± SEM; AUC = area-under-the-curve
P < 0.05 versus Rifampicin/Placebo-Placebo;
P < 0.05 versus Rifampicin/Placebo-Placebo;
P < 0.05 versus Placebo-Ketamine
Table 2.
Descriptive analysis of the neurocognitive data
| Rifampicin/Placebo- Placebo |
Placebo- Ketamine |
Rifampicin- Ketamine |
|
|---|---|---|---|
| Neurocognitive Index ¶ | |||
| 0 min | 105.6 ± 1.9 | 104.3 ± 4.1 | 104.6 ± 2.4 |
| 120 min | 104.3 ± 4.1 | 77.6 ± 4.1*§ | 83.8 ± 11.3*§ |
| 300 min | 104.6 ± 2.4§ | 101.0 ± 2.4§ | 98.1 ± 2.7§ |
| Memory ¶ | |||
| 0 min | 101.3 ± 5.2 | 104.7 ± 5.0 | 106.6 ± 4.5 |
| 120 min | 88.9 ± 6.1 | 55.5 ± 5.7* | 65.1 ± 4.7* |
| 300 min | 90.7 ± 5.3§ | 96.1 ± 4.1§ | 93.8 ± 5.5§ |
| Psychomotor Speed ¶ | |||
| 0 min | 108.2 ± 5.2 | 108.7 ± 4.8 | 107.8 ± 5.8 |
| 120 min | 112.6 ± 7.0 | 86.8 ± 5.2*§ | 90.6 ± 3.9*§ |
| 300 min | 117.0 ± 5.8§ | 113.9 ± 5.2§ | 114.8 ± 5.9§ |
| Reaction Time ¶ | |||
| 0 min | 97.4 ± 3.8 | 95.9 ± 5.3 | 90.9 ± 4.4 |
| 120 min | 88.6 ± 3.6 | 78.8 ± 4.8*§ | 83.8 ± 4.8*§ |
| 300 min | 91.5 ± 4.2 | 93.1 ± 3.6 | 88.6 ± 2.6 |
| Complex Attention ¶ | |||
| 0 min | 104.0 ± 4.4 | 99.4 ± 4.3 | 102.9 ± 3.3 |
| 120 min | 97.2 ± 3.7 | 77.3 ± 7.5*§ | 85.8 ± 5.4*§ |
| 300 min | 91.3 ± 2.9§ | 94.4 ± 7.5§ | 88.2 ± 4.1§ |
| Cognitive Flexibility ¶ | |||
| 0 min | 116.3 ± 3.2 | 112.6 ± 4.5 | 114.8 ± 3.3 |
| 120 min | 110.1 ± 4.0 | 89.5 ± 7.3*§ | 94.2 ± 6.3*§ |
| 300 min | 106.4 ± 4.0§ | 107.6 ± 10.9§ | 104.8 ± 3.7§ |
Values are mean ± SEM;
Significant main treatment, time and time×treatment effects at P < 0.05.
Post-hoc analysis:
Treatment: P < 0.01 versus Rifampicin/Placebo-Placebo (at 120 min);
Time: P < 0.05 versus t = 0.
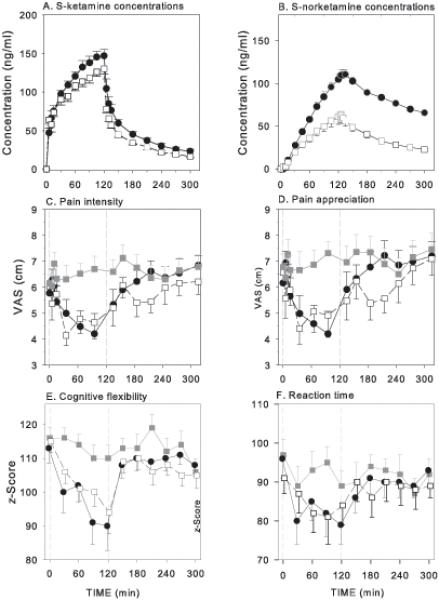
The population averages are given in Fig. 2. Based on the area-under-the-curves (Table 1), S-ketamine produced antinociception to a greater extent than placebo (Rifampicin/Placebo-Placebo). No difference in area-under-the-curve was observed for antinociception between Placebo-Ketamine and Rifampicin-Ketamine. As determined from the measurement at the end of infusion, drug high was reduced in the subjects pretreated with rifampicin (Rifampicin-Ketamine) compared to those treated with placebo (Placebo-Ketamine; Table 1). S-ketamine produces cognitive impairment greater than placebo (Rifampicin/Placebo-Placebo) for all measures at t = 120 min (difference ranging between 17 and 24%, except for reaction time where the differences ranged from 5 to 12%) with no difference between treatment groups Placebo-Ketamine and Rifampicine-Ketamine. Most indices showed a decline over time, possibly caused by fatigue. An exception is psychomotor speed, which showed an increase over time, which may be related to a learning effect. The results of the full battery are given in Table 2, the results of the short battery in Figure 1. These latter data were used in the PK-PD analysis.
