

Abstract
Nitric oxide (NO), a key regulator of cardiovascular function, is synthesized from L-arginine and oxygen by the enzyme nitric oxide synthase (NOS). This reaction requires tetrahydrobiopterin (BH4) as a cofactor. BH4 is synthesized from guanosine triphosphate (GTP) by GTP cyclohydrolase I (GTPCH) and recycled from 7,8-dihydrobiopterin (BH2) by dihydrofolate reductase. Under conditions of low BH4 bioavailability relative to NOS or BH2, oxygen activation is “uncoupled” from L-arginine oxidation, and NOS produces superoxide (O−2) instead of NO. NOS-derived superoxide reacts with NO to produce peroxynitrite (ONOO−), a highly reactive anion that rapidly oxidizes BH4 and propagates NOS uncoupling. BH4 depletion and NOS uncoupling contribute to overload-induced heart failure, hypertension, ischemia/reperfusion injury, and atrial fibrillation. L-arginine depletion, methylarginine accumulation, and S-glutathionylation of NOS also promote uncoupling. Recoupling NOS is a promising approach to treating myocardial and vascular dysfunction associated with heart failure.
Keywords: Nitric oxide, Nitric oxide synthase, Uncoupling, Superoxide, Tetrahydrobiopterin, Dihydrobiopterin, Sepiapterin, Sapropterin, GTP cyclohydrolase I, Dihydrofolate reductase, Endothelial cell, Cardiomyocyte, Hypertension, Heart failure, Atrial fibrillation, Ischemia, Reperfusion, Vasodilation, L-Arginine, Monomethylarginine, Asymmetric dimethylarginine, S-Glutathionylation, Peroxynitrite, Oxidative stress, NADPH oxidase
Introduction
Synthesis of the gaseous signaling molecule nitric oxide (NO) by the nitric oxide synthase (NOS) family of enzymes is a key mechanism of cardiovascular homeostasis. Three NOS isoforms, neuronal (nNOS), inducible (iNOS), and endothelial (eNOS), catalyze the reaction of molecular oxygen with the amino acid substrate L-arginine to produce L-citrulline and NO [1]. During this reaction, electrons donated by nicotinamide adenine dinucleotide phosphate (NADPH) at the carboxy-terminal reductase domain of NOS are passed to the heme catalytic center of the oxidase domain, where activation of molecular oxygen is “coupled” to NO synthesis by two successive monooxygenations of L-arginine [2]. The cofactor 6R-5,6,7,8-tetrahydrobiopterin (BH4) is required for these reactions; in its absence, electron flow to molecular oxygen becomes “uncoupled” from L-arginine oxidation, resulting in production of superoxide (O−2) instead of NO [3–6]. The combination of increased oxidative stress and impaired NO signaling resulting from NOS uncoupling has been implicated in the pathogenesis of a wide range of disease states, including atherosclerosis [7], hypertension [8, 9] and diabetes [10]. Advances in understanding the role of NO signaling in myocardial function have encouraged recent research into whether NOS uncoupling contributes to the pathogenesis of several aspects of heart failure. In this review, we will discuss current progress in this field, focusing on key studies that have identified BH4 bioavailability as a critical biochemical determinant of NOS uncoupling.
BH4 Synthesis is a Determinant of NOS Uncoupling
BH4 as a NOS Cofactor
Tetrahydrobiopterin was initially recognized as a cofactor required for enzymatic hydroxylation of phenylalanine, tyrosine, and tryptophan. Subsequently, the identity of the vasodilator known as “endothelial-derived relaxing factor” was discovered to be NO [11, 12], and BH4 also was found to be essential for NOS-mediated NO synthesis [13, 14]. As an NOS cofactor, BH4 performs both structural and biochemical functions. Structurally, BH4 stabilizes the active NOS homodimer [15, 16] and induces conformational changes that promote catalytic activity [17]. Biochemically, the donation of an electron by BH4 to produce a transient BH4• + radical is required for the oxidation of L-arginine to L-citrulline [18–20] and associated formation of a ferrous iron-NO complex at the NOS heme catalytic center [1, 2]. In the final step of NO synthesis, BH4• + recaptures an electron from this ferrous iron-NO complex, allowing release of gaseous NO [18, 21]. Therefore, BH4 is not consumed during NOS catalysis, but is necessary for both NO synthesis and release. However, BH4 is not required for the initiating step of NO synthesis, which consists of reduction of the heme iron by NADPH-derived electrons so that molecular oxygen may be bound and activated [20]. In the absence of BH4, NADPH consumption and oxygen activation proceed, but are no longer “coupled” to subsequent BH4-dependent L-arginine oxygenation [3–5]. As a result, activated oxygen is released directly from the heme catalytic center as superoxide in a process termed “NOS uncoupling” [3–5]. The addition of increasing concentrations of BH4 effectively reverses NOS uncoupling by proportionally reducing superoxide production and restoring NO synthesis [5]. De novo BH4 synthesis is a central mechanism by which sufficient cellular BH4 concentrations are maintained to support coupled NOS activity.
BH4 Biosynthesis
BH4 is a heterocyclic nitrogenous molecule derived from guanosine-5′-triphosphate (GTP). The enzymes GTP cyclohydrolase I (GTPCH), 6-pyruvoyl tetrahydrobiopterin synthase (PTPS), and sepiapterin reductase (SPR) catalyze successive steps in BH4 biosynthesis [22]. In most mammalian tissue, GTPCH-regulated conversion of GTP to 7,8-dihydroneopterin triphosphate is the rate-limiting step in this pathway. As a result, BH4 concentrations correlate strongly with GTPCH expression [23]. Transcription-level regulation is primarily responsible for increased BH4 synthesis in endothelial cells [24] and cardiomyocytes [25] in response to proinflammatory cytokine signaling. However, GTPCH also may be regulated post-transcriptionally by protein–protein interaction with GTPCH feedback regulatory protein (GFRP), which mediates feedback inhibition of GTPCH by BH4 [26, 27]. In contrast, disruption of GTPCH-GFRP binding may explain phosphorylation-dependent upregulation of GTPCH activity in response to endothelial shear stress [28, 29]. However, the importance of GFRP regulation may be overshadowed by transcription-level regulation of GTPCH, as evidenced by stable intracellular BH4 concentrations following GFRP overexpression or knockdown in endothelial cells [23].
