

Abstract
A portion of HIV-infected patients under therapy with protease inhibitors (HIV PIs) concomitantly consume or abuse alcohol leading to hepatic injury. The underling mechanisms are not known. We hypothesize that HIV PI aggravates alcohol-induced liver injury through an endoplasmic reticulum (ER) stress mechanism. To address this, we treated mice and primary mouse and human hepatocytes (PMH and PHH respectively) with alcohol and the two HIV PIs: ritonavir and lopinavir. In mice, ritonavir and lopinavir (15 mg/kg body weight each) induced mild ER stress and inhibition of Sarco/ER Calcium-ATPase (SERCA) without significant increase in serum ALT levels. However, a single dose of alcohol by gavage (5g/kg body weight) plus the two HIV PIs caused a greater than 5-fold increase in serum ALT, a synergistic increase in alcohol-induced liver lipid accumulation and ER stress response, and a decrease of SERCA. Mice treated with chronic HIV PIs and alcohol developed moderate liver fibrosis. In PMH, the HIV drugs plus alcohol also inhibited SERCA expression and increased expression of GRP78, CHOP, SREBP1c and phosphorylated JNK2, which were accompanied by a synergistic increase in cell death compared to alcohol or the HIV drugs alone. In PHH, ritonavir and lopinavir or alcohol alone treatment increased mRNA of spliced Xbp1 and decreased SERCA, which were accompanied by reduced levels of intracellular calcium. Alcohol combined with the HIV drugs significantly reduced intracellular calcium levels and potentiated cell death, which was comparable to the cell death caused by the SERCA inhibitor-thapsigargin. Our findings suggest the possibility that HIV PIs potentiate alcohol-induced ER stress and injury through modulation of SERCA and maintaining calcium homeostasis should be a therapeutic aim for a better care of HIV patients.
Introduction
Excessive alcohol consumption continues to be a leading cause of chronic liver disease worldwide1, 2. Chronic alcohol-induced liver disease (ALD) includes a spectrum of liver diseases, from fatty liver or simple steatosis, alcoholic hepatitis, to hepatic fibrosis or cirrhosis3. A growing list of primary and secondary risk factors for ALD has been identified. Primary factors can be alcohol metabolite-acetalaldehyde4, oxidative stress from mitochondrial malfunction and cytochrome P450IIE1 (CYP2E1)5, increased endotoxin and inflammatory TNFα6, centrilobular hypoxia7, impaired one carbon metabolism8, 9, impaired innate and adaptive immunity3, and epigenetic alterations8, 10. Secondary factors can be malnutrition or complications with diabetes, obesity, gender difference, smoking, or HCV/HIV infection11-13. Alcohol-induced unfolded protein response (UPR) in the endoplasmic reticulum (ER) has evolved as an important factor contributing to alcoholic fatty liver and injuries 14-20. Potential causes for alcohol-induced ER stress are directly or indirectly related to alcohol metabolism, which include but may not be limited to: toxic acetaldehyde that forms protein adducts, increased homocysteine/homocysteine thiolactone that modifies proteins, oxidative stress that disturbs oxidative protein folding, alterations of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) ratio that causes epigenetic modifications of ER stress response components, and disturbance of calcium homeostasis14-20. In addition, interactions between the primary factors and secondary factors may determine severity of liver injury from alcohol-induced ER stress. For instance, the Sarco/ER Calcium-ATPase (SERCA), which regulates calcium store and ER homeostasis, has recently been identified to be a key factor involved in the complex obesity-induced ER stress and fatty liver injury21, 22. It is not known whether SERCA also play a role in interactions between the primary and secondary risk factors for alcohol-induced stress and liver injury.
HIV protease inhibitors (HIV PIs) have been used in the highly active antiretroviral combination therapy (HAART) that dramatically decreases the mortality rate of HIV-infected patients in western countries23, 24. However, HIV PI-induced hepatotoxicity or lipodystrophy has emerged as an important potential complication of HAART. Lipid dysregulation in hepatocytes and macrophages has been associated with HIV PIs, most commonly with a single administration of full doses24-27. Mechanisms that contribute to the side effects by HIV PIs in the liver are not well understood. Recent evidence suggests that HIV PIs induce ER stress response and promote liver injury28-31. For instance, at therapeutic concentrations (i.e. single PI at 5–50 μM), most HIV PIs, individually or combined, were found to increase the levels of ER stress markers such as active sterol regulatory element-binding proteins (SREBPs), X box-binding protein 1 (XBP-1), activating transcription factor 4 (ATF-4), C/EBP homologous protein (CHOP) and caspase-12, and increase apoptosis in macrophages and rat hepatocytes28-30. A few possible cellular stress events, such as depletion of ER calcium store and impaired lipid synthesis in the ER by HIV PIs are proposed to activate the UPR28-30. In addition, other ER stress causing factors such as alcohol or hepatitis C virus (HCV) and B virus (HBV) may aggravate HIV PI-induced ER stress and liver injury. Indeed, portion of HIV-infected patients consume or abuse alcohol12, 13, 32, which complicates the outcome of HAART. Thus, in order to provide new strategies for a better caring of HIV-infected patients, it is necessary to investigate possible synergism between HIV PI and alcohol with respect to ER stress and liver injury. In this study, we examined ER stress response and cell death in mice and primary mouse and human hepatocytes and found that calcium homeostasis is a potential target of the interaction between alcohol and HIV drugs that worsens liver injury.
Methods
Animal experiments
Male C57BL/6 mice from the Jackson Laboratory were used. For acute alcohol feeding, the animals were fasted for 6 hours and gavaged with a liquid diet containing alcohol (Dyets, Inc., Bethlehem, PA) at a dose of 5 g/kg body weight (bw), or with an isocaloric control diet. In some experiments, HIV protease inhibitor-ritonavir (RIT, 15 mg/kg bw) or ritonavir combined with lopinavir (LOP, 15 mg/kg bw) were mixed with the alcohol liquid diet and delivered into the mice through gavage. The mice were sacrificed 20-24 hours after the alcohol and/or drug administrations. For chronic treatment, mice were fed the liquid diet at 3 g alcohol/kg bw for the first week, and at 5 g alcohol/kg bw for the next four weeks. RIT plus LOP (15 mg/kg bw each) were i.p. injected into mice every other day from the second week to the fifth week. All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals approved by the local animal care committee.
Liver pathological parameters
Serum alanine aminotransferase (ALT) and liver lipid extraction and analysis were described previously14, 30, 33. For Oil Red O staining, liver tissues were embedded in O.C.T., snap frozen, sectioned at 5μm, and mounted on glass slides. The tissues on the slides were fixed in 10% formalin and stained with an Oil Red O isopropanol solution (Electron Microscopy Sciences, Hatfield, PA).
HPLC detection of HIV protease inhibitors
RIT and LOP in mouse plasma were extracted using the solid phase Sep-Pak Plus C18 cartridge from Waters (Waters, Milford, MD). The cartridge was conditioned with 1 ml of methanol followed by 1 ml of water, loaded with an aliquot of plasma (250 μl), and incubated at room temperature for 30 minutes. The cartridge was then washed with 1 ml of water, followed by 1 ml of methanol-water solution (30:70, vol/vol). The drugs were eluted with 1 ml of methanol, dried by blowing with nitrogen gas, and then dissolved in a mobile solution (250 μl) containing acetonitrile mixed with NaH2PO4 (20 mM, pH 6) at 60:40 (vol:vol). The mixture contained 0.025% triethylamine. An aliquot of the drug extracts (50 μl) with 1 μg of RIT or LOP (added as internal control) was injected onto a C18 reverse-phase HPLC column. The drugs were monitored spectrophotometrically at 210 nm. The drug quantity was calculated based on total area under the curve of HPLC spectrum for each drug.
