

Abstract
During pathogenesis of diabetes, pancreatic islets are exposed to high levels of cytokines and other inflammatory mediators that induce deterioration of insulin-producing beta cells. Macrophage migration inhibitory factor (MIF) plays a key role in the onset and development of several immunoinflammatory diseases and also controls apoptotic cell death. Because the occurrence of apoptosis plays a pathogenetic role in beta cell death during type 1 diabetes development and MIF is expressed in beta cells, we explored the influence of MIF deficiency on cytokine-induced apoptosis in pancreatic islets. The results indicated clearly that elevated MIF secretion preceded C57BL/6 pancreatic islets death induced by interferon (IFN)-γ + tumour necrosis factor (TNF)-α + interleukin (IL)-1β. Consequently, MIF-deficient [MIF-knock-out (KO)] pancreatic islets or islet cells showed significant resistance to cytokine-induced death than those isolated from C57BL/6 mice. Furthermore, upon exposure to cytokines pancreatic islets from MIF-KO mice maintained normal insulin expression and produced less cyclooxygenase-2 (COX-2) than those from wild-type C57BL6 mice. The final outcome of cytokine-induced islet apoptosis in islets from wild-type mice was the activation of mitochondrial membrane pore-forming protein Bcl-2-associated X protein and effector caspase 3. In contrast, these apoptotic mediators remained at normal levels in islets from MIF-KO mice suggesting that MIF absence prevented initiation of the mitochondrial apoptotic pathway. Additionally, the protection from apoptosis was also mediated by up-regulation of prosurvival kinase extracellular-regulated kinase 1/2 in MIF-KO islets. These data indicate that MIF is involved in the propagation of pancreatic islets apoptosis probably via nuclear factor-κB and mitochondria-related proteins.
Keywords: apoptosis, diabetes, IL-1β, macrophage migration inhibitory factor (MIF), pancreatic islets
Introduction
Type 1 diabetes (T1D) is characterized by the progressive loss of beta cells due to autoimmune attack and consequent inflammation. Although the induction of autoimmunity involves the adaptive immune system, innate effector cells (monocytes/macrophages, dendritic cells, natural killer cells, natural killer T cells and γδ T cells) are important in priming or promoting autoimmune response [1]. All these islet-infiltrating as well as resident cells produce cytokines [tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-17, IL-18, interferon (IFN)-γ] and other proinflammatory mediators (reactive oxygen and nitrogen species) that induce dysfunction of beta cells, and ultimately apoptosis (reviewed in [2–4]).
Under the influence of cytokines, pancreatic islets follow two pathways of apoptotic death that are either nuclear factor (NF)-κB- [5–7] or mitochondrial-driven [7,8]. NF-κB is an important transcription factor that induces production of cytokines (such as IL-1β), death receptors, chemokines, prostaglandins and nitric oxide (NO) that can provoke beta cell death in either autocrine or paracrine fashion [6,7]. Prostaglandin E2 (PGE2) is implicated in cytokine-mediated beta cell dysfunction and diabetes development, as selective inhibition of cyclooxygenase-2 (COX-2) (the key enzyme for prostaglandin biosynthesis) attenuates the development of immunoinflammatory diabetes induced by multiple low doses of streptozotocin [9] and protects rat islets from cytokine-induced inhibition of glucose-stimulated insulin secretion [10,11]. NO is another proinflammatory mediator that is thought to be detrimental for beta cell function and survival in the inflammatory settings during T1D, as it has been demonstrated by the anti-diabetogenic efficacy of specific antagonists of inducible NO synthase (iNOS) in several models of T1D [12]. The final outcome of NF-κB-driven apoptotic pathway is similar to the mitochondrial pathway, as it includes mitochondrial permeabilization and caspase activation leading ultimately to cell death [13,14]. Mitochondrial pathway is governed by the Bcl-2 family of proteins that includes Bcl-2-associated X protein (BAX) – a mitochondrial membrane pore-forming protein. The leakage of pro-apoptotic proteins into cytoplasm initiates a downstream cascade of apoptotic events that leads eventually to activation of effector caspase 3 and cleavage of vital cell proteins [14].
Conversely, one of the molecules that play a crucial role in preserving beta cells from various detrimental stimuli is extracellular signal-regulated kinase 1/2 (ERK1/2). Activation of this kinase seems to sustain beta cell survival and/or growth in response to glucose or serum [15,16].
One of the potentially deleterious molecules for beta cell function is macrophage migration inhibitory factor (MIF). MIF is a pluripotent cytokine involved in microbe eradication, promotion of inflammation (through up-regulation of proinflammatory cytokines and prostaglandins), regulation of glucocorticoid action and glucose metabolism [17–19]. Because of its role in propagating inflammation, MIF is involved in the pathogenesis of several inflammatory conditions, including T1D [20]. Evidence indicates that disease incidence in non-obese diabetic (NOD) mice treated with recombinant MIF is highly increased, while MIF deficiency or inhibition protects mice from streptozotocin-induced diabetes [20–22]. Apart from evident production in various immune cells, MIF is also produced by beta cells and localized within insulin secretory granules [23].