BH4: NOS Stoichiometry
By increasing cellular BH4 concentrations, endothelial GTPCH overexpression improves coupled NOS activity in both cells [30] and mouse tissue [10]. Conversely, GTPCH knockdown reduces cellular BH4 concentrations, impairs coupled NOS activity, and increases superoxide production [31]. Furthermore, the ratio of BH4 to eNOS protein correlates strongly with NOS-dependent superoxide production across a range of GTPCH and eNOS expression levels [31]. These findings suggest that absolute cellular BH4 concentrations influence NOS coupling insofar as they reflect the ratio of BH4 to NOS protein. This concept may explain why transgenic overexpression of eNOS (thereby reducing the BH4:eNOS ratio) increases aortic and cardiac superoxide production in mice [32] and accelerates plaque formation in the apolipoprotein E–knockout model of atherosclerosis [33]. Restoration of BH4:eNOS stoichiometry by pharmacological BH4 supplementation [33] or concomitant overexpression of transgenic GCH1 [32] attenuates superoxide production and disease pathogenesis. The clinical relevance of GTPCH expression is illustrated by the association of a relatively common GCH1 polymorphism with reduced BH4 production, reduced renal excretion of NO metabolites and evidence of NOS uncoupling in ex vivo vessels [34, 35]. These studies taken together demonstrate that GTPCH expression and activity play an important role in determining cellular BH4, the ratio of BH4 to NOS protein, and NOS uncoupling.
BH4 Recycling is a Determinant of NOS Uncoupling
BH4 Oxidation
BH4 may be oxidized to 7,8-dihydrobiopterin (7,8-BH2) or quinoid-dihydrobiopterin (q-BH2) by two distinct mechanisms. BH4 undergoes oxidation to q-BH2 as a result of participating as a cofactor in the enzymatic hydroxylation of phenylalanine, tyrosine, or tryptophan. In the vascular endothelium, BH4 is not consumed by participating in NOS catalysis; instead, the primary mechanism of BH4 consumption is direct oxidation by cellular oxidants. The most abundant product of such oxidation reactions at physiological pH is 7,8-dihydrobiopterin (BH2), which competes with reduced BH4 for binding sites at the NOS oxidase domain [36]. However, because BH2 lacks the ability to supply reductive electrons required for L-arginine oxygenation, the displacement of BH4 by BH2 results in NOS uncoupling [5]. In a physiological setting, high levels of cellular oxidative stress contribute to NOS uncoupling by reducing the abundance of BH4 relative to BH2, thereby decreasing the proportion of NOS protein bound by catalytically active BH4. Thus, NOS uncoupling is influenced by the stoichiometric ratio of BH4 to BH2 as well as the ratio of BH4 to NOS protein [5]. In patients with coronary artery disease, both tissue BH4 concentrations and BH4 to BH2 ratios correlate with superoxide production and acetylcholine-mediated vasodilation [37].
BH4 Recycling
Dihydrofolate reductase (DHFR) is the enzyme responsible for recycling 7,8-BH2 back to catalytically-active BH4 using reductive electrons from NADPH [38]. Pharmacological DHFR inhibition (methotrexate treatment) or short interfering RNA knockdown of DHFR expression both result in significantly reduced levels of BH4 relative to BH2 [39•, 40]. Although DHFR inhibition does not alter total cellular biopterin concentrations, this reduction in the ratio of BH4 to BH2 is sufficient to increase superoxide production and inhibit L-arginine to L-citrulline conversion, a measure of coupled NOS activity [39•, 40]. Alternately, a similar effect may be achieved by GTPCH knockdown, which decreases cellular concentrations of all biopterins without altering the ratio of BH4 to BH2 [31, 39•, 40]. Concomitant DHFR and GTPCH knockdown increases superoxide production compared to knockdown of either gene alone [39•]. This finding demonstrates that NOS uncoupling is determined by BH4 stoichiometry relative to both BH2 and NOS. The functional interdependence of these ratios may explain why the effects of DHFR inhibition on NOS coupling are significantly stronger in mice with low tissue concentrations of BH4 due to reduced basal GTPCH expression [41]. BH4 recycling may also play a key role in BH4 transport, which is sensitive to DHFR inhibition in vivo [42]. Furthermore, injection of mice with equal parts 6S-BH4 and 6R-BH4 results in tissue accumulation of primarily 6R-BH4 [42]. These findings suggest that the primary mechanism of BH4 transport across the plasma membrane is via oxidation to BH2, an achiral compound, and subsequent reduction to bioactive 6R-BH4 within the cell.
BH4 Oxidation is a Cause and Consequence of NOS Uncoupling
Superoxide derived from uncoupled NOS or other mechanisms is a critical source of cellular oxidative stress, including BH4 oxidation. Direct BH4 oxidation by superoxide is relatively slow [43], but BH4 is rapidly oxidized to BH2 by peroxynitrite (ONOO−) [44, 45], a highly reactive anion produced by the reaction of superoxide with NO [46]. Peroxynitrite also causes significant cellular damage by nitrating amino acid side chain thiol and hydroxyl groups, thereby disrupting enzymatic function. Consequently, the implications of NOS uncoupling include 1) reduced de novo NO production; 2) sequestration of bioactive NO by superoxide anions via peroxynitrite formation; 3) superoxide and peroxynitrite-mediated cellular damage; and 4) peroxynitrite-mediated oxidation of BH4 to BH2, resulting in further propagation of NOS uncoupling. Therefore, NOS uncoupling is a self-reinforcing biochemical state driven by BH4 oxidation (Fig. 1). BH4 oxidation due to uncoupled eNOS-dependent oxidative stress was observed both in vitro [31] and in vivo [32]. In these experiments, uncoupling was induced by increasing eNOS expression under conditions of stable GTPCH expression, effectively reducing the ratio of BH4 to eNOS [31, 32]. With all other variables held constant, the resulting oxidation of BH4 to BH2 can only be attributed to superoxide production by uncoupled NOS.
Fig. 1.
