

Abstract
Activity-dependent regulation of calcium dynamics in neuronal cells can play significant roles in the modulation of many cellular processes such as intracellular signalling, neuronal activity and synaptic plasticity. Among many calcium influx pathways into neurons, the voltage-dependent calcium channel (VDCC) is the major source of calcium influx, but its modulation by synaptic activity has still been under debate. While the metabotropic glutamate receptor (mGluR) is supposed to modulate L-type VDCCs (L-VDCCs), its reported actions include both facilitation and suppression, probably reflecting the uncertainty of both the molecular targets of the mGluR agonists and the source of the recorded calcium signal in previous reports. In this study, using subtype-specific knockout mice, we have shown that mGluR5 induces facilitation of the depolarization-evoked calcium current. This facilitation was not accompanied by the change in single-channel properties of the VDCC itself; instead, it required the activation of calcium-induced calcium release (CICR) that was triggered by VDCC opening, suggesting that the opening of CICR-coupled cation channels was essential for the facilitation. This facilitation was blocked or reduced by the inhibitors of both L-VDCCs and InsP3 receptors (InsP3Rs). Furthermore, L-VDCCs and mGluR5 were shown to form a complex by coimmunoprecipitation, suggesting that the specific functional coupling between mGluR5, InsP3Rs and L-VDCCs played a pivotal role in the calcium-current facilitation. Finally, we showed that mGluR5 enhanced VDCC-dependent long-term potentiation (LTP) of synaptic transmission. Our study has identified a novel mechanism of the interaction between the mGluR and calcium signalling, and suggested a contribution of mGluR5 to synaptic plasticity.
Key points
While the metabotropic glutamate receptor (mGluR) is supposed to modulate L-type voltage-dependent calcium channels (L-VDCCs), its reported actions include both facilitation and suppression, and thus the modulation of L-VDCCs by synaptic activity has still been under debate.
In this study, using acute hippocampal slices of subtype-specific knockout mice, we have shown that mGluR5 induces facilitation of the depolarization-evoked calcium current.
This facilitation was not accompanied by the change in single-channel properties of the L-VDCC itself, but required the activation of calcium-induced calcium release that was triggered by L-VDCC opening.
L-VDCCs and mGluR5 were shown to form a complex by coimmunoprecipitation, suggesting that the specific functional coupling between mGluR5, InsP3 receptors and L-VDCCs played a pivotal role in the calcium-current facilitation.
Our study has identified a novel mechanism of the interaction between the mGluR and calcium signalling, and suggested a contribution of mGluR5 to synaptic plasticity.
Introduction
The metabotropic glutamate receptor (mGluR) is a G-protein-coupled receptor encoded by eight different genes, Grm1 to Grm8, and is involved in various aspects of neuronal functions (Masu et al. 1991; Riedel, 1996). The receptors (mGluR1 to mGluR8) encoded by these genes are classified into three groups (group I, II and III) based on their sequence similarities and signal transduction pathways (Nakanishi, 1994; Conn & Pin, 1997). Group I mGluRs consist of mGluR1 and mGluR5 and are coupled to Gq/11-protein to activate the production of InsP3 (Abe et al. 1992). Among a wide range of possible targets of group I mGluRs, the voltage-dependent calcium channel (VDCC) may play the principal role in the control of intracellular calcium dynamics. It is well established that neuronal depolarization triggers large neuron-wide calcium influx through VDCCs (Tsien et al. 1988; Jaffe et al. 1992). Especially, L-VDCCs have the largest conductance and the slowest inactivation kinetics. Although many studies have addressed a possible contribution of group I mGluRs to L-VDCC modulation, the results have been largely controversial. The application of mGluR agonists has been reported to either reduce (Sayer et al. 1992; Sahara & Westbrook, 1993) or increase (Mironov & Lux, 1992; Chavis et al. 1996; Topolnik et al. 2009) a calcium influx through L-VDCCs in many brain regions. There seem to be at least two main reasons for this discrepancy. The first is the use of the non-specific mGluR agonist (±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid (t-ACPD). Although t-ACPD was originally discovered as an agonist for phosphoinositide-coupled mGluRs, now it is known to be non-selective among the mGluR groups (Niswender & Conn, 2010), and additional involvement of Gi/o-coupled group II/III mGluRs in the ACPD effects has been suggested. The second is the uncertainty in the source of calcium influx. Many previous reports do not discriminate among the sources of calcium influx, resulting in the contamination by non-L-type VDCCs (N-type, P/Q-type, R-type and T-type). Even though several reports have utilized L-VDCC antagonists to verify the origin of calcium influx, they still ignore the contribution of calcium-generating processes that are secondary to VDCC activation, such as calcium-induced calcium release (CICR) (Simpson et al. 1995). In order to understand the mechanisms underlying the modulation of L-VDCCs by group I mGluRs, it is essential to precisely identify and separate the components that might contribute to the observed effects.
In this study, we set out to investigate the interaction between group I mGluRs and calcium signalling in CA1 pyramidal cells of the mouse hippocampus. Using subtype-specific knockout (KO) mice, we found that the activation of mGluR5 facilitated a calcium influx triggered by depolarization. This facilitation was not accompanied by the change in single-channel properties of the L-VDCC itself; instead, it was dependent on CICR, suggesting that the opening of CICR-coupled surface cation channels was associated with the facilitation. Furthermore, we showed that VDCC-induced long-term potentiation (LTP) was enhanced by mGluR5 activation. These results represented a previously unidentified mechanism for the mGluR-dependent modulation of VDCC-mediated signalling, and demonstrated a possible mechanism for the involvement of mGluR5 in synaptic plasticity.
Methods
Animals
This research was approved by the Animal Care and Experimentation Committee of University of Tokyo, and all experiments were performed according to the guidelines laid down by the Committee. C57BL/6J mice (6–11 weeks old; male) were used in all experiments except those in which mGluR5 KO (Lu et al. 1997) mice (6–11 weeks old; male) were used, which had been backcrossed to C57BL/6N mice for more than 10 generations. In the experiments using mGluR5KO mice, their littermate wild-type (WT) mice were used as controls. Mice were deeply anaesthetized with halothane and decapitated. The brains were quickly removed and 400 μm hippocampal slices were prepared acutely with a tissue slicer (Kato et al. 2009). In this study, we used a minimum number of mice that were required to draw the conclusions and tried to minimize their suffering as much as possible.