Figure 2.
A and B. Effect of placebo (closed symbols) and rifampicin (open symbols) pretreatment (600 mg po per day for five days) on S-ketamine (A) and S-norketamine (B) concentrations during a following a 2-h S-ketamine infusion (from t = 0 to 120 min; dose = 20 mg/h). Values are mean ± SEM. Data are from Noppers et al.9
C – F. Average responses of the influence of rifampicin or placebo pretreatment on pain intensity (C), pain appreciation (D), cognitive flexibility (E) and reaction time (F). The responses were measured during and 3-h following a 2-h S-ketamine infusion of 20 mg/h from t = 0 to t = 120 min. Grey squares are the placebo infusion data following a 5-day pretreatment with placebo or rifampicin (rifampicin/placebo or placebo/placebo); Black circles are the ketamine infusion data following a 5-day pretreatment with placebo; Open squares are the ketamine infusion data following a 5-day pretreatment with rifampicin. Values are mean ± SEM. VAS = visual analogue score.
Pharmacokinetic-Pharmacodynamic Analysis
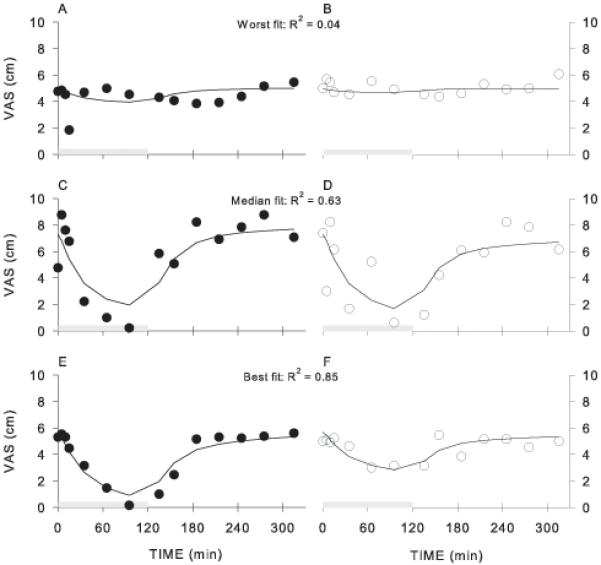
An initial analysis was performed in which the S-norketamine contribution to S-ketamine effect was constrained to behave in a similar direction as S-ketamine (e.g., ketamine and norketamine are both analgesic or produce both drug high). This yielded no contribution of norketamine to effect in any of the tested end-points (i.e., FN = 0). Since we observed that in some of the end-points the rifampicin-ketamine data following infusion remained below the pharmacodynamic data (e.g., pain intensity and pain appreciation, Fig 1C and D), any constraint on FN was removed and FN was allowed to have values causing an effect in the same as well opposite direction as S-ketamine. Examples of best, median and worst data fits for two end-points are given in Fig. 3 for pain intensity. The population pharmacodynamic parameter estimates are given in Table 3. Goodness of fit plots for all end-points are given in Figure 4. Overall, the data were adequately described by the linear model. For pain intensity and pain appreciation the value of FN* indicates an effect of S-norketamine opposite to that of S-ketamine (Table 3). For the cognitive end-points (cognitive flexibility and reaction time) no contribution of S-norketamine to effect could be estimated.
Figure 3.

Examples of data fits from three subjects showing worst (A and B), median (C and D) and best (E and F) data fits for the effect of S-ketamine on pain intensity following rifampicin (A, C and E) or placebo (B, D and F) pretreatment. VAS = visual analogue score.
Table 3.
