

Abstract
Autologous stem cell transplantation (ASCT) and novel therapies have improved overall survival of patients with multiple myeloma; however, most patients relapse and eventually succumb to their disease. Evidence indicates that residual cancer cells contaminate autologous grafts and may contribute to early relapses after ASCT. Here, we demonstrate that ex vivo treatment with an oncolytic poxvirus called myxoma virus results in specific elimination of human myeloma cells by inducing rapid cellular apoptosis while fully sparing normal hematopoietic stem and progenitor cells (HSPCs). The specificity of this elimination is based on strong binding of the virus to myeloma cells coupled with an inability of the virus to bind or infect CD34+ HSPCs. These two features allow myxoma to readily identify and distinguish even low levels of myeloma cells in complex mixtures. This ex vivo MYXV treatment also effectively inhibits systemic in vivo engraftment of human myeloma cells into immunodeficient mice and results in efficient elimination of primary CD138+ myeloma cells contaminating patient hematopoietic cell products. We conclude that ex vivo myxoma treatment represents a safe and effective method to selectively eliminate myeloma cells from hematopoietic autografts prior to reinfusion.
Keywords: Ex vivo Purging, Myxoma Virus, Viral Oncolytics, Multiple Myeloma, Autologous hematopoietic stem cell transplant
Introduction
Multiple myeloma (MM) is a clonal plasma cell malignancy that is most prevalent in adults over the age of 65 and accounts for 10-15% of newly diagnosed hematopoietic cancers with patients losing an average of 17 years of life expectancy per diagnosis(1). While recent clinical advances have improved prognosis for MM patients, life expectancy at diagnosis remains 2-5 years with a 5-year survival rate of only 34%(2, 3). Currently, the standard of care for MM patients is treatment with high dose chemotherapy followed by autologous stem cell transplant (ASCT). In North America alone, ASCT is used to treat approximately 5000 MM patients annually. This treatment results in improved rates of disease remission as well as significantly prolonged event-free survival time compared to patients treated with conventional chemotherapy(4, 5). However, despite the improved prognosis associated with myeloablative chemotherapy followed by ASCT, the treatment is generally not curative and a large majority of all MM patients will suffer from relapsed disease.
The malignant cells which cause relapse are thought to originate from two sources: residual MM cells which escape ablative therapy in bone marrow niches, and low levels of MM cells that contaminate the autografts. To date, the relative impact of each of these two sources in causing MM relapse remains unclear; however, several observations suggest that residual plasma cell contamination plays a significant role. First, it has been repeatedly reported that MM patients with lower levels of autograft contamination achieve greater benefit from transplant than patients with higher levels of contamination(6-8). Second, patients undergoing syngeneic transplant, using uncontaminated bone marrow from an identical twin donor, routinely achieve better rates of complete remission, improved long-term survival and possibly even complete cures(9-11). These data, particularly the syngeneic transplant studies, support the conclusion that even low levels of MM cells contaminating the autograft can play a significant role in disease relapse and suggest that ex vivo manipulation of the autograft prior to infusion to remove all contaminating malignant cells, a process known as purging(12), could improve MM patient outcomes.
Proposed MM purging procedures must meet two important criteria: 1) they must effectively remove all contaminating cancer cells from the grafts; and, 2) they must fully spare the normal hematopoietic stem/progenitor cells (HSPCs) in the autograft allowing for successful reconstitution of the patient’s hematopoietic system. Several purging methods have been explored in ASCT(13-16), including a recent study focusing on ex vivo culture conditions that favor survival of HSPCs(17). For MM, most of the focus has been placed on CD34+ stem cell enrichment(18-20) which can reduce the level of MM contamination within the graft by 2-3 logs(20). Unfortunately, clinical trials have demonstrated that this CD34 based purging does not improve clinical outcomes for MM patients(19, 21). The results of these trials were initially interpreted as proof that myeloma relapse was primarily caused by residual disease persisting in the patient following ablative chemotherapy; however, subsequent molecular studies have demonstrated that low levels of contaminating CD138+ MM cells remain in ASCT samples even after multiple rounds of CD34+ cell enrichment(22-24). Moreover, CD34+ malignant MM clones have been identified in patients which calls into questions the utility of CD34 enrichment in these patients(25, 26). Together, these data suggest that CD34+ stem cell enrichment might fail to improve MM patient prognosis because disease-causing MM cells remain in the autografts following positive CD34+ cell selection of peripheral blood stem cells. Therefore, alternative means of ex vivo purging must be explored(12).
Previously, our laboratory has demonstrated that a rabbit specific oncolytic poxvirus called myxoma virus (MYXV) can eliminate primary acute myeloid leukemia cells from primary human bone marrow samples while sparing normal HSPCs(27). MYXV is an attractive virotherapeutic to target and eliminate human cancer cells for several reasons. First, the virus does not elicit detectable disease in any non-rabbit species, including humans or severely immunocompromised mice(28, 29). Second, the ex vivo therapeutic application of MYXV is not dependent on expression of transgenes or addition of chemotherapeutic agents, and requires only a brief ex vivo incubation of the graft with MYXV prior to transplant, thus making it an attractive strategy for clinical administration that minimally deviates from standard ASCT clinical practice (27, 30). Due to our previous success using MYXV to purge primary human acute myeloid leukemia cells, the virus’s safety for the engraftment of normal human HSPCs, and the high rate of MM relapse after AHCT, we hypothesized that ex vivo MYXV treatment might represent an improved method for clinical elimination of MM cells contaminating patient autografts samples prior to reinfusion.
Materials and Methods
Cells and reagents
U266 (ATCC# TIB-196), RPMI-8266 (ATCC# CCL-155), MM.1S (ATCC# CRL-2974) and HuNS1 (ATCC# CRL-8644) human myeloma cells as well as HL60 acute myeloid leukemia cells (ATCC# CCL-240) were obtained from ATCC and were maintained below 2×106 cells/ml in RPMI media supplemented with 1x pen/strep, 2mM L-glutamine, and 20% FBS. The following antibodies were used: HLA-A,B,C-APC, CD45-PE, CD45-FitC, PARP, HLA-A2.1-PE (BD Bioscience), caspase 3 and cleaved caspase 3 (Cell Signaling), B actin (Ambion). Clinical grade heparin (1000 USP U/ml) was a kind gift from Dr. Alexandra Lucas. Primary MM cells were obtained by patient donation under the approval of the University of Florida Institutional Review Board.