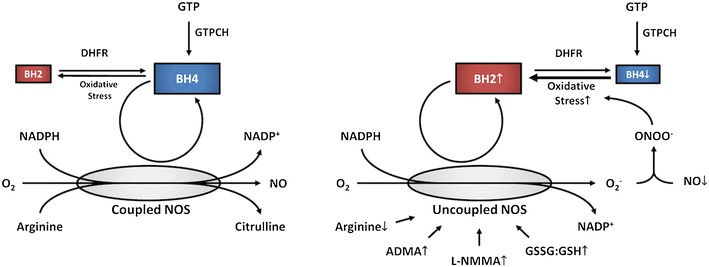
BH4 synthesis, recycling, and oxidation as determinants of NOS uncoupling. Left To produce nitric oxide (NO), nitric oxide synthase (NOS) enzymes require the substrates L-arginine and molecular oxygen (O2) and the cofactors tetrahydrobiopterin (BH4), reduced nicotinamide adenine diphosphate (NADPH), heme (not pictured), flavin mononucleotide (FMN, not pictured), and flavin adenine dinucleotide (FAD, not pictured). Under normal conditions, BH4 bioavailability is maintained by 1) de novo synthesis from guanosine triphosphate (GTP), in which the rate-limiting step is catalyzed by GTP cyclohydrolase (GTPCH) and 2) dihydrofolate reductase (DHFR)-mediated recycling of 7,8-dihydrobiopterin (BH2), the primary product of nonenzymatic BH4 oxidation. Right “Uncoupled” NOS is characterized by production of superoxide (O−2). NOS uncoupling is promoted by reduced BH4 bioavailability relative to either BH2 or NOS protein. In turn, O−2 produced by uncoupled NOS reacts with NO, forming peroxynitrite (ONOO−), a highly reactive anion that rapidly oxidizes BH4. Therefore, a state of NOS uncoupling is stabilized by self-propagating oxidative stress. In addition to this primary BH4-mediated cycle, additional mechanisms have been shown to promote uncoupling, including reduced arginine bioavailability, high levels of oxidized glutathione (GSSG) relative to reduced glutathione (GSH), or increased concentrations of the endogenous NOS inhibitors L-N-monomethylarginine (L-NMMA) and asymmetric dimethylarginine (ADMA)
Cellular superoxide is also produced by a number of other mechanisms, including NADPH oxidase [8, 47, 48•], xanthine oxidase [49], and mitochondrial respiration [50]. During disease pathogenesis, oxidative stress from one or more of these sources could contribute to BH4 oxidation, thereby initiating or maintaining NOS uncoupling [8, 48•]. Additional superoxide resulting from initial BH4 oxidation and NOS uncoupling would promote a cycle of cellular damage, further BH4 oxidation and biochemical stabilization of uncoupled NOS activity (Fig. 1). Interruption of this feed-forward process is a promising strategy for the treatment of a wide range of cardiovascular disease states caused or exacerbated by NOS uncoupling.
Roles of NOS in Cardiac Physiology and Pathophysiology
Coronary Blood Flow and Myocardial Function
NO maintains vascular tone by diffusing to the smooth muscle cells in the vascular wall and activating soluble guanylate cyclase to produce cyclic GMP, which in turn modulates calcium influx and smooth muscle contractility. This pathway is the primary mechanism underlying endothelium-dependent vasodilation in response to shear stress or cholinergic stimulation [51]. In the myocardium, NO signaling is therefore an important regulator of both basal blood flow and induced vessel dilation [52]. However, cardiomyocytes also express functional eNOS [53], nNOS [54, 55], and, after cytokine stimulation, iNOS [56]. The major functions of endogenous NOS activity in the myocardium include modulation of cholinergic and β-adrenergic responsiveness [55, 57], stretch-induced Ca2+ handling in cardiomyocytes [58], and whole-heart contractility in response to volume loading [59]. BH4 bioavailability plays an important role in supporting myocardial NO production, as evidenced by increased basal heart rate and β-adrenergic responsiveness in mice with reduced GTPCH expression [60]. Further discussion of the complexities of subcellular NOS targeting and physiological roles in the myocardium are available elsewhere [61, 62].
Overload-induced Heart Failure
One of the first assessments of NOS uncoupling in the context of heart failure was conducted using transaortic constriction in mice as a model of chronic ventricular pressure overload [63]. In the weeks following aortic constriction, untreated wild-type mice develop cardiac hypertrophy, structural remodeling, increased myocardial superoxide production, and, ultimately ventricular dysfunction [63]. Recoupling of NOS via BH4 supplementation reverses these effects and improves cardiac function at 9 weeks of constriction, but similar effects were also achieved by complete knockout of eNOS [63]. These findings suggest that oxidant stress due to eNOS uncoupling is a central mechanism underlying overload-induced heart failure. Follow-up studies using the same model showed that BH4 supplementation can also treat established cardiac dysfunction (4 weeks of transaortic constriction), significantly reversing superoxide production, and improving left ventricular function by 9 weeks after introduction of cardiac overload [64]. Importantly, these studies showed that reversal of cardiac dysfunction cannot be achieved with tetrahydroneopterin or tempol, compounds that exhibit antioxidant properties but are unable to recouple NOS [63, 64].
Hypertension and Associated Cardiac Dysfunction
The importance of eNOS as a regulator of systemic blood pressure is well-established [65], and the net impact of all three NOS isoforms in maintaining cardiovascular homeostasis was recently assessed in triple knockout mice [66]. Studies in mice with hypertension due to treatment with deoxycorticosterone acetate (DOCA) demonstrated that NOS uncoupling is a critical molecular mechanism involved in the development of hypertension [8, 9]. Aortas harvested from DOCA-treated mice demonstrate evidence of BH4 oxidation, increased superoxide production, and reduced L-arginine to L-citrulline conversion [8]. These effects are eNOS-specific and responsive to BH4 treatment [8] or overexpression of GTPCH [9]. BH4 supplementation also has been shown to recouple NOS and reverse endothelial dysfunction associated with pulmonary hypertension [67] or following infusion with angiotensin II [68] or ACTH [69]. In addition to influencing coronary blood supply, systemic hypertension also determines cardiac loading and represents an important risk factor associated with heart failure. In subsequent in vivo studies, DOCA treatment was shown to result in significant left ventricular diastolic dysfunction associated with BH4 oxidation and NOS uncoupling in the myocardium [70•]. Ventricular diastolic dysfunction can be reversed by treatment with BH4, but not the smooth muscle–targeted vasodilator hydralazine, indicating the therapeutic importance of recoupling NOS as opposed to normalizing blood pressure alone [70•].