Pharmacodynamic model parameters
| θ (95% ci) |
ω2 (95% ci) |
ν2 (95% ci) |
|
|---|---|---|---|
| Pain Intensity | |||
| FK (cm.(ng/ml)−1) | −3.80 × 10−2 (−6.10×10−2 – −2.63×10−2) |
1.26 × 10−2 (1.16×10−2 – 1.35×10−2) |
2.0 × 10−4 (4.6×10−4 – 35.4×10−4) |
| FN* | −0.824 (−1.34 – −0.30) |
5.12 × 10−4(4.32×10−4 – 5.92×10−4) | - |
| Y0 (cm) | 6.11 (5.36 – 6.80) |
1.46 (0.36 – 1.66) |
- |
| t½ke0 (min) | 11.8 (11.4 – 21.2) |
- | a- |
| ε | 1.28 (0.78 – 1.78) |
||
| Pain Appreciation | |||
| FK (cm.(ng/ml)−1) | −4.35×10−2 (2.01×10−2 – 6.69×10−2) |
1.30×10−2 (1.20×10−2 – 1.40×10−2) |
3.76×10−4 (0.88×10−4 – 6.64×10−4) |
| FN* | −0.785 (−1.19 – −0.38) |
4.95×10−4 (0.1 ×10−4 – 8.9×10−4) |
- |
| Y0 (cm) | 6.55 (5.75 – 7.35) |
1.95 (0.1 – 4.29) |
- |
| t½ke0 (min) | 10.0 (6.1 – 13.9) |
- | - |
| ε | 1.71 ± 0.40 (1.00 – 2.42) |
||
| Cognitive Flexibility | |||
| FK (cm.(ng/ml)−1) | −0.245 (−0.34 – −0.14) |
3.12×10−2 (0.1×10−2 – 6.0×10−2) |
5.72×10−3 (0.2×10−3 – 10.9×10−3) |
| FN* | 0.00 ± 0.00 (−) |
- | - |
| Y0 (cm) | 113.0 (108.5 – 117.5) |
4.17×10−3 (1.7×10−3 – 6.7×10−3) |
5.35×10−4 (0.1×10−4 – 13.7×10−4) |
| t½ke0 (min) | 0.0* (−) |
- | - |
| ε | 0.976 (0.62 – 1.33) |
||
| Reaction Time | |||
| FK (cm.(ng/ml)−1) | −0.166 (−0.22 – −0.10) |
8.66×10−3 (0.1×10−3 – 19.9×10−3) |
- |
| FN* | 0.00 ± 0.00 (−) |
4.06×10−2 (0.1×10−2 – 11.3×10−2) |
- |
| Y0 (cm) | 92.0 (84.6 – 99.4) |
2.01×10−4 (0.1×10−4 – 19.0×10−4) |
1.21 (1.18 – 1.24) |
| t½ke0 (min) | 2.4 (0.1 – 6.4) |
- | - |
| ε | 62.4 (44.4 – 80.4) |
ci = confidence interval.
ε is a residual error term;
FK is the parameter that describes the contribution of ketamine to total effect;
FN* is the fraction of FK that describes the contribution of norketamine to total effect;
ν2 is interoccasion variability (in the log-domain);
θ is the typical parameter value;
t½ke0 is the blood-effects-site equilibration half-life;
Y0 is baseline value;
ω2 is the between-subject variability (in the log-domain).
no hysteresis between blood-concentration and effect observed.
Figure 4.
Goodness of fit plots for pain intensity (A), pain appreciation (B), cognitive flexibility (C) and reaction time (D). Individual predicted values are plotted against the observed values. The grey lines are the lines of identity.
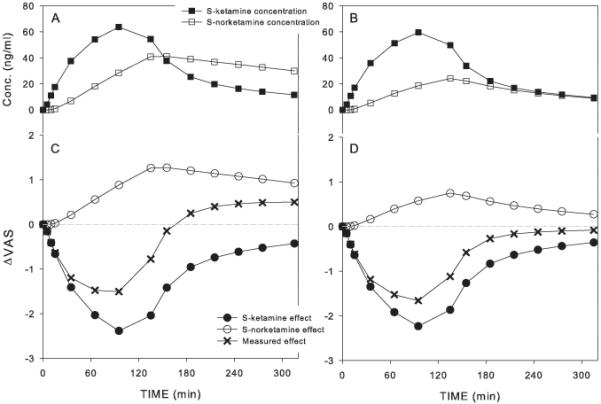
As an example we will further discuss pain intensity. For pain intensity the S-ketamine contribution FK is −0.038 cm.(ng/ml)−1. This indicates that at an effect-site S-ketamine concentration of 100 ng/ml, the effect due to just ketamine will be a 3.8 cm decrease in VAS. The S-norketamine contribution FN is +0.03 (= FK × FN* = −0.824 × −0.038) cm.(ng/ml)−1, which indicates that at a S-norketamine concentration of 50 ng/ml (assuming that this is the S-norketamine effect-site concentration that coincides with an effect site S-ketamine concentration of 100 ng/ml in short-term infusion paradigms), the contribution of just S-norketamine is +1.5 cm VAS increase resulting in a total VAS change of −2.3 cm (= −3.8 + 1.5 cm). In Figure 5 the relative contributions of S-ketamine and S-norketamine to the changes in VAS score and their sum (the measured response) are simulated using the model parameters of Table 3 for the two test conditions (placebo pretreatment, Panels A and C; and rifampicin pretreatment, Panels B and D). It shows the negative effect of norketamine on the change in VAS (relative to S-ketamine’s effect) with hyperalgesia following S-ketamine infusion when S-norketamine levels are high (Panels A and C). When S-norketamine levels are relatively low (Panels B and D) the negative effect on analgesia is less and no hyperalgesia is observed following the 2-h S-ketamine infusion.