MYXV and viral infections
vMyx-GFP has been previously described(31). Unless indicated, infections were carried out by exposing cells to vMyx-GFP at a multiplicity of infection (MOI) of 10 for one hour in PBS + 10% FBS in a humidified chamber at 37°C and 5% CO2. Mock-treated cells were incubated in PBS + 10% FBS containing no virus under the same incubation conditions. Treatment with inactivated virus was performed using the same incubation conditions but with inactivated vMyx-GFP prepared by exposing virus to UV light for two hours (UV inactivated) or incubating virus at 55°C for two hours (heat inactivated). Fluorescently tagged MYXV virions (vMyx-M093L-Venus) were created by fusing the GFP variant ‘Venus’ protein in frame to the N-terminus of the M093 open reading frame as previously described(32).
Analysis of MYXV infection in cultured myeloma cells
To measure initiation of early viral gene expression, human myeloma cells were analysed 24 hours after infection with vMyx-GFP for expression of GFP using flow cytometry. To measure completion of the viral replication cycle and production of new infectious progeny virus, myeloma cells were harvested at the indicated time points, pelleted and frozen. After harvesting, infectious virus was released by sequential freeze-thaw and the amount of virus in each sample was determined as previously described(33). To measure the binding of MYXV to the cell surface, myeloma cells were exposed to vMyx-M093L-Venus at MOI=10 for one hour at 37°C. Cells were then washed four times with PBS + 10% FBS. The contents of the resulting pellet (virus) as well as the supernatant from the last wash (wash) were then acid precipitated using trichloroacetic acid (final concentration 30%). Samples were resuspended in Laemmli buffer, separated on a 15% acrylamide gel, and transferred to PVDF membrane. The presence of viral proteins derived from MYXV virions was then analysed by standard immunoblot analysis using an αMYXV rabbit polyclonal serum(34).
Myeloma cell viability and proliferation assays
Human myeloma cells were mock-treated or infected with vMyx-GFP as above. For viability studies, 1×105 myeloma cells were plated in triplicate into 96-well plates. Twenty-four hours after treatment, cellular ATP generation, which correlates with cell viability, was measured using the commercial MTT assay (Pierce) as per the manufacturers recommended procedure. For cell proliferation studies, 1×104 myeloma cells were mock- or vMyx-GFP treated and plated in triplicate into 6 well dishes. Cell density was analysed every 24 hours by manually counting trypan blue excluding cells using a hemocytometer.
Multiple myeloma xenografts
For in vivo systemic engraftment studies, NOD/Scid/IL2Rγ−/− (NSG) mice were sublethally irradiated using 175 cGy total body irradiation from a Cs137 source. Within twenty-four hours after irradiation, mice were injected through the tail vein with 100ul PBS + 10% FBS containing 1×106 MM cells which had been either mock-treated or infected with vMyx-GFP at MOI=10. Prophylactic antibiotics were administered to animals in the drinking water for two weeks after transplantation to prevent opportunistic bacterial infection. Two to six weeks after transplantation, mice were euthanized and bone marrow was harvested from the right hind femur using a tuberculin syringe, stained with antibodies against human HLA-A,B,C and analyzed using flow cytometry. Data is presented as ‘level of engraftment’ which corresponds to the percent of HLA-A,B,C+ cells in the bone marrow of each mouse. Mice displaying any level of HLA-A,B,C+ cells were scored as engrafted (●) while mice without any detectable HLA-A,B,C+ cells were scored as non-engrafted (○). All animal experiments were carried out under approved University of Florida IACUC protocol# 2010-5023.
Mixing experiments with human myeloma into normal donor bone marrow samples
Fresh human bone marrow aspirates were obtained commercially from Lonza (Basal, Switzerland). Bone marrow mononuclear cells were then enriched over a Ficoll gradient using a clinical Sepax device (Biosafe Inc.) as per manufacturer’s recommendations. CD34+ human cells were fractionated from Sepax purified normal bone marrow aspirates using the CD34+ microbead separation kit (Miltenyi Biotec) as per manufacturer’s recommendations. Cells were then separated on an autoMACS pro separator (Miltenyi Biotec) as per manufacturer’s recommendations. The relative purity of the fractionated population was confirmed after separation using flow cytometry. The total number of fractionated cells was determined after separation using a hemocytometer. 1×105 CD34+ cells were then mixed with 1×106 U266 myeloma cells. U266 cells could be distinguished from normal hematopoietic cells based on their expression of HLA-A2.1 and CD138 as well as their lack of expression of CD45 and CD34.
Results
MYXV infects and kills human MM cell lines in vitro
The therapeutic potential of oncolytic viruses in vivo frequently correlates with their ability to selectively infect and kill target cancer cells in vitro. We therefore tested whether MYXV infected and killed four established human MM cell lines (U266, HuNS1, MM.1S and RPMI-8266) in vitro. Each cell line was incubated with vMyx-GFP, which expresses GFP from a viral synthetic early/late promoter, at a multiplicity of infection (MOI) of 10. After 24 hours of insubation, all four cell lines displayed significant numbers of GFP+ cells (Figure 1A) demonstrating that MYXV efficiently initiated infections in all four cell lines. Additionally, treatment with vMyx-GFP significantly reduced the cellular viability, as measured by the MTT mitochondrial function assay, of all four cell lines (Figure 1B) and completely abrogated subsequent cellular proliferation in culture (Figure 1C).
Figure 1. MYXV infects and kills multiple human MM cell lines in vitro.

(A) Each of the four human MM cell lines tested was infected with vMyx-GFP at MOI=10. 24 hours later, the percent of cells expressing GFP was quantified using flow cytometry. (B) Each MM cell line was mock-treated or infected with vMyx-GFP and the cellular viability was measured 24 hours after infection using the commercial MTT assay. (C) Each MM cell line was mock-treated or infected with vMyx-GFP and the number of trypan blue excluding cells was determined at the indicated times using a hemocytometer.