Both pro-apoptotic and anti-apoptotic effects were observed in the circumstances of excessive concentration of MIF [24–30]. Regarding the impact of MIF on beta cell death, our recent results confirm that innate MIF deficiency conveys a resistance to palmitic acid-induced apoptosis through inactivation of the caspase 3-dependent pathway [30]. In addition, cultured MIF-deficient [MIF-knock-out (KO)] islets exhibit a better survival rate both after transplantation and in vitro compared to wild-type (WT) islets [31]. As we have shown previously that in vivo MIF inhibition or innate mif gene deficiency ameliorates streptozotocin-induced T1D development in C57BL/6 mice [22,32], the aim of this study was to investigate the in vitro role of MIF in cytokine-induced apoptosis of pancreatic islets by using MIF-deficient (MIF-KO) islets. In addition, we compared the expression of proinflammatory mediators, pro-apoptotic and pro-survival molecule(s) in the islets obtained from WT and MIF-KO mice under cytokine stimulation.
Methods
Animals
The generation of homozygous mif gene-deficient (MIF-KO) mice (background: C57BL/6) has been described elsewhere [33]. The mice were further bred using homozygous MIF-KO animals and kept under standard conditions (non-specific pathogen-free) in the Animal Facility at the Institute for Biological Research ‘Sinisa Stankovic’, along with their WT C57BL/6 counterparts. The experiments were approved by the Ethic Committee for Animal Experimentation at Belgrade University and conducted in accordance with local and international legislation regarding the wellbeing of laboratory animals. Male mice (10 weeks old) were killed by cervical dislocation and pancreata were removed aseptically.
Isolation of pancreatic islets and islet cells and treatments
Pancreatic islets were isolated by collagenase V (Sigma-Aldrich, St Louis, MO, USA) digestion technique followed by handpicking [34]. Before performing the experiments, islets were cultured overnight in RPMI-1640 medium containing 10% fetal calf serum (FCS) (PAA Chemicals, Pasching, Austria), 10 mM HEPES, 5 µM β-mercaptoethanol, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 IU/ml penicillin and 100 µg/ml streptomycin (all from Sigma-Aldrich) in a humidified (5% CO2, 95% air) atmosphere at 37°C.
Pancreatic islets were cultured in six-well non-adhesive culture plates (Sardstedt, Numbrecht, Germany) in 2 ml of medium.
Pancreatic islet cells were obtained after enzymatic digestion of pancreatic islets with 0·025% trypsin and 0·04% ethylenediamine tetraacetic acid (EDTA) in phosphate-buffered saline (PBS) (all from Sigma-Aldrich). After 10 min of incubation at 37°C with occasional shaking, disrupted islets were washed three times by centrifugation at 350 g in PBS + 5% FCS. Cells were counted by trypan blue exclusion test.
Pancreatic islets or islet cells were treated with mouse IFN-γ (R&D, Minneapolis, MN, USA), mouse TNF-α (R&D) and rat IL-1β (R&D), 10 ng/ml each.
Cell viability assay
In order to assess the viability of pancreatic islets or islet cells, we used the mitochondrial-dependent reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) to formazan [35]. At the end of the appropriate treatments, islet cell culture supernatants were removed from the plates and MTT solution (1 mg/ml) was applied. Alternatively, pancreatic islets were collected into tubes, spun down, supernatants removed and the cell pellet dissolved in the MTT solution. Incubation with MTT lasted for 30 min at 37°C. Dimethylsulphoxide (DMSO) (Zorka Farma, Belgrade, Serbia) was added to the pellet to dissolve the formazan crystals. The absorbance was measured at 570 nm, with a correction at 690 nm, using automated microplate reader (LKB 5060-006; LKB, Vienna, Austria).
Determination of cytokine and nitrite levels in culture media
MIF concentration in the supernatants of cultured pancreatic islets was measured by sandwich enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's instructions. Paired MIF antibodies specific for mouse MIF [mouse monoclonal immunoglobulin (Ig)G1, κ as capture antibody, goat polyclonal IgG with biotin as detection antibody] were purchased from US Biological (Swampscott, MA, USA). The results were calculated using standard curve which was made on the basis of known concentrations of recombinant mouse MIF (R&D).
Nitrite accumulation, an indicator of NO production, was measured in cell culture supernatants using the Griess reagent. The supernatants (50 µl) were mixed with an equal volume of Griess reagent (a mixture at 1:1 of 0·1% naphthylethylenediamine dihydrochloride and 1% sulphanilamide in 5% H3PO4) (both purchased from Sigma-Aldrich) and the absorbance at 570 nm was measured in a microplate reader (LKB). The nitrite concentration was calculated from a NaNO2 standard curve. The data obtained from triplicates are presented as µM of nitrite.
Caspase 3 assay
The activity of caspase 3 was determined in cultures using the caspase 3 DEVD-R110 Fluorimetric and Colorimetric Assay Kit (Biotium, Hayward, CA, USA), according to the manufacturer's protocol. The ability of cell lysates to cleave the specific caspase 3 substrate was quantified fluorimetrically using an excitation wavelength of 485 nm and emission wavelength of 535 nm with a microplate reader (Chameleon, Hidex, Turku, Finland). The results are expressed as amount of substrate conversion, as deduced from standard curve generated according to known concentrations of the dye R110.