Figure 5.
Pharmacokinetic-pharmacodynamic (PK-PD) simulation showing the relative contribution of S-ketmaine and S-norketamine to measured effect. A and C. Simulated pharmacokinetic (A) and pharmacodynamic data (C) assuming placebo pretreatment. B and D. Simulated pharmacokinetic (B) and pharmacodynamic data (D) assuming rifampicin pretreatment. VAS = visual analogue score.
The blood-effect-site equilibration half-life (t½ke0) ranged from 0 min (cognitive flexibility) to 11.8 min (pain intensity). For cognitive flexibility no hysteresis between arterial plasma concentrations and effect was estimated indicating that the effect instantaneously followed arterial plasma concentrations. The value of t½ke0 averaged across all end-points was 6.1 min.
Sensitivity of Pharmacodynamic Data in Response to Variations in Pharmacokinetics
The re-analysis of the pharmacodynamic data using variations in pharmacokinetic parameters (by setting the parameters at both endpoints of their 95% confidence intervals) showed that parameter FN* was most sensitive to changes in ketamine clearance and volume of norketamine’s peripheral compartment. Variations in FN* ranged from −1.2 to −0.6 (compare to value of −0.8 observed in the analysis, see Table 3), with less than 4 points change in objective function.
Discussion
Ketamine causes many side effects,16 including nausea/vomiting, hypertension, psychotropic (psychedelic) effects and cognitive impairment. Knowledge on the contribution of norketamine to ketamine analgesia and any of these side effects is of importance as it may lead to further drug development or adaptation of dosing regimens aimed at optimizing analgesia while minimizing side effects. Our current study was aimed at quantifying S-norketamine contribution to S-ketamine analgesia and S-ketamine cognitive effects. The descriptive analysis indicates that S-ketamine produced greater analgesia, psychotropic effects (drug high) and impairment of cognition than placebo (Tables 1 and 2), in agreement with earlier studies on racemic ketamine.17,18 As expected, the PK-PD analysis of the S-ketamine data, using a linear additive model of the S-ketamine and S-norketamine contribution, enabled estimation of the S-norketamine contribution. For pain intensity and pain appreciation a negative rather than a positive contribution to effect was observed (negative meaning an effect opposing the direction of the S-ketamine effect). The magnitude of these opposing effects is not easily quantified as they depend on the pertaining S-ketamine and S-norketamine concentrations. To visualize their relative contributions to measured (simulated) effect, we performed PK-PD simulations and plotted the magnitude of S-ketamine and S-norketamine effect versus time in Figure 5 for two conditions: placebo (Fig. 5 A and C) and rifampicin (Fig. 5 B and D) pretreatment. This simulation shows that following S-ketamine infusion, when S-norketamine concentrations exceed S-ketamine concentrations the VAS response is hyperalgesic (Fig. 5C). This observation is realistic and in close agreement with earlier studies on the effect of ketamine on pain responses in healthy volunteers and chronic pain patients.6,19-21 When S-norketamine concentrations are relatively low as occurs after rifampicin pretreatment the VAS-response is reduced an no hyperalgesia is observed (Fig. 5 B and D).
There are various observation that ketamine under specific circumstances is associated with pain facilitation.6,19-23 In volunteers ketamine has a dose-dependent antinociceptive effect on experimental nociceptive pain, but pain responses following infusion were perceived as more painful compared to pretreatment responses.21 In agreement with these findings, Mitchell described a cancer patient that developed severe hyperalgesia and allodynia directly following treatment with ketamine.19 Recently we showed that endogenous modulation of pain (using the Conditioning Pain Modulation paradigm) displayed pain facilitation following a 1-h infusion with S-ketamine.20 These findings together with our current observations indicate that ketamine may be anti-analgesic and produce pain facilitatory effects, especially when ketamine concentrations are low and norketamine concentrations are elevated, as occurs following a short-term infusion.