Whole-cell calcium-current recordings
The external solution contained (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.0 NaH2PO4, 26.2 NaHCO3 and 11 glucose. The internal solution contained (in mm): 122.5 caesium gluconate, 17.5 CsCl, 8 NaCl, 10 Hepes, 0.2 EGTA, 2 Mg-ATP and 0.3 Na3-GTP (pH 7.2; 290–310 mosmol l-1). The ATP-regenerating internal solution contained (in mm): 105 caesium gluconate, 17.5 CsCl, 8 NaCl, 10 Hepes, 0.2 EGTA, 2 Mg-ATP, 2 Na2-ATP, 0.3 Na3-GTP, 20 phosphocreatine and 50 U ml-1 creatine phosphokinase (pH 7.2; 290–310 mosmol l-1). In the experiments examining the effect of GDP-βS, Na3-GTP was replaced by 1 mm GDP-βS in both the internal solution and the ATP-regenerating internal solution. Acute hippocampal slices were continuously superfused at a rate of 1.7–1.9 ml min−1 with the external solution saturated with 95% O2 and 5% CO2 in a submersion-type recording chamber. All the experiments were performed at 25 ± 2°C. The connections between the CA1 and CA3 regions were kept intact. Whole-cell patch-clamp recordings were made from CA1 pyramidal cells with the blind technique. The tips of recording electrodes were filled with the internal solution and then backfilled with the ATP-regenerating internal solution. The resistance of glass electrodes was 3.5–6.5 MΩ. After forming the whole-cell configuration, 25 mm TEA and 1 μm TTX were added to the external solution to block voltage-dependent potassium and sodium channels, respectively. The membrane potential of the recorded cells was held at −80 mV immediately after forming the whole-cell configuration, and was changed to −45 mV after starting the perfusion of TEA and TTX. Membrane potential values were not corrected for the liquid-junction potential. The calcium current was calculated using the leak subtraction method. Briefly, two 5 mV hyperpolarizing pulses and one 10 mV depolarizing pulse (200 ms duration each) were delivered to the cells every 10 s. The sums of the holding currents during these three pulses were calculated, and the average values of the last 50 ms were used as the calcium-current amplitude. Recordings were made using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). The signal was filtered at 4 kHz, digitized at 10 kHz and stored on a personal computer equipped with Clampex 9 (Molecular Devices). The series resistance and input resistance were monitored by applying 5 mV hyperpolarizing pulses throughout the experiments. The series resistance was not compensated in order to maintain the signal-to-noise ratio as high as possible. The data from the cells that deviated from any of the following criteria were discarded: first, the resting membrane potential must be more negative than −60 mV at the beginning of whole-cell recordings; second, the input resistance must be higher than 120 MΩ at the beginning of whole-cell recordings; third, the series resistance must not change by more than 20% throughout the experiment; finally, the calcium current must not exhibit spike-like activity during the recording period. The facilitation ratio was calculated using the averaged value from 4 to 6 min after the start of dihydroxyphenylglycine (DHPG) perfusion. All the experiments in this paper were conducted as described above, unless otherwise mentioned.
Glutamate iontophoresis
Sharp glass electrodes for iontophoresis (30–70 MΩ) were filled with 250 mm sodium glutamate solution (pH 8.0). The braking (+10 nA) and ejection (−80 to −200 nA; 10–20 ms) currents were provided to the electrode by using Clampex 9. The position of iontophoresis electrodes was determined in a blind manner: an ejection current was applied at 1 Hz, and the electrode was moved forward in the stratum radiatum until the response to the glutamate ejection was observed in the whole-cell recording. The amplitude and duration of the ejection current were then adjusted so that the response of about 100 pA was obtained. Then, the ejection frequency was reduced to 0.1 Hz, and after establishing the stable recording of responses to glutamate iontophoresis, 10 mm TEA, 1 μm TTX and 50 μm d-(–)-2-amino-5-phosphonovaleric acid (d-APV; an NMDA receptor antagonist) were added to the external solution, and calcium currents were recorded along with the iontophoretic response. In order to prevent an immediate effect of iontophoretically applied glutamate on calcium currents, glutamate was ejected after each depolarizing pulse. At the end of experiments, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), a non-NMDA-receptor antagonist, was applied to verify that the iontophoretic response was mediated by AMPA receptors. Only the data of the experiments in which CNQX blocked more than 80% of the iontophoretic response in amplitude were accepted for analysis.
Synaptic activation of mGluRs
The solutions were the same as those described in the whole-cell calcium-current recording section, except the following two conditions: first, 5 mm QX-314 chloride was added to both the internal solution and ATP-regenerating internal solution, and the concentration of CsCl was reduced by 5 mm; second, the external solution contained a cocktail of receptor antagonists (20 μm CNQX, 50 μm d-APV, 100 μm picrotoxin (an A-type GABA-receptor (GABAAR) antagonist), 2 μm CGP55845 (a B-type GABA-receptor (GABABR) antagonist), 1 μm atropine (a muscarinic ACh-receptor (mAChR) antagonist) and 10 μm mecamylamine (a nicotinic ACh-receptor (nAChR) antagonist)) instead of TTX. A bipolar tungsten stimulating electrode was placed in the stratum radiatum close to the cell layer. To avoid direct stimulation of the dendrites of the recorded cells, the stimulating electrode was placed in the position more than 500 μm distant from the recording electrode. In each recorded cell, the intensity of the stimulus was adjusted to evoke EPSCs of about 200 pA before application of CNQX. After recording baseline calcium currents for 5 min, tetanic stimulation (100 Hz, 1 s) was applied immediately before depolarizing pulses. This procedure was repeated by applying stimulation with ×1, ×2 and ×3 intensities at a 5 min interval. The slow EPSC component that overlapped with the calcium current was subtracted using Excel software.
Single-channel recordings of L-VDCCs
The external solution contained (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 NaH2PO4, 26.2 NaHCO3 and 11 glucose. The internal solution contained (in mm): 110 mm BaCl2, 10 mm Hepes, 1 μm TTX and 1 μm BayK-8644 (pH 7.2), an L-VDCC activator. The resistance of glass recording electrodes was 2.5–5.0 MΩ. Cell-attached patch-clamp recordings were made from CA1 pyramidal cells with the blind technique. Depolarization (90 mV, 500 ms) was applied to the patched membrane at a 10 s interval. The signal was filtered at 0.6–1.0 kHz, digitized at 10 kHz and stored on a personal computer. Single-channel events were detected from the stable part of the traces during depolarizing pulses, using Clampfit 9 (Molecular Devices). In some experiments, 20 μm nifedipine was perfused at the end of experiments, which confirmed that the single-channel events originated in the L-VDCC on the patched membrane (data not shown).