In vitro studies with primary mouse hepatocytes
Primary mouse hepatocytes were provided by the Cell Culture Core (USC Research Center for Liver Disease) and the cell culturing was described previously33. The hepatocytes were treated with 15 μg/ml of RIT and/or LOP with or without alcohol (85 mM) for 24 hours. Vehicle (DMSO, <0.5%; v/v) was used as control. The treated hepatocytes were washed with cold PBS and were either stained for cell death or scratched off for protein extraction. The cells were doubly stained with Sytox green (Molecular Probes, Eugene) and Hoechst 33258 dye (Sigma) for 30 minutes at 37°C. The cell death count (combination of necrosis and apoptosis) was previously described30, 33.
In vitro studies with primary human hepatocytes
Primary human hepatocytes (PHH) were freshly isolated from alcohol- or drug-free patients and were cold preserved in 24-well plates overlaid with collagen or in suspension, which were shipped by CellzDirect to Dr. Shelly Lu who allocated us a portion of the PHH enough for each set of experiments. Upon arrival, the original media were replaced with a fresh DMEN medium containing gentamicin (0.1% v/v), dexamethasone (0.01%, v/v), fetal bovine serum (5.5%, v/v), L-glutamine (1%, v/v), HEPES (15 mM), and insulin (0.1% v/v). The PHH in the fresh medium were incubated in a humidified incubator at 37C for 8-12 hours to acclimatize, after which, the medium was replaced with a serum-free DMEN medium containing penicillin-streptomycin (0.5%, v/v), dexamethasone (10 ppm), ITS (1%, v/v), L-glutamine (1%, v/v), and HEPES (15 mM). The PHH were then treated with RIT (5-15 μg/ml) and LOP (10-15 μg/ml) and/or alcohol (35-85 mM), tunicamycin (3 μg/ml), thapsigargin (6 μg/ml), or homocysteine thiolactone (1 mM) for 24 hours. DMSO was used as control. After the treatments, the PHH were washed with cold PBS and were either scratched off for protein and RNA extraction or stained for cell death. Portion of the treated PHH were used for evaluation of intracellular Ca2+ using Fluo-4 (Molecular Probe) according to the manufacture protocol. The intracellular Ca2+ levels were monitored with Microplate Fluorescence Reader (Molecular Devices Corp.) after addition of thapsigargin (5 μM).
Immunoblotting and RT-PCR
Methods for protein extraction and analysis and for mRNA extraction and RT-PCR analysis, antibodies for ER stress markers, and the specific primers for spliced Xbp1 were reported previously14, 30, 33. Antibodies for SERCA were from Santa Cruz (sc-271669). The intensity of protein bands on the Western blots were quantified with the NIH software, ImageJ, after the blots of protein samples were scanned into TIF files.
Confocal fluorescence microscopy
The hepatocytes were fixed in cold acetone for 10 min and washed in PBS three times. The fixed cells were blocked with PBS containing 5% of BSA and 2% of serum for secondary anti-bodies for 30 min, washed 3 times in PBS, incubated with anti-GRP78 antibodies in PBS containing 1.5 % BSA for 60 min, and washed three times with PBS. Antibody binding was detected with FITC-conjugated secondary antibodies (Southern Biotechnology Associates, Birmingham, AL). Microfilaments of the cells were stained with rhodamine-phalloidin (1:100 dilutions, 30 min). The stained cells were examined by using a Nikon PCM2000 confocal laser-scanning microscope.
Statistical Analysis
Values are expressed as means ± s.e.m. unless otherwise indicated. Statistical analyses were performed using the Student's t test or ANOVA for comparison of multiple groups. P < 0.05 or less was considered significant.
Results
1. HIV protease inhibitor- and alcohol-induced ER stress and liver injury
Single dose alcohol administration increased serum ALT levels by two fold in mice (Fig. 1A). Neither HIV protease inhibitor (HIV PI) ritonavir (RIT) alone nor RIT combined with lopinavir (LOP) or with alcohol had significant effects on ALT values. However, more than five-fold increase in ALT was detected in mice treated with alcohol plus the two HIV drugs. The acute alcohol treatment induced a moderate increase in liver triglycerides and a slight increase in liver cholesterol (Fig. 1B and 1C). Similar to their effects on ALT, RIT and LOP synergistically increased alcohol-induced lipid accumulation (Fig. 1B and 1C), which was confirmed histologically with Oil Red O staining (Fig. 1D). To know whether alcohol affected the metabolism or bioavailability of the two HIV drugs, we extracted RIT and LOP from plasma of the treated mice, and detected the drugs with HPLC. The retention times for RIT and LOP were 17.5±0.2 and 19.6±0.2 minutes respectively (Fig. 2). Alcohol did not affect the plasma RIT level significantly. Interestingly, alcohol increased the LOP level, which was further increased in response to an increased alcohol dose (Fig. 2D).
Figure 1.
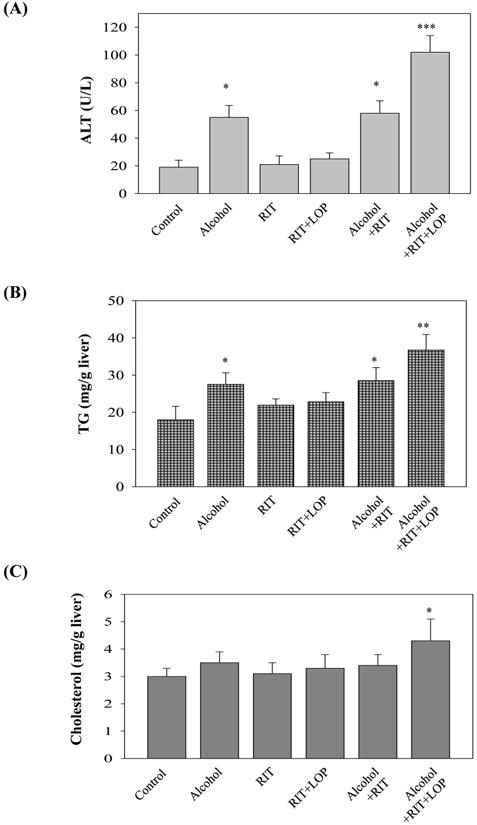
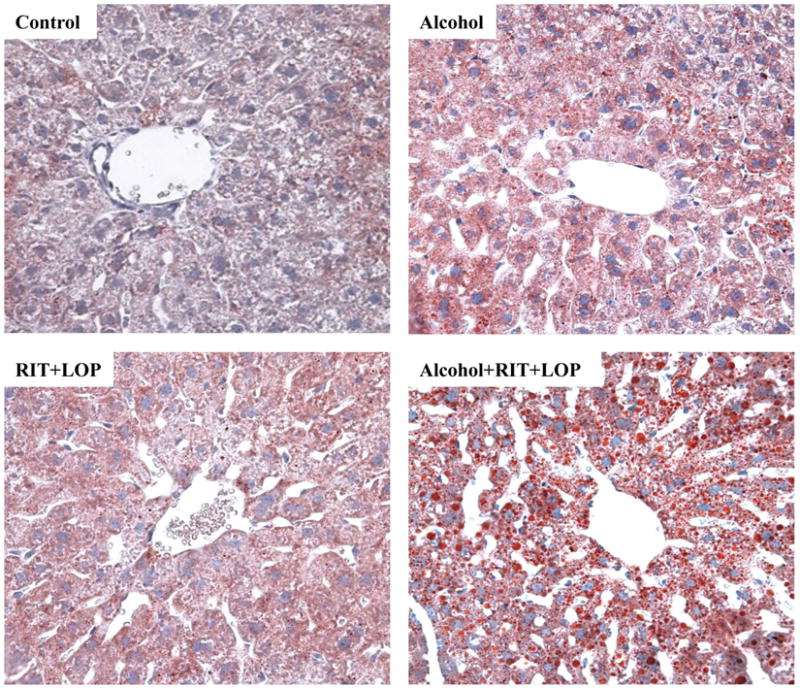
Liver injury in mice gavaged with alcohol and/or HIV protease inhibitors (HIV PIs). RIT, ritonavir (15 mg/kg body weight); LOP, lopinavir (15 mg/kg body weight); (A) Serum alanine aminotransferase (ALT); (B) Liver triglycerides; (C) Liver cholesterol; *p<0.05; **p<0.01; ***p<0.005 compared to control, n=5; (D) Synergistic effects of HIV protease inhibitors on alcohol-induced fat accumulation in the liver of mice gavaged with a liquid alcohol diet alone or mixed with HIV protease inhibitors: RIT and LOP. The red lipid droplets in liver tissue were revealed by Oil Red O staining (200×).