It has been argued that the hyperalgesic effects from N-methyl-d-asparate (NMDA) receptor antagonists are related to activation of metabotropic or non-NMDA ionotropic glutamate receptors activated by excitatory amino acids released from spinal or supraspinal sites or are related to a rebound increase in NMDA receptor activity following the rapid decrease in ketamine concentration.6,19-23 Our data indicate that norketamine may be an additional contributor to the hyperalgesic or anti-analgesic effects of ketamine. One possible mechanism of the excitatory behavior of norketamine on pain responses may be activation of excitatory receptors (other than the excitatory glutamate receptors) such as the σ-, κ- and muscarinic receptors.24 For example, known agonists of the σ-receptor include the NMDA receptor-antagonists phencyclidine and ketamine, and σ1-receptor activation has been associated with pronociceptive and psychotomimetic responses.25 Assuming higher affinity and intrinsic activity of norketamine for the σ-receptor compared to ketamine, can explain that when norketamine concentrations are relatively low (as occurs in the rifampicin treatment group) relatively more analgesia will be present (Fig. 5) compared to a condition in which the norketamine concentrations are relatively higher. Our data are consistent in that they suggest that norketamine acts at a receptor system associated with excitatory responses, including hyperalgesia, and psychotomimetic side effects, possibly the σ-receptor. However, no human data are available on the activity of norketamine at the σ-receptor or any of excitatory receptor system and further studies are warranted to better understand our observations. The absence of effect of variations in norketamine concentration on cognitive function suggests absence of involvement of norketamine in these ketamine-related effects. However, the changes in cognition were large and variable (Fig. 2). We therefore may have missed subtle changes in cognition related to norketamine.
The PK-PD model that we applied did not make a distinction between S-ketamine and S-norketamine onset/offset times (t½ke0). The blood-effect-site equilibration half-lifes of the two compounds were assumed to be similar as reliable estimates of ketamine’s t½ke0 and that of its metabolite are not available and separate estimations were not possible from the data. The estimated values of t½ke0 ranged from 0 (absence hysteresis between plasma concentration and effect) to 11.8 min (overall mean = 6.1 min; Table 3). There are just two earlier studies that report estimates of ketamine’s t½ke0. Schüttler et al.26 showed no hysteresis between S-ketamine plasma concentration and changes in the electroencephalogram. Similarly, Herd et al.27 estimated a values of t½ke0 of 11 s in a pediatric population during induction and recovery from general anesthesia (end-point arousal and recall memory) using racemic ketamine. These data together with ours point towards a rapid onset/offset of S-ketamine’s effect following a short-term infusion paradigm.
In the current study we did assess the pharmacodynamics of S-ketamine without obtaining S-ketamine and S-norketamine pharmacokinetic data. Instead, we relied on previously obtained pharmacokinetics in a similar group of volunteers that received a similar pretreatment with rifampicin.11 The use of simulated pharmacokinetic data in PK-PD modeling studies has been applied with success before when we modeled the effect of opioids on the control of breathing and recently on naloxone reversal of opioid-induced respiratory depression.28,29 The main reason for not obtaining ketamine pharmacokinetic data is that frequent blood sampling from an arterial-line can cause arousal and stress, which may interfere with obtaining reliable data such as pain responses and cognition. A second issue is that the ethics committee of our institution has a restrictive policy regarding the use of arterial-lines when reliable pharmacokinetic data is available from earlier studies.30 We performed a post-hoc re-analysis of the data to assess the sensitivity of the pharmacodynamics on variations in plasma concentrations of S-ketamine and S-norketamine. The results indicate that it is very unlikely that the finding of a negative contribution of norketamine to effect is caused by differences in model predicted and absence of measured ketamine and norketamine concentrations. However, we agree that the lack of pharmacokinetic data is a potential drawback of our study; we do believe, however, that taken the quality of our pharmacokinetic data set that our approach is valid and allows reliable assessment of the relevant pharmacodynamics model parameters.
Our results are surprising in light of previous animal studies showing that norketamine has significant antinociceptive properties.7-10 Our findings are similar to the observations with morphine and its active metabolite M6G.1,2 While rodent data showed that M6G produces potent analgesia at already low plasma concentrations, in humans M6G-induced analgesia occurs only at high plasma concentrations. It is though that the low M6G potency in humans is related to its very slow passage across the blood-brain barrier.1,2 Apart from the evident species differences, the discrepant norketamine data in humans and animals remain unexplained. Possibly different NMDA receptor subtypes in humans compared to rodents may be held responsible for the observed absence of norketamine effect.
The observation from our PK-PD study that S-norketamine anti-analgesic effects opposite to its parent and co-NMDA receptor antagonist is an intriguing finding. While it may explain some of the observations made in human studies on the development of pain facilitation following ketamine infusion,6,19-21 we believe that one has to be careful with the interpretation of these data derived from “complex” PK-PD modeling using simulated pharmacokinetic data. Further proof is required before we can conclude that norketamine has a negative contribution to ketamine-induced analgesia and side effects. A careful conclusion at present is that norketamine contribution to ketamine analgesia is limited and that we cannot exclude a small anti-analgesic effect from norketamine.