Coimmunoprecipitation
Hippocampi were dissected out from adult mice and homogenized in a lysis buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% NP-40 and a protease inhibitor cocktail (Complete mini EDTA-free, Roche Applied Science, Tokyo, Japan). The suspension was centrifuged at 20,000 g for 5 min, and the supernatant was incubated overnight with a rabbit polyclonal antibody against mGluR5 (no. 06-451, Upstate Biotechnology, Lake Placid, NY, USA). The immune complex was then precipitated using Protein G-Sepharose (GE healthcare, Hino, Japan), and washed three times with the lysis buffer. The bound proteins were eluted and subjected to Western-blot analysis using the following antibodies: anti-mGluR5 (51-69996, BD Pharmingen, Franklin Lakes, NJ, USA), anti-Homer (sc-8921, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-calcium channel antibodies (α1c subunit, C1603, Sigma-Aldrich, Tokyo, Japan).
Experiments on VDCC-dependent LTP
The solutions were the same as those described in the whole-cell calcium-current recording section, except that 50 μm d-APV and 100 μm picrotoxin were added to the external solution instead of TEA and TTX. To prevent epileptiform activity propagating from the CA3 region, a cut was made between the CA1 and CA3 regions. Whole-cell patch-clamp recordings were made from CA1 pyramidal cells with the blind technique. The membrane potential was held at −80 mV throughout the experiment. For recording the baseline response, electrical stimuli were delivered at 0.1 Hz through a bipolar tungsten stimulating electrode placed in the stratum radiatum. The stimulus intensity was adjusted to evoke EPSCs of about 100 pA. VDCC-dependent LTP was induced by repeated application of depolarizing pulses, as described in the previous report (Kato et al. 2009). Briefly, the cell was depolarized repetitively to +10 mV for 1 s 20 times at a 6 s interval in the presence of d-APV. The stimulation of afferent fibres was interrupted during the depolarizing pulses. The potentiation ratio was calculated as the ratio of the averaged value from 26 to 30 min after the conditioning to that during the baseline period.
Statistical analysis
All values are expressed as the mean ± SEM. Statistical analysis was performed using Student's t test for unpaired data, except the analysis comparing the values in the same cells, to which a paired t test was applied. The experiments using mutant mice were performed in a blind manner.
Drugs
Drugs used in this study were DHPG, d-APV, CNQX, CGP55845, QX-314, nifedipine, thapsigargin, cyclopiazonic acid (CPA) (Tocris Bioscience, Bristol, UK), GDP-βS, picrotoxin, atropine, mecamylamine, H89, PKI 6-22 amide, Rp-cAMPS (Sigma-Aldrich), BayK-8644, chelerythrine, Bis I, Bis V, caffeine, 2-APB, TMB-8 (Calbiochem, San Diego, USA) and TTX (Sankyo, Osaka, Japan). In the experiments with H89, PKI 6–22 amide, thapsigargin, CPA and caffeine, hippocampal slices were incubated in an interface-type recovery chamber with the drugs for at least 1 h before transferring them to the recording chamber, and were perfused with the Ringer solution containing each drug throughout the experiments. In the experiments with Bis I, Bis V, chelerythrine, barium, 2-APB, TMB-8 and nifedipine, drug application was started in the recording chamber and the incubation time ranged from 15 to 60 min before DHPG application.
Results
Calcium-current facilitation by mGluR5 activation
We tested the effect of DHPG, a group I mGluR-selective agonist, on calcium currents in the CA1 pyramidal cells of the mouse hippocampus. The group I mGluR includes mGluR1 and mGluR5, and the CA1 pyramidal cells have been shown to express mGluR5 predominantly (Shigemoto et al. 1993). Thus, we evaluated the specific action of DHPG by comparing the effects between mGluR5 KO and WT mice (Fig. 1A). Calcium currents were evoked by applying depolarizing pulses to the voltage-clamped pyramidal cells in the presence of TTX and TEA. These currents were carried by calcium, as replacing external calcium with magnesium abolished the majority of the evoked currents (77.9 ± 0.8% reduction 8–10 min after the substitution: Supplementary Fig. S1A). Application of 5 μm DHPG for 5 min significantly increased the calcium current in WT mice (135.6 ± 5.3%, n= 10, P < 0.0001), but this effect was completely absent in mGluR5 KO mice (102.2 ± 1.5%, n= 7, P= 0.1993; WT vs. KO: P < 0.0001). There was no difference in amplitudes of the baseline calcium current between the genotypes (WT: 41.1 ± 2.9 pA; KO: 35.9 ± 2.6 pA, P= 0.2050). The facilitation observed in WT mice disappeared after withdrawal of the drug (115.3 ± 3.5%, 10 min after the start of washout), suggesting that the effect of DHPG was reversible. In most experiments in this paper, we applied DHPG within 20 min after the formation of the whole-cell configuration; however, whenever we waited for longer periods before its application, we observed smaller facilitation of calcium currents (data not shown). This washout effect suggested the involvement of cytoplasmic second messengers and/or other intracellular components in this facilitation process.
Figure 1. mGluR5-dependent facilitation of calcium currents.
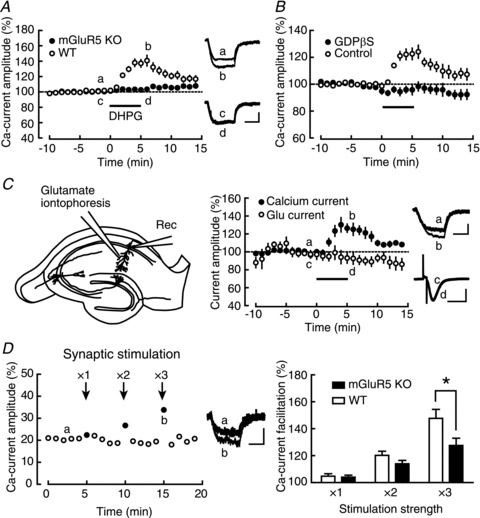
A, left, comparison of the DHPG (a group I-mGluR agonist; horizontal bar)-induced calcium-current facilitation between WT (open circles; n= 10) and mGluR5 KO (filled circles; n= 7) mice. Right, averaged calcium-current traces recorded at the times indicated by a–d. B, comparison of the DHPG-induced calcium-current facilitation in the presence (filled circles; n= 8) and absence (open circles; n= 7) of intracellular GDP-βS, a G-protein inhibitor. C, left, schematic diagram of the glutamate-iontophoresis experiment. Middle, comparison of calcium-current (filled circles) and glutamate-response (open circles) amplitudes during DHPG application (n= 5). Right, averaged traces of calcium currents (a and b) and glutamate responses (c and d) recorded at the times indicated by a–d. D, left, representative example of the calcium-current facilitation induced by tetanic stimulation (100 Hz, 1 s; arrows) of Schaffer collaterals. The number above each arrow indicates how many times stronger the stimulation intensity was, compared to the baseline stimulation intensity. Middle, representative traces of calcium currents recorded at the times indicated by a and b are shown. Right, summary of the synaptic stimulation-induced calcium-current facilitation in WT (open columns; n= 8) and mGluR5 KO (filled columns; n= 9) mice. *P < 0.05. Calibration: 20 pA, 100 ms for calcium currents and 50 pA, 100 ms for glutamate responses.
