

Abstract
Dexamethasone has been found to reduce the incidence of high-altitude pulmonary oedema. Mechanisms explaining this effect still remain unclear. We assessed the effect of dexamethasone using established cell lines, including rat alveolar epithelial cells (AEC), pulmonary artery endothelial cells (RPAEC) and alveolar macrophages (MAC), in an environment of low oxygen, simulating a condition of alveolar hypoxia as found at high altitude. Inflammatory mediators and ion transporter expression were quantified. Based on earlier results, we hypothesized that hypoxic conditions trigger inflammation. AEC, RPAEC and MAC, pre-incubated for 1 h with or without dexamethasone (10−7 mol/l), were subsequently exposed to mild hypoxia (5% O2, or normoxia as control) for 24 h. mRNA and protein levels of cytokine-induced neutrophil chemoattractant-1, monocyte chemoattractant protein-1 and interleukin-6 were analysed. mRNA expression and functional activity of the apical epithelial sodium channel and basolateral Na+/K+-ATPase were determined using radioactive marker ions. In all three types of pulmonary cells hypoxic conditions led to an attenuated secretion of inflammatory mediators, which was even more pronounced in dexamethasone pretreated samples. Function of Na+/K+-ATPase was not significantly influenced by hypoxia or dexamethasone, while activity of epithelial sodium channels was decreased under hypoxic conditions. When pre-incubated with dexamethasone, however, transporter activity was partially maintained. These findings illustrate that long-term hypoxia does not trigger an inflammatory response. The ion transport across apical epithelial sodium channels under hypoxic conditions is ameliorated in cells treated with dexamethasone.
Keywords: cytokines and chemokines, dexamethasone, ion channels, lung oedema
Introduction
Alveolar hypoxia has been shown to induce injury within the lung parenchyma including epithelial lung damage, capillary leakage and oedema formation [1]. Conditions potentially imparting states of alveolar hypoxia include exposure to high altitudes, lung diseases, bulbar cerebral injuries and overdoses of narcotic agents (alcohols, barbiturates, opioids).
In particular, high-altitude pulmonary oedema (HAPE) is a high permeability pulmonary oedema caused by increased pulmonary capillary pressure leading to a protein-rich oedema fluid [2]. Elevated pulmonary capillary pressure is induced most probably by hypoxic pulmonary vasoconstriction. Pulmonary vasodilators are efficient in treating and preventing HAPE, suggesting that elevated pulmonary artery pressure is indeed a crucial pathophysiological mechanism [3,4]. Administration of corticosteroids such as dexamethasone has been found to reduce the incidence of HAPE [5,6]. Mechanisms explaining this effect are still not thoroughly clear: besides a decrease of the systolic pulmonary artery pressure, amelioration of hypoxia-induced impairment of fluid clearance has been suggested as the underlying mechanism of the dexamethasone effect [6]. Moreover, there is a known cross-link between hypoxia and inflammation pathways [7]. Proinflammatory proteins such as interleukin (IL)-6 and C-reactive protein (CRP) have been found to be increased in response to high-altitude exposure [8]. Attenuation in proinflammatory cytokine expression by corticosteroids might therefore also be an underlying factor for the preventing effect of dexamethasone.
In inflammatory processes, chemokines such as cytokine-induced neutrophil chemoattractant protein-1 (CINC-1) [9], monocyte chemoattractant protein-1 (MCP-1) [10] and IL-6 [11] are among the most important inflammatory mediators orchestrating, directly or indirectly, the effector cell migration to the location of tissue injury, where an elevated expression of adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1), mediate effector–target cell interaction in the respiratory and vascular compartment of the lung [12,13] (Fig. 1), an interplay between alveolar epithelial cells (AEC), endothelial cells (rat pulmonary artery endothelial cells, RPAEC) and alveolar macrophages (MAC).
Fig. 1.
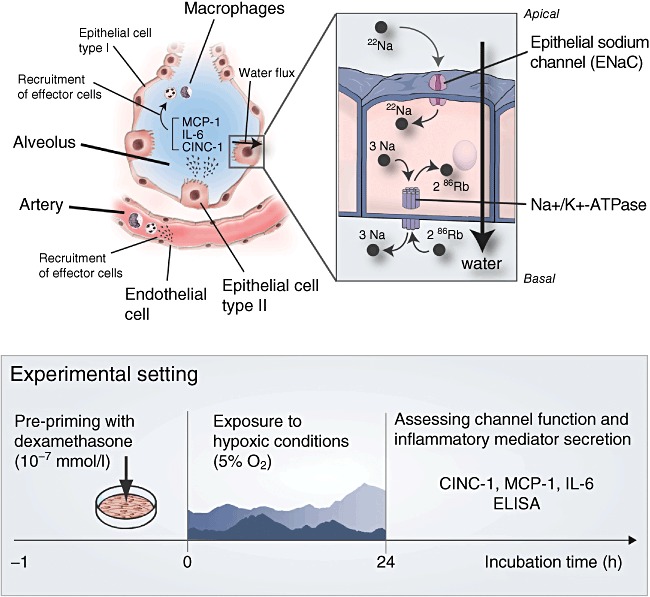
Schematic drawing of an alveolus in state of early inflammation. Effector cell recruitment by inflammatory mediators from pulmonary epithelial and endothelial cells is illustrated (left side). Water transport through ion channels located on the alveolar epithelial cell apical surface and basolateral membrane (right side) is impaired. Sodium enters the cell via apical epithelial sodium channels (ENaC). On the basolateral interface, sodium is excreted via Na+/K+-ATPase, while potassium enters the cell. Reabsorption of sodium goes along with the reabsorption of Cl–, which generates an osmotic driving force for the transepithelial movement of water. Radioactive marker ions (22Na+ for Na+ and 86Rb for K+) were used to measure the ion channel turnover. CINC-1, cytokine-induced neutrophil chemoattractant-1; MCP-1, monocyte chemoattractant protein-1; IL, interleukin.