MS #201201040 Final box summary.
What we already know about this topic
Ketamine is metabolized to norketamine, and since both of these compounds block n-methyl-d-aspartate (NMDA) receptors and produce analgesia in animals, both are speculated to contribute to analgesia from ketamine administration
What this article tells us that is new
In a study of 12 healthy volunteers who received an inducer of ketamine metabolism or placebo on separate occasions to alter the ketamine / norketamine ratio, modeling of responses to S-ketamine administration suggested a mild antagonism of analgesia from ketamine by norketamine, rather than a supplement
These data, if confirmed in more direct ways, suggest that pain facilitation which is sometimes observed after ketamine administration ends, may reflect action of the norketamine metabolite
Acknowledgments
Support: This study is supported in part by TREND (Trauma RElated Neuronal Dysfunction), a non-profit consortium of academic hospitals, technical research groups and companies focused on the study of Complex Regional Pain Syndrome type 1 (The Hague, The Netherlands); also supported by National Institutes of Health, Bethesda, MD, grant K24DA00417.
Appendix 1: Cognition tests
The CNS Vital Signs cognition tests have been described in full elsewhere.11 In short:
Symbol digit coding: The test consists of serial presentations of screens, each of which contains a bank of 8 symbols above and 8 empty boxes below. The subject types in the number that corresponds to the symbol that is highlighted. Each time the test is administered, the program randomly chooses eight new symbols to match to the eight digits Scoring is the number of correct responses generated in 2 minutes.
Stroop Test: A. The test has three parts. A. The words RED, YELLOW, BLUE and GREEN (printed in black) appear at random on the screen. The subject has to press a button as the word appears. B. The words RED, YELLOW, BLUE and GREEN appear on the screen printed in color. The subject has to press a button when the color of the word matches the meaning of the word. C. The words RED, YELLOW, BLUE and GREEN appear on the screen printed in color. The subject is asked to press a button when the color and word meaning do not match. Each test generates a separate reaction time score (test A generates a simple reaction time, tests B and C complex reaction times), which combined give an indication of information processing speed. The value of the Stroop reaction time is on average 120 ms longer than the complex reaction time generated in part B of the test (range 78-188 ms). Part C also generates an error score. The test requires about 4 minutes.
Shifting attention: In shifting attention test subjects are instructed to match geometric objects either by shape or color. The test measures the ability to shift from one instruction to another quickly and accurately. Three figures appear on the screen, one on top and two on the bottom. The top figure is either a square or a circle. The bottom figures are a square and a circle. These figures are either red or blue; the colors are mixed randomly. The subject is asked to match one of the bottom figures to the top figure, either by color or by shape. The rules of the matching change at random. This goes on for 90 seconds. The goal is to make as many correct matches as possible. The scores generated by SAT are: correct matches, errors and response time in ms.
Finger tapping: The test generates relevant data about fine motor control, which is based on motor speed as well as kinesthetic and visual-motor ability. The subjects press the space bar with their index finger as many times as they can in 10 s; this test is performed 3 times with the right index finger and 3 times with the left index finger. The score is the average number of taps.
Continuous performance: This test is a measure of vigilance or sustained attention over time. The subject is asked to respond to a target stimulus, e.g. the letter B, but not to any other letter, by pressing the space bar. In 5 min, the test presents 200 letters; 40 of the letters are the target B, 160 are non-targets (any other letter). The stimuli are presented at random, although the target stimulus is only appears 8 times during each minute of the test. The scores generated are: correct matches, commission errors (pressing when no B is shown, e.g., impulsive responding) and omission errors (not pressing when a B appears, e.g., inattention).
Immediate and delayed verbal memory: This is an adaptation of the Rey Auditory Verbal Learning Test. Fifteen words are presented, one by one, on the screen. A new word is presented every two seconds. The subject is asked to remember these words. Then a list of thirty words is presented. The fifteen target words are mixed randomly among 30 words of which 15 new words. When the subject recognizes a word from the original list, he or she presses the space bar. This is a recognition test, however, not a test of recall. After finishing the other tests, a delayed recognition test is performed. The 15 targets remain the same for the delayed memory testing; the 15 distractors are different between the immediate and delayed challenges.
Immediate and delayed visual memory: This test is the same as the verbal memory test, but instead of words geometric figures are used.
These tests generate scores on 5 separate domains: memory, psychomotor speed, reaction time, complex attention and cognitive flexibility.
The Memory domain is calculated from the correct scores of the verbal and visual (immediate and delayed) memory tests.
Psychomotor speed is derived from number of tabs in the finger tapping test and number of correct answers in the symbol digit coding tests.