Figure 2.
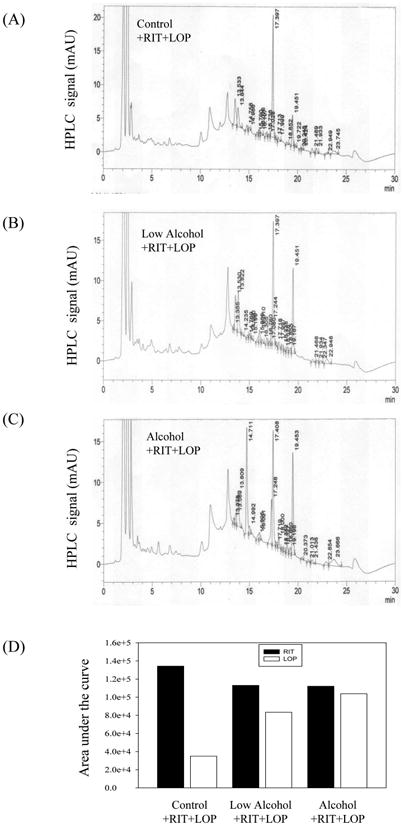
Effect of alcohol on HIV protease inhibitor levels in mouse plasma. HPLC spectra of ritonavir (RIT) and lopinavir (LOP) from plasma of mice gavaged with control diet plus RIT and LOP (A), half doses of alcohol diet plus RIT and LOP (B), and alcohol diet plus RIT and LOP (C); (D) Quantitation of the HIV drugs in the mouse plasma. HPLC analysis was run twice and the representative spectra were shown.
Previous studies demonstrated that either HIV PIs or alcohol feeding at high concentrations induced ER stress in cell and animal models14, 15, 30, 33. To know if ER stress was induced in response to acute treatments with the HIV drugs and/or alcohol at reduced doses, we examined expression of major ER stress markers in the liver of the treated mice. Compared to the vehicle control, RIT combined with LOP induced moderate ER stress response as indicated by increased expression of GRP78 and GRP94, IRE1 and ATF6 in mouse liver (Fig. 3A&B). Single doses of alcohol gavage (5g/kg) did not induced apparent ER stress response. In alcohol plus RIT and LOP, alcohol synergistically increased the HIV drug-induced GRP78 and ratio of spliced sXbp1 to Xbp1 (Fig. 3A&B). In addition, RIT and LOP inhibited protein expression of the Sarco/ER Calcium-ATPase (SERCA) which was further inhibited in the presence of alcohol. CYP2E1 was not significantly affected in response to any of these acute treatments (Fig. 3A&B).
Figure 3.
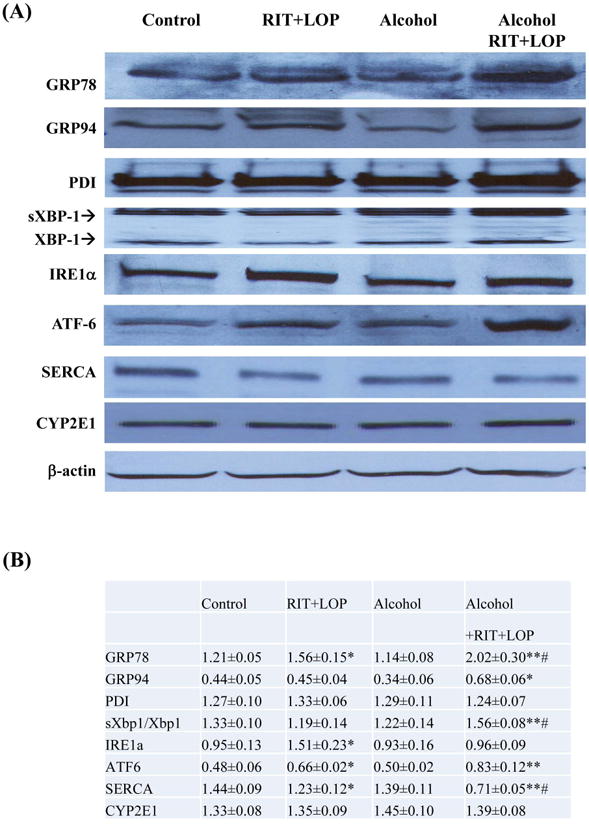
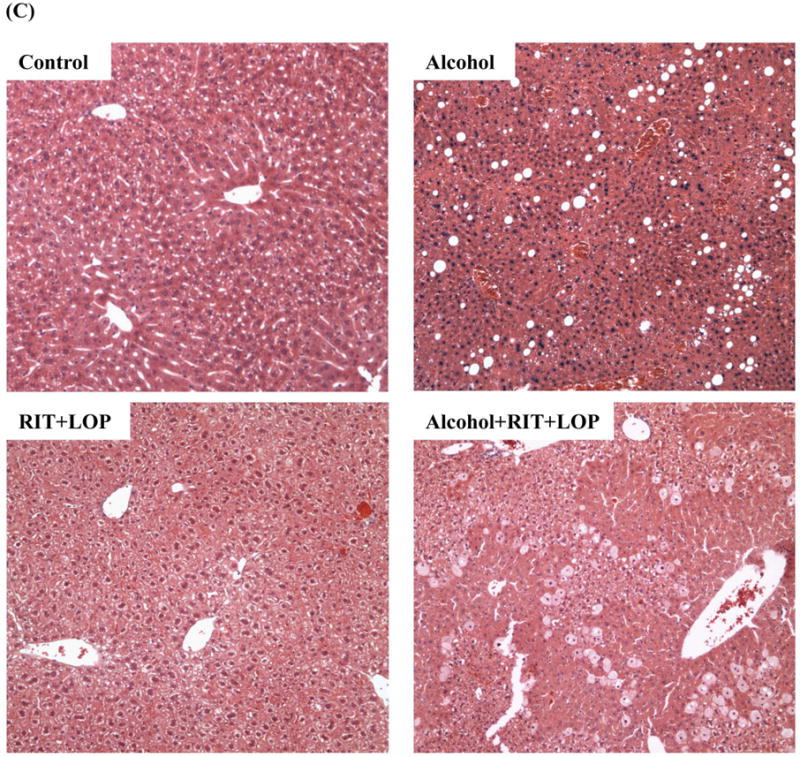
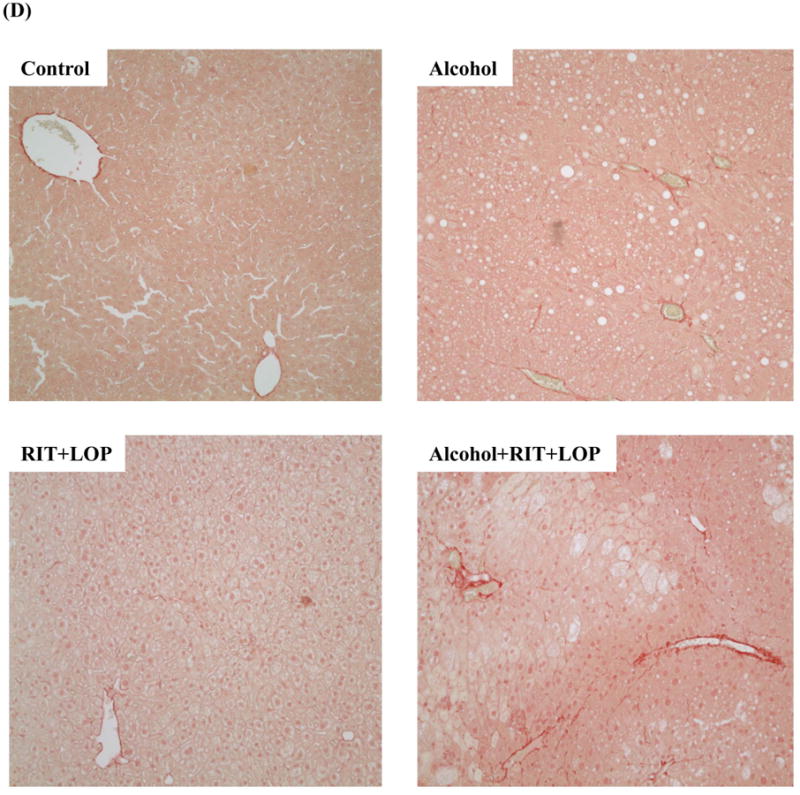
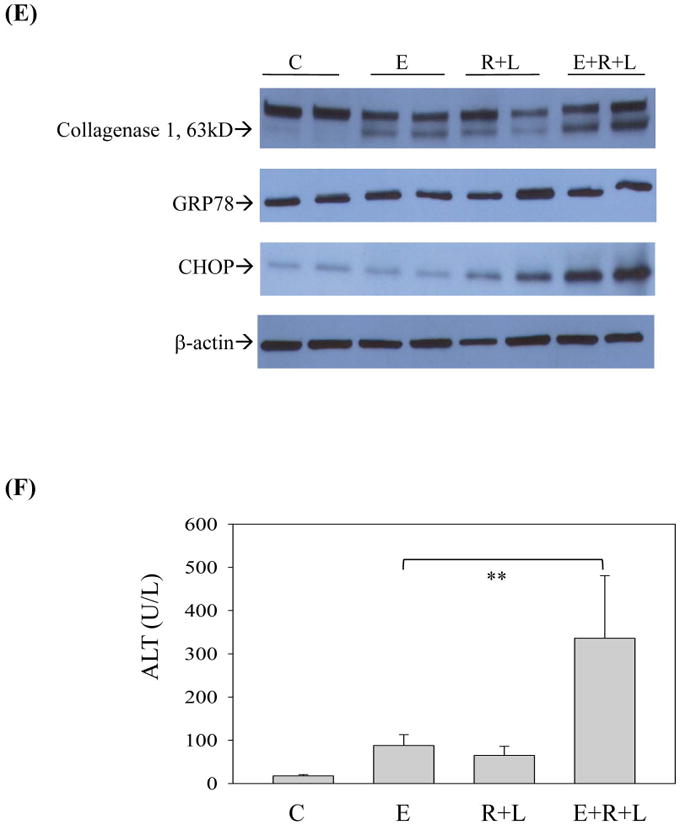
HIV protease inhibitors and/or alcohol induce endoplasmic reticulum stress and injury in mouse liver. (A) Immunoblots of proteins from liver. RIT, ritonavir; LOP, lopinavir; GRP78 and 94, glucose-regulated protein 78 and 94; PDI, protein disulfide isomerase; XBP1, X box-binding protein 1; sXBP1, spliced Xbp1; IRE1α, inositol requiring enzyme 1; ATF-6, activating transcription factor 6; SERCA, the Sarco/ER Calcium-ATPase; CYP2E1, cytochrome P450IIE1. (B) Protein quantity normalized with β-actin. *p<0.05 and **p<0.01 compared to control; #p<0.05 