In this study, we hypothesized that dexamethasone treatment before and during exposure to hypoxic conditions attenuates inflammatory mediator synthesis in AEC, RPAEC and MAC in vitro and enhances alveolar water clearance in AEC. The study aims at a clearer understanding of the effect of dexamethasone on alveolar water clearance and the secretion of proinflammatory mediators under long-term hypoxic conditions found at high altitudes. Insights regarding the effect of dexamethasone in alveolar hypoxia may have implications for the future treatment of HAPE.
Material and methods
Experimental design
Conditions at high altitude were simulated exposing AEC, RPAEC and MAC to 5% hypoxia during 24 h. After pre-incubation with either dexamethasone or NaCl containing cell culture medium for 1 h, cells were exposed for 24 h to hypoxia or normoxia in a cell incubator (Bioblock, Ittigen, Switzerland) with adjustable O2 levels. Oxygen concentration was reached by flushing nitrogen through the exposure chamber and monitored continuously by an oxygen sensor. During hypoxic conditions, oxygen and carbon dioxide concentrations were both 5%, whereas under normoxic control conditions, oxygen was 21% and CO2 was 5%. Temperature was kept constant at 37°C during all experiments. After 24-h exposure to the respective atmosphere, supernatants and/or cells were collected immediately without reoxygenation.
Alveolar epithelial cells (L2)
The L2 cell line (CCL 149; American Type Culture Collection, Rockville, MD, USA) was used as representative of rat alveolar epithelial cells [14], cultured in Dulbecco's modified eagle medium (DMEM; Invitrogen AG, Basel, Switzerland), enriched with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cells were grown in uncoated 35 × 10 mm plates (Corning Inc., Corning, NY, USA) to a confluent monolayer. Before performing the experiments, the medium was changed to DMEM/1% FBS.
Rat pulmonary artery endothelial cells (RPAEC)
Rat pulmonary artery endothelial cells, kindly provided by Dr Roscoe Warner (Department of Pathology, University of Michigan, Ann Arbor, MI, USA), were cultured in DMEM with 10% FBS, 1% penicillin/streptomycin and 1% HEPES. Prior to the experiments, medium was changed to DMEM/1% FBS.
Rat alveolar macrophages (MAC)
The rat alveolar macrophage cell line CRL-2192 was obtained from the American Type Culture Collection. Alveolar macrophages were cultured in nutrient mixture F-12 Ham (Ham's F-12; Invitrogen Corp., Carlsbad, CA, USA), completed with 15% FBS, 5% penicillin/streptomycin (10 000 U/l) (Invitrogen Corp.) and 5% HEPES (Invitrogen Corp.). The cells were grown to confluence. At day 3 cells were centrifuged for 5 min at 250 g, and experiments were performed.
All three types of cells are represented by well-established pulmonary cell lines.
Dexamethasone treatment
Dexamethasone (Mephamesone-4®; Mepha Pharma AG, Aesch, Switzerland) was dissolved in NaCl and diluted in culture medium DMEM/1%FBS to a concentration of 10−7 mol/l. In order to investigate the effect of dexamethasone on the secretion of inflammatory mediators and ion channel function under normoxic and hypoxic conditions, cells were pre-incubated with dexamethasone-containing medium 1 h before exposure to hypoxia/normoxia for 24 h (NaCl served as negative control). Cells therefore underwent a total of 25 h of exposure to dexamethasone before measurements.
Cytotoxicity and viability tests
To determine possible cytotoxicity mediated by dexamethasone and/or hypoxia, we measured lactate dehydrogenase concentrations (LDH) (Promega, Madison, WI, USA) in cell supernatants. The cell's viability was monitored by performing 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) assays [15].
Enzyme-linked immunoabsorbent assay (ELISA)
Sandwich ELISAs were performed according to the manufacturer's protocol. The concentration of the chemokines CINC-1 (R&D Systems Europe Ltd, Abingdon, UK), MCP-1 (BD Biosciences, San Diego, CA, USA) and IL-6 (R&D Systems Europe Ltd) were measured. Endothelial cells did not express any IL-6 protein. Cell-based ICAM-1 ELISA was performed on AEC and RPAEC, as described previously [16].
RNA extraction, reverse transcriptase and real-time PCR
After collection of the supernatants, cell lysate was harvested using lysing buffer provided in the RNeasy® Mini Kit (Qiagen, Basel, Switzerland). RNA was isolated according to the manufacturer's protocol. After determination of the RNA amount (NanoDrop ND 1000; NanoDrop Technologies, Wilmington, DE, USA), reverse transcription from RNA to cDNA was performed (GeneAmp 9700 system; Applied Biosystems, Branchburg, NJ, USA). The TaqMan real-time PCR system 7500 Fast (Applied Biosystems) was used to amplify and quantify the targeted molecules simultaneously. The following primers were designed: CINC-1, MCP-1, IL-6, ICAM-1, α-ENaC and α- Na+/K+-ATPase [17]. Samples were normalized to the housekeeping gene 18S (all primers from Microsynth, Balgach, Switzerland; labelled TaqMan probes were from Roche Applied Science; for details see Table S8).
Stimulation of AEC with dimethyloxalyl glycine (DMOG) instead of hypoxia
After pre-incubation with either dexamethasone or NaCl for 1 h, AEC were incubated over a time-period of 24 h with DMOG (Sigma-Aldrich, Hamburg, Germany) at a concentration of 1 mM [18] instead of hypoxia (5%).