The domain score for Reaction time is made up by combining the three reaction time scores (A, B and C) of the Stroop test.
The domain score for Complex attention is generated by adding the number of errors in the complex performance test, the shifting attention test and the Stroop test.
The domain score for Cognitive flexibility is generated by taking the number of the correct responses on the shifting attention test and subtracting the number of errors on the shifting attention and Stroop tests.
Footnotes
Attribution: The work should be attributed to the Department of Anesthesiology at Leiden University Medical Center
www.trialregister.nl, date last accessed is March 30, 2012.
Summary Statement: Norketamine has an effect opposite to that of ketamine on pain relief.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Skarke C, Darimont J, Schmidt H, Geisslinger G, Lötsch J. Analgesic effects of morphine and morphine-6-glucuronide in a transcutaneous electrical pain model in healthy volunteers. Clin Pharmacol Ther. 2003;73:107–21. doi: 10.1067/mcp.2003.5. [DOI] [PubMed] [Google Scholar]
- 2.Romberg R, Olofsen E, Sarton E, den Hartigh J, Taschner PE, Dahan A. Pharmacokinetic-pharmacodynamic modeling of morphine-6-glucuronide-induced analgesia in healthy volunteers: absence of sex differences. Anesthesiology. 2004;100:120–33. doi: 10.1097/00000542-200401000-00021. [DOI] [PubMed] [Google Scholar]
- 3.Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain; A review. Anesth Analg. 2003;97:1108–16. doi: 10.1213/01.ANE.0000081061.12235.55. [DOI] [PubMed] [Google Scholar]
- 4.Bell R, Eckleston C, Kalso E. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database Syst Rev. 2003:CD003351. doi: 10.1002/14651858.CD003351. [DOI] [PubMed] [Google Scholar]
- 5.Nesher N, Serovian I, Marouani N, Chazan S, Weinbroum AA. Ketamine spares morphine consumption after transthoracic lung and heart surgery without adverse hemodynamic effects. Pharmacol Res. 2008;58:38–44. doi: 10.1016/j.phrs.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E, Olofsen E. S(+)-ketamine effect on experimental pain and cardiac output: A population pharmacokinetic-pharmacodynamic modeling study in healthy volunteers. Anesthesiology. 2009;111:892–903. doi: 10.1097/ALN.0b013e3181b437b1. [DOI] [PubMed] [Google Scholar]
- 7.Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol. 1997;333:99–104. doi: 10.1016/s0014-2999(97)01116-3. [DOI] [PubMed] [Google Scholar]
- 8.Holtman JR, Jr, Crooks PA, Johnson-Hardy JK, Hojomat M, Kleven M, Wala EP. Effects of norketamine enantiomers in rodent models of persistent pain. Pharmacol Biochem Behav. 2008;90:675–85. doi: 10.1016/j.pbb.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–9. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- 10.Shimoyama M, Shimoyama N, Gorman AL, Elliott KJ, Inturrisi CE. Oral ketamine is antinociceptive in the rat formalin test: Role of the metabolite, norketamine. Pain. 1999;81:85–93. doi: 10.1016/s0304-3959(98)00269-3. [DOI] [PubMed] [Google Scholar]
- 11.Noppers I, Olofsen E, Niesters M, Aarts L, Mooren R, Dahan A, Kharasch E, Sarton E. Effect of rifampicin on S-ketamine and S-norketamine plasma concentrations in healthy volunteers after intravenous S-ketamine administration. Anesthesiology. 2011;114:1435–45. doi: 10.1097/ALN.0b013e318218a881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swartjes M, Morariu A, Niesters M, Aarts L, Dahan A. Non-selective and NR2B-selective NMDA receptor antagonists produce antinociception and long-term relief of allodynia in acute and neuropathic pain. Anesthesiology. 2011;115:165–74. doi: 10.1097/ALN.0b013e31821bdb9b. [DOI] [PubMed] [Google Scholar]
- 13.Olofsen E, Romberg R, Bijl H, Mooren R, Engbers F, Kest B, Dahan A. Alfentanil and Placebo Analgesia: No Sex Differences Detected in Models of Experimental Pain. Anesthesiology. 2005;103:130–9. doi: 10.1097/00000542-200507000-00020. [DOI] [PubMed] [Google Scholar]