Ex vivo MYXV treatment inhibits in vivo systemic engraftment of MM cell lines into NSG mice
To determine if ex vivo MYXV treatment could inhibit systemic engraftment of MM cells in vivo,four human MM cell lines were mock-treated or treated with vMyx-GFP ex vivo and then transplanted intravenously (IV) into sublethally irradiated immunodeficient NSG mice. Two to six weeks after transplant, the animals were analyzed for human MM in femurs. Engrafted human MM cells were readily observed in the bone marrow of mice transplanted with mock-treated RPMI-8266 cells (9/10 mice positive, average human myeloma engraftment in mouse bone marrow = 6.2%), MM.1S cells (10/10 mice positive, average = 75.7%), HuNS1 cells (10/10 mice positive, average = 34.5%), and U266 cells (9/10 mice positive, average = 35.0%) demonstrating that all four MM cell lines effectively xenografted into NSG mouse bone marrow (Figure 2). In contrast, no human MM cells were detected in the bone marrow of mice transplanted with MYXV-treated RPMI-8266, HuNS1, or U266 cells, and were only observed at a very low level (0.1%) in 1 of the 10 mice injected with MYXV-treated MM.1S cells. Additionally, mice transplanted with MYXV-treated cells did not develop any symptoms of clinical MM, which were routinely observed two weeks after injection of mock-treated HuNS1 and five weeks after injection of mock-treated U266 cells (our unpublished observations).
Figure 2. MYXV inhibits systemic engraftment of human MM cell lines into immunodeficient NSG mice.
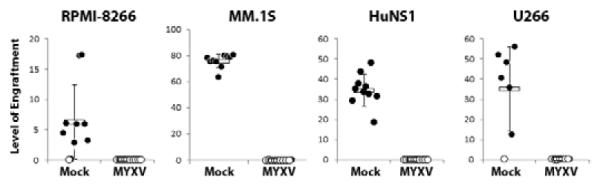
Each human MM cell line was mock-treated or treated with MYXV at MOI=10 (n=10) and then injected IV into NSG mice. Two weeks (HuNS1) or six weeks (RPMI-8266, MM.1S, U266) after injection, bone marrow was harvested from each mouse, stained with antibodies against human HLA-A,B,C, and analyzed using flow cytometry. Data is presented as ‘level of engraftment’, which corresponds to the percent of HLA-A,B,C+ cells in the bone marrow of each mouse. Mice displaying any level of HLA-A,B,C+ cells were scored as engrafted (●) while mice without any detectable HLA-A,B,C+ cells were scored as non-engrafted (○).
MYXV specifically eliminates MM cells while sparing normal hematopoietic stem cells
One of the major challenges associated with autograft purging is specifically eliminating contaminating cancer cells while sparing the normal HSPCs required for reconstitution of the patient’s hematopoietic system. To determine if MYXV treatment distinguished human MM cells from normal HSPCs we therefore treated mixtures of U266 cells and primary human HSPCs with vMyx-GFP and then analyzed GFP expression in both populations of cells after 24 hours. In these experiments, U266 cells can be distinguished from HSPCs based on expression of the surface markers HLA-A,B,C, HLA-A2.1, CD45, and CD34 (U266 cells are HLA-A,B,C+/HLA-A2.1+/CD45−/CD34− while human HSPCs are HLA-A,B,C+/HLA-A2.1−/CD45+/CD34+). Expression of GFP, indicating the initiation of viral infection, was observed in a significant percentage of HLA-A2.1+ U266 cells (45.9%). In contrast, expression of GFP was observed in less than 5% of the CD34+ cell population (Figure 3A).
Figure 3. MYXV eliminates human MM cells while sparing normal primary human hematopoietic stem cells.
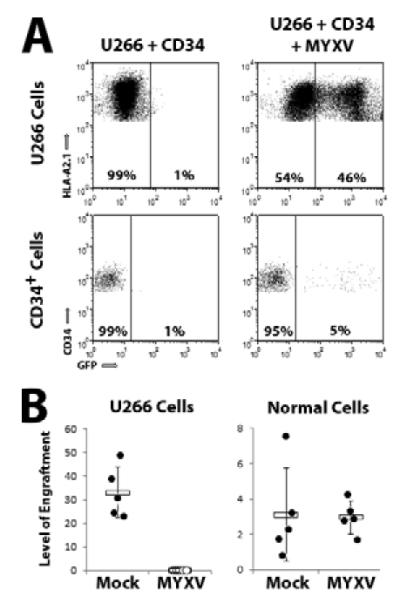
(A) U266 cells were mixed at a 10:1 ratio with donor human bone marrow derived CD34+ HSPCs. Mixtures were then mock-treated or infected with vMyx-GFP at MOI=10 for 24 hours and expression of GFP was analyzed using flow cytometry. Events shown are gated on live HLA-A2.1+ (Top) or CD34+ (Bottom) cells. Inset number indicates the percent of GFP− and GFP+ cells. (B) U266 cells were mixed at a 10:1 ratio with purified human bone marrow derived CD34+ HSPCs. Mixtures were then mock-treated or infected with vMyx-GFP at MOI=10 and injected IV into NSG mice. Six weeks after injection, bone marrow was harvested from each mouse, stained with antibodies against human HLA-A,B,C, human HLA-A2.1, and human CD45 and analyzed using flow cytometry. U266 cells were identified as staining HLA-A,B,C+/HLA-A2.1+/CD45− while progeny derived from normal HSPCs were identified as staining HLA-A,B,C+/HLA-A2.1−/CD45+. Data is presented as ‘level of engraftment’ which corresponds to the percent of HLA-A,B,C+/HLA-A2.1+ or HLA-A,B,C+/CD45+ cells in the bone marrow of each mouse. Mice displaying any level of positive cells were scored as engrafted (●) while mice without any detectable positive cells were scored as non-engrafted (○).
To confirm that MYXV treatment functionally discriminated MM cells from normal HSPCs, mixtures of U266 cells and primary human CD34+ HSPCs were mock-treated or treated with vMyx-GFP and then transplanted into sublethally irradiated NSG mice. Six weeks after transplant, engraftment of both U266 cells and repopulating human leukocytes derived from HSPCs were analyzed using flow cytometry. In mice transplanted with mock-treated mixtures, co-engraftment of both U266 cells as well as HSPCs was readily observed (Figure 3B). In contrast, mice injected with MYXV-treated mixtures failed to display detectable engraftment of U266 cells, while exhibiting engraftment of HSPCs at levels identical to mock-treated samples. Together, these results indicate that MYXV effectively discriminates between MM cells and normal HSPCs found in the same graft sample, and purges the MM cells while sparing the normal HSPCs allowing efficient hematopoiesis.