compared between RIT+LOP and alcohol+RIT+LOP. (C), Liver tissues stained with H&E (100×); (D) Fibrotic changes in liver tissues stained with Sirius Red (200×); (E) Immunoblots of liver proteins; (F), Serum ALT values; C, pair-fed control; E, alcohol feeding; R+L, injected with RIP and LOP; E+R+L, alcohol feeding plus RIT and LOP injection; **p<0.01.
Chronic HIV drugs worsened alcoholic fatty liver injury (Fig. 3C) and induced moderate liver fibrotic changes, which were confirmed by Sirius Red staining and by immunoblotting with anti-collagenase I antibodies (Fig. 3D&E). The chronic treatment induced ER stress (Fig 3E) and increased ALT by 15 fold compared to control (Fig. 3F).
2. HIV PI- and alcohol-induced ER stress and death in primary mouse hepatocytes
To further investigate the synergistic effects of alcohol on the two HIV drugs-induced stress and injury, primary mouse hepatocytes (PMH) were isolated for this purpose. Figure 4A demonstrates that the RIT and LOP treatment increased GRP78 expression in the hepatocytes, but alcohol alone did not increase GRP78. However, using more sensitive in situ immunoblotting with fluorescent antibodies, we were able to detect enhanced GRP78 expression in the PMH in response to the HIV drugs as well as to alcohol (Fig. S1). The GRP78 expression was remarkably increased in the combination treatment with RIT, LOP and alcohol (Fig. S1 and Fig. 4B). Consistent with the in vivo study with mice, expression of SERCA in PMH was also inhibited by RIT and LOP, and alcohol alone did not affect SERCA significantly (Fig. 4B). The mature SREBP1 that regulates triglyceride synthesis was increased in the presence of RIT, LOP and alcohol despite that neither the HIV drugs nor acute alcohol alone increased SREBP1 activation (Fig. 4B). CHOP and phosphorylated JNK2 that mediate ER stress-caused cell death were induced by the HIV drugs and alcohol. Correspondingly, only moderate cell death was observed in the hepatocytes treated with either alcohol or the two HIV drugs (Fig. 4C and 4D). A synergistic increase of cell death was detected in the hepatocytes in response to the drug and alcohol combination treatment.
Figure 4.