To confirm the involvement of the canonical mGluR5-mediated signal-transduction pathway, we next examined whether G-proteins were involved in this facilitation by including the G-protein inhibitor GDP-βS in the internal solution (Fig. 1B). GDP-βS caused gradual decrease in the calcium-current amplitude (data not shown), and it took about 30 min after the formation of the whole-cell configuration until the current amplitude stabilized. In order to adjust the timing of the DHPG application, in control experiments, we kept recording calcium currents without any manipulations for about 30 min before starting the DHPG application, which may have resulted in smaller potentiation in this set of experiments than that in the other control experiments. Intracellular application of 1 mm GDP-βS completely blocked the DHPG-induced facilitation of calcium currents (GDP-βS: 97.9 ± 2.3%, n= 8, P= 0.3991; control: 124.2 ± 5.8%, n= 7, P= 0.0061). There was a significant difference between GDP-βS and control conditions (P= 0.0004). This result suggested the involvement of G-proteins in the calcium-current facilitation, supporting the idea that G-protein-coupled receptors such as mGluRs were involved in this facilitation.
In the control experiments shown in Fig. 1A and B, we always observed DHPG-induced increase in the input resistance of the recorded cells (Fig. 1A: 147.7 ± 3.0%, P= 0.0025; Fig. 1B: 133.1 ± 2.3%, P= 0.0171). This observation pointed to the possibility that DHPG might have increased the recorded calcium current just because the increase in input resistance resulted in a decrease of the current loss through the cell membrane (Williams & Mitchell, 2008). To rule out this possibility, we measured the AMPA receptor (AMPAR)-mediated current evoked by glutamate iontophoresis onto the dendrite as a control and tested whether DHPG had any effects on the evoked current (Fig. 1C). In order to exclude the contribution of the NMDA receptor (NMDAR)-mediated current and to prevent the induction of NMDAR-dependent synaptic plasticity, 50 μm d-APV was added to the external solution. Although DHPG significantly facilitated the calcium current, no change was observed in the AMPAR-mediated current simultaneously recorded in the same cells (calcium current: 127.2 ± 6.3%, P= 0.0126; AMPAR current: 93.2 ± 6.4%, P= 0.3459, n= 5). The smaller facilitation in calcium currents compared to Fig. 1A may have been caused by the washout of the facilitation mechanism in the recorded neurons due to the additional time needed for searching the optimal position for iontophoretic glutamate application. There was a significant difference between the calcium current and AMPAR-mediated current (P= 0.0347). This result strongly argued against the contribution of the passive membrane properties to the facilitation, and confirmed the specific effect of DHPG on the calcium current.
We next tested whether synaptic activation of mGluR5 instead of the drug application could facilitate calcium currents (Fig. 1D). Stimulating electrodes were placed in the stratum radiatum to stimulate Schaffer collaterals for evoking synaptic release of glutamate. In this set of experiments, TTX was excluded from the external solution to allow electrical stimulation of Schaffer collaterals and 5 mm QX-314 was included in the recording pipette instead to block the generation of action potentials in the recorded neuron. The cocktail of receptor blockers was also applied to the external solution to exclude the possible contribution of other receptors (AMPAR, NMDAR, GABAAR, GABABR, mAChR and nAChR: see Methods). Tetanic stimulation (100 Hz, 1 s) at various intensities was first applied, and then, the depolarizing pulse for evoking a calcium current was delivered 15 ms after to maximize mGluR activation at the time of calcium-current recording. As shown in a representative example (Fig. 1D, left), tetanic stimulation caused facilitation of the calcium currents. The facilitation was observed only in the calcium current that was recorded immediately after tetanic stimulation, and the facilitating effect disappeared within 10 s after tetanic stimulation. To confirm that this facilitation was mediated by mGluR5, we also performed the same experiments in mGluR5 KO mice and compared the results with those in WT mice (Fig. 1D, right). The facilitation of calcium currents was observed both in WT (×1: 104.8 ± 1.8%, P= 0.0328; ×2: 120.2 ± 3.1%, P= 0.0003; ×3: 147.7 ± 6.7%, P= 0.0002, n= 8) and KO (×1: 104.0 ± 1.4%, P= 0.0238; ×2: 114.0 ± 2.5%, P= 0.0006; ×3: 127.6 ± 5.4%, P= 0.0010, n= 9) mice at all stimulation intensities. However, the facilitation in WT was larger than that in KO mice at the highest stimulation intensity (×1: P= 0.7538; ×2: P= 0.1368; ×3: P= 0.0344), supporting the idea that the calcium-current facilitation induced by synaptic stimulation was at least partially dependent on mGluR5.