Western blot
Pancreatic islets (groups of 100) were disrupted in the lysis buffer containing 62·5 mM Tris-HCl (pH 6·8 at 25°C), 2% w/v sodium dodecyl sulphide (SDS), 10% glycerol, 50 mM dithiothreitol (DTT) and 0·01% w/v bromophenol blue (all from Sigma-Aldrich). The concentration of proteins was measured using Bradford method. All samples (50 µg of each) were loaded onto a 15% SDS-polyacrylamide gel and electrophoresed. After electrotransferring to polyvinylidene difluoride membranes at 5 mA/cm2, using a semi-dry blotting system (Semi-Dry Transfer Unit; GE Healthcare, Chalfont St Giles, UK), the blots were blocked with 5% w/v bovine serum albumin (BSA) in PBS-Tris (PBST) buffer (80 mM Na2HPO4; 20 mM NaH2PO4; 100 mM NaCl; 0·1% Tween-20) (all obtained from Sigma-Aldrich) and probed with specific mouse IgG1antibody for mouse p-IκB (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 1:1000 dilution in 1% blocking buffer or rabbit anti-mouse IgG p-p44/42 (ERK1/2), rabbit anti-mouse IgG β-actin (both from Cell Signaling Technology Inc., Danver, MA, USA), rabbit anti-mouse IgG BAX (1:100) (eBioscience) or rabbit anti-mouse IgG COX-2 (1:200) (SantaCruz Biotechnology Inc.), followed by incubation with secondary sheep anti-mouse IgG horseradish peroxidase (HRP) antibody at 1:2500 dilution or donkey anti-rabbit IgG HRP antibody at 1:10000 dilution (GE Healthcare) in 1% blocking buffer. Detection was performed by colorimetric reaction with tetramethylbenzidine (TMB) (Sigma-Aldrich) specific for membranes. Protein production was calculated by Scion Image Alpha version 4·0.3·2 (Scion Corporation, Frederick, MD, USA).
RNA isolation, reverse transcription and polymerase chain reaction (PCR)
Total RNA was isolated from pancreatic islets (groups of 80) with TRIzol reagent (Genosys, Woodlands, TX, USA), according to the manufacturer's instructions. RNA was reverse-transcribed using Moloney leukaemia virus reverse transcriptase and random hexamers (Pharmacia, Uppsala, Sweden). PCR amplification of cDNA with primers specific for the gene in question and β-actin as a housekeeping gene was carried out in a Mastercycler Gradient thermal cycler (Eppendorf, Hamburg, Germany) as follows: 30 s of denaturation at 95°C, 30 s of annealing at 58°C and 30 s of extension at 72°C. For each gene, preliminary experiments were conducted to ascertain that amplification of cDNA was in the linear range under the respective cycling conditions. Primer pair sequences are given in Table 1. The PCR products were visualized by electrophoresis through 2·5% agarose gels containing ethidium bromide and gels were photographed by GelDoc (Biorad, Hercules, CA, USA). Results were analysed by densitometry using Kodak 1D 3·6 software and the expression of target gene was calculated relative to β-actin mRNA expression. Relative BAX and insulin mRNA expression was determined by real-time PCR (Applied Biosystems, Woolston, UK) using SYBRGreen PCR master mix (Applied Biosystems) as follows: 10 min at 50°C for dUTP activation, 10 min at 95°C for initial denaturation of cDNA followed by 40 cycles (15 s of denaturation at 95°C and 60 s for primer annealing and chain extension step at 60°C). The efficiency of PCR reaction for target cDNAs was similar to that for β-actin (Eβ-actin = 94·92%; Einsulin = 90·08%; EBAX = 90·25%).
Table 1.
Primer pairs sequences
| Gene | Primer pairs | PCR product (bp) | GenBank acc. no. |
|---|---|---|---|
| β-actin | 5′-TCCTTCTTGGGTATGG-3′ | 358 | NM_007393·3 |
| 5′-ACGCAGCTCAGTAACAG-3′ | |||
| COX-2 | 5′-GTGAATGCCACCTTCATCC-3′ | 343 | NM_008969·3 |
| 5′-CCATCTTTCCAGAGGTCTTGA-3′ | |||
| iNOS | 5′-AAGTCAAATCCTACCAAAGTGA-3′ | 409 | NM_010927·3 |
| 5′-CCATAATACTGGTTGATGAACT-3′ | |||
| BAX | 5′-TGAAGACAGGGGCCTTTTTG-3′ | 140 | NM_007527 |
| 5′-AATTCGCCGGAGACACTCG-3′ | |||
| Insulin | 5′-CCATCAGCAAGCAGGT-3′ | 184 | NM_008386 |
| 5′-GGGTGTGTAGAAGAAGCCA-3′ | |||
| β-actin | 5′-GGACCTGACAGACTACC-3′ | 337 | NM_007393·2 |
| 5′-GGCATAGAGGTCTTTACGG-3′ |
BAX, Bcl-2-associated X protein; COX-2, cyclooxygenase 2; iNOS, inducible nitric oxide synthase; PCR, polymerase chain reaction; bp, base pairs.
The expression of BAX and insulin was calculated according to the formula 2–(Ct-Ctactin), where Ct is the cycle threshold of the gene of interest and Ctactin is the cycle threshold value of housekeeping gene (β-actin) (last three primer pairs in Table 1). Data were analysed quantitatively using SDS version 2·1 software (Applied Biosystems).
Statistical analysis
To analyse the significance of the differences between various treatments, analysis of variance (anova) was used followed by the Student–Newman–Keuls test for multiple comparisons or Student's t-test, as appropriate. A P-value less than 0·05 was considered significant. The statistical package used was statistica version 6·0 (StatSoft Inc., Tulsa, OK, USA).