- 14.Gualtieri CT, Johnson LG. Reliability and validity of a computerized neurocognitive test battery, CNS Vital Signs. Arch Clin Neuropsychol. 2006;21:623–43. doi: 10.1016/j.acn.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Beal BL, Sheiner LB, Boeckman AJ, Bauer RJ. NONMEM User’s Guide. Icon development Solutions; Ellicott City, Maryland, USA: 1989-2009. [Google Scholar]
- 16.Noppers I, Niesters M, Aarts L, Smith T, Sarton E, Dahan A. Ketamine for treatment of chronic non-cancer pain. Exp Opin Pharmacother. 2010;11:2417–29. doi: 10.1517/14656566.2010.515978. [DOI] [PubMed] [Google Scholar]
- 17.Pomeroll-Clotet E, Honey GD, Murray GK, Corlett PR, Absalom AR, Lee M, McKenna PJ, Bullmore ET, Fletcher PC. Psychological effects of ketamine in healthy volunteers: Phenomenological study. Br J Psychiatry. 2006;189:173–9. doi: 10.1192/bjp.bp.105.015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passie T, Karts M, Wiese B, Emrich HM, Schneider U. Effects of different subanesthetic doses of (S)-ketamine on neuropsychology, psychopathology, and state of consciousness. Neuropsychobiol. 2005;51:226–33. doi: 10.1159/000085724. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell AC. Generalized hyperalgesia and allodynia following abrupt cessation of subcutaneous ketamine infusion. Palliat Medicine. 1999;13:427–8. doi: 10.1191/026921699667559279. [DOI] [PubMed] [Google Scholar]
- 20.Niesters M, Dahan A, Swartjes M, Noppers I, Fillingim R, Aarts L, Sarton E. Effect of Ketamine on Endogenous Pain Modulation in Healthy Volunteers. Pain. 152:656–63. doi: 10.1016/j.pain.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Sigtermans M, Noppers I, Sarton E, Bauer M, Mooren R, Olofsen E, Dahan A. An observational study on the effect of S(+)-ketamine on chronic versus experimental acute pain in Complex Regional Pain Syndrome type 1 patients. Eur J Pain. 2010;14:302–7. doi: 10.1016/j.ejpain.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt AP, Tort AB, Silveira PP, Böhmer AE, Hansel G, Knorr L, Schallenberger C, Dalmaz C, Elisabetsky E, Crestana RH, Lara DR, Souza DO. The NMDA antagonist MK-801 induces hyperalgesia and increases CSF excitatory amino acids in rats: Reversal by guanosine. Pharmacol Biochem Behav. 2009;91:549–53. doi: 10.1016/j.pbb.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Guan Y, Tereyama R, Dubner R, Ren K. Plasticity in excitatory amino acid receptor-mediated descending pain modulation and inflammation. J Pharmacol Exp Ther. 2002;300:513–20. doi: 10.1124/jpet.300.2.513. [DOI] [PubMed] [Google Scholar]
- 24.Hustveit O, maurset A, Oue I. Interaction of the chiral forms of ketamine with opioid, phencyclidine, sigma and muscarinic receptors. Pharmacol Toxicol. 1995;77:355–9. doi: 10.1111/j.1600-0773.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- 25.Maurice T, Katz JL. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schüttler J, Stanski DR, White PF, Trevor AJ, Horai Y, Verotta D, Sheiner LB. Pharmacodynamic modeling of the EEG effects of ketamine and its enantiomers in man. J Pharmacokinet Biopharm. 1987;15:241–53. doi: 10.1007/BF01066320. [DOI] [PubMed] [Google Scholar]
- 27.Herd DW, Anderson BJ, Keene NA, Holford NHG. Investigating the pharmacodynamics of ketamine in children. Ped Anesth. 2008;18:36–42. doi: 10.1111/j.1460-9592.2007.02384.x. [DOI] [PubMed] [Google Scholar]
- 28.Romberg R, Olofsen E, Sarton E, Teppema L, Dahan A. Pharmacodynamic effect of morphine-6-glucuronide versus morphine on hypoxic and hypercapnic breathing in healthy volunteers. Anesthesiology. 2003;99:788–98. doi: 10.1097/00000542-200310000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Olofsen E, van Dorp E, Teppema L, Aarts L, Smith TW, Dahan A, Sarton E. Naloxone reversal of morphine- and morphine-6-glucuronide-induced respiratory depression in healthy volunteers: a mechanism-based pharmacokinetic-pharmacodynamic modeling study. Anesthesiology. 2010;112:1417–27. doi: 10.1097/ALN.0b013e3181d5e29d. [DOI] [PubMed] [Google Scholar]
- 30.Olofsen E, Mooren R, van Dorp E, Aarts L, Smith T, den Hartigh J, Dahan A, Sarton E. Arterial and venous pharmacokinetics of morphine-6-glucuronide and impact of sample site on pharmacodynamic parameter estimates. Anesth Analg. 2010;111:626–32. doi: 10.1213/ANE.0b013e3181e5e8af. [DOI] [PubMed] [Google Scholar]