22Na influx studies
Sodium influx through ENaC was measured with the method established by Clerici et al. [19]. Cells were rinsed twice and pre-incubated at 37°C for 20 min in buffered sodium-free medium. Thereafter cells were incubated with 22Na for 6 min, and uptake was stopped by washing the cell monolayer. Events were detected by a liquid scintillation counter (Tri-carb 2900TR; Packard, Bloomington, IL, USA). To estimate the portion of ENaC in total ion flux 22Na uptake inhibited by amiloride (100 µM; Sigma-Aldrich) was also measured.
86Rubidium influx studies
86Rubidium (86Rb), serving as surrogate for potassium [20], was used to quantify ion transport via Na+/K+-ATPase. Radioactive counts were measured using Tri-carb 2900TR (Packard). The portion of 86Na uptake inhibited by ouabain was also measured reflecting the portion of Na+/K+-ATPase of the total 86Na flux (4 mM; Sigma-Aldrich).
Statistics
Data are summarized as medians (quartiles). Matlab Software (MATLAB 2008R; Mathworks, Natick, MA, USA) and spss (SPSS Inc., Chicago, IL, USA) was used to perform all statistical analyses. Linear regression analysis was performed to assess the influence of hypoxia, dexamethasone and changes in viability/MTT (independent variables) on inflammatory mediator levels (dependent variable) (see Tables S2–5 for detailed statistical tables given in the Supporting information). Spearman's rank correlation analysis was performed to compare inflammatory mediator protein concentration to their related mRNA levels. The influence of hypoxia, dexamethasone treatment or viability/MTT (independent variables) on ion fluxes (dependent variable) was also analysed using linear regression (see Tables S6–7 for detailed statistical tables given in the Supporting information). All experiments were performed at least three times each with four independent samples per group.
Results
Inflammatory response in AEC upon exposure hypoxic conditions
After 24 h of exposure to hypoxic conditions, both CINC-1 (−39%, P < 0·001, R2: 0·568) and MCP-1 (−42%, P < 0·001, R2: 0·757) protein expressions were decreased (Fig. 2). This was not observed for IL-6 protein or ICAM-1 expression levels (Fig. S1), which remained unchanged. Dexamethasone incubation prior to hypoxia was found to decrease CINC-1 (−56%, P < 0·001, R2: 0·568) and MCP-1 (−82%, P < 0·001, R2: 0·787) protein levels even more compared to mere hypoxia treatment. Exposure to dexamethasone alone under normoxic conditions decreased CINC-1 (−32%, P < 0·001, R2: 0·568), MCP-1 (−82%, P < 0·001, R2: 0·757) and IL-6 protein expression (−36%, P = 0·004, R2: 0·380). No changes in ICAM-1 protein levels were observed. Measured mRNA expression of CINC-1, MCP-1 and IL-6 (Fig. 2) correlated well with observed inflammatory mediator protein levels (Table S1), except for ICAM-1, where a decrease upon exposure to hypoxic conditions and dexamethasone was found on mRNA (−33%, P = 0·010, R2: 0·126), but not on protein level.
Fig. 2.
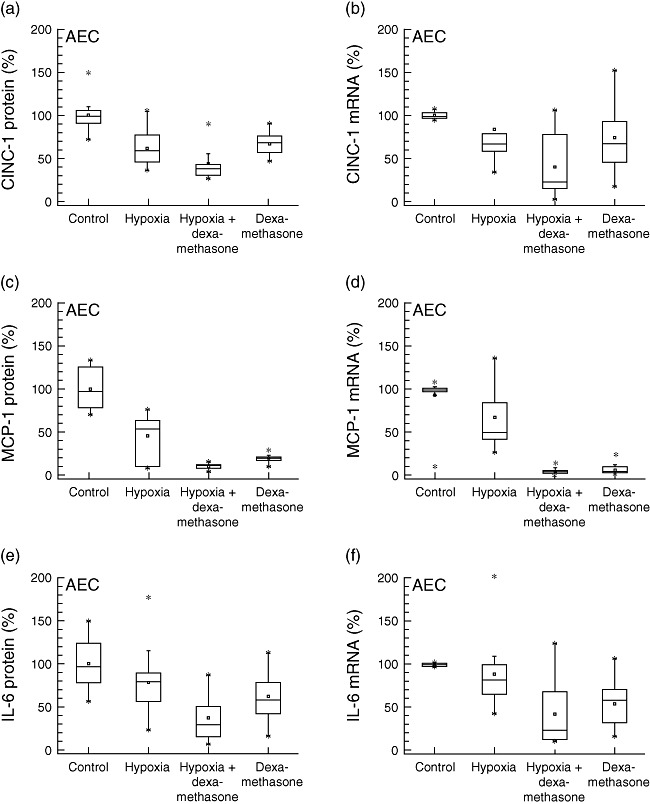
Alveolar epithelial cells (AEC). Production of inflammatory mediators from alveolar epithelial cells type II (L2, AEC) under hypoxic and normoxic conditions, with or without dexamethasone pretreatment: protein concentrations in supernatants (left) and mRNA expression levels (right). Hypoxia decreased cytokine-induced neutrophil chemoattractant-1 (CINC-1) and monocyte chemoattractant protein-1 (MCP-1) expression (P < 0·001), but not interleukin (IL)-6 protein secretion (P = 0·08). Addition of dexamethasone under hypoxic conditions attenuated CINC-1 and MCP-1 levels even more (P < 0·001).
lnflammatory mediator expression in RPAEC after exposure to hypoxia
Inflammatory response upon hypoxic conditions and influence of dexamethasone was also evaluated in RPAEC, mimicking the response of endothelial cells from the vascular compartment in vitro (Fig. 3). In contrast to results from AEC, a decrease was found only in CINC-1 protein levels after 24 h exposure to hypoxia (−35%, P < 0·001, R2: 0·772). No significant changes were measured regarding mRNA levels of all cytokines and protein expression of MCP-1 and ICAM-1. IL-6 protein was not detectable in supernatants of RPAEC. Similar to results from AEC, addition of dexamethasone attenuated MCP-1 protein and mRNA expression under normoxic (−47%, P < 0·001, R2: 0·575) and hypoxic conditions (−52%, P < 0·001, R2: 0·575). ICAM-1 mRNA (−44%, P = 0·002, R2: 0·317), but not protein levels, were decreased slightly after exposure to dexamethasone alone under normoxic conditions (Fig. 3).