Results
MIF deficiency protects pancreatic islets from cytokine-induced dysfunction
To determine the causal relationship between MIF and pancreatic islet death, we treated pancreatic islets isolated from C57BL/6 mice (WT) with a combination of cytokines (TNF-α + IL-1β + IFN-γ). After 6 h of cultivation, cytokine-stimulated pancreatic islets secreted higher levels of MIF compared to non-treated islets (Fig. 1a). Along with elevated MIF, a prominent decrease in islet cells and pancreatic islets viability was observed after 24 h and 48 h of culture with cytokines (Fig. 1b,c), suggesting a potential role of MIF in the propagation of pancreatic islet death. In contrast, islet cells from MIF-KO mice were fully protected from the deleterious effects of these cytokines as determined by MTT assay after 24 h of incubation (Fig. 1b). In addition, pancreatic islets from MIF-KO mice displayed similar features regarding viability (Fig. 1c), and showed a completely normal function depicted by intact insulin expression (Fig. 1d). Based on these results, it is reasonable to assume that MIF participates in cytokine induced loss of viability of insulin-producing cells.
Fig. 1.
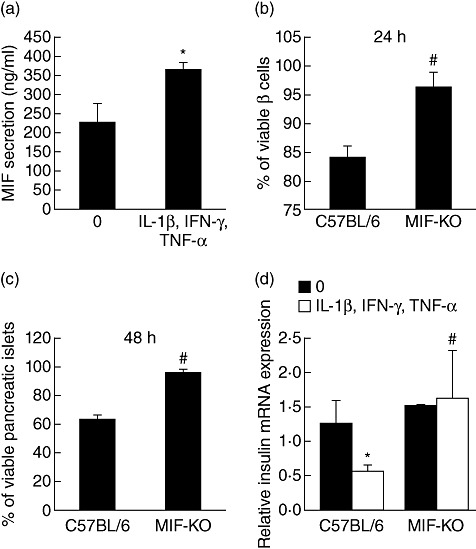
Macrophage migration inhibitory factor (MIF) is a mediator of islet dysfunction and death. Pancreatic islets from C57BL/6 mice were incubated in groups of 20, in the presence or absence of cytokine mixture (interleukin-1β + interferon-γ + tumour necrosis factor-α, 10 ng/ml each) for 24 h and MIF concentration from cell-free supernatants was determined by enzyme-linked immunosorbent assay (a). Islet cells (1 × 104/well) (b) or pancreatic islets (groups of 20) (c) isolated from C57BL/6 and MIF-knock-out (KO) mice were incubated in the presence or absence of cytokine mixture and their viability was assessed by MTT assay after 24 h or 48 h, respectively. Data are presented as % viable cells or islets compared to untreated cells or islets (100%). (d) Relative insulin mRNA expression was determined by real-time polymerase chain reaction in C57BL/6 and MIF-KO islets either left untreated or treated with cytokine mixture for 6 h. Results from three experiments were presented as mean ± standard deviation. *P < 0·05 cytokine-treated versus untreated (0). #P < 0·05 MIF-KO versus C57BL/6 islet cells or islets.
Production of proinflammatory mediators in stimulated MIF-KO islets
Because proinflammatory cytokines trigger NF-κB activation and subsequent expression of proinflammatory mediators such as COX-2 and iNOS [6], we determined their production upon exposure to cytokines in both types of islets. Concordant with previous findings [3], cytokine-provoked WT islets contained higher levels of phosphorylated IκB (p-IκB) and therefore of more active NF-κB transcription factor. In contrast, p-IκB content in MIF-KO islets was down-regulated significantly after cytokine stimulation (Fig. 2a). In addition, cytokine-stimulated COX-2 expression and production were suppressed in MIF-KO islets in contrast to WT islets (Fig. 2b,c). However, iNOS expression and NO production in stimulated MIF-KO islets were enhanced further compared to WT islets (Fig. 2d,e).
Fig. 2.
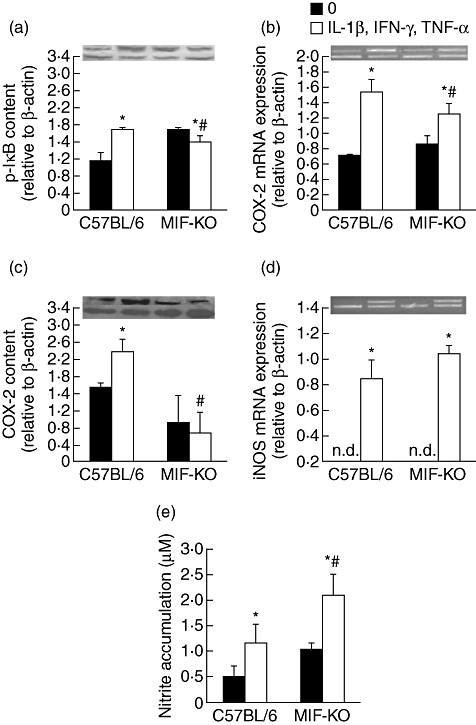
Macrophage migration inhibitory factor (MIF) absence confers resistance of islets through impairment of nuclear factor-κB and cyclooxygenase (COX)-2 activation. Pancreatic islets (in groups of 100) from C57BL/6 or MIF-knock-out (KO) mice were incubated in the presence or absence of cytokine mixture (interleukin-1β + interferon-γ + tumour necrosis factor-α, 10 ng/ml each) for determination of p-IκB protein content by Western blot after 24 h (a), COX-2 mRNA expression by polymerase chain reaction (PCR) after 6 h (b), COX-2 content by Western blot after 24 h (c), inducible nitric oxide synthase mRNA expression after 6 h (d) or nitric oxide production after 48 h (e); n.d. = not detectable. Data are presented as mean ± standard deviation from three separate experiments. *P < 0·05 cytokine-treated versus untreated (0); #P < 0·05 MIF-KO versus C57BL/6 islets. Photographs of PCR amplicons and Western blot are displayed above the adequate graphs; lower lane: β-actin, upper lane: gene or protein in question.