Ischemia/Reperfusion Injury
Cardiac ischemia followed by reperfusion results in significant BH4 oxidation, NOS uncoupling, endothelial dysfunction in coronary arterioles, and impaired coronary blood flow [71, 72]. BH4 supplementation partially inhibits superoxide production and restores NO production in isolated rat hearts [71]. Treatment with BH4 or the BH4 precursor sepiapterin also restores endothelium-dependent vasorelaxation in isolated coronary arterioles and improves ventricular function following ischemia/reperfusion injury [72–74]. However, the ability of BH4 to restore NO production decreases after 30 min and is absent after 90 min of ischemia in isolated rat hearts, suggesting that mechanisms in addition to BH4 depletion also contribute to NOS uncoupling induced by prolonged ischemia [71].
Atrial Fibrillation
Atrial NO synthesis is reduced in a porcine model of atrial fibrillation [75], and atrial production of superoxide and peroxynitrite is correlated with risk of developing atrial fibrillation in patients undergoing cardiac surgery [76]. In a recent study pairing data from a goat model with data from patients receiving cardiac surgery, NADPH oxidase was found to be an important source of atrial superoxide production in patients with postoperative atrial fibrillation and in goats after 2 weeks of induced fibrillation [48•]. In contrast, BH4 depletion and atrial superoxide production by uncoupled NOS are increased in patients with permanent atrial fibrillation and goats subjected to 6 months of induced fibrillation [48•]. These data suggest that initial oxidative stress from NADPH oxidase may ultimately lead to permanent atrial fibrillation by depleting BH4 and establishing a steady state of NOS uncoupling.
Additional Mechanisms of NOS Uncoupling
L-Arginine Bioavailability
The role of L-arginine bioavailability in determining NOS uncoupling was first illustrated by experiments in nNOS-transfected cells that demonstrated increased peroxynitrite-mediated cellular damage under conditions of L-arginine depletion [77]. Subsequent studies in isolated nNOS and eNOS enzymes suggest that the effect of L-arginine depletion depends on NOS isoform, as well as on the presence of BH4. In isolated, BH4-replete nNOS, superoxide production is significantly increased in the absence of L-arginine [78]. This finding suggests that despite the presence of BH4, nNOS ejects activated oxygen as superoxide with greater frequency when L-arginine substrate is not available for further reactions. In the absence of BH4, superoxide production by nNOS is maximal and independent of L-arginine availability [78]. In isolated, BH4-depleted eNOS enzyme, however, superoxide production is in fact stimulated by L-arginine levels [79]. These results suggest that in the absence of BH4, the introduction of L-arginine may induce conformational changes in eNOS that promote electron flow and increase superoxide production if BH4 is not available to couple oxygen activation to L-arginine oxidation. This interpretation is supported by the observation that L-arginine dose-dependently increases NADPH consumption by BH4-depleted eNOS [79].
Accumulation of Methylarginines
Methylated L-arginine species, including L-N-monomethylarginine (L-NMMA) and ω-N G,N G-asymmetric dimethylarginine (ADMA), are produced by post-translational methylation of L-arginine residues by protein methyltransferases and liberated by subsequent proteolysis [80]. ADMA and L-NMMA may therefore accumulate due to increased methyltransferase activity [81] and/or inhibition of dimethylarginine dimethylaminohydrolase (DDAH), the enzyme responsible for ADMA catabolism [82]. Both L-NMMA and ADMA inhibit NOS-mediated NO production by competing with L-arginine to bind at the NOS catalytic site [83]. Under conditions of BH4 depletion, however, L-NMMA increases nNOS-mediated superoxide production and both L-NMMA and ADMA increase NADPH consumption and superoxide production by eNOS [78, 79]. In conjunction with BH4 depletion, accumulation of methylarginines may therefore contribute to NOS uncoupling in the context of endothelial or myocardial dysfunction. In support of this hypothesis, a study of patients undergoing coronary artery bypass surgery demonstrated that increased plasma ADMA concentrations correlate with reduced vasodilation and increased superoxide production in ex vivo vessels [84].
S-Glutathionylation
S-glutathionylation involves the binding of a glutathione tripeptide to the thiol group of an available cysteine residue. In a recent study, S-glutathionylation of two critical eNOS cysteine residues (C689 and C908) significantly inhibited NO production and increased superoxide production [85••]. Furthermore, increased glutathionylation in the aortic rings of oxidant-treated or spontaneously hypertensive rats impaired eNOS-dependent vasorelaxation following acetylcholine stimulation [85••]. The authors of this study speculate that superoxide production following S-glutathionylation occurs at the reductase domain, rather than at the heme catalytic center [85••], although further experimentation is needed to conclusively differentiate these two potential mechanisms of superoxide production.
Recoupling NOS as a Therapeutic Approach
L-Arginine Supplementation
L-arginine supplementation has been pursued as a potential approach to restoring NOS-mediated NO production in patients with cardiovascular disease states characterized by endothelial dysfunction. However, completed clinical trials have largely failed to show beneficial effects, and in some cases demonstrated evidence of harm. For example, 3 g of L-arginine per day delivered orally for 6 months resulted in impaired flow-mediated vasodilation, reduced vascular compliance, and low plasma and urinary nitrites and nitrates in a cohort study of patients with peripheral arterial disease [86]. In addition, patients receiving L-arginine also scored lower on treadmill assessments of exercise capacity compared to placebo-treated control patients. Similarly, treatment with 3 g of L-arginine three times daily for 6 months failed to produce a significant effect on ejection fraction or vascular stiffness in a study of patients who suffered myocardial infarction [87]. The group receiving L-arginine treatment did, however, exhibit a higher death rate than the control group leading to closure of enrollment due to safety concerns [87]. Can the general lack of efficacy of L-arginine supplementation be ascribed to the failure to also replete BH4? Based on available biochemical data [78, 79], supplementation of L-arginine under conditions of BH4 depletion is not only insufficient to recouple NOS, but in fact exacerbates NOS uncoupling. Therefore, administration of L-arginine alone would not be expected to improve endothelial function in disease states characterized by BH4 depletion.