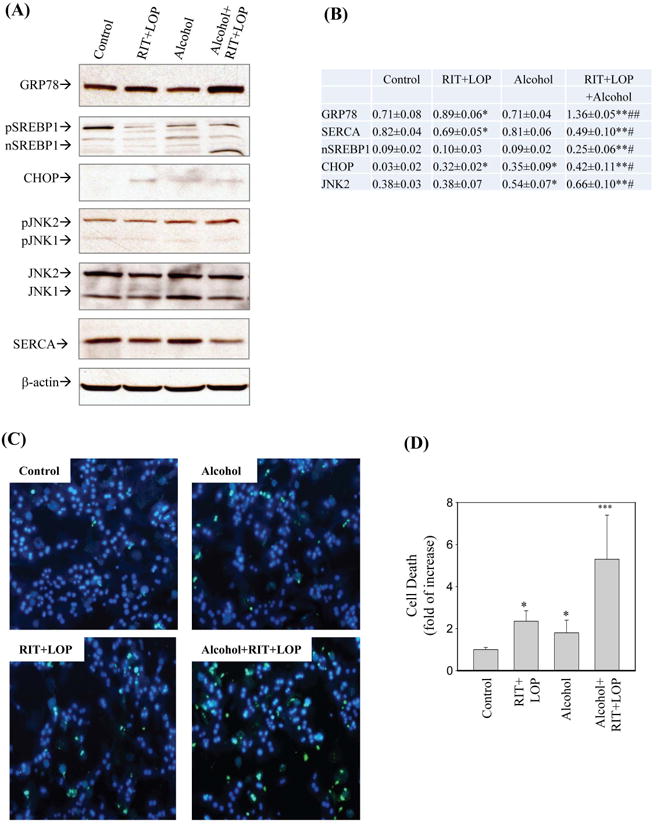
Endoplasmic reticulum stress and cell death in primary mouse hepatocytes treated with HIV protease inhibitors and/or alcohol. (A) Immunoblots of proteins; SREBP1, sterol regulatory element binding protein1; pSREPB, SREBP1 precursor; nSREBP1, mature SREBP1 (68kD); CHOP, CCAAT/enhancer binding protein (C/EBP) homologous protein; JNK, c-Jun N-terminal protein kinase; pJNK, phosphorylated JNK. (B) Protein quantity normalized with β-actin. *p<0.05 and **p<0.01 compared to control; #p<0.05 and ##p<0.01 compared between RIT+LOP and alcohol+RIT+LOP. (C) Cell morphological changes under fluorescence microscopy after double-stained with Syntax green and Hoechst blue. Green spots indicate dead cells and blue spots indicate live cells. (D) Quantitation of cell death relative to control; *p<0.05; ***p<0.001.
3. Effects of HIV PIs on alcohol-induced ER stress in primary human hepatocytes
The above observed effects of HIV drugs on alcohol-induced ER stress and injury in animals and animal liver cells prompted us to ask whether similar effects occur in primary human hepatocytes (PHH). In PHH, tunicamycin increased GRP78 expression from 8 hours to 24 hours after treatment, indicative of a positive ER stress response (Fig. 5A and 5B). Neither alcohol nor RIT and LOP treatments induced a significant increase of GRP78. However, GRP78 was increased significantly in response to alcohol and the HIV drugs. To confirm the ER stress response in PHH, we measured the unconventional splicing of Xbp1 at more treatment combinations: tunicamycin as positive control, low and high doses of RIT and LOP, low and high doses of alcohol, and homocysteine thiolactone that homocysteinylates proteins and mediates chronic alcohol-induced ER stress14, 33, the HIV drugs plus alcohol, or the HIV drugs plus homocysteine thiolactone (Fig. 5C). As expected, tunicamycin increased the ratio of the spliced Xbp1 (sXbp1) to Xbp1 mRNA, indicating an increased expression of sXbp1. Homocysteine thiolactone nearly eliminated the Xbp1 precursor and induced robust expression of sXbp1. RIT and LOP induced moderate Xbp1 splicing at low or high doses. Alcohol alone at low or high doses did not induce significant Xbp1 splicing. However, alcohol at 85 mM significantly increased RIT and LOP-induced Xbp1 splicing (Fig. 5C).
Figure 5.
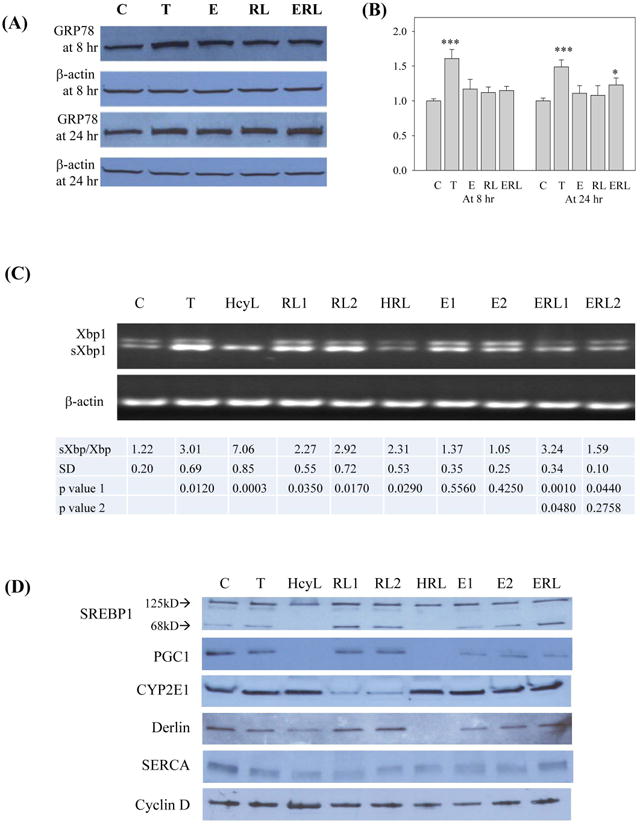
Effects of HIV protease inhibitors and alcohol on ER stress response in primary human hepatocytes. (A) GRP78 expression in response to the HIV drug and/or alcohol; C, control; T, tunicamycin (3 μg/ml) used as ER stress positive control; E, alcohol; RL, ritonavir and lopinavir; ERL, alcohol plus RL. (B) Quantitation of GRP78 expression after normalized with β-actin; *p<0.05 and ***p<0.005 compared to control. (C) RT-PCR of mRNA showing shifting from Xbp1 to spliced Xbp1 (sXbp1) upon ER stress; HcyL, homocystein thiolactone (1 mM); RL, ritonavir and lopinavir at 15 μg/ml (RL1) and 15 μg/ml (RL2); HRL, homocystein thiolactone (1mM) plus RL at 5 μg/ml; ERL1 and ERL2, RIT and LOP (10 μg/ml) plus alcohol at 85 mM and 35 mM respectively. Quantitation of ratio of sXbp1 to Xbp1 is shown under the gels. Comparisons were made between control and treatments (p value 1) or between RL1 and ERL1 or ERL2 (p value 2). (D) Immunoblots of ER stress markers; PGC1, PPARγ coactivator1; SERCA, the Sarco/ER Calcium-ATPase; Detection of cyclin D indicated that the cells were viable in the presence of HcyL.
Alcohol-induced ER stress has been linked to impaired lipogenesis in animal models14, 15. To know if the linkage occurs in PHH, we examined expression of SREBP1c as well as PGC1 that regulates lipid oxidation in the liver (Fig. 5D and Table S1). RIT and LOP but not alcohol or tunicamycin increased expression of the mature SREBP1. PGC1 was not affected by RIT and LOP. However, PGC1 was inhibited by alcohol or tunicamycin and was inhibited to a much greater extent in response to alcohol plus the two HIV drugs. Interestingly, homocysteine thiolactone inhibit both SREBP1 and PGC1, indicating that the human hepatocytes may be more sensitive to protein homocysteienylation than mouse hepatocytes. In addition, CYP2E1 was inhibited by the HIV drugs and the inhibition was reduced in the presence of alcohol in the human cells. Derlin that regulates ER stress-associated protein degradation (ERAD) was not affected by the drugs but was inhibited by tunicamycin, homocysteine thiolactone, or alcohol (Fig. 5D and Table S1).