Fig. 3.
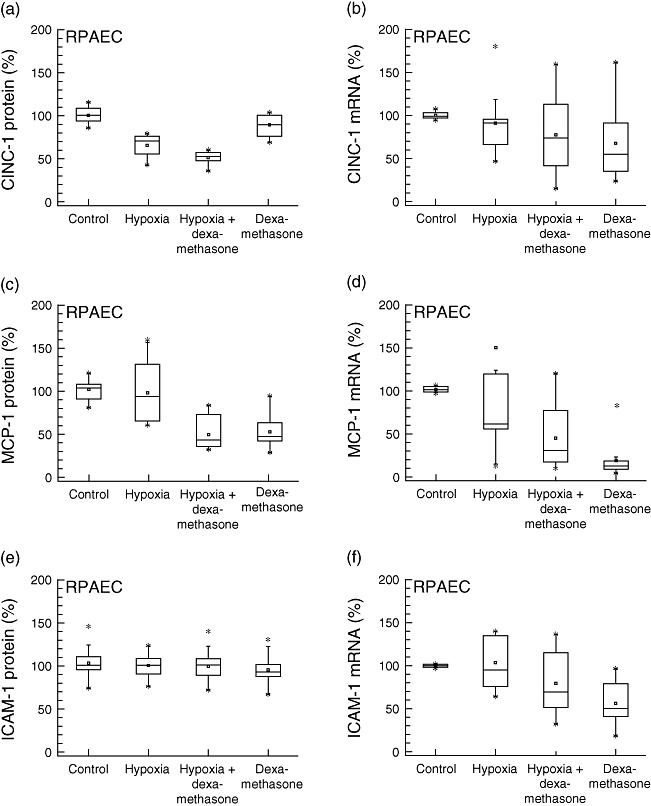
Rat pulmonary artery endothelial cells. Production of inflammatory mediators from rat pulmonary endothelial cells (RPAEC) under hypoxic and normoxic conditions, with or without dexamethasone pretreatment: protein concentrations in supernatants (left) and mRNA expression levels (right). Hypoxia decreased cytokine-induced neutrophil chemoattractant-1 (CINC-1) (P < 0·001), but not monocyte chemoattractant protein-1 (MCP-1) or interleukin (IL)-6 protein expression. Dexamethasone attenuated MCP-1 secretion under normoxic and hypoxic conditions (P < 0·001). ICAM-1, intercellular adhesion molecule-1.
Inflammatory mediator expression in MAC after exposure to hypoxia
Effector cell response upon long-term incubation for 24 h under hypoxic conditions of 5% oxygen was studied in alveolar macrophages. An attenuated expression of MCP-1 protein (−32%, P < 0·001, R2: 0·851) and mRNA (−33%, P = 0·031, R2: 0·524) was found after exposure to hypoxia (Fig. 4). Addition of dexamethasone provoked an even more pronounced decrease in MCP-1 protein levels (−91%, P < 0·001, R2: 0·851). CINC-1 and IL-6 protein concentrations were below detection levels. Hypoxia attenuated ICAM-1 mRNA expression (−65%, P < 0·001, R2: 0·919). Dexamethasone incubation prior to hypoxia was found to decrease ICAM-1 (−80%, P < 0·001, R2: 0·919) protein levels even more compared to hypoxia treatment alone.
Fig. 4.
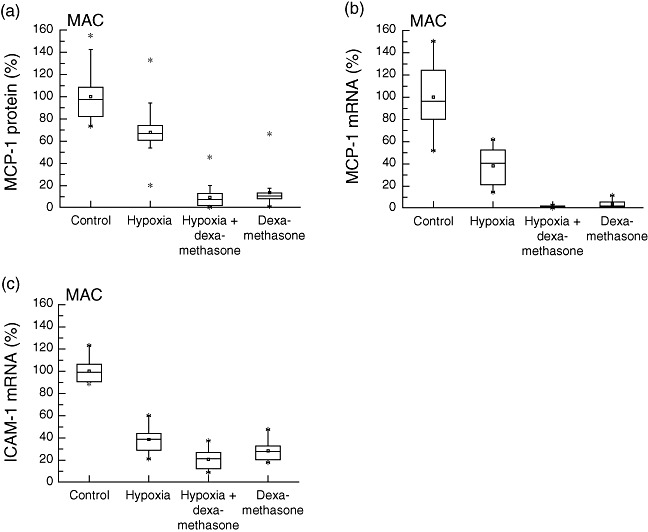
Alveolar macrophages. Production of inflammatory mediators from alveolar macrophages (MAC) under hypoxic and normoxic conditions, with or without dexamethasone pretreatment: both hypoxic conditions and dexamethasone treatment decreased monocyte chemoattractant protein-1 (MCP-1) protein concentration (P < 0·001), as well as MCP-1 and intercellular adhesion molecule-1 (ICAM-1) mRNA levels (P < 0·05) in alveolar macrophages. Cytokine-induced neutrophil chemoattractant-1 and interleukin (IL)-6 protein was not detectable in supernatants.