BH4 Supplementation
BH4 supplementation as a therapeutic approach promises the possibility of improving endothelial dysfunction while reducing the risk of hypotension associated with therapies aimed at downstream NO signaling targets. Several physiological experiments have demonstrated improved eNOS-dependent vasodilation following BH4 infusion in individuals with hypertension [88], hypercholesterolemia [89], diabetes [90], or a history of smoking [91]. Early small trials also demonstrated that oral BH4 administration improved forearm blood flow in hypercholestrolemic patients [92] and reduced blood pressure in patients with poorly controlled hypertension [93]. However, these studies were limited in scope, largely due to the instability of BH4 preparations for oral delivery. In 2007, the U.S. Food and Drug Administration approved use of sapropterin, a thermo- and photo-stable, orally available preparation of synthetic 6R-BH4 (trade name: Kuvan [manufactured by Biomarin Pharmaceutical, Inc., Novato, CA]) for the treatment of phenylketonuria (PKU). PKU is a metabolic disorder resulting from a loss-of-function mutation in the BH4-dependent enzyme phenylalanine hydroxylase that is characterized by accumulation of plasma phenylalanine and potentially severe neurological complications. Large phase 3 trials in this patient population demonstrated that sapropterin was well-tolerated and significantly reduced plasma phenylalanine levels by 1 week of therapy [94, 95]. The efficacy and safety profiles demonstrated in these trials resulted in high interest in sapropterin as a potential treatment for cardiovascular disease characterized by endothelial dysfunction. In a study of patients with sickle cell disease, initial results suggested some improvement in reactive forearm vasodilation following sustained treatment with sapropterin. However, trials in patients with systemic or pulmonary hypertension failed to demonstrate significant efficacy, and full data on these and most other registered studies have yet to be published (Table 1).
Table 1.
Clinical trials of oral sapropterin
| Disease state | Intervention | Duration, wk | Results | Clinical trial ID |
|---|---|---|---|---|
| Systemic hypertension | Oral sapropterin, 5 mg/kg, BID | 8 | No statistically significant effect on SBP vs placebo (http://www.bmrn.com/) | NCT00325962 |
| Pulmonary arterial hypertension | Oral sapropterin, dose escalation every 2 wks from 2.5 to 5 to 10 mg/kg/d + 2 d of 20 mg/kg BID | 6 | No statistically significant effects vs placebo (http://www.bmrn.com/) | NCT00435331 |
| Sickle cell disease | Oral sapropterin, dose escalation every 4 wks from 2.5 to 5 to 10 to 20 mg/kg/d | 16 | Improvement of RH-PAT at 8, 12, and 16 wks of treatment. Sapropterin was well tolerated (http://www.bmrn.com/) | NCT00445978 |
| Coronary artery disease | 400 mg/d or 700 mg/d | 2–6 | No statistically significant difference in clinical end points or in dilation or NOS coupling in ex vivo vessels | NCT00423280, see reference [96••] |
| Peripheral arterial disease | 400 mg BID | 24 | Not reported to date (Start date: Dec 2006; est. completion: Jan 2009) | NCT00403494 |
| Systolic hypertension | Oral sapropterin, 5 mg/kg, BID | 8 | Not reported (Start date: Dec 2008) | NCT00802893 |
| Chronic kidney disease | Oral sapropterin, 400 mg BID + 400 mg vitamin C BID for the second 6 wks | 12 | Not reported to date (Start date: May 2008; est. completion: Aug 2009) | NCT00625820 |
| All-cause endothelial dysfunction | Oral sapropterin, 5 mg/kg oral ± 500 mg vitamin C BID | 2 | Not reported to date (Start date: Sept 2007; est. completion: March 2009) | NCT00532844 |
| Liver cirrhosis, portal hypertension | Oral sapropterin, dose escalation weekly from 5 to 10 mg/kg/d | 2 | Ongoing (Start date: Oct 2011; est. completion: Jan 2013) | NCT01456286 |
Synthetic 6R-BH4, Trade name: Kuvan (manufactured by Biomarin Pharmaceutical Inc., Novato, CA)
BID twice daily; SBP systolic blood pressure; RH-PAT reactive hyperemia-peripheral arterial tonometry (a measurement of vasodilation following temporary constriction of the forearm); NOS nitric oxide synthase; est. estimated.
Despite this general lack of published clinical trial data, a recent report on sapropterin treatment in patients with coronary artery disease provides some mechanistic insight into the limitations of oral BH4 supplementation as a therapeutic approach to treating cardiovascular disease [96••]. In this study, oral sapropterin treatment failed to improve brachial flow-mediated vasodilation, aortic or carotid distensibility, or acetylcholine-induced vasodilation in ex vivo saphenous vein rings [96••]. Plasma and saphenous vein tissue BH4 concentrations were higher in patients receiving sapropterin treatment compared to placebo-treated control patients, but a concomitant increase in BH2 concentrations yielded no significant improvement in the ratio of BH4 to oxidized biopterin species, conversion of L-arginine to L-citrulline, or superoxide production [96••]. Furthermore, exogenous BH4 added to whole blood was rapidly oxidized and incubation of ex vivo saphenous vein rings with exogenous BH4 paradoxically reduced the ratio of BH4 to oxidized biopterins due to accumulation of BH2 [96••]. The ability of BH4 to recouple NOS in patients with cardiovascular disease may therefore be limited by BH4 oxidation, BH2 accumulation, and failure to improve BH4:BH2 ratios. This limitation also has recently been recognized in an animal model of chronic pressure overload-induced heart failure. Following increasing doses of BH4, assessment of myocardial biopterin levels showed that BH2 rose linearly, but BH4 plateaued at higher doses, resulting in BH4:BH2 ratios returning back toward baseline values [97•].
Indirect Preservation of BH4
Given these limitations of direct BH4 administration, therapeutic interventions aimed at improving or preserving endogenous BH4 bioavailability may represent a viable alternative. For example, the beneficial effects of statins may depend in part on their ability to increase BH4 bioavailability, as has been demonstrated both in vitro [98] and in patients [99]. By improving BH4 recycling and/or reducing oxidant stress, ascorbic acid [44] and 5-methyl-tetrahydrofolate [100] also have been shown to improve BH4 bioavailability and coupled NOS activity.