Direct modification of the L-VDCC is not involved in the calcium-current facilitation
To elucidate molecular mechanisms underlying the calcium-current facilitation, we next examined a possible contribution of protein kinase A (PKA) and protein kinase C (PKC) to the facilitation, both of which were reported to upregulate the L-VDCC through direct phosphorylation of the channel (Flockerzi et al. 1986; Stea et al. 1995). Especially, PKC has been suggested to mediate the mGluR-induced facilitation of calcium signalling in hippocampal interneurons (Topolnik et al. 2009). Therefore, we first tested the effects of the PKC inhibitors chelerythrine and Bis I on the calcium-current facilitation (Fig. 2A); however, we did not observe any change in the DHPG-induced calcium-current facilitation in the presence of 2 μm chelerythrine (chelerythrine: 139.7 ± 5.6%, n= 8, P= 0.0002; control: 136.1 ± 3.8%, n= 7, P= 0.0001; chelerythrine vs. control: P= 0.6132) nor 1 μm Bis I (Bis I: 145.6 ± 5.5%, n= 7, P= 0.0002; Bis V (an inactive Bis I analogue) control: 145.4 ± 6.7%, n= 7, P= 0.0005; Bis I vs. Bis V: P= 0.9822). We next tested the effects of the PKA inhibitors PKI, Rp-cAMPS and H89 (Fig. 2B). Again, we did not observe any change in the DHPG-induced calcium-current facilitation by 100 μm intracellular PKI 6–22 amide (PKI: 150.2 ± 7.5%, n= 9, P= 0.0002; control: 148.3 ± 5.7%, n= 6, P= 0.0004; PKI vs. control: P= 0.8443), bath-applied 1 μm membrane-permeant myristoylated PKI 14–22 (PKI: 137.8 ± 3.0%, n= 7, P= 0.0001; control: 140.4 ± 7.3%, n= 6, P= 0.0027; PKI vs. control: P= 0.7564) nor 1 mm intracellular Rp-cAMPS (Rp-cAMPS: 140.1 ± 6.1%, n= 6, P= 0.0012; control: 138.9 ± 4.2%, n= 7, P < 0.0001; Rp-cAMPS vs. control: P= 0.8806). However, in the presence of 10 μm H89, the facilitation was significantly decreased (H89: 122.8 ± 5.0%, n= 11, P= 0.0010; control: 149.0 ± 6.9%, n= 12, P < 0.0001; H89 vs. control: P= 0.0061). Because H89 is a drug with many reported side effects (Murray, 2008), especially the side effect causing the depletion of intracellular calcium stores (Lahouratate et al. 1997: see below), we concluded that neither PKC nor PKA seemed to work downstream of mGluR5.
Figure 2. Calcium-current facilitation is independent of both PKC and PKA.
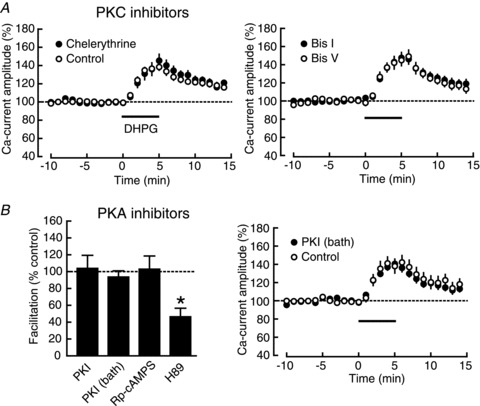
A, left, DHPG (horizontal bar)-induced calcium-current facilitation in the presence (filled circles; n= 8) and absence (open circles; n= 7) of chelerythrine, a PKC blocker. Right, DHPG-induced calcium-current facilitation in the presence of Bis I, a PKC inhibitor (filled circles; n = 7) and Bis V, an inactive analogue of Bis I (open circles; n= 7). B, left, summary of the DHPG-induced calcium-current facilitation in the presence of PKA inhibitors, intracellularly applied PKI 6–22 (PKI: n= 9; control: n= 6), bath-applied myristoylated PKI 14–22 (PKI: n= 7; control: n= 6), Rp-cAMPS (Rp-cAMPS: n= 6; control: n= 7) and H89 (H89: n= 11; control: n= 12). The facilitation ratio was normalized to that in the control experiment for each drug. *P < 0.05. Right, the time course of the DHPG-induced calcium-current facilitation in the presence (filled circles) and the absence (open circles) of bath-applied myristoylated PKI 14–22.
The results so far make it less likely that the calcium-current facilitation is mediated by the modulation of channel properties of VDCCs through phosphorylation. Therefore, we next tested this possibility directly by analysing single-channel properties of VDCCs. Considering its long inactivation time constant, the L-VDCC should be the major VDCC that contributes to the calcium current in our recording conditions (Tsien et al. 1988: see Fig. 5A). In a previous report, t-ACPD has been shown to facilitate the single-channel barium current of L-VDCCs in the cerebellum (Chavis et al. 1996). We thus tested whether the same mechanism contributed to the mGluR5 action we observed in this study. Figure 3A shows a representative example, in which DHPG was applied during the single-channel recording of the barium current through L-VDCCs. In contrast to the report by Chavis et al. (1996), we did not observe any change in the current amplitude (baseline: 1.04 ± 0.03 pA; DHPG: 1.07 ± 0.03 pA, P= 0.5203, n= 8), the open probability (baseline: 0.0606 ± 0.0086; DHPG: 0.0636 ± 0.0164, P= 0.8738) or the median of open time (baseline: 5.10 ± 0.50 ms; DHPG: 5.66 ± 0.65 ms, P= 0.5057) (Fig. 3B). The single-channel current amplitude was similar to that of L-VDCCs at the membrane potential of +10 to +20 mV in the previous report (Church & Stanley, 1996). Taken together, these results argued against the direct modulation of channel properties of the L-VDCC by mGluR5 activation, suggesting that the facilitation mechanism was different from that reported in the previous studies.
Figure 5. mGluR5 acts specifically on L-VDCC-evoked CICR.
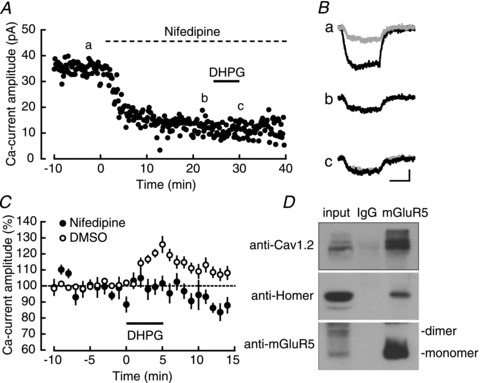
A, a representative experiment examining the effect of DHPG (horizontal bar) in the presence of nifedipine (an L-VDCC blocker; dotted horizontal bar). B, averaged calcium currents recorded at the times indicated by a–c in A. The trace b is superimposed (a grey trace) on the traces a and c. Calibration: 10 pA, 100 ms. C, the DHPG-induced calcium-current facilitation in the presence (filled circles; n= 9) and the absence (open circles; n= 9) of nifedipine. D, coimmunoprecipitation of mGluR5 with Cav1.2, Homer and mGluR5.
Figure 3. Single-channel recordings of L-VDCCs.