BAX and caspase 3 activities are suppressed in stimulated MIF-KO islets
Mitochondria-associated proteins usually mediate terminal events in cytokine-induced apoptosis of beta cells in pancreatic islets [13,14]. Therefore, we have measured BAX expression and production in both types of islets and found that this pore-forming molecule is up-regulated highly in cytokine-stimulated WT islets (Fig. 3a,b). In contrast, BAX expression and production remained at a similar level in both untreated and cytokine-treated MIF-KO islets. The executor apoptotic molecule, caspase 3, was also suppressed significantly in MIF-KO islets after cytokine treatment judging by its potential to convert the substrate R110 (Fig. 3c).
Fig. 3.
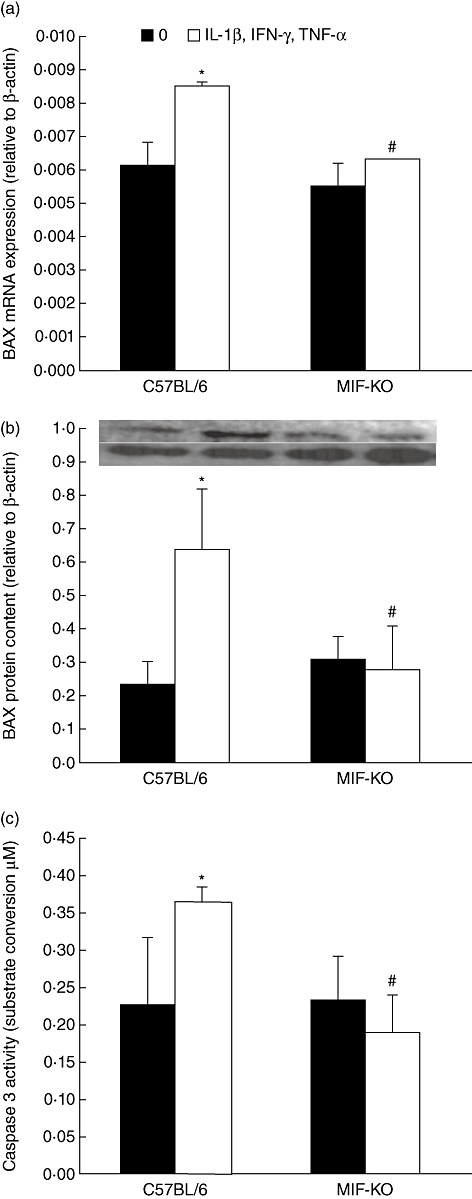
Macrophage migration inhibitory factor (MIF) absence protects islets through down-regulation of mitochondrial apoptotic pathway. Pancreatic islets from C57BL/6 or MIF-knock-out (KO) mice were incubated in the presence or absence of cytokine mixture (interleukin-1β + interferon-γ + tumour necrosis factor-α, 10 ng/ml each) for determination of BAX mRNA expression (in groups of 100) by real-time polymerase chain reaction after 6 h (a) or BAX protein content after 24 h (b), or caspase 3 activity (in groups of 10) after 24 h (c). Photograph of the blot is displayed above the adequate graph; lower lane: β-actin, upper lane: BAX protein. Data are presented as mean ± standard deviation from three experiments. *P < 0·05 cytokine-treated versus untreated (0); #P < 0·05 MIF-KO versus C57BL/6 islets.
MIF-KO islets enhance the content of ERK1/2 upon cytokine stimulation
To determine the activation of prosurvival molecule ERK1/2 in beta cells, we have measured the quantity of phosphorylated forms of p44 (ERK1) and p42 (ERK2) in pancreatic islets of both WT and MIF-KO mice. The obtained results indicate that activation of ERK1/2 is suppressed significantly in WT islets upon cytokine treatment, while MIF-KO islets up-regulated the content of p-ERK1/2 (Fig. 4). The change in activation of ERK1/2 was related inversely to the induction of apoptosis in islets of both strains, thus indicating that ERK1/2 plays a role in preservation of beta cells.
Fig. 4.