MYXV elimination of human MM cells requires direct virion binding
We recently discovered that the ability of MYXV to purge acute myeloid leukemia cells necessitates viral binding to the cell surface(35). We therefore tested whether MYXV also required direct adsorption of the virus particles in order to eliminate MM cells. U266 cells were treated with vMyx-GFP in either the presence of absence of soluble heparin, which inhibits poxvirus binding to mammalian cells by coating the positively charged virion ((36) and our unpublished observations). After 24 hours, a high percentage of U266 cells incubated in the absence of heparin displayed GFP expression while virtually no GFP+ cells could be identified in samples incubated in the presence of heparin (Figure 4A). The presence of heparin during infection also largely abrogated the ability of MYXV to reduce cellular viability (data not shown) and prevent subsequent cellular proliferation (Figure 4B). Finally, to determine if virion binding was required for MYXV to inhibit engraftment of MM cells in vivo, U266 cells were incubated with vMyx-M093L-Venus in either the presence of absence of soluble heparin and then transplanted IV into NSG mice. Consistent with our previous results, mice transplanted with mock-treated U266 cells displayed a high number of human cells engrafted in the bone marrow six weeks following transplant while these cells could not be detected in mice transplanted with ex vivo MYXV-treated cells. In contrast, engrafted human cells were readily detectable able in the bone marrow of mice transplanted with cells treated with MYXV in the presence of soluble heparin (Figure 4C) suggesting that effective purging requires MYXV binding to the target MM cancer cell.
Figure 4. MYXV-based killing of human MM cells in vitro requires virion binding.
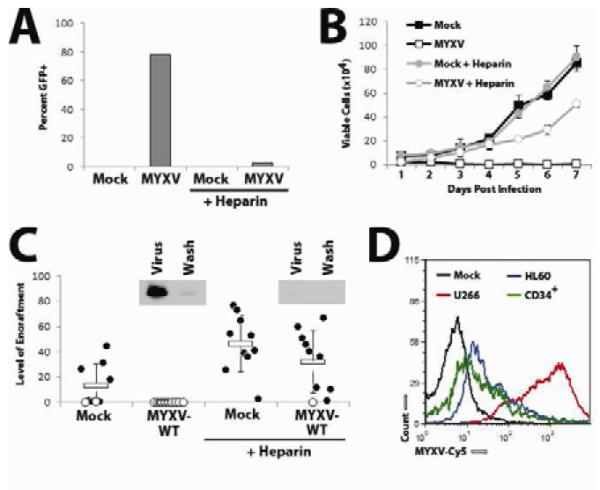
(A) U266 cells were infected with vMyx-GFP at MOI=10 in either the presence of absence of 5% soluble heparin. 24 hours later, the percent of cells expressing GFP was quantified using flow cytometry. (B) U266 cells were infected with vMyx-GFP at MOI=10 in either the presence of absence of 5% soluble heparin and the number of trypan blue excluding cells was determined at the indicated times using a hemocytometer. (C) U266 cells were mock-treated or treated with vMyx-GFP in either the presence or absence of 5% soluble heparin and then injected IV into NSG mice. Six weeks after injection, bone marrow was harvested from each mouse, stained with antibodies against human HLA-A,B,C and analyzed using flow cytometry. Data is presented as ‘level of engraftment’ which corresponds to the percent of HLA-A,B,C+ cells in the bone marrow of each mouse. Mice displaying any level of HLA-A,B,C+ cells were scored as engrafted (●) while mice without any detectable HLA-A,B,C+ cells were scored as non-engrafted (○). Inset depicts the binding of MYXV virions in the presence or absence of soluble heparin as detected by immunoblot using an αMYXV polyclonal antiserum. (D) U266 cells (red), HL60 cells (blue), or normal CD34+ stem cells (green) were incubated with vMYXV-Cy5 for one hour and then washed extensively. Cy5 florescence (indicating virion binding) was then analyzed using flow cytometry and compared to mock-treated cells (black).
MYXV does not bind to normal human HSPCs
Due to this result, we hypothesized that MYXV might spare normal human HSPCs due to a failure of these cells to support virion binding. To test this, primary human BM was mixed with U266 cells or HL60 cells, an acute myeloid leukemia cell line which is unable to support MYXV binding (our unpublished observations), and then incubated with vMyx-M093L-Venus virions. After washing, the amount of Venus florescence on U266 cells, HL60 cells, or primary CD34+ HSPCs was analyzed using flow cytometry (Figure 4D). While the MYXV-purgeable U266 cells displayed a high level of Venus florescence, corresponding to efficient MYXV virion binding, only low levels of Venus florescence were detected on the MYXV-nonpurgable HL60 cells and CD34+ HSPCs supporting the hypothesis that MYXV spares normal HSPCs because these cells do not support MYXV virion binding.
MYXV kills human MM cells by rapidly inducing apoptosis
Frequently, oncolytic viruses eradicate their targets through direct lytic replication; we therefore asked whether this mechanism was responsible for MYXV-based elimination of MM cells. Each of our four human MM cell lines was incubated with MYXV at MOI=1 and the replication of new infectious viral progeny was measured using single-step viral growth analysis. Surprisingly, we observed that even though MYXV effectively initiated infection and killed all four MM cell lines, no new viral progeny were created during this process (Figure 5A). Additionally, treatment of human MM cells with replication incompetent MYXV which had been UV-inactivated reduced cellular viability and proliferation comparably to treatment with live virus (Figure 5B and 5C). In contrast, treatment with heat-inactivated MYXV did not affect MM cell viability or proliferation.
Figure 5. MYXV kills human MM cells in vitro by inducing rapid apoptosis.