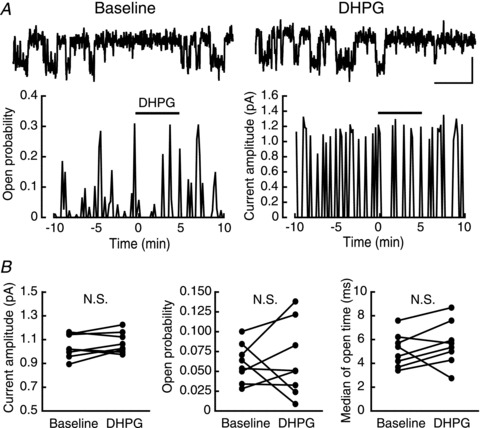
A, top, representative sweeps of single-channel currents of the L-VDCC before (left) and during (right) DHPG application. Bottom, representative plots of single-channel open probability (left) and current amplitude (right) in the experiment examining the effect of the DHPG application (horizontal bar). B, summary of the DHPG-induced change in single-channel current amplitude (left), open probability (middle) and median of open time (right) (n= 8). Calibration: 1 pA, 50 ms.
Requirement of CICR for the mGluR5-induced calcium-current facilitation
The lack of the change in single-channel properties of VDCCs raised the possibility that mGluR5 might have facilitated calcium currents not through the change in the VDCC itself, but by affecting the secondary process that was evoked by VDCC activation, such as CICR. CICR is a phenomenon of calcium release from intracellular stores mediated by InsP3 receptors (InsP3Rs) and ryanodine receptors (RyRs) localized in the membrane of the sarcoplasmic/endoplasmic reticulum (Simpson et al. 1995), the primary stores of intracellular calcium. The plasma membrane and the sarcoplasmic/endoplasmic reticulum have been reported to contact each other (Gardiner & Grey, 1983; Takeshima et al. 2000), and the L-VDCC has a direct interaction with the ryanodine receptor (RyR) on the sarcoplasmic reticulum (Nakai et al. 1996), suggesting that the L-VDCC is a well-suited candidate for the trigger for CICR. In order to examine the contribution of CICR to the calcium-current facilitation, we interfered with this process pharmacologically, using three mechanistically independent means, as follows (Fig. 4A). First, we tested the effects of thapsigargin and CPA, blockers of sarcoplasmic/endoplasmic-reticulum calcium ATPase (SERCA). Because SERCA is required for the loading of calcium into the endoplasmic reticulum, its prolonged blockade causes the depletion of intracellular calcium stores. As shown in Fig. 4A, the DHPG-induced calcium-current facilitation was significantly suppressed in both 1 μm thapsigargin (thapsigargin: 129.8 ± 2.8%, n= 10, P < 0.0001; DMSO control: 148.1 ± 2.7%, n= 9, P < 0.0001; thapsigargin vs. DMSO control: P= 0.0002) and 15 μm CPA (CPA: 139.9 ± 4.1%, n= 9, P < 0.0001; DMSO control: 157.0 ± 6.6%, n= 9, P < 0.0001; CPA vs. DMSO control: P= 0.0464). Second, we tested the effect of caffeine, an agonist of RyRs, on the facilitation (Fig. 4B). Although the significant effects of the SERCA blockers supported the contribution of CICR to the facilitation, the remaining facilitation might have resulted from incomplete depletion of the calcium store. Therefore, we aimed at thorough depletion by releasing stored calcium actively through RyRs. Caffeine (20 mm) completely blocked the DHPG-induced facilitation of calcium currents (caffeine: 86.4 ± 2.7%, n= 8, P= 0.0023; control: 143.6 ± 7.9%, n= 7, P= 0.0010; caffeine vs. control: P < 0.0001) (Fig. 4A and B), further supporting the role of calcium stores in the facilitation. A high concentration of caffeine is reported to reduce VDCC currents in the muscle blastomere (Nakajo et al. 1999), but there was no significant difference in the baseline calcium-current amplitude between the two groups (caffeine: 35.9 ± 2.7 pA; control: 31.7 ± 3.5 pA, P= 0.3532). Third, we examined the effect of the substitution of barium for the extracellular calcium. CICR is inhibited by this manipulation because it requires an initial increase in the intracellular calcium concentration, and barium was unable to trigger CICR (Nagasaki & Kasai, 1984). For this purpose, we substituted 1.3 mm BaCl2 and 2.5 mm MgCl2 for 2.5 mm CaCl2 and 1.3 mm MgSO4 in the external solution. In this solution DHPG failed to facilitate barium currents (BaCl2: 98.7 ± 1.2%, n= 7, P= 0.3142; control: 132.7 ± 2.3%, n= 7, P < 0.0001; BaCl2 vs. control: P < 0.0001) (Fig. 4A). Taken together, the results of these three sets of experiments clearly indicated the requirement of CICR for the DHPG-induced calcium-current facilitation. This conclusion well explains the result of the H89 experiment (Fig. 2B), considering its calcium-depleting side effect. Because CICR itself is not supposed to contribute to the currents in whole-cell recordings, this result suggests the involvement of CICR-coupled surface channels, such as transient receptor potential canonical (TRPC) channels (Minke & Cook, 2002).
Figure 4. InsP3R-dependent facilitation of calcium currents.
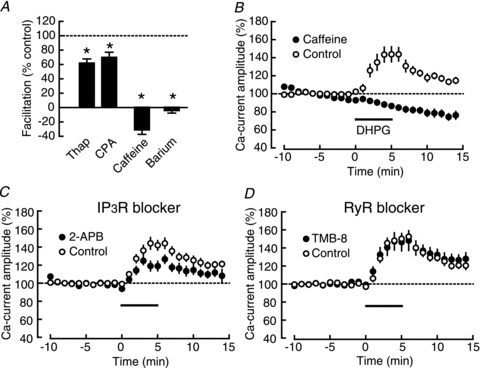
A, summary of the DHPG-induced calcium-current facilitation in the presence of calcium store-depleting drugs, thapsigargin (thap: n= 10; DMSO control: n= 9), CPA (CPA: n= 9; DMSO control: n= 9), caffeine (caffeine: n= 9; control: n= 8) and BaCl2 (BaCl2: n= 7; CaCl2 control: n= 7). *P < 0.05. B, time course of the DHPG (horizontal bar)-induced calcium-current facilitation in the presence (filled circles) and the absence (open circles) of caffeine. C, the DHPG-induced calcium-current facilitation in the presence (filled circles; n= 9) and the absence (open circles; n= 8) of 2-APB, an InsP3R inhibitor. D, the DHPG-induced calcium-current facilitation in the presence (filled circles; n = 7) and the absence (open circles; n= 7) of TMB-8, a RyR inhibitor.