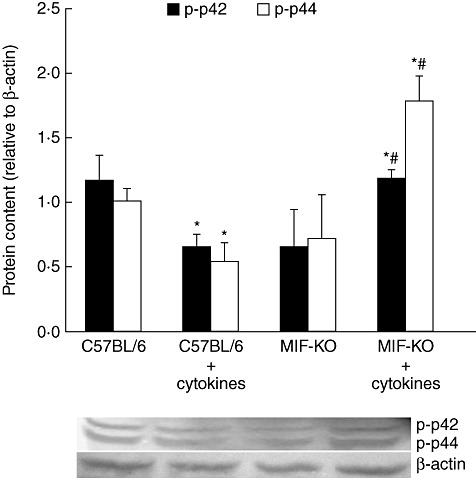
Prosurvival p-extracellular-regulated kinase (ERK)1/2 is up-regulated in cytokine-stimulated macrophage migration inhibitory factor knock-out (MIF-KO) islets. Pancreatic islets (in groups of 100) from C57BL/6 or MIF-KO mice were incubated in the presence or absence of cytokine mixture (interleukin-1β + interferon-γ + tumour necrosis factor-α, 10 ng/ml each) for determination of phosphorylated ERK subunits (p-p42 and p-p44) after 24 h. Data are presented as mean ± standard deviation from two experiments. *P < 0·05 cytokine-treated versus untreated (0); #P < 0·05 MIF-KO versus C57BL/6 islets.
Discussion
This paper shows that endogenous MIF is indispensable for cytokine-induced pancreatic cell dysfunction in vitro. The inability of IL-1β, TNF-α and IFN-γ to sufficiently activate NF-κB-driven production of COX-2 and the mitochondrial apoptotic pathway in MIF-KO islets proves to be a dominant role of MIF in the islet apoptotic process during inflammation.
Beta cells are highly susceptible to cytokine-induced damage due to very low levels of anti-oxidant protection [36]. In addition to succumbing to external effects of cytokines, beta cells themselves can produce cytokines that operate in both autocrine and paracrine fashion and further promote apoptosis [37,38]. MIF is one of the cytokines produced by beta cells [23], and is known to induce apoptosis in several cell types [24–27], including beta cells [30]. Our results indicate that MIF secretion precedes the occurrence of apoptosis in cytokine-stimulated C57BL/6 islets, suggesting its potential role in triggering the apoptotic pathway. This is corroborated by the fact that the absence of MIF confers resistance to cytokine-induced apoptosis of islets and preserves normal insulin expression. Although MIF presence within insulin granules is essential for proper glucose-stimulated insulin release ([23] and our unpublished results), in the circumstances of its excessive concentration, MIF becomes a molecule with autodestructive features. This concentration-dependent dichotomic feature is not attributed solely to the functions of MIF. For example, under physiological circumstances IL-1β plays an important role in the daily maintenance of beta cell mass and function, whereas the long-term and pathologically elevated levels of islet IL-1β associated with inflammation of the islet lead to decreased beta cell function and beta cell mass in diabetes [39].
Under cytokine stimulus, pancreatic islets undergo NF-κB activation and subsequent production of proinflammatory mediators COX-2 and NO [6]. Increase in COX-2 activity augments PGE2 synthesis that may lead to loss in functional beta cell mass and impairment of beta cell function [40]. Additionally, inhibition of COX-2 was shown to preserve beta cell function and increase basal insulin secretion [41]. Therefore, impaired activation of NF-κB and down-regulated COX-2 expression and production in cytokine-stimulated MIF-KO islets could account for the observed enhanced viability of these islets and normal insulin expression. However, Heitmeier et al. [42] showed that selective inhibition of COX-2 activity does not protect islets from cytokine-induced beta cell dysfunction and islet degeneration. Moreover, they showed that islet production of PGE2 does not mediate cytokine-induced inhibitory and destructive effects. Even so, inefficient activation of mitochondria-related apoptotic proteins could account solely for MIF-KO islet resistance to apoptosis.
In contrast to COX-2, iNOS expression and NO production were fully preserved in stimulated MIF-KO islets, suggesting that the absence of MIF could not interfere with this pathway. However, NO was unable to promote damage to MIF-KO islets. Some reports indicate that NO has a less relevant role for cytokine-induced beta cell death in humans and mice compared to rats. Thus, islets obtained from an iNOS-KO mouse are only partially protected against death induced by IL-1β + IFN-γ[43].
Apart from inducing apoptosis indirectly via COX-2-meditated action, cytokines also directly trigger mitochondria-associated pro-apoptotic proteins that depend largely upon the co-ordinated action of BAX and caspase 3 [14]. Our data suggest that MIF absence impairs activation of both BAX production and caspase 3. This coincides with our previous finding, where MIF-KO islets were exposed to detrimental effect of palmitic acid [30]; therefore it could be assumed that apart from triggering NF-κB apoptotic pathway, MIF also mediates apoptosis of islets through the mitochondria-dependent pathway.
Beta cell survival and growth is often associated with increased phosphorylation of ERK1/2 from a family of mitogen-activated kinases [15,16]. Our results substantiate these findings, as cytokine-induced apoptosis coincided with decreased activation of both subunits of ERK. We demonstrate currently that the absence of MIF may confer islet resistance to cytokines through up-regulation of prosurvival signals mediated by ERK.
Although, in physiological conditions, MIF regulates beta cell function, during immunoinflammatory events the pancreatic islets secrete high amounts of MIF that may mediate islet apoptosis by inducing an inflammatory vicious cycle, on one hand, and by promoting NF-κB and/or mitochondria-dependent apoptotic pathways on the other hand. These data further highlight MIF as a key pathogenic molecule in autoimmune diabetogenesis and ideal therapeutic target, for its prevention and reversal.