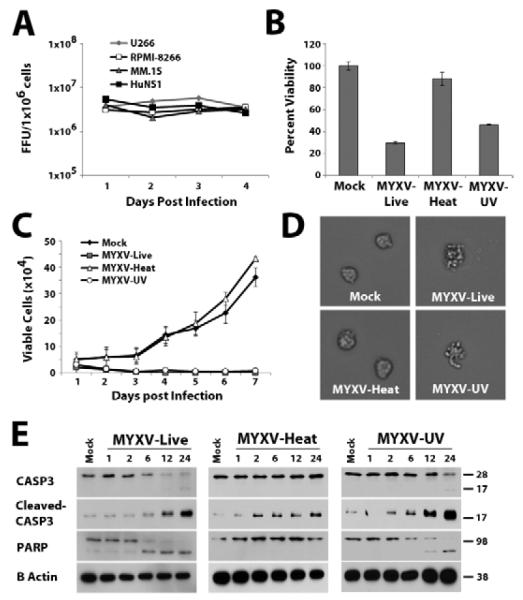
(A) Each human MM cell line was infected with vMyx-GFP at MOI=1. At the indicated times cells were harvested and frozen. After all time points had been collected, cells were lysed using repeated freeze-thaw and the amount of infectious virus in each sample was quantitated using foci formation on BSC40 cells. (B) U266 cells were either mock-treated or infected with live vMyx-GFP, heat-inactivated vMyx-GFP, or UV inactivated vMyx-GFP at an apparent MOI=10. After 24 hours cellular ATP generation, which correlates with the number of living cells, was measured using the commercial MTT assay. (C) U266 cells were mock-treated or infected with live-, heat- or UV-inactivated vMyx-GFP as above and the number of trypan blue excluding cells was determined at the indicated times using a hemocytometer. (D) U266 cells were mock-treated or infected with live-, heat- or UV-inactivated vMyx-GFP as above and then cellular morphology was observed 24 hours after infection using a Leica DMI 6000B light microscope. (E) U266 cells were mock-treated or infected with live-, heat- or UV-inactivated vMyx-GFP as above. Cells were harvested at the indicated time points and whole cell lystates were assayed with the indicated antibodies via immunoblot.
Since our data suggested that MYXV kills human MM cells via a mechanism that is not dependent on lytic viral replication, we next determined the cause of MYXV-induced MM cell death. A phenotypic examination of MM cells treated with MYXV revealed that these cells rapidly displayed membrane blebbing, a process frequently associated with apoptosis (Figure 5D). Additionally, U266 cells treated with either live- or UV-inactivated MYXV displayed cleavage of the apoptotic effector, caspase-3, as well as cleavage of PARP, a marker for late stage apoptosis (Figure 5E). We therefore conclude that MYXV kills human MM cells by inducing rapid cellular apoptosis that is triggered so quickly that it aborts the virus replication cycle prior to the generation of progeny virus.
MYXV infects primary human CD138+ myeloma cells contaminating patient bone marrow
Immortalized cell lines frequently exhibit phenotypes significantly different from primary cancer cells analyzed directly ex vivo. We therefore tested whether MYXV targeted and eliminated primary CD138+ MM cells contaminating primary patient bone marrow samples. Three bone marrow samples from different myeloma patients were mock treated or incubated with vMyx-M093L-Venus (which expresses the GFP variant ‘venus’ protein fused in-frame to the major structural protein M093, thus causing the virion particle to incorporate high levels of Venus that can be detected by flow cytometry). Immediately after viral adsorption, primary CD138+ MM cells from all three patient samples exhibited Venus florescence comparable to U266 cells (Figure 6A Top) indicating that MYXV bound to primary CD138+ MM cells and established myeloma cell lines at similar levels. Consistent with our previous results, virtually no Venus florescence was observed in the CD34+ HSPC compartment from the same patient samples (Figure 6B Top). After 24 hours, high numbers of CD138+ MM cells could still be found in all three mock treated samples, whereas virtually no contaminating CD138+ cells remained in any patient sample which had been treated with MYXV (Figure 6A Bottom). Significantly, the few CD138+ cells that still persisted after 24 hours displayed high levels of Venus fluorescence indicating they were undergoing active MYXV infection. In contrast, the number of CD34+ stem cells was unaffected by MYXV treatment and relatively few of these cells displayed any evidence of viral infection (Figure 6B Bottom). Unfortunately, our repeated attempts to engraft primary human MM samples into NSG mice resulted in little or no stable engraftment even in mock-treated controls, and so we were unable to document the in vivo efficacy of MYXV treatment at inhibiting engraftment of primary myeloma. However, our data clearly demonstrates that MYXV infects and eliminates U266 cells and primary CD138+ MM cells in vitro with similar efficacy in the context of myeloma patient bone marrow samples.
Figure 6. MYXV effectively eliminates CD138+ cells from primary MM patient samples.
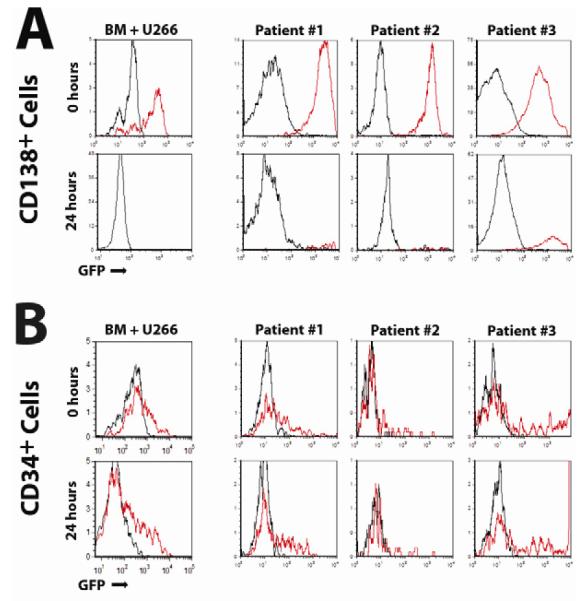
(A) Primary BM from three MM patients was mock-treated or incubated with vMyx-M093L-Venus at MOI=10 for one hour, stained with antibodies against either CD138 (A) or CD34 (B) and then washed extensively. Virus produced by vMYX-M093L-Venus incorporates high levels of Venus into the virion and can be used to quantify virion binding by flow cytometry. Venus florescence from mock-treated (black line) as well as vMyx-M093L-Venus treated (red line) samples was then analyzed using flow cytometry at 0 hours and 24 hours after adsorption. Data from normal bone marrow mixed with U266 cells is shown as a comparison.