Specific functional coupling between the mGluR5, InsP3R and L-VDCC
Next, we tried to determine which type of receptors, either InsP3Rs or RyRs, was responsible for this observed effect. We used 2-APB, a blocker of InsP3Rs (Fig. 4C), and TMB-8, a blocker of RyRs (Fig. 4D). Although we did not observe any effect of 10 μm TMB-8 on the calcium-current facilitation (TMB-8: 147.2 ± 10.0%, n= 7, P= 0.0033; control: 150.2 ± 3.6%, n= 7, P < 0.0001; TMB-8 vs. control: P= 0.7840), 30 μm 2-APB significantly decreased it (2-APB: 121.5 ± 4.7%, n= 9, P= 0.0018; control: 143.4 ± 5.7%, n= 8, P= 0.0001; 2-APB vs. control: P= 0.0104). These results suggested the contribution of InsP3Rs, but not RyRs, to the signalling downstream of mGluR5. This conclusion is consistent with the report showing that mGluR5, Homer and InsP3Rs constitute a protein complex (Brakeman et al. 1997; Kato et al. 1998; Tu et al. 1998). Considering the tight interaction between L-VDCCs and the sarcoplasmic reticulum (Nakai et al. 1996), it is reasonable to assume that mGluR5, InsP3Rs and L-VDCCs form a large protein complex, which enables efficient functional coupling between each component. To test this hypothesis, we examined the specificity of the mGluR5 action on L-VDCC-coupled CICR. Figure. 5A and B shows a representative experiment, in which DHPG was applied after blocking the L-VDCC component of calcium currents with 20 μm nifedipine (blockade 20 min after starting the perfusion: 62.4 ± 2.3% of baseline (n= 10); Supplementary Fig. S1B). As shown in Fig. 5C, DHPG did not have a facilitatory effect on the nifedipine-insensitive component of the calcium current (nifedipine: 98.8 ± 3.2%, n= 9, P= 0.7190; DMSO control: 121.9 ± 3.1%, n= 9, P= 0.0001; nifedipine vs. DMSO: P < 0.0001). This result confirmed the specific action of mGluR5 on L-VDCC-coupled CICR and suggested the functional coupling between mGluR5, InsP3Rs and L-VDCCs. Finally, we directly tested the physical interaction between L-VDCCs and mGluR5 by coimmunoprecipitation experiments (Fig. 5D), and found that mGluR5 was coimmunoprecipitated with Cav1.2, a major L-VDCC subunit in the hippocampus, and the scaffolding protein Homer (Brakeman et al. 1997; Kato et al. 1998), which was reported to bridge many proteins including mGluR5, InsP3Rs and TRPC channels (Tu et al. 1998; Yuan et al. 2003). These results strongly suggest the formation of a large protein complex that facilitates the functional coupling between mGluR5, L-VDCCs and InsP3Rs.
Facilitation of VDCC-dependent LTP by mGluR5 activation
Finally, we asked whether this mechanism of mGluR5-dependent calcium-current amplification had any roles in synaptic plasticity. We have previously shown that LTP is induced by applying repeated depolarizing pulses to the voltage-clamped cells (Kato et al. 2009). Because this LTP has been shown to depend on L-VDCCs, we examined whether this type of LTP was augmented by the DHPG-induced calcium-current facilitation. The calcium-current facilitation was dependent on mGluR5 activation (Fig. 1A), and thus, we compared the effects of DHPG on this type of LTP between WT and mGluR5 KO mice (Fig. 6). As shown in Fig. 6A, L-VDCC-dependent LTP was significantly facilitated by the application of DHPG in WT mice (DHPG: 144.9 ± 3.7%, n= 14, P < 0.0001; control: 122.8 ± 6.4%, n= 15, P= 0.0030; DHPG vs. control: P= 0.0066). However, this facilitating effect was completely absent in mGluR5 KO mice (DHPG: 124.0 ± 9.4%, n= 11, P= 0.0295; control: 123.0 ± 6.6%, n= 11, P= 0.0057; DHPG vs. control: P= 0.9318). These results supported the mGluR5-dependent facilitation of L-VDCC-dependent LTP. Although the detailed analysis of calcium currents during depolarizing pulses was difficult because the experiments were conducted in the absence of TTX and TEA to record synaptic responses, it seemed to be reasonable to conclude that the DHPG-induced enhancement of LTP specifically observed in WT mice was caused by the calcium-current facilitation observed in this study.
Figure 6. Facilitation of VDCC-dependent LTP by mGluR5 activation.
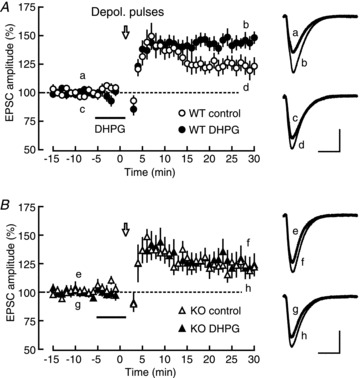
A, left, depolarizing pulse (open arrow; Depol. pulses)-induced LTP in the presence (filled circles; n= 14) and the absence (open circles; n= 15) of DHPG in WT mice. Right, averaged EPSC traces recorded at the times indicated by a–d in the left panel. B, left, depolarizing pulse-induced LTP in the presence (filled triangle, n= 11) and the absence (open triangle, n= 11) of DHPG in mGluR5 KO mice. Right, averaged EPSC traces recorded at the times indicated by e–h in the left panel. Calibration: 50 pA, 20 ms.
Discussion
The present study has shown that the group I mGluR agonist facilitates calcium signalling through previously unidentified mechanisms. Our results have several implications for the molecular mechanisms underlying this facilitation. First, this facilitation was abolished by the genetic deletion of mGluR5 or G-protein inhibitors, suggesting the involvement of the mGluR5 signalling pathway (Abe et al. 1992). Second, inhibitors of either PKA or PKC did not block the calcium-current facilitation, suggesting that these kinases were not likely to contribute to it. Third, the facilitation was not accompanied by the change in single-channel properties of L-VDCCs, ruling out the direct modification of L-VDCCs. Fourth, the perturbation of CICR reproducibly blocked the facilitation, suggesting the requirement of intracellular calcium-channel activation. Fifth, the blocker of InsP3Rs, but not RyRs, decreased the facilitation, suggesting the role of CICR mediated by InsP3Rs. Finally, the L-VDCC antagonist completely blocked the facilitation, suggesting the specific requirement of L-VDCCs as a calcium source for CICR. Taken together, our results suggest that mGluR5 activation facilitates calcium signalling not through the change in L-VDCC properties per se, but through the activation of CICR-coupled surface channels that is triggered by L-VDCC activation.