4. Inhibition of SERCA and cell death promotion by HIV PIs and alcohol in primary human hepatocytes
SERCA was inhibited by tunicamycin and by homocysteine thiolactone to a greater extent (Fig. 5D and Table S1). RIT and LOP but not alcohol alone significantly inhibited SERCA. SERCA remained inhibited by the combined treatment with the two HIV drugs and alcohol. It is known that SERCA sequesters cytosolic Ca2+ in the ER and inhibition of SERCA leads to discharges of ER calcium34. To test this, we incubated the human hepatocytes with vehicle control, alcohol, RIT and LOP, or alcohol combined with RIT and LOP, and then measured ER calcium discharges upon stimulation by the specific SERCA inhibitor: thapsigargin. Figure 6 demonstrates that intracellular calcium levels in the human cells were reduced slightly by alcohol treatment and significantly by RIT and LOP. The pre-incubation with alcohol and the two HIV drugs reduced remarkably the intracellular calcium. Correspondingly, alcohol or the HIV drugs-induced disturbance of intracellular calcium homeostasis promoted cell death (Fig. 7). The cell death was synergistically increased in the PHH treated with alcohol plus the two HIV drugs, which was comparable to the cell death induced by thapsigargin, suggesting a specific involvement of impaired calcium homeostasis.
Figure 6.
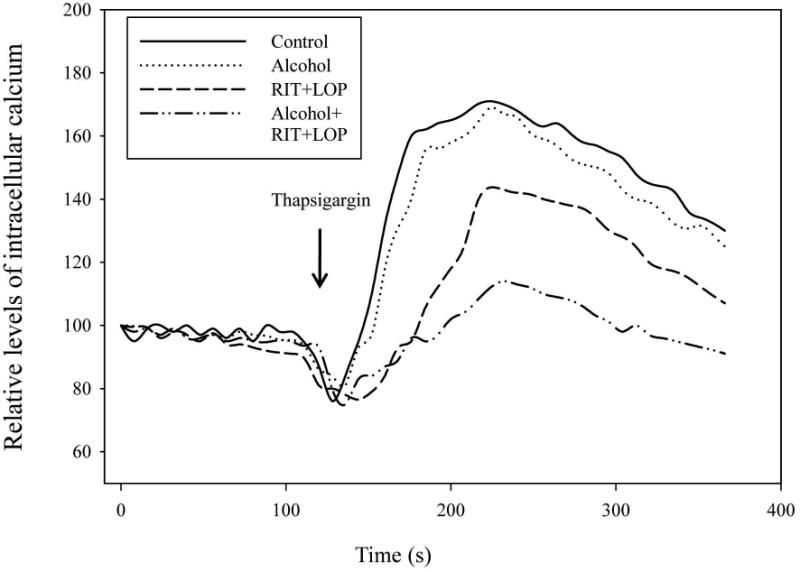
Effects of alcohol and HIV protease inhibitors on thapsigargin-stimulated discharges of intracellular Ca2+ stores in human primary hepatocytes. See Materials and Methods for details. Calcium levels are expressed as % of vehicle control at time zero.
Figure 7.
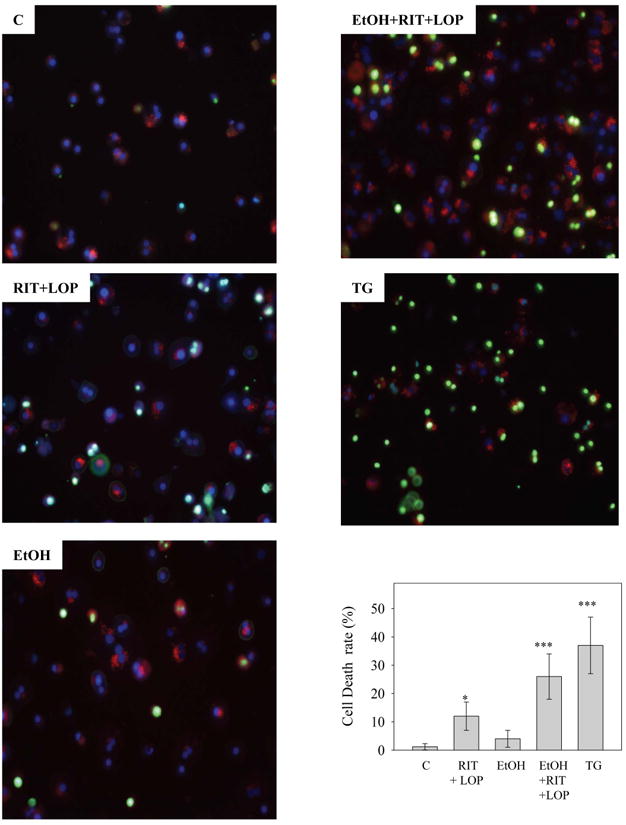
Synergistic effects of HIV protease inhibitors and alcohol on cell death in primary human hepatocytes. The hepatocytes were doubly stained with Sytox green and Hoechst blue dyes after incubation with vehicle DMSO (C), ritonavir and lopinavir (RIT+LOP), alcohol (EtOH), or thapsigargin (TG, 6 μg/ml) for 24 hours. Dead cells (combination of apoptosis and necrosis) were stained green. Graph depicts cell death rate. *p<0.05 and ***p<0.005 compared to control.
Discussion
In this study we tested effects of HIV protease inhibitors and alcohol on hepatic injury in animals and in both animal and human liver cells. Acute alcohol administration by gavage caused moderate liver injury which is consistent with studies reported previously30. Alcohol synergistically increased hepatotoxicity in the presence of ritonavir and lopinavir. Our results are clinical relevant. First, the HIV drugs induced little injury in mouse and human hepatocytes. This implies that the drugs themselves, at proper doses and without a concomitant alcohol consumption, may not be a significant problem, considering their dramatic curing potential for HIV-infected patients under HAART. Second, ritonavir is usually used to boost the efficacy of other HIV PIs23. To comply with the practice, we tested the injurious effects by using a combination of ritonavir and lopinavir, which is the most popular regimen applied to the patients23, 35. Third, the amount of drugs used in this study is compatible with the concentrations that restrain HIV effectively and have been used by others in both animal experiments and clinical practices. A minimum dose of ritonavir at 2.1 μg/ml has been shown to inhibit different strains of HIV-1 in the human T cell line MT4 system23, 36, 37. Considering that the liver is the primary site for drug metabolism and is usually exposed to high concentrations of drugs, it is reasonable to use higher doses in liver cells than in peripheral cells in drug toxicity tests. In fact, a range of concentrations between 0 and 100 μM were used by others28, 29. For the in vivo studies, we adopted a safety conversion factor 10 in converting the known minimum dose in human to an equivalent dose in rodent (mouse). The equivalent dose of ritonavir for the mouse was calculated as 12.5 mg/kg body weight. Thus, the single dose of HIV PI at 15 mg/kg body weight was well justified for the in vivo experiments and is clinically relevant.
Alcohol plus the two HIV drugs robustly induced a synergistic increase in serum ALT levels, which indicated an aggravated hepatic injury. The underlying mechanisms are likely multi-fold. The metabolisms of the HIV drugs and alcohol might mutually be affected since both HIV drugs and alcohol are metabolized in the liver by the cytochrome P450 enzyme system4, 12, 23, 28, 32. On one hand, alcohol induced moderate CYP2E1 expression (Fig. 5D), which is consistent with observations by others5. On the other hand, the drugs slightly inhibited CYP2E1 expression in mice (Fig. 3A) and significantly reduced CYP2E1 expression in the human hepatocytes (Fig. 5D). In the presence of alcohol, the amount of lopinavir in mouse plasma was nearly doubled in the drug combination treatment (Fig. 2). This may be a result of drug-drug or drug-alcohol interactions because in a regimen of drug combination, ritonavir boosts the bioavailability of other drugs23, 38, 39, and alcohol and the drugs appeared to have opposite effects on CYP2E1 expression. In addition, since the increase of lopinair was also alcohol dose dependent, the drug-drug interactions may not be the only cause. Conceivably, alcohol may potentiate the boosting effects of ritonavir on lopinavir and increased drug exposure and subsequent hepatotoxicity.