Acknowledgments
This work was supported by the Project of Ministry of Education and Science, Republic of Serbia (no. 173013). The authors thank Dr Djordje Miljkovic (Institute for Biological Research ‘Sinisa Stankovic’, University of Belgrade, Serbia) for useful comments, Dr Yousef Al-Abed (Feinsten Institute for Medical Research, Manhasset, NY, USA) for kindly providing us with breeding stock of MIF-KO mice and Dr Diana Bugarski (Institute for Medical Research, University of Belgrade, Serbia) for COX-2 antibody.
Disclosure
There are no conflicts of interest.
References
- 1.Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD. A role for innate immunity in type 1 diabetes? Diabetes Metab Res Rev. 2003;19:89–100. doi: 10.1002/dmrr.341. [DOI] [PubMed] [Google Scholar]
- 2.Kaminitz A, Stein J, Yaniv I, Askenasy N. The vicious cycle of apoptotic β-cell death in type 1 diabetes. Immunol Cell Biol. 2007;85:582–9. doi: 10.1038/sj.icb.7100093. [DOI] [PubMed] [Google Scholar]
- 3.Pirot P, Cardozo AK, Eizirik DL. Mediators and mechanisms of pancreatic beta-cell death in type 1 diabetes. Arq Bras Endocrinol Metabol. 2008;52:156–65. doi: 10.1590/s0004-27302008000200003. [DOI] [PubMed] [Google Scholar]
- 4.Davi G, Falco A, Patrono C. Lipid peroxidation in diabetes mellitus. Antioxid Redox Signal. 2005;7:256–68. doi: 10.1089/ars.2005.7.256. [DOI] [PubMed] [Google Scholar]
- 5.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 6.Cardozo AK, Heimberg H, Heremans Y, et al. A comprehensive analysis of cytokine induced and nuclear factor-κB-dependent genes in primary rat pancreatic beta-cells. J Biol Chem. 2001;276:48879–86. doi: 10.1074/jbc.M108658200. [DOI] [PubMed] [Google Scholar]
- 7.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54:S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 8.Gurzov EN, Eizirik DL. Bcl-2 proteins in diabetes: mitochondrial pathways of β-cell death and dysfunction. Trends Cell Biol. 2011;21:424–31. doi: 10.1016/j.tcb.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Tabatabaie T, Waldon AM, Jacob JM, Floyd RA, Kotake Y. COX-2 inhibition prevents insulin-dependent diabetes in low-dose streptozotocin-treated mice. Biochem Biophys Res Commun. 2000;273:699–704. doi: 10.1006/bbrc.2000.2959. [DOI] [PubMed] [Google Scholar]
- 10.Persaud SJ, Burns CJ, Belin VD, Jones PM. Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes. 2004;53:S190–2. doi: 10.2337/diabetes.53.2007.s190. [DOI] [PubMed] [Google Scholar]
- 11.Han X, Chen S, Sun Y, Nadler JL, Bleich D. Induction of cyclooxygenase-2 gene in pancreatic beta-cells by 12-lipoxygenase pathway product 12-hydroxyeicosatetraenoic acid. Mol Endocrinol. 2002;16:2145–54. doi: 10.1210/me.2001-0300. [DOI] [PubMed] [Google Scholar]
- 12.Sandler S, Andersson AK, Barbu A, et al. Novel experimental strategies to prevent the development of type 1 diabetes mellitus. Ups J Med Sci. 2000;105:17–34. doi: 10.1517/03009734000000053. [DOI] [PubMed] [Google Scholar]
- 13.Ortis F, Pirot P, Naamane N, et al. Induction of nuclear factor-kappaB and its downstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role in pancreatic beta cells. Diabetologia. 2008;51:1213–25. doi: 10.1007/s00125-008-0999-7. [DOI] [PubMed] [Google Scholar]
- 14.Mehmeti I, Lenzen S, Lortz S. Modulation of Bcl-2-related protein expression in pancreatic beta cells by pro-inflammatory cytokines and its dependence on the antioxidative defense status. Mol Cell Endocrinol. 2011;332:88–96. doi: 10.1016/j.mce.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Costes S, Broca C, Bertrand G, et al. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: a key role in glucose-mediated pancreatic beta-cell survival. Diabetes. 2006;55:2220–30. doi: 10.2337/db05-1618. [DOI] [PubMed] [Google Scholar]
- 16.Schuppin GT, Pons S, Hügl S, et al. A specific increased expression of insulin receptor substrate 2 in pancreatic beta-cell lines is involved in mediating serum-stimulated beta-cell growth. Diabetes. 1998;47:1074–85. doi: 10.2337/diabetes.47.7.1074. [DOI] [PubMed] [Google Scholar]
- 17.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cvetkovic I, Stosic-Grujicic S. Neutralization of macrophage migration inhibitory factor-novel approach for the treatment of immunoinflammatory disorders. Int Immunopharmacol. 2006;6:1527–34. doi: 10.1016/j.intimp.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor–glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol. 2007;21:1267–80. doi: 10.1210/me.2007-0065. [DOI] [PubMed] [Google Scholar]
- 20.Stosic-Grujicic S, Stojanovic I, Nicoletti F. MIF in autoimmunity and novel therapeutic approaches. Autoimmun Rev. 2009;8:244–9. doi: 10.1016/j.autrev.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 21.Bojunga J, Kusterer K, Bacher M, Kurek R, Usadel KH, Renneberg H. Macrophage migration inhibitory factor and development of type-1 diabetes in non-obese diabetic mice. Cytokine. 2003;21:179–86. doi: 10.1016/s1043-4666(03)00076-0. [DOI] [PubMed] [Google Scholar]