Discussion
We describe a novel method to eliminate human MM cells from autologous HSPC samples ex vivo, using an oncolytic virus, MYXV, which selectively targets MM cells but does not bind or infect normal human CD34+ stem cells. MM disease relapse following ASCT may arise from two sources, residual disease sequestered in bone niches that failed to be eliminated by the myeloablative chemotherapy and/or low levels of MM cells that contaminate the HSPC graft. However, previous studies have demonstrated that MM patient prognosis does correlate with the level of contamination in stem cell product used for transplant(6-8) and that patients treated with syngeneic transplants from identical twin donors achieve considerably better rates of disease remission and prolonged survival(9-11). This strongly indicates that clinical outcomes will be improved if a method can be developed that quantitatively eliminates all contaminating MM cells from autologous HSPC samples. These observations have led to a variety of other methods being explored for purging MM cells from autograft samples, most notably positive selection of CD34+ HSPCs. Unfortunately, CD34+ positive selection based purging strategies have not proven clinically effective(18, 19). While the reason for these failures is not fully known, it has been demonstrated that CD34+ stem cell selection is unable to completely eliminate all MM cells contaminating ASCT products(22). Additionally, patient samples can contain a CD34+ clonal cell population related to the patient MM that cannot be eliminated by CD34+ enrichment. These data suggest that, while MM purging in general might be clinically beneficial, CD34+ stem cell enrichment is unable to completely eradicate disease-causing MM cells from HSPC samples. Alternatively, it is possible that the major cause of myeloma relapse following ASCT is residual disease persisting in the patient following ablative chemotherapy. Therefore, novel MM purging strategies that are not based on positive CD34+ stem cell selection, particularly procedure also capable of treating residual disease, are needed.
Our data demonstrates the effectiveness of oncolytic MYXV as a selective purging agent for human MM cells both in vitro and in vivo. Several other groups have recently published purging strategies for various malignancies based on other oncolytic viruses, including: adenovirus, vesicular stom atitis virus, herpes simplex virus, and reovirus [reviewed in(37, 38)]. While many of these viral treatments were effective in vitro, to date only three have demonstrated purging efficacy in vivo(39-41). Unfortunately, all three of these previously described viral purging strategies required the stem cell transplant graft to be incubated ex vivo for long periods of time, and then treated with cytotoxic chemotherapeutics prior to transplant, limiting applicability to clinical translation. In contrast, the MYXV-based ex vivo purging strategy described here demonstrates excellent efficacy against human MM both in vitro and in vivo, did not harm the engraftment potential of normal HSPCs, and required minimal ex vivo manipulation of the autograft prior to transplant. Therefore, this study represents the first report of an effective viral ex vivo purging strategy that demonstrates efficacy at preventing relapse in in vivo models and can be easily translated to the clinic. Additionally, the absence of documented cases of MYXV infection in any non-rabbit species, including humans or mice, suggests that MYXV possesses an excellent safety profile for in vivo use.
Interestingly, while MYXV treatment had a profound effect on long term MM growth both in vitro and in vivo, evidence of direct viral infection, as measured by GFP expression from an encoded transgene, was only observed in 50-70% of treated cells (Figure 1). This could be related to the limitations in detecting GFL fluorescence or to the ability of MYXV to eliminate MM cells through a mechanism independent of viral replication. For example, epitopes on the MYXV virion may act as functional ligands that trigger the rapid activation of a cell surface death receptor on MM cells, such as those of the TNF-receptor superfamily. This second direct-killing hypothesis is supported by the observation that U266 cells treated with MYXV at MOI’s as low as 0.1 universally undergo apoptosis and fail to engraft into NSG mice (data not shown). Importantly, the observed MYXV-induced reductions in cellular viability and levels of direct infection were similar in both the four established human MM cell lines as well as all three primary patient samples, and these levels of MM cell depletion translated into significant clinical benefit in vivo in terms of the ability to prevent engraftment of the MYXV-treated MM cell lines. These data collectively suggest that ex vivo MYXV treatment will be efficacious in reducing relapse rates of MM caused by infusion of contaminated ASCT samples.
It is believed that residual MM cells persisting in the patient bone marrow despite myeloablative chemotherapy also play a major role in patient disease relapse. Interestingly, systemic injection of oncolytic vaccinia virus has been shown to decrease levels of established MM in humans(42), while injection of oncolytic measles virus bound to MM carrier cells has been shown to effectively treat established MM in murine models(43, 44). Additionally, it has been demonstrated that treatment with a variety of oncolytic viruses can result in elimination of residual cancer cells from therapy-resistant niches by redirecting the immune system to more effectively recognize tumor antigens(45, 46). While still untested, it should be noted that because of the potent MM cell death induced by cell contact with this virus, the MYXV-based purging strategy described here also has the potential to treat established MM which is resistant to current therapy through all these mechanisms. Alternatively, systemic injection of MYXV might also be used to directly treat residual myeloma that has resisted the primary myeloablative chemotherapy. In our preliminary experiments, such treatments can have promising efficacy in mouse models of pre-established myeloma (our unpublished observations). Significant issues, such as developing an optimal strategy to deliver virus to all potential myeloma niches in vivo, remain to be addressed. Despite these limitations, however, we propose that MYXV based virotherapy, either as an ex vivo MYXV-based purging protocol or possibly augmented by systemic viral injection, has the potential to treat both MM contaminated autografts as well as in vivo residual MM which persists following ablative therapy. Interestingly, since UV-inactivated and live-MYXV showed similar in vitro efficacy at inducing apoptosis in MM cells, it is possible that completely replication-incompetent virus could also be used to eliminate MM cells contaminating the ASCT grafts. This would create an even safer clinical purging agent and alleviate any ethical concerns associated with injection of live virus.