Activation of hypoxic-inducible factor attenuates inflammatory mediator expression
Additional experiments in AEC were performed using DMOG to investigate whether activation of hypoxia-inducible factor-1α (HIF-1α) attenuates inflammatory mediator expression comparable to exposure to hypoxia (5% O2). An attenuation of CINC-1 protein expression of −44% was measured after incubation with DMOG (P < 0·001, R2: 0·934) (Fig. 5). In the DMOG and dexamethasone groups, CINC-1 protein expression was even more decreased (−59%, P < 0·001, R2: 0·934). Stimulation with DMOG also attenuated MCP-1 protein secretion by −89% compared to control (P < 0·001; R2: 0·863). Incubation of both dexamethasone and DMOG decreased MCP-1 protein by −80% (P < 0·001; R2: 0·863). No differences were observed between samples incubated with DMOG and DMOG in the presence of dexamethasone.
Fig. 5.
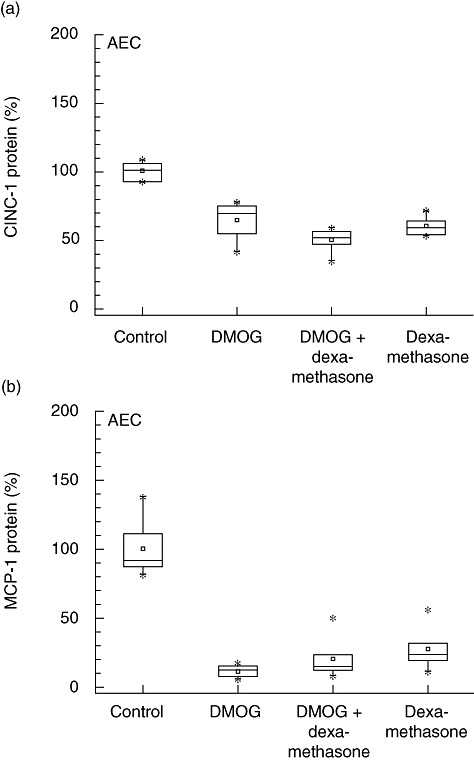
Activation of hypoxic-inducible factor attenuates inflammatory mediator expression. Similar as in experiments using exposure to hypoxia (5% oxygen), inflammatory mediator secretion is attenuated (P < 0·001) in alveolar epithelial cells (AEC) stimulated with 1 mM N-(methoxyoxoacetyl)-glycine methyl ester [dimethyloxalyl glycine (DMOG), an inhibitor of prolyl-4-hydroxylase]. CINC-1, cytokine-induced neutrophil chemoattractant-1; MCP-1, monocyte chemoattractant protein-1.
Influence of hypoxia and dexamethasone on ion transporter expression and activity
To assess the influence of dexamethasone on sodium transport, mRNA expression of the basolateral sodium/potassium ATPase and the apical epithelial sodium channels was measured (Figs 1 and 6). In addition, the influence of hypoxia and dexamethasone on inhibitor-sensitive (reflecting ENaC and Na+/K+-ATPase function) and the inhibitor-insensitive portion of the total sodium transport was determined in 22Na and 86Rb influx studies. Total 22Na flux was decreased significantly upon hypoxic conditions (−42%, P < 0·001, R2: 0·415) (Fig. 6). When cells were pre-incubated with dexamethasone before exposure to hypoxic conditions, however, Na+ influx was partially maintained (−30% compared to control; P = 0·001; R2: 0·415). Amiloride inhibited on average 45% of total 22Na flux (−45%, P < 0·001, R2: 0·415) which, in turn, reflects the portion of ENaC in total 22Na flux. Amiloride-insensitive 22Na flux was not influenced significantly by dexamethasone or hypoxia.
Fig. 6.
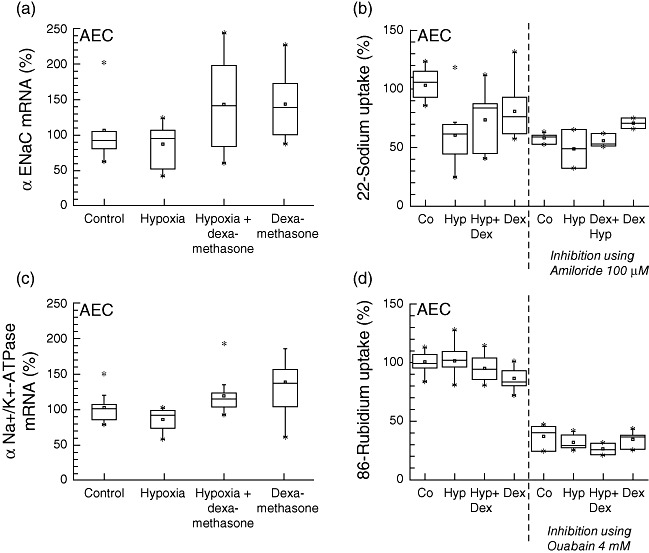
Ion channel expression and function. Expression of apical epithelial sodium channel (αENaC) and basolateral Na+/K+-ATPase (mRNA-expression, left) with corresponding actual channel turnover measured by radioactive tracer ions (amiloride-sensitive 22Na uptake for ENaC function, ouabain-sensitive 86rubidium uptake for Na+/K+-ATPase function, right). Total 22Na flux was decreased after exposure to hypoxia (P < 0·001). When pre-incubated with dexamethasone, 22Na flux was partially maintained (P = 0·001). In contrast, no increased 86rubidium uptake upon dexamethasone treatment was observed. Co, control, Hyp, hypoxia, Dex, dexamethasone.
Pre-incubation with dexamethasone increased mRNA expression of Na+/K+-ATPase slightly in comparison to normoxic control after 24 h (+36%, P = 0·021, R2: 0·268) (Fig. 6). In experiments using 86Rb as marker of ion flux, no increased Na+/K+-ATPase activity was observed upon dexamethasone treatment, in contrast to results from mRNA assays. Addition of dexamethasone under normoxic conditions even marginally decreased total 86Rb flux (−14%, P < 0·001, R2: 0·900). Ouabain impaired 86Rb uptake on average by −68% (P < 0·001, R2: 0·900), reflecting the portion of Na+/K+-ATPase in total 86Rb flux. However, oubain-insensitive 86Rb uptake was decreased by −10% in the presence of dexamethasone and hypoxia (−11%, P = 0·017, R2: 0·582).