- 22.Cvetkovic I, Al-Abed Y, Miljkovic D, et al. Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology. 2005;146:2942–51. doi: 10.1210/en.2004-1393. [DOI] [PubMed] [Google Scholar]
- 23.Waeber G, Calandra T, Roduit R, et al. Insulin secretion is regulated by the glucose-dependent production of islet beta cell macrophage migration inhibitory factor. Proc Natl Acad Sci USA. 1997;94:4782–7. doi: 10.1073/pnas.94.9.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor JA, Zhu Q, Irwin B, et al. Null mutation in macrophage migration inhibitory factor prevents muscle cell loss and fibrosis in partial bladder outlet obstruction. Am J Physiol Renal Physiol. 2006;291:F1343–53. doi: 10.1152/ajprenal.00144.2006. [DOI] [PubMed] [Google Scholar]
- 25.Yao K, Shida S, Selvakumaran M, et al. Macrophage migration inhibitory factor is a determinant of hypoxia-induced apoptosis in colon cancer cell lines. Clin Cancer Res. 2005;11:7264–72. doi: 10.1158/1078-0432.CCR-05-0135. [DOI] [PubMed] [Google Scholar]
- 26.Chagnon F, Metz CN, Bucala R, Lesur O. Endotoxin-induced myocardial dysfunction: effects of macrophage migration inhibitory factor neutralization. Circ Res. 2005;96:1095–102. doi: 10.1161/01.RES.0000168327.22888.4d. [DOI] [PubMed] [Google Scholar]
- 27.Inácio AR, Ruscher K, Leng L, Bucala R, Deierborg T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow Metab. 2011;31:1093–106. doi: 10.1038/jcbfm.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lue H, Thiele M, Franz J, et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26:5046–59. doi: 10.1038/sj.onc.1210318. [DOI] [PubMed] [Google Scholar]
- 29.Taranto E, Xue JR, Morand EF, Leech M. Modulation of expression and cellular distribution of p21 by macrophage migration inhibitory factor. J Inflamm (Lond) 2009;6:24–34. doi: 10.1186/1476-9255-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saksida T, Stosic-Grujicic S, Timotijevic G, Sandler S, Stojanovic I. Macrophage migration inhibitory factor deficiency protects pancreatic islets from palmitic acid-induced apoptosis. Immunol Cell Biol. 2011 doi: 10.1038/icb.2011.89. doi: 10.1038/icb.2011.89. [DOI] [PubMed] [Google Scholar]
- 31.Toso C, Serre-Beinier V, Emamaullee J, et al. The role of macrophage migration inhibitory factor in mouse islet transplantation. Transplantation. 2008;86:1361–9. doi: 10.1097/TP.0b013e31818bdbef. [DOI] [PubMed] [Google Scholar]
- 32.Stosic-Grujicic S, Stojanovic I, Maksimovic-Ivanic D, et al. Macrophage migration inhibitory factor (MIF) is necessary for progression of autoimmune diabetes mellitus. J Cell Physiol. 2008;215:665–75. doi: 10.1002/jcp.21346. [DOI] [PubMed] [Google Scholar]
- 33.Fingerle-Rowson G, Petrenko O, Metz CN, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–59. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rydgren T, Sandler S. Efficacy of 1400 W, a novel inhibitor of inducible nitric oxide synthase, in preventing interleukin-1beta-induced suppression of pancreatic islet function in vitro and multiple low-dose streptozotocin-induced diabetes in vivo. Eur J Endocrinol. 2002;147:543–51. doi: 10.1530/eje.0.1470543. [DOI] [PubMed] [Google Scholar]
- 35.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Acharya JD, Ghaskadbi SS. Islets and their antioxidant defense. Islets. 2010;2:225–35. doi: 10.4161/isl.2.4.12219. [DOI] [PubMed] [Google Scholar]
- 37.Ribaux P, Ehses JA, Lin-Marq N, et al. Induction of CXCL1 by extracellular matrix and autocrine enhancement by interleukin-1 in rat pancreatic beta-cells. Endocrinology. 2007;148:5582–90. doi: 10.1210/en.2007-0325. [DOI] [PubMed] [Google Scholar]
- 38.Eizirik DL, Mandrup-Poulsen T. A choice of death – the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44:2115–33. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 39.Donath MY, Böni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab. 2010;21:261–7. doi: 10.1016/j.tem.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Robertson RP. Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes. 1998;47:1379–83. doi: 10.2337/diabetes.47.9.1379. [DOI] [PubMed] [Google Scholar]
- 41.Tran PO, Gleason CE, Poitout V, Robertson RP. Prostaglandin E(2) mediates inhibition of insulin secretion by interleukin-1 beta. J Biol Chem. 1999;274:31245–8. doi: 10.1074/jbc.274.44.31245. [DOI] [PubMed] [Google Scholar]
- 42.Heitmeier MR, Kelly CB, Ensor NJ, et al. Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. J Biol Chem. 2004;279:53145–51. doi: 10.1074/jbc.M410978200. [DOI] [PubMed] [Google Scholar]
- 43.Liu D, Pavlovic D, Chen MC, Flodstrom M, Sandler S, Eizirik DL. Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS–/–. Diabetes. 2000;49:1116–22. doi: 10.2337/diabetes.49.7.1116. [DOI] [PubMed] [Google Scholar]