Significantly, we demonstrate that when normal primary CD34+ human HSPCs are admixed with human MM cells, the resulting mixture causes engraftment of both normal HSPCs and MM cells in the bone marrow of NSG mice. However, in stark contrast, when these same mixtures are treated ex vivo with MYXV, only the normal HSPCs successfully engraft into the recipients while the contaminating MM cells are selectively purged. This specificity appears to be due to the selective binding of MYXV to MM cells but not to primary CD34+ HSPCs. Interestingly, poxviruses like MYXV are extremely promiscuous in their binding to relatively conserved glycosaminoglycans, effectively attaching to a broad spectrum of mammalian cells(47).
In conclusion, our data demonstrates that ex vivo MYXV treatment of autografts contaminated with MM is safe and effective in preclinical models of human MM. Results from this work support the translation of this novel purging method to the clinic.
Acknowledgements
We thank Sherin Smallwood for preparing the IACUC and IRB protocols, Dorothy Smith for preparing virus stocks for the study, and Amy Meacham and Elizabeth Wise for assisting with animal experiments. This study was supported by start-up funding to G.M. from the University of Florida-College of Medicine, NIH-R01 CA138541 and the Florida Department of Health Bankhead Coley Research Program Team Science grant 1BT02, and a research grant to E.B. from STOP! Children’s Cancer, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure Statement: Authors declare no financial interests
References
- 1.Ludwig H, Bolejack V, Crowley J, et al. Survival and years of life lost in different age cohorts of patients with multiple myeloma. J Clin Oncol. 2008;28(9):1599–605. doi: 10.1200/JCO.2009.25.2114. [DOI] [PubMed] [Google Scholar]
- 2.Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood. 2008;111(5):2521–6. doi: 10.1182/blood-2007-08-104984. [DOI] [PubMed] [Google Scholar]
- 3.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attal M, Harousseau JL, Stoppa AM, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335(2):91–7. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 5.Bjorkstrand B, Gahrton G. High-dose treatment with autologous stem cell transplantation in multiple myeloma: past, present, and future. Semin Hematol. 2007;44(4):227–33. doi: 10.1053/j.seminhematol.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Gertz MA, Witzig TE, Pineda AA, Greipp PR, Kyle RA, Litzow MR. Monoclonal plasma cells in the blood stem cell harvest from patients with multiple myeloma are associated with shortened relapse-free survival after transplantation. Bone Marrow Transplant. 1997;19(4):337–42. doi: 10.1038/sj.bmt.1700670. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Perez R, Garcia-Sanz R, Gonzalez D, et al. The detection of contaminating clonal cells in apheresis products is related to response and outcome in multiple myeloma undergoing autologous peripheral blood stem cell transplantation. Leukemia. 2000;14(8):1493–9. doi: 10.1038/sj.leu.2401862. [DOI] [PubMed] [Google Scholar]
- 8.Vogel W, Kopp HG, Kanz L, Einsele H. Myeloma cell contamination of peripheral blood stem-cell grafts can predict the outcome in multiple myeloma patients after high-dose chemotherapy and autologous stem-cell transplantation. J Cancer Res Clin Oncol. 2005;131(4):214–8. doi: 10.1007/s00432-004-0635-y. [DOI] [PubMed] [Google Scholar]
- 9.Bashey A, Perez WS, Zhang MJ, et al. Comparison of twin and autologous transplants for multiple myeloma. Biol Blood Marrow Transplant. 2008;14(10):1118–24. doi: 10.1016/j.bbmt.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gahrton G, Svensson H, Bjorkstrand B, et al. Syngeneic transplantation in multiple myeloma - a case-matched comparison with autologous and allogeneic transplantation. European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1999;24(7):741–5. doi: 10.1038/sj.bmt.1701975. [DOI] [PubMed] [Google Scholar]
- 11.Bensinger WI, Demirer T, Buckner CD, et al. Syngeneic marrow transplantation in patients with multiple myeloma. Bone Marrow Transplant. 1996;18(3):527–31. [PubMed] [Google Scholar]
- 12.Alvarnas JC, Forman SJ. Graft purging in autologous bone marrow transplantation: a promise not quite fulfilled. Oncology (Williston Park) 2004;18(7):867–76. discussion 76-8, 81, 84. [PubMed] [Google Scholar]
- 13.Motta MR, Mangianti S, Rizzi S, et al. Pharmacological purging of minimal residual disease from peripheral blood stem cell collections of acute myeloblastic leukemia patients: preclinical studies. Exp Hematol. 1997;25(12):1261–9. [PubMed] [Google Scholar]
- 14.Strauss G, Westhoff MA, Fischer-Posovszky P, et al. 4-hydroperoxy-cyclophosphamide mediates caspase-independent T-cell apoptosis involving oxidative stress-induced nuclear relocation of mitochondrial apoptogenic factors AIF and EndoG. Cell Death Differ. 2008;15(2):332–43. doi: 10.1038/sj.cdd.4402272. [DOI] [PubMed] [Google Scholar]
- 15.Spyridonidis A, Schmidt M, Bernhardt W, et al. Purging of mammary carcinoma cells during ex vivo culture of CD34+ hematopoietic progenitor cells with recombinant immunotoxins. Blood. 1998;91(5):1820–7. [PubMed] [Google Scholar]
- 16.Gee A, Moss T, Mansour V, et al. Large-scale immunomagnetic separation system for the removal of tumor cells from bone marrow. Prog Clin Biol Res. 1992;377:181–7. [PubMed] [Google Scholar]
- 17.Yang H, Robinson SN, Nieto Y, et al. Ex vivo graft purging and expansion of autologous blood progenitor cell products from patients with multiple myeloma. Cancer Res. 2011;71(14):5040–9. doi: 10.1158/0008-5472.CAN-11-0842. [DOI] [PubMed] [Google Scholar]
- 18.Ho J, Yang L, Banihashemi B, et al. Contaminating tumour cells in autologous PBSC grafts do not influence survival or relapse following transplant for multiple myeloma or B-cell non-Hodgkin’s lymphoma. Bone Marrow Transplant. 2009;43(3):223–8. doi: 10.1038/bmt.2008.318. [DOI] [PubMed] [Google Scholar]
- 19.Bourhis JH, Bouko Y, Koscielny S, et al. Relapse risk after autologous transplantation in patients with newly diagnosed myeloma is not related with infused tumor cell load and the outcome is not improved by CD34+ cell selection: long term follow-up of an EBMT phase III randomized study. Haematologica. 2007;92(8):1083–90. doi: 10.3324/haematol.10535. [DOI] [PubMed] [Google Scholar]