Assessment of viability after exposure to hypoxic conditions and dexamethasone
Viability of the cells after exposure to hypoxic conditions and dexamethasone treatment was monitored by performing MTT assays (Fig. 7). No relation between changes in MTT and inflammatory mediator expression was found for AEC, RPAEC or MAC. 86Rb and 22Na influx studies were not influenced by changes in MTT.
Fig. 7.
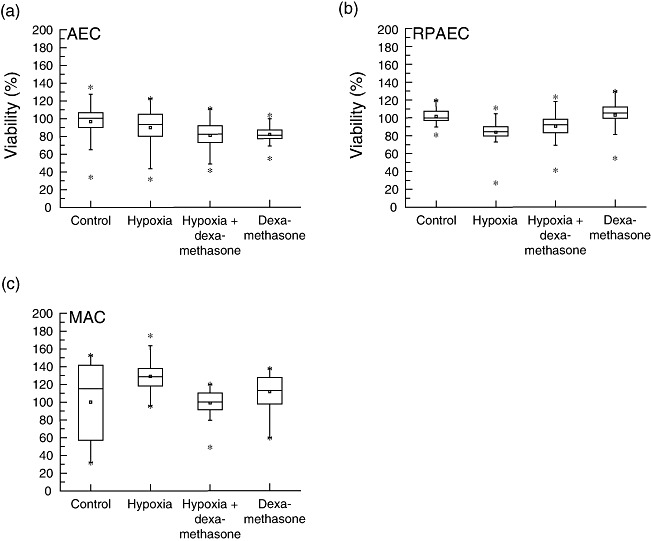
Viability. Alveolar epithelial cells (AEC) rat pulmonary artery endothelial cells (RPAEC), and alveolar macrophages (MAC) were pretreated with dexamethasone (or not) and exposed to 5% oxygen (or 21% as control) for 24 h. Dimethylthiozol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assays were performed: no influences of variations in viability on inflammatory mediator expression or ion fluxes were found.
Discussion
With regard to the formation of hypoxia-induced pulmonary oedema, the role of inflammation has been the subject of numerous investigations; however, studies under isolated conditions (i.e. accurately controlled oxygen concentration, timing and extent of reoxygenation) remain scarce [8,12,21,22]. In this study, we evaluated the inflammatory response in pulmonary epithelial and endothelial cells as well as in alveolar macrophages after 24 h of hypoxia (5% oxygen; simulating conditions at high altitudes). Hypoxia induced down-regulation of inflammatory mediators, which was even more accentuated in the presence of dexamethasone. As a marker of water flux, the activity of sodium transport in alveolar epithelial cells was determined. Under hypoxia, sodium ion transport was impaired in AEC, but transport activity of ENaC could be maintained when cells were pre-incubated with dexamethasone.
The influence of hypoxia on inflammation is discussed controversially in the literature: in previous studies, hypoxia has been shown to promote inflammation by activation of the inflammatory transcription nuclear factor (NF)-κB by prolonging neutrophil survival [23] and by induction of Toll-like receptors [24]. Raised levels of MCP-1 protein in macrophages have been described in recent work by Chao et al. [25] and Madjdpour et al. [26] after exposure to conditions of low oxygen. Chao et al. [25] demonstrated that alveolar macrophage-derived MCP-1 plays a crucial role in the initiation of systemic inflammation under hypoxic conditions, where exposure of primary culture of rat alveolar macrophages to various oxygen concentrations (0%, 5%, 10%, and 15%) induced an almost 20-fold increase of MCP-1 expression. These macrophages, however, were exposed to hypoxia for only 30 min. In the present in vitro data we show that after 24 h exposure, mild hypoxia does not necessarily induce inflammation in pulmonary cells. In our study, even an attenuating effect of hypoxia was found for all three types of pulmonary cells. This was underlined by additional experiments using DMOG, in which down-regulation of MCP-1 and CINC-1 protein expression has also been found comparable to the experiments with hypoxia (5% oxygen). All these observations suggest that HIF-1α might mediate anti-inflammatory effects, which would be in good accordance with findings from previous studies [27–29]. However, from our data we cannot exclude that hypoxia may trigger inflammation at a very early phase [26]. Also further in vitro and in vivo studies under controlled conditions are required to ultimately clarify the role, mechanisms and dynamics of inflammation in hypoxia in general and the pathogenesis of hypoxia-induced oedema.
Epithelial sodium channels have been identified as the major pathway for apical sodium entry in AEC, thereby controlling fluid clearance from the alveolar space. In close interplay with apical sodium channels, basolateral Na+/K+-ATPase is a key player involved in transepithelial sodium transport by alveolar cells, and ensures efficient vectorial sodium transport [30,31]. In the present study, 5% oxygen did not influence mRNA expression of Na+/K+-ATPase and ENaC significantly. Comparisons with other studies are delicate, as exposure time, concentration of oxygen and cell types are different [32–34]. Because injury-induced changes in the mRNA expression of sodium transporters might not necessarily be correlated with impaired function, in our experimental setting sodium transport was determined additionally using radioactive marker ions. Sodium transport was decreased significantly in the presence of hypoxia, and was maintained when cells were pretreated with dexamethasone prior to hypoxic conditions. A similar observation was made in the work of Güney et al. [34]: in freshly prepared AEC of type II character the authors did not find a difference in mRNA expression of ENaC in hypoxia or with dexamethasone exposure, while the impaired capacity of the epithelial sodium channel was influenced positively by dexamethasone [34]. A study from Mairbäurl et al. described inhibition of the amiloride-sensitive portion of the active sodium transport to 55% upon exposure to 5% oxygen in primary cultured adult rat alveolar epithelial cell monolayers [35]. In our study, mRNA expression of Na+/K+-ATPase was up-regulated by dexamethasone. Activity, however, remained unchanged upon exposure to hypoxia and/or dexamethasone. Several factors might account for the differences in findings among different studies, whereas the concentration of oxygen (5% versus 1·5%), exposure time (24 h versus 48 h) and extent of reoxygenation might be of utmost importance.