Conclusions
Reduced BH4 concentrations relative to cellular BH2 and NOS protein result in the uncoupling of oxygen activation from NO production and promote the generation of superoxide anions instead. Oxidative stress due to uncoupled NOS activity in turn leads to oxidation of BH4 to BH2 and further propagation of NOS uncoupling. NOS uncoupling and BH4 oxidation thus represent a self-perpetuating cycle that ultimately results in impaired NO signaling in addition to cellular damage and inflammation due to increased oxidative stress (Fig. 1). Restoration of BH4 bioavailability therefore represents a promising approach to recoupling NOS for the treatment of cardiovascular disease. However, early clinical trials of oral synthetic BH4 (sapropterin) thus far generally have failed to demonstrate efficacy in the treatment of diseases characterized by endothelial dysfunction (Table 1). These disappointing results may result from failure to improve the ratio of BH4 to BH2 [96••], highlighting the need for further research on cellular and systemic BH4 oxidation, trafficking and recycling. The clinical efficacy of improving BH4 bioavailability also may depend on other biochemical determinants of NOS uncoupling, including cellular concentrations of L-arginine, methylarginines, and oxidized glutathione. Future studies are therefore needed to understand how BH4 bioavailability and these factors interact with one another to determine NOS activity at biochemical, cellular, and systemic levels. Resulting advances could lead to significant improvement in the treatment of the wide range of cardiovascular disease states in which NOS uncoupling is a central pathogenic mechanism.
Acknowledgments
Disclosures
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Daff S. NO synthase: structures and mechanisms. Nitric Oxide. 2010;23(1):1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Stuehr DJ, Kwon NS, Nathan CF, et al. N omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J Biol Chem. 1991;266(10):6259–6263. [PubMed] [Google Scholar]
- 3.Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95(16):9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273(40):25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 5.Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362(Pt 3):733–739. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM. Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem. 1992;267(34):24173–24176. [PubMed] [Google Scholar]
- 7.Hattori Y, Hattori S, Wang X, et al. Oral administration of tetrahydrobiopterin slows the progression of atherosclerosis in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2007;27(4):865–870. doi: 10.1161/01.ATV.0000258946.55438.0e. [DOI] [PubMed] [Google Scholar]
- 8.Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117(8):1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 10.Alp NJ, Mussa S, Khoo J, et al. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112(5):725–735. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 13.Kwon NS, Nathan CF, Stuehr DJ. Reduced biopterin as a cofactor in the generation of nitrogen oxides by murine macrophages. J Biol Chem. 1989;264(34):20496–20501. [PubMed] [Google Scholar]
- 14.Tayeh MA, Marletta MA. Macrophage oxidation of L-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. J Biol Chem. 1989;264(33):19654–19658. [PubMed] [Google Scholar]
- 15.Baek KJ, Thiel BA, Lucas S, Stuehr DJ. Macrophage nitric oxide synthase subunits. Purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J Biol Chem. 1993;268(28):21120–21129. [PubMed] [Google Scholar]
- 16.Klatt P, Schmidt K, Lehner D, et al. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. EMBO J. 1995;14(15):3687–3695. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crane BR, Arvai AS, Ghosh DK, et al. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science (New York, NY) 1998;279(5359):2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 18.Wei CC, Wang ZQ, Hemann C, Hille R, Stuehr DJ. A tetrahydrobiopterin radical forms and then becomes reduced during Nomega-hydroxyarginine oxidation by nitric-oxide synthase. J Biol Chem. 2003;278(47):46668–46673. doi: 10.1074/jbc.M307682200. [DOI] [PubMed] [Google Scholar]
- 19.Hurshman AR, Krebs C, Edmondson DE, Huynh BH, Marletta MA. Formation of a pterin radical in the reaction of the heme domain of inducible nitric oxide synthase with oxygen. Biochemistry. 1999;38(48):15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]
- 20.Wei CC, Wang ZQ, Wang Q, et al. Rapid kinetic studies link tetrahydrobiopterin radical formation to heme-dioxy reduction and arginine hydroxylation in inducible nitric-oxide synthase. J Biol Chem. 2001;276(1):315–319. doi: 10.1074/jbc.M008441200. [DOI] [PubMed] [Google Scholar]
- 21.Wei CC, Wang ZQ, Tejero J, et al. Catalytic reduction of a tetrahydrobiopterin radical within nitric-oxide synthase. J Biol Chem. 2008;283(17):11734–11742. doi: 10.1074/jbc.M709250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. doi: 10.1042/0264-6021:3470001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tatham AL, Crabtree MJ, Warrick N, et al. GTP cyclohydrolase I expression, protein, and activity determine intracellular tetrahydrobiopterin levels, independent of GTP cyclohydrolase feedback regulatory protein expression. J Biol Chem. 2009;284(20):13660–13668. doi: 10.1074/jbc.M807959200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katusic ZS, Stelter A, Milstien S. Cytokines stimulate GTP cyclohydrolase I gene expression in cultured human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18(1):27–32. doi: 10.1161/01.ATV.18.1.27. [DOI] [PubMed] [Google Scholar]
- 25.Kasai K, Hattori Y, Banba N, et al. Induction of tetrahydrobiopterin synthesis in rat cardiac myocytes: impact on cytokine-induced NO generation. Am J Physiol. 1997;273(2 Pt 2):H665–H672. doi: 10.1152/ajpheart.1997.273.2.H665. [DOI] [PubMed] [Google Scholar]
- 26.Harada T, Kagamiyama H, Hatakeyama K. Feedback regulation mechanisms for the control of GTP cyclohydrolase I activity. Science (New York, NY) 1993;260(5113):1507–1510. doi: 10.1126/science.8502995. [DOI] [PubMed] [Google Scholar]
- 27.Milstien S, Jaffe H, Kowlessur D, Bonner TI. Purification and cloning of the GTP cyclohydrolase I feedback regulatory protein, GFRP. J Biol Chem. 1996;271(33):19743–19751. doi: 10.1074/jbc.271.33.19743. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Rezvan A, Salerno JC, et al. GTP cyclohydrolase I phosphorylation and interaction with GTP cyclohydrolase feedback regulatory protein provide novel regulation of endothelial tetrahydrobiopterin and nitric oxide. Circ Res. 2010;106(2):328–336. doi: 10.1161/CIRCRESAHA.109.210658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du J, Wei N, Xu H, et al. Identification and functional characterization of phosphorylation sites on GTP cyclohydrolase I. Arterioscler Thromb Vasc Biol. 2009;29(12):2161–2168. doi: 10.1161/ATVBAHA.109.194464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai S, Alp NJ, McDonald D, et al. GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovasc Res. 2002;55(4):838–849. doi: 10.1016/S0008-6363(02)00460-1. [DOI] [PubMed] [Google Scholar]