- 20.Abonour R, Scott KM, Kunkel LA, et al. Autologous transplantation of mobilized peripheral blood CD34+ cells selected by immunomagnetic procedures in patients with multiple myeloma. Bone Marrow Transplant. 1998;22(10):957–63. doi: 10.1038/sj.bmt.1701473. [DOI] [PubMed] [Google Scholar]
- 21.Stewart AK, Vescio R, Schiller G, et al. Purging of autologous peripheral-blood stem cells using CD34 selection does not improve overall or progression-free survival after high-dose chemotherapy for multiple myeloma: results of a multicenter randomized controlled trial. J Clin Oncol. 2001;19(17):3771–9. doi: 10.1200/JCO.2001.19.17.3771. [DOI] [PubMed] [Google Scholar]
- 22.Lemoli RM, Fortuna A, Motta MR, et al. Concomitant mobilization of plasma cells and hematopoietic progenitors into peripheral blood of multiple myeloma patients: positive selection and transplantation of enriched CD34+ cells to remove circulating tumor cells. Blood. 1996;87(4):1625–34. [PubMed] [Google Scholar]
- 23.Rasmussen T, Bjorkstrand B, Andersen H, Gaarsdal E, Johnsen HE. Efficacy and safety of CD34-selected and CD19-depleted autografting in multiple myeloma patients: a pilot study. Exp Hematol. 2002;30(1):82–8. doi: 10.1016/s0301-472x(01)00758-5. [DOI] [PubMed] [Google Scholar]
- 24.Voena C, Locatelli G, Castellino C, et al. Qualitative and quantitative polymerase chain reaction detection of the residual myeloma cell contamination after positive selection of CD34+ cells with small- and large-scale Miltenyi cell sorting system. Br J Haematol. 2002;117(3):642–5. doi: 10.1046/j.1365-2141.2002.03448.x. [DOI] [PubMed] [Google Scholar]
- 25.Szczepek AJ, Bergsagel PL, Axelsson L, Brown CB, Belch AR, Pilarski LM. CD34+ cells in the blood of patients with multiple myeloma express CD19 and IgH mRNA and have patient-specific IgH VDJ gene rearrangements. Blood. 1997;89(5):1824–33. [PubMed] [Google Scholar]
- 26.Takishita M, Kosaka M, Goto T, Saito S. Cellular origin and extent of clonal involvement in multiple myeloma: genetic and phenotypic studies. Br J Haematol. 1994;87(4):735–42. doi: 10.1111/j.1365-2141.1994.tb06732.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim M, Madlambayan GJ, Rahman MM, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23(12):2313–7. doi: 10.1038/leu.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenner F, Ratcliffe FN. Myxomatosis. Cambridge University Press; Cambridge, UK: 1965. [Google Scholar]
- 29.Stanford MM, McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin Biol Ther. 2007;7(9):1415–25. doi: 10.1517/14712598.7.9.1415. [DOI] [PubMed] [Google Scholar]
- 30.Rahman MM, Madlambayan GJ, Cogle CR, McFadden G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev. 2010;21(2-3):169–75. doi: 10.1016/j.cytogfr.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston JB, Barrett JW, Chang W, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J Virol. 2003;77(10):5877–88. doi: 10.1128/JVI.77.10.5877-5888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan W, McFadden G. Characterization of a Recombinant Myxoma Virus Expressing Venus Protein Fused to a Virion-specific Protein M093. 2012.
- 33.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010 doi: 10.1002/9780471729259.mc14a01s17. Chapter 14:Unit 14A 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macen JL, Upton C, Nation N, McFadden G. SERP1, a serine proteinase inhibitor encoded by myxoma virus, is a secreted glycoprotein that interferes with inflammation. Virology. 1993;195(2):348–63. doi: 10.1006/viro.1993.1385. [DOI] [PubMed] [Google Scholar]
- 35.Madlambayan G, Bartee E, Manbok K, et al. Acute Myeloid Leukemia Purging by Myxoma Virus Depends on Cell Binding and Not Permissiveness to Infection. Leukemia Research. 2011 doi: 10.1016/j.leukres.2012.01.020. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol. 1998;72(2):1577–85. doi: 10.1128/jvi.72.2.1577-1585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thirukkumaran CM, Russell JA, Stewart DA, Morris DG. Viral purging of haematological autografts: should we sneeze on the graft? Bone Marrow Transplant. 2007;40(1):1–12. doi: 10.1038/sj.bmt.1705668. [DOI] [PubMed] [Google Scholar]
- 38.Bais S, Bartee E, Rahman M, McFadden G, Cogle CR. Oncolytic Virotherapy for Hematological Malignancies. Advances in Virology. 2011 doi: 10.1155/2012/186512. Accepted, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lillo R, Ramirez M, Alvarez A, et al. Efficient and nontoxic adenoviral purging method for autologous transplantation in breast cancer patients. Cancer Res. 2002;62(17):5013–8. [PubMed] [Google Scholar]
- 40.Wagner LM, Guichard SM, Burger RA, et al. Efficacy and toxicity of a virus-directed enzyme prodrug therapy purging method: preclinical assessment and application to bone marrow samples from neuroblastoma patients. Cancer Res. 2002;62(17):5001–7. [PubMed] [Google Scholar]
- 41.Garcia-Sanchez F, Pizzorno G, Fu SQ, et al. Cytosine deaminase adenoviral vector and 5-fluorocytosine selectively reduce breast cancer cells 1 million-fold when they contaminate hematopoietic cells: a potential purging method for autologous transplantation. Blood. 1998;92(2):672–82. [PubMed] [Google Scholar]
- 42.Kawa A, Arakawa S. The effect of attenuated vaccinia virus AS strain on multiple myeloma; a case report. Jpn J Exp Med. 1987;57(1):79–81. [PubMed] [Google Scholar]
- 43.Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther. 2010;18(6):1155–64. doi: 10.1038/mt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng KW, Dogan A, Vrana J, et al. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol. 2009;84(7):401–7. doi: 10.1002/ajh.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prestwich RJ, Errington F, Diaz RM, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20(10):1119–32. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19(6):1008–16. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3(3):201–13. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]