Although numerous studies have investigated different aspects of hypoxia and high-altitude pulmonary oedema, it remains controversial as to whether the inflammatory response upon alveolar hypoxia is simply a secondary event to an oedema of hydrostatic genesis (hypoxic pulmonary vasoconstriction; impaired fluid clearance due to decreased sodium and water transport [6]) or one of the causative factors in alveolar oedema formation (such as in acute respiratory distress syndrome) [36]. The present study was performed with a special focus on in vitro evaluation of changes possibly observed in HAPE; the findings might also play an important role in acute lung injury (ALI) and/or acute respiratory distress syndrome (ARDS). The role and time-point of inflammation with regard to oedema formation might, however, not necessarily be the same, because of the substantial differences in the pathogenesis of ALI/ARDS [37] and HAPE [22]. In conclusion, cells at the alveolar barrier do not seem to be involved in proinflammatory actions under prolonged hypoxic conditions. Whether or not the in vitro observed positive effect of dexamethasone of maintaining ENaC function in hypoxic conditions contributes to improved fluid reabsorption in vivo in situations such as HAPE remains the subject of further investigations.
Acknowledgments
We kindly acknowledge financial support from the University Research Priority Program ‘Integrative Human Physiology’ at the University of Zurich [Zurich Center for Integrative Human Physiology (ZIHP)]. We thank L. Reyes, F. Aigner, P. Joerg, S. Gammeter, A. Faes and C. Kalberer for technical support. This study was supported by a research grant from the Zurich Center for Integrative Human Physiology.
Disclosure
The authors declare that they have no competing financial interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Intercellular adhesion molecule-1 (ICAM-1) expression in alveolar epithelial cells (AEC). AEC were pretreated with dexamethasone (or not) and exposed to 5% oxygen (or 21% as control) for 24 h. Cell-based enzyme-linked immunosorbent assay (ELISA) was performed (a) or mRNA determined (b).
Table S1.TaqMan primers and probes.
Table S2. Linear regression on expression of inflammatory mediator protein expression in alveolar epithelial cells (AEC).
Table S3. Linear regression on expression of inflammatory mediator protein expression in rat pulmonary artery endothelial cells (RPAEC).
Table S4. Linear regression on expression of inflammatory mediator protein expression in alveolar macrophages (MAC).
Table S5. Linear regression on expression of inflammatory mediator expression after stimulation with dimethyloxalyl glycine (DMOG).
Table S6. Linear regression regarding influence of hypoxia and dexamethasone on 22sodium uptake.
Table S7. Linear regression regarding influence of hypoxia and dexamethasone on 86rubidium uptake.
Table S8. Correlation analysis on mRNA expression and measured inflammatory mediator protein levels in alveolar epithelial cells (AEC).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Sulkowska M. Morphological studies of the lungs in chronic hypobaric hypoxia. Pol J Pathol. 1997;48:225–34. [PubMed] [Google Scholar]
- 2.Bartsch P, Mairbaurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101–10. doi: 10.1152/japplphysiol.01167.2004. [DOI] [PubMed] [Google Scholar]
- 3.Oelz O, Maggiorini M, Ritter M, et al. Nifedipine for high altitude pulmonary oedema. Lancet. 1989;2:1241–4. doi: 10.1016/s0140-6736(89)91851-5. [DOI] [PubMed] [Google Scholar]
- 4.Bärtsch P, Maggiorini M, Ritter M, Noti C, Vock P, Oelz O. Prevention of high altitude pulmonary edema by nifedipine. N Engl J Med. 1991;325:1284–9. doi: 10.1056/NEJM199110313251805. [DOI] [PubMed] [Google Scholar]
- 5.Maggiorini M, Brunner-La Rocca HP, Peth S, et al. Both tadalafil and dexamethasone may reduce the incidence of high-altitude pulmonary edema: a randomized trial. Ann Intern Med. 2006;145:497–506. doi: 10.7326/0003-4819-145-7-200610030-00007. [DOI] [PubMed] [Google Scholar]
- 6.Mairbäurl H. Role of alveolar epithelial sodium transport in high altitude pulmonary edema (HAPE) Respir Physiol Neurobiol. 2006;151:178–91. doi: 10.1016/j.resp.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–65. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartmann G, Tschop M, Fischer R, et al. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine. 2000;12:246–52. doi: 10.1006/cyto.1999.0533. [DOI] [PubMed] [Google Scholar]
- 9.Haddad E-B, McCluskie K, Birrell MA, et al. Differential effects of ebselen on neutrophil recruitment, chemokine, and inflammatory mediator expression in a rat model of lipopolysaccharide-induced pulmonary inflammation. J Immunol. 2002;169:974–82. doi: 10.4049/jimmunol.169.2.974. [DOI] [PubMed] [Google Scholar]
- 10.Bless NM, Huber-Lang M, Guo R-F, et al. Role of CC chemokines (macrophage inflammatory protein-1ß, monocyte chemoattractant protein-1, RANTES) in acute lung injury in rats. J Immunol. 2000;164:2650–9. doi: 10.4049/jimmunol.164.5.2650. [DOI] [PubMed] [Google Scholar]
- 11.Saito F, Tasaka S, Inoue K, et al. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell Mol Biol. 2008;38:566–71. doi: 10.1165/rcmb.2007-0299OC. [DOI] [PubMed] [Google Scholar]