- 31.Crabtree MJ, Tatham AL, Al-Wakeel Y, et al. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. J Biol Chem. 2009;284(2):1136–1144. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- 32.Bendall JK, Alp NJ, Warrick N, et al. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97(9):864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 33.Ozaki M, Kawashima S, Yamashita T, et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest. 2002;110(3):331–340. doi: 10.1172/JCI15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Rao F, Zhang K, et al. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest. 2007;117(9):2658–2671. doi: 10.1172/JCI31093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antoniades C, Shirodaria C, Van Assche T, et al. GCH1 haplotype determines vascular and plasma biopterin availability in coronary artery disease effects on vascular superoxide production and endothelial function. J Am Coll Cardiol. 2008;52(2):158–165. doi: 10.1016/j.jacc.2007.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am J Physiol Heart Circ Physiol. 2008;294(4):H1530–H1540. doi: 10.1152/ajpheart.00823.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antoniades C, Shirodaria C, Crabtree M, et al. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation. 2007;116(24):2851–2859. doi: 10.1161/CIRCULATIONAHA.107.704155. [DOI] [PubMed] [Google Scholar]
- 38.Curtius HC, Heintel D, Ghisla S, et al. Tetrahydrobiopterin biosynthesis. Studies with specifically labeled (2H)NAD(P)H and 2H2O and of the enzymes involved. Eur J Biochem. 1985;148(3):413–419. doi: 10.1111/j.1432-1033.1985.tb08855.x. [DOI] [PubMed] [Google Scholar]
- 39.Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem. 2009;284(41):28128–28136. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugiyama T, Levy BD, Michel T. Tetrahydrobiopterin recycling, a key determinant of endothelial nitric-oxide synthase-dependent signaling pathways in cultured vascular endothelial cells. J Biol Chem. 2009;284(19):12691–12700. doi: 10.1074/jbc.M809295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crabtree MJ, Hale AB, Channon KM. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic Biol Med. 2011;50(11):1639–1646. doi: 10.1016/j.freeradbiomed.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawabe K, Wakasugi KO, Hasegawa H. Tetrahydrobiopterin uptake in supplemental administration: elevation of tissue tetrahydrobiopterin in mice following uptake of the exogenously oxidized product 7,8-dihydrobiopterin and subsequent reduction by an anti-folate-sensitive process. J Pharmacol Sci. 2004;96(2):124–133. doi: 10.1254/jphs.FP0040280. [DOI] [PubMed] [Google Scholar]
- 43.Vasquez-Vivar J, Whitsett J, Martasek P, Hogg N, Kalyanaraman B. Reaction of tetrahydrobiopterin with superoxide: EPR-kinetic analysis and characterization of the pteridine radical. Free Radic Biol Med. 2001;31(8):975–985. doi: 10.1016/S0891-5849(01)00680-3. [DOI] [PubMed] [Google Scholar]
- 44.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278(25):22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 45.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87(4):1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263(3):681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 47.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97(8):1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reilly SN, Jayaram R, Nahar K, et al. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2011;124(10):1107–1117. doi: 10.1161/CIRCULATIONAHA.111.029223. [DOI] [PubMed] [Google Scholar]
- 49.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 52.Seddon M, Melikian N, Dworakowski R, et al. Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation. 2009;119(20):2656–2662. doi: 10.1161/CIRCULATIONAHA.108.822205. [DOI] [PubMed] [Google Scholar]
- 53.Balligand JL, Kobzik L, Han X, et al. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J Biol Chem. 1995;270(24):14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- 54.Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96(2):657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105(25):3011–3016. doi: 10.1161/01.CIR.0000019516.31040.2D. [DOI] [PubMed] [Google Scholar]
- 56.Balligand JL, Ungureanu-Longrois D, Simmons WW, et al. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J Biol Chem. 1994;269(44):27580–27588. [PubMed] [Google Scholar]
- 57.Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci USA. 1993;90(1):347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petroff MG, Kim SH, Pepe S, et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3(10):867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 59.Prendergast BD, Sagach VF, Shah AM. Basal release of nitric oxide augments the Frank-Starling response in the isolated heart. Circulation. 1997;96(4):1320–1329. doi: 10.1161/01.CIR.96.4.1320. [DOI] [PubMed] [Google Scholar]
- 60.Adlam D, Herring N, Douglas G, et al. Regulation of beta-adrenergic control of heart rate by GTP-cyclohydrolase 1 (GCH1) and tetrahydrobiopterin. Cardiovasc Res. 2012;93(4):694–701. doi: 10.1093/cvr/cvs005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93(5):388–398. doi: 10.1161/01.RES.0000088351.58510.21. [DOI] [PubMed] [Google Scholar]
- 62.Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol. 2012;52(2):341–350. doi: 10.1016/j.yjmcc.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 63.Takimoto E, Champion HC, Li M, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115(5):1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moens AL, Takimoto E, Tocchetti CG, et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117(20):2626–2636. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang PL, Huang Z, Mashimo H, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377(6546):239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 66.Nakata S, Tsutsui M, Shimokawa H, et al. Spontaneous myocardial infarction in mice lacking all nitric oxide synthase isoforms. Circulation. 2008;117(17):2211–2223. doi: 10.1161/CIRCULATIONAHA.107.742692. [DOI] [PubMed] [Google Scholar]
- 67.Khoo JP, Zhao L, Alp NJ, et al. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation. 2005;111(16):2126–2133. doi: 10.1161/01.CIR.0000162470.26840.89. [DOI] [PubMed] [Google Scholar]