Our results support that the ER stress mediates the potentiation effects by alcohol. The ER is an essential organelle for protein and lipid synthesis and modifications, drug metabolism and Ca2+ homeostasis. General attenuation of protein synthesis upon ER stress will affect the efficiency of enzymes metabolizing alcohol and drugs. Evidence is accumulating that either excessive alcohol consumption or high doses of HIV PIs induce ER stress and injury in cell and animal models14, 15, 28-33. In the present study, especially for first time with the human hepatocytes, we demonstrated an occurrence of ER stress in response to alcohol combined with the HIV drugs at practical concentrations. First, although increased expression of GRP78 was not readily detected in response to either alcohol or the HIV drugs alone, alcohol combined with the drugs induced significant GRP78 increase. Second, ER stress has been associated with impaired lipid metabolism in both alcoholic and nonalcoholic liver disease14, 15, 40 and we detected an association of lipid accumulation with an increased expression of SREBP1c and a decreased expression of PGC1 in the presence of alcohol and HIV drugs. Third, alcohol-induced hyperhomocysteinemia is a well-established phenomenon in animals and patients8, 14, 41. We observed a robust unconventional splicing of Xbp1 in the human cells challenged with homocysteine thiolactone that is interchangeable with homocysteine. With respect to what ER stress components are involved, we propose that disturbance of Ca2+ homeostasis is a major mechanism for the activation of ER stress by HIV PIs and alcohol. Previously, a number of possible cellular stress signals, such as increased cholesterol in ER membranes, deprivation of glucose, inhibition of ER associated protein degradation (ERAD), and depletion of ER calcium store were suggested to initiate the UPR by the HIV drugs28-30. For instance, in macrophages, ritonavir significantly up-regulates CD36 and LDLR expression, promoting foam cell formation by disrupting ABCA1-mediated cholesterol efflux28, 29, 42. The glucose transporter isoform GluT4 was also selectively inhibited by HIV PIs in macrophage43. However, in hepatocytes treated with alcohol and the HIV drugs, we did not detect any apparent changes of LDLR and GluT2 (Data not shown), excluding the involvement of cholesterol or glucose in activating the ER stress. With respect to ERAD, it was reported that ritonavir inhibited chymotrypsin-like activity while enhance trypsin-like activity in T cells44. However, in the human hepatocytes in this study, Derlin that regulates ERAD was not changed (Fig. 5D, lane ERL), ruling out the involvement of ERAD in the ER stress activation by alcohol and the drugs. Interestingly, SERCA that mainly regulates ER Ca2+ store was inhibited by ritonavir and lopinavir in mice as well as in primary mouse and human hepatocytes. Although we have not examined expression of SERCA mRNA in this study, the inhibition of SERCA was quite specific in the human cells since the inhibition was similar to the pharmacological ER stress-induced agent-thapsigargin (Fig. 5D). Further, the intracellular calcium in the human hepatocytes was reduced after incubation with the two HIV drugs or with alcohol (albeit to a less extent). The calcium level was reduced further after incubation with both alcohol and the drugs. Parallel to the calcium reduction, cell death due to the ER stress in the human liver cells was synergistically increased in response to alcohol and the drugs. Therefore, the HIV drugs may interact with SERCA, disturbing calcium homeostasis and causing ER stress and injury. The drugs may also affect the function of CYP2E1, delaying alcohol degradation. An increased alcohol exposure induces oxidative stress and/or hyperhomocysteinmia, which disturb SERCA further and exacerbate the HIV drug-induced ER stress leading to hepatic steatosis and fibrosis (Fig. 8).
Figure 8.
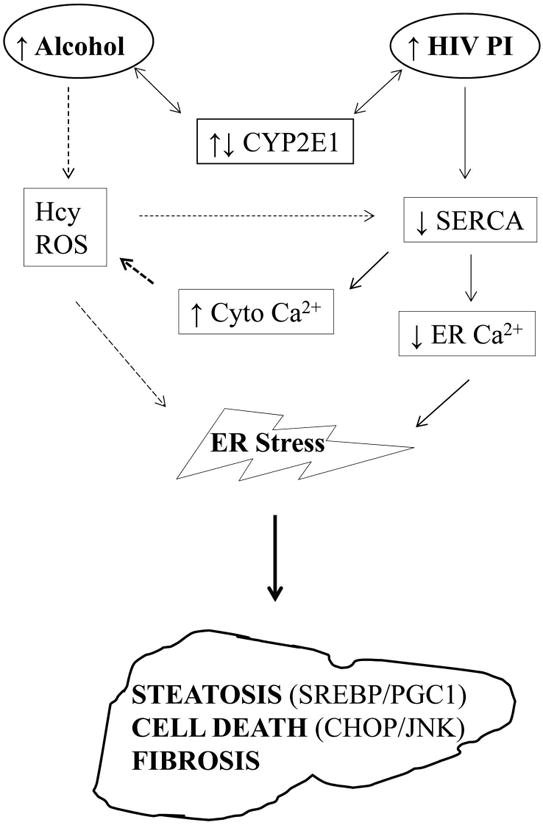
Interplay between alcohol and HIV protease inhibitors on hepatic endoplasmic reticulum stress and injury. HIV PI, HIV protease inhibitor; CYP2E1, cytochrome P450 2E1; Hcy, homocysteine; ROS, reactive oxygen species; SERCA, the Sarco/ER Calcium-ATPase; Cyto Ca2+, cytoplasmic calcium; ER Ca2+, calcium within the ER. ↑, level is up; ↓, level is down. Solid lines indicate potential direct effects and dotted lines indicate potential indirect effects.
Supplementary Material
Fig. S1 Confocal fluorescence image of hepatocytes double-stained with rhodamine-phalloidin (red) and anti-GRP78 antibody conjugated with FITC (green). Stressed or dying cells contained increased green fluorescence. RIT, ritonavir; LOP, lopinavir.
Acknowledgments
This work is supported by US NIH/NIAAA grants R01AA018846 and R01AA018612 (to CJ). We thank CellzDirect™ and Dr. Shelly Lu for providing primary human hepatocytes and the USC Research Center for Liver Diseases for technical support. MS is currently a gastrointestinal endoscopist & hepatologist at Toho University Hospital, Tokyo, Japan. MF is a post-graduate medical research trainee in Ji's lab. EK, MS and MF contributed equally to this study.