- 12.Beck-Schimmer B, Schimmer RC, Madjdpour C, Bonvini JM, Pasch T, Ward PA. Hypoxia mediates increased neutrophil and macrophage adhesiveness to alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;25:780–7. doi: 10.1165/ajrcmb.25.6.4433. [DOI] [PubMed] [Google Scholar]
- 13.Meyer S, Z'Graggen BR, Blumenthal S, et al. Hypoxia attenuates effector–target cell interaction in the airway and pulmonary vascular compartment. Clin Exp Immunol. 2007;150:358–67. doi: 10.1111/j.1365-2249.2007.03495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas WH, Kaighn ME. Clonal isolation of differentiated rat lung cells. In Vitro. 1974;10:230–7. doi: 10.1007/BF02615237. [DOI] [PubMed] [Google Scholar]
- 15.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 16.Madjdpour C, Oertli B, Ziegler U, Bonvini JM, Pasch T, Beck-Schimmer B. Lipopolysaccharide induces functional ICAM-1 expression in rat alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol. 2000;278:L572–9. doi: 10.1152/ajplung.2000.278.3.L572. [DOI] [PubMed] [Google Scholar]
- 17.Rahman MS, Gandhi S, Otulakowski G, Duan W, Sarangapani A, O'Brodovich H. Long-term terbutaline exposure stimulates α1-Na+-K+-ATPase expression at posttranscriptional level in rat fetal distal lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2010;298:L96–L104. doi: 10.1152/ajplung.00158.2009. [DOI] [PubMed] [Google Scholar]
- 18.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clerici C, Friedlander G, Amiel C. Impairment of sodium-coupled uptakes by hydrogen peroxide in alveolar type II cells: protective effect of d-alpha-tocopherol. Am J Physiol Lung Cell Mol Physiol. 1992;262:L542–8. doi: 10.1152/ajplung.1992.262.5.L542. [DOI] [PubMed] [Google Scholar]
- 20.Nimigean CM. A radioactive uptake assay to measure ion transport across ion channel-containing liposomes. Nat Protocols. 2006;1:1207–12. doi: 10.1038/nprot.2006.166. [DOI] [PubMed] [Google Scholar]
- 21.Kubo K, Hanaoka M, Hayano T, et al. Inflammatory cytokines in BAL fluid and pulmonary hemodynamics in high-altitude pulmonary edema. Respir Physiol. 1998;111:301–10. doi: 10.1016/s0034-5687(98)00006-1. [DOI] [PubMed] [Google Scholar]
- 22.Maggiorini M, Melot C, Pierre S, et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 2001;103:2078–83. doi: 10.1161/01.cir.103.16.2078. [DOI] [PubMed] [Google Scholar]
- 23.Walmsley SR, Print C, Farahi N, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–15. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE. 2007;2:e1364. doi: 10.1371/journal.pone.0001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao J, Donham P, van Rooijen N, Wood JG, Gonzalez NC. Monocyte chemoattractant protein-1 released from alveolar macrophages mediates the systemic inflammation of acute alveolar hypoxia. Am J Respir Cell Mol Biol. 2011;45:53–61. doi: 10.1165/rcmb.2010-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madjdpour C, Jewell UR, Kneller S, et al. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284:L360–7. doi: 10.1152/ajplung.00158.2002. [DOI] [PubMed] [Google Scholar]
- 27.Sitkovsky MV, Lukashev D, Apasov S, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 28.Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186:4367–74. doi: 10.4049/jimmunol.0903617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Rosenberger P, Schwab JM, Mirakaj V, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 30.Clerici C, Matthay MA. Hypoxia regulates gene expression of alveolar epithelial transport proteins. J Appl Physiol. 2000;88:1890–6. doi: 10.1152/jappl.2000.88.5.1890. [DOI] [PubMed] [Google Scholar]
- 31.Dagenais A, Denis C, Vives M-F, et al. Modulation of α-ENaC and α1-Na+-K+-ATPase by cAMP and dexamethasone in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L217–L30. doi: 10.1152/ajplung.2001.281.1.L217. [DOI] [PubMed] [Google Scholar]
- 32.Planes C, Friedlander G, Loiseau A, Amiel C, Clerici C. Inhibition of Na-K-ATPase activity after prolonged hypoxia in an alveolar epithelial cell line. Am J Physiol. 1996;271:L70–8. doi: 10.1152/ajplung.1996.271.1.L70. [DOI] [PubMed] [Google Scholar]
- 33.Planes C, Escoubet B, Blot-Chabaud M, Friedlander G, Farman N, Clerici C. Hypoxia downregulates expression and activity of epithelial sodium channels in rat alveolar epithelial cells. Am J Respir Cell Mol Biol. 1997;17:508–18. doi: 10.1165/ajrcmb.17.4.2680. [DOI] [PubMed] [Google Scholar]
- 34.Güney S, Schuler A, Ott A, et al. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1332–L8. doi: 10.1152/ajplung.00338.2006. [DOI] [PubMed] [Google Scholar]
- 35.Mairbäurl H, Mayer K, Kim K-J, Borok Z, Bärtsch P, Crandall ED. Hypoxia decreases active Na transport across primary rat alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2002;282:L659–L65. doi: 10.1152/ajplung.00355.2001. [DOI] [PubMed] [Google Scholar]
- 36.Basnyat B, Murdoch DR. High-altitude illness. Lancet. 2003;361:1967–74. doi: 10.1016/S0140-6736(03)13591-X. [DOI] [PubMed] [Google Scholar]
- 37.Matthay MA, Zimmerman GA, Esmon C, et al. Future research directions in acute lung injury: summary of a national Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167:1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.