- 68.Kase H, Hashikabe Y, Uchida K, Nakanishi N, Hattori Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J Hypertens. 2005;23(7):1375–1382. doi: 10.1097/01.hjh.0000173520.13976.7d. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Pang T, Earl J, et al. Role of tetrahydrobiopterin in adrenocorticotropic hormone-induced hypertension in the rat. Clin Exp Hypertens. 2004;26(3):231–241. doi: 10.1081/CEH-120030232. [DOI] [PubMed] [Google Scholar]
- 70.Silberman GA, Fan TH, Liu H, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121(4):519–528. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dumitrescu C, Biondi R, Xia Y, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci USA. 2007;104(38):15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tiefenbacher CP, Chilian WM, Mitchell M, DeFily DV. Restoration of endothelium-dependent vasodilation after reperfusion injury by tetrahydrobiopterin. Circulation. 1996;94(6):1423–1429. doi: 10.1161/01.CIR.94.6.1423. [DOI] [PubMed] [Google Scholar]
- 73.Verma S, Maitland A, Weisel RD, et al. Novel cardioprotective effects of tetrahydrobiopterin after anoxia and reoxygenation: identifying cellular targets for pharmacologic manipulation. J Thorac Cardiovasc Surg. 2002;123(6):1074–1083. doi: 10.1067/mtc.2002.121687. [DOI] [PubMed] [Google Scholar]
- 74.Yamashiro S, Noguchi K, Matsuzaki T, et al. Beneficial effect of tetrahydrobiopterin on ischemia-reperfusion injury in isolated perfused rat hearts. J Thorac Cardiovasc Surg. 2002;124(4):775–784. doi: 10.1067/mtc.2002.124393. [DOI] [PubMed] [Google Scholar]
- 75.Cai H, Li Z, Goette A, et al. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. 2002;106(22):2854–2858. doi: 10.1161/01.CIR.0000039327.11661.16. [DOI] [PubMed] [Google Scholar]
- 76.Antoniades C, Demosthenous M, Reilly S, et al. Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J Am Coll Cardiol. 2012;59(1):60–70. doi: 10.1016/j.jacc.2011.08.062. [DOI] [PubMed] [Google Scholar]
- 77.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA. 1996;93(13):6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cardounel AJ, Xia Y, Zweier JL. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J Biol Chem. 2005;280(9):7540–7549. doi: 10.1074/jbc.M410241200. [DOI] [PubMed] [Google Scholar]
- 79.Druhan LJ, Forbes SP, Pope AJ, et al. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry. 2008;47(27):7256–7263. doi: 10.1021/bi702377a. [DOI] [PubMed] [Google Scholar]
- 80.Leiper J, Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat Rev Drug Discov. 2011;10(4):277–291. doi: 10.1038/nrd3358. [DOI] [PubMed] [Google Scholar]
- 81.Boger RH, Sydow K, Borlak J, et al. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. 2000;87(2):99–105. doi: 10.1161/01.RES.87.2.99. [DOI] [PubMed] [Google Scholar]
- 82.Forbes SP, Druhan LJ, Guzman JE, et al. Mechanism of 4-HNE mediated inhibition of hDDAH-1: implications in no regulation. Biochemistry. 2008;47(6):1819–1826. doi: 10.1021/bi701659n. [DOI] [PubMed] [Google Scholar]
- 83.Cardounel AJ, Cui H, Samouilov A, et al. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem. 2007;282(2):879–887. doi: 10.1074/jbc.M603606200. [DOI] [PubMed] [Google Scholar]
- 84.Antoniades C, Shirodaria C, Leeson P, et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J. 2009;30(9):1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 85.Chen CA, Wang TY, Varadharaj S, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson AM, Harada R, Nair N, Balasubramanian N, Cooke JP. L-arginine supplementation in peripheral arterial disease: no benefit and possible harm. Circulation. 2007;116(2):188–195. doi: 10.1161/CIRCULATIONAHA.106.683656. [DOI] [PubMed] [Google Scholar]
- 87.Schulman SP, Becker LC, Kass DA, et al. L-arginine therapy in acute myocardial infarction: the Vascular Interaction With Age in Myocardial Infarction (VINTAGE MI) randomized clinical trial. JAMA. 2006;295(1):58–64. doi: 10.1001/jama.295.1.58. [DOI] [PubMed] [Google Scholar]
- 88.Higashi Y, Sasaki S, Nakagawa K, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15(4 Pt 1):326–332. doi: 10.1016/S0895-7061(01)02317-2. [DOI] [PubMed] [Google Scholar]
- 89.Stroes E, Kastelein J, Cosentino F, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99(1):41–46. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia. 2000;43(11):1435–1438. doi: 10.1007/s001250051551. [DOI] [PubMed] [Google Scholar]
- 91.Heitzer T, Brockhoff C, Mayer B, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86(2):E36–E41. doi: 10.1161/01.RES.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 92.Cosentino F, Hurlimann D, Delli Gatti C, et al. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart. 2008;94(4):487–492. doi: 10.1136/hrt.2007.122184. [DOI] [PubMed] [Google Scholar]
- 93.Porkert M, Sher S, Reddy U, et al. Tetrahydrobiopterin: a novel antihypertensive therapy. J Hum Hypertens. 2008;22(6):401–407. doi: 10.1038/sj.jhh.1002329. [DOI] [PubMed] [Google Scholar]
- 94.Levy HL, Milanowski A, Chakrapani A, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet. 2007;370(9586):504–510. doi: 10.1016/S0140-6736(07)61234-3. [DOI] [PubMed] [Google Scholar]
- 95.Trefz FK, Burton BK, Longo N, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr. 2009;154(5):700–707. doi: 10.1016/j.jpeds.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 96.•• Cunnington C, Van Assche T, Shirodaria C, et al. Systemic and vascular oxidation limits efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012, [Epub ahead of print]. This study pairs a randomized controlled trial of two doses of oral BH4 in patients with coronary artery disease with ex vivo and in vitro studies of BH4 oxidation, transport, and effects on NOS coupling. The authors demonstrated that oral BH4 supplementation may be limited by a failure to improve vascular BH4:BH2 ratios. [DOI] [PMC free article] [PubMed]
- 97.Moens AL, Ketner EA, Takimoto E, et al. Bi-modal dose-dependent cardiac response to tetrahydrobiopterin in pressure-overload induced hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51(4):564–569. doi: 10.1016/j.yjmcc.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hattori Y, Nakanishi N, Akimoto K, Yoshida M, Kasai K. HMG-CoA reductase inhibitor increases GTP cyclohydrolase I mRNA and tetrahydrobiopterin in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23(2):176–182. doi: 10.1161/01.ATV.0000054659.72231.A1. [DOI] [PubMed] [Google Scholar]
- 99.Antoniades C, Bakogiannis C, Leeson P, et al. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation. 2011;124(3):335–345. doi: 10.1161/CIRCULATIONAHA.110.985150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antoniades C, Shirodaria C, Warrick N, et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114(11):1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]