References
- 1.Gunzerath L, Hewitt BG, Li TK, Warren KR. Alcohol research: past, present, and future. Ann N Y Acad Sci. 2011;1216:1–23. doi: 10.1111/j.1749-6632.2010.05832.x. [DOI] [PubMed] [Google Scholar]
- 2.Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223–33. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- 3.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–85. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29(4):245–254. [PMC free article] [PubMed] [Google Scholar]
- 5.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;(83):519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 6.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275(4 Pt 1):G605–11. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 7.Thurman RG, Ji S, Lemasters JJ. Alcohol-induced liver injury. The role of oxygen. Recent Dev Alcohol. 1984;2:103–17. [PubMed] [Google Scholar]
- 8.Halsted CH, Medici V. Aberrant hepatic methionine metabolism and gene methylation in the pathogenesis and treatment of alcoholic steatohepatitis. Int J Hepatol. 2012;2012:959746. doi: 10.1155/2012/959746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu SC, Martínez-Chantar ML, Mato JM. Methionine adenosyltransferase and S-adenosylmethionine in alcoholic liver disease. J Gastroenterol Hepatol. 2006;21(3):S61–4. doi: 10.1111/j.1440-1746.2006.04575.x. [DOI] [PubMed] [Google Scholar]
- 10.Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32(9):1525–34. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 11.McClain CJ, Barve SS, Barve A, Marsano L. Alcoholic liver disease and malnutrition. Alcohol Clin Exp Res. 2011;35(5):815–20. doi: 10.1111/j.1530-0277.2010.01405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int. 2012;2012:216450. doi: 10.1155/2012/216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szabo G, Zakhari S. Mechanisms of alcohol-mediated hepatotoxicity in human-immunodeficiency-virus-infected patients. World J Gastroenterol. 2011;17(20):2500–6. doi: 10.3748/wjg.v17.i20.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124(5):1488–99. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 15.Esfandiari F, Villanueva JA, Wong DH, French SW, Halsted CH. Chronic ethanol feeding and folate deficiency activate hepatic endoplasmic reticulum stress pathway in micropigs. Am J Physiol Gastrointest Liver Physiol. 2005;289(1):G54–63. doi: 10.1152/ajpgi.00542.2004. [DOI] [PubMed] [Google Scholar]
- 16.Nishitani Y, Matsumoto H. Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Lett. 2006;580(1):9–14. doi: 10.1016/j.febslet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Esfandiari F, Medici V, Wong DH, Jose S, Dolatshahi M, Quinlivan E, et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology. 2010;51(3):932–41. doi: 10.1002/hep.23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howarth DL, Vacaru AM, Tsedensodnom O, Mormone E, Nieto N, Costantini LM, et al. Alcohol Disrupts Endoplasmic Reticulum Function and Protein Secretion in Hepatocytes. Alcohol Clin Exp Res. 2012;36(1):14–23. doi: 10.1111/j.1530-0277.2011.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galligan JJ, Fritz KS, Tipney H, Smathers RL, Roede JR, Shearn CT, et al. Profiling impaired hepatic endoplasmic reticulum glycosylation as a consequence of ethanol ingestion. J Proteome Res. 2011;10(4):1837–47. doi: 10.1021/pr101101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55(3):673–82. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–31. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco(endo) plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci U S A. 2010;107(45):19320–5. doi: 10.1073/pnas.1012044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flexner C. HIV-Protease Inhibitors. N Engl J Med. 1998;338(18):1281–92. doi: 10.1056/NEJM199804303381808. [DOI] [PubMed] [Google Scholar]
- 24.Tejerina F, Bernaldo de Quirós JC. Protease inhibitors as preferred initial regimen for antiretroviral-naive HIV patients. AIDS Rev. 2011;13(4):227–33. [PubMed] [Google Scholar]
- 25.Senise JF, Castelo A, Martínez M. Current treatment strategies, complications and considerations for the use of HIV antiretroviral therapy during pregnancy. AIDS Rev. 2011;13(4):198–213. [PubMed] [Google Scholar]
- 26.Reust CE. Common Adverse Effects of Antiretroviral Therapy for HIV Disease. Am Fam Physician. 2011;83(12):1443–51. [PubMed] [Google Scholar]
- 27.Merwat SN, Vierling JM. HIV infection and the liver: the importance of HCV-HIV coinfection and drug-induced liver injury. Clin Liver Dis. 2011;15(1):131–52. doi: 10.1016/j.cld.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Parker RA, Flint OP, Mulvey R, Elosua C, Wang F, Fenderson W, et al. Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol. 2005;67(6):1909–19. doi: 10.1124/mol.104.010165. [DOI] [PubMed] [Google Scholar]
- 29.Zhou H, Gurley EC, Jarujaron S, Ding H, Fang Y, Xu Z, et al. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291(6):G1071–80. doi: 10.1152/ajpgi.00182.2006. [DOI] [PubMed] [Google Scholar]
- 30.Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54(1):229–39. doi: 10.1002/hep.24368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brüning A. Analysis of nelfinavir-induced endoplasmic reticulum stress. Methods Enzymol. 2011;491:127–42. doi: 10.1016/B978-0-12-385928-0.00008-0. [DOI] [PubMed] [Google Scholar]
- 32.Samet JH, Horton NJ, Meli S, Freedberg KA, Palepu A. Alcohol consumption and antiretroviral adherence among HIV-infected persons with alcohol problems. Alcohol Clin Exp Res. 2004;28:572–577. doi: 10.1097/01.alc.0000122103.74491.78. [DOI] [PubMed] [Google Scholar]
- 33.Ji C, Deng Q, Kaplowitz N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40(2):442–51. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- 34.Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87(7):2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan D, Walmsley S. Lopinavir plus ritonavir: a novel protease inhibitor combination for HIV infections. Expert Rev Anti Infect Ther. 2007;5(1):13–28. doi: 10.1586/14787210.5.1.13. [DOI] [PubMed] [Google Scholar]
- 36.Danner SA, Carr A, Leonard JM, Lehman LM, Gudiol F, Gonzales J, et al. A short-term study of the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. European-Australian Collaborative Ritonavir Study Group. N Engl J Med. 1995;333(23):1528–33. doi: 10.1056/NEJM199512073332303. [DOI] [PubMed] [Google Scholar]
- 37.Justesen US. Protease inhibitor plasma concentrations in HIV antiretroviral therapy. Dan Med Bull. 2008;55(4):165–85. [PubMed] [Google Scholar]
- 38.Foisy MM, Yakiwchuk EM, Hughes CA. Induction effects of ritonavir: implications for drug interactions. Ann Pharmacother. 2008;42(7):1048–59. doi: 10.1345/aph.1K615. [DOI] [PubMed] [Google Scholar]
- 39.Youle M. Overview of boosted protease inhibitors in treatment-experienced HIV-infected patients. J Antimicrob Chemother. 2007;60(6):1195–205. doi: 10.1093/jac/dkm364. [DOI] [PubMed] [Google Scholar]
- 40.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277(32):29342–7. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 41.Blasco C, Caballería J, Deulofeu R, Lligoña A, Parés A, Lluis JM, et al. Prevalence and mechanisms of hyperhomocysteinemia in chronic alcoholics. Alcohol Clin Exp Res. 2005;29(6):1044–8. doi: 10.1097/01.alc.0000169265.36440.ee. [DOI] [PubMed] [Google Scholar]
- 42.Dressman J, Kincer J, Matveev SV, Guo L, Greenberg RN, Guerin T, et al. HIV protease inhibitors promote atherosclerotic lesion formation independent of dyslipidemia by increasing CD36-dependent cholesteryl ester accumulation in macrophages. J Clin Investig. 2003;111:389–397. doi: 10.1172/JCI16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murata H, Hruz PW, Mueckler M. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS. 2002;16:859–863. doi: 10.1097/00002030-200204120-00005. [DOI] [PubMed] [Google Scholar]
- 44.Andre P, Groettrup M, Klenerman P, de Giuli R, Booth BL, Jr, Cerundolo V, et al. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation and T cell responses. Proc Natl Acad Sci USA. 1998;95:13120–24. doi: 10.1073/pnas.95.22.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Confocal fluorescence image of hepatocytes double-stained with rhodamine-phalloidin (red) and anti-GRP78 antibody conjugated with FITC (green). Stressed or dying cells contained increased green fluorescence. RIT, ritonavir; LOP, lopinavir.