Validity of the proposed molecular mechanisms
Even though we included caesium, TTX and TEA in our solutions to block most of the voltage-dependent channels, additional care should be taken to interpret the properties of the current recorded in the whole-cell configuration. First, the increase in the recorded currents might be attributable to the reduction in a leak current, rather than real change in depolarization-evoked currents (Williams & Mitchell, 2008). However, this is quite unlikely in our study, because we did not observe any effect of DHPG on the AMPA current that was evoked by glutamate iontophoresis (Fig. 1C). If there is any change of membrane properties, it should affect the AMPA current evoked on the distal dendrites more seriously than the current deriving from L-VDCCs that localized mainly in the proximal region of pyramidal cells (Westenbroek et al. 1990). Second, the depolarization-evoked current might contain current components originating from channels other than VDCCs. It is known that some K+ channels cannot be blocked completely by caesium and TEA. However, considering almost thorough disappearance of the depolarization-evoked current in nominally Ca2+-free external solution (Supplementary Fig. S1A), the contribution of voltage-dependent K+ channels to the evoked current is negligible in our experimental conditions, ruling out the possibility that DHPG directly closes K+ channels. The large decrease in the depolarization-evoked current by nifedipine also confirmed that the recorded current was mediated by the L-VDCC (Fig. 5A; Supplementary Fig. S1B). However, these results do not exclude possible contribution of the channels that are activated secondarily by increased intracellular Ca2+ that enters through VDCCs. Although the DHPG-induced facilitation of the depolarization-evoked current is dependent on the L-VDCC (Fig. 5C), the lack of change in the L-VDCC single-channel properties (Fig. 3) suggests that it is the channels activated secondarily, but not the L-VDCC itself, that are modulated by DHPG. The candidates include Ca2+-activated K+ channels, Ca2+-activated Cl− channels and CICR-coupled TRPC channels; however, considering the effects of various CICR blockers (Fig. 4), the TRPC channel is likely to mediate the current facilitation. This idea is supported by the reports showing the direct (Kiselyov et al. 1999) and Homer-mediated (Kiselyov et al. 1998; Yuan et al. 2003) interaction between InsP3Rs and TRPC channels, and we confirmed here the interaction of L-VDCCs and mGluR5 with Homer (Fig. 5D). In accord with this hypothesis, 2-APB, which we used as an InsP3R blocker, also interferes with the TRPC channels (Xu et al. 2005), and therefore, the blockade of the current facilitation by 2-APB (Fig. 4C) may be mediated not only by its action on InsP3Rs but also on TRPC channels. The lack of specific blockers for TRPC channels hampered us from directly testing this possibility, but it would be interesting to study the facilitation mechanism further using subtype-specific TRPC KO mice in future (Freichel et al. 2005).
There still remains an argument related to the contribution of VDCCs to the facilitation. Although the lack of change in the single-channel properties in the presence of DHPG (Fig. 3) argues against any change in the channel properties of VDCCs, it is still possible that the current facilitation is mediated by increase in the number of L-VDCCs. However, it is rather unlikely based on the following three reasons. First, we did not observe any increase in the number of channels in the patched membrane during single-channel recordings in the presence of DHPG. Second, the rapid time course of the facilitation induced by synaptic stimulation is hard to explained by the change in surface expression of the channels, which would have taken much more time (Herlitze et al. 2003). Third, the requirement of InsP3Rs for the facilitation could not be explained by simple increase in the number of L-VDCCs. Taken together, these results support the idea that the primary target of DHPG is not the VDCC itself, but the channels that are activated downstream of the VDCC.
We have shown that synaptic stimulation can be substituted for DHPG in inducing the calcium-current facilitation (Fig. 1D). Although the time course of the synaptically induced facilitation is apparently faster than that of the DHPG-induced facilitation, it is likely attributed to fast reuptake of synaptically released glutamate and slow penetration and washout of DHPG in slice preparations. The decrease of the synaptically induced facilitation in mGluR5 KO mice confirms its dependency on mGluR5. However, the genetic ablation of mGluR5 blocked only half of this facilitation, suggesting the existence of other factors required for the synaptically induced facilitation. We propose three possible mechanisms. First, mGluR1, the other subtype of the group I mGluR, might also contribute to the calcium current facilitation. However, this is quite unlikely considering the complete disappearance of the facilitation by the application of DHPG, which is an agonist for both mGluR1 and mGluR5, in mGluR5 KO mice (Fig. 1A). The predominant expression of mGluR5 over mGluR1 in CA1 hippocampal pyramidal cells also makes it unlikely that mGluR1 contributes to the facilitation (Shigemoto et al. 1993). Second, insufficient clamping of the membrane potential of recorded neurons during strong synaptic stimulation might have contributed to the remaining facilitation. In spite of the pharmacological blockade of the major receptors by the blocker cocktail (Fig. 1D), we still observed remaining synaptic responses at stronger stimulation intensities, whose source we did not seek further. These remaining excitatory inputs might have caused poorer space-clamping in distal dendrites (Williams & Mitchell, 2008), resulting in regenerating activation of L-VDCCs (Schiller et al. 1997). However, considering biased distribution of L-VDCCs toward the soma (Westenbroek et al. 1990), the contribution of distal L-VDCCs should be relatively small. Third, stronger synaptic stimulation might have released other neurotransmitters and/or neuromodulators including noradrenaline, serotonin and dopamine, which may have contributed to the facilitation. Indeed, there are reports on the modulation of VDCCs by β-adrenergic (Gray & Johnston, 1987) and 5-HT (Nakamura et al. 2000) receptors. Further studies will be required to test whether these neuromodulators contribute to the synaptically induced facilitation of calcium currents.
Some of our results are inconsistent with the two previous studies that argue that the mGluR facilitates calcium signalling through direct modulation of L-VDCCs. First, Topolnik et al. (2009) studied the effect of DHPG on action potential-evoked calcium signalling using calcium imaging in hippocampal interneurons, and suggested PKC-dependent long-term facilitation of calcium influx through L-VDCCs, which is incompatible with the PKC-independent transient facilitation in our study. Second, Chavis et al. (1996) studied the effect of t-ACPD on L-VDCC currents in cerebellar granule cells. In contrast to our results, they observed the facilitation of barium currents through L-VDCCs even in the absence of extracellular calcium, which was mediated by increase in single-channel open probability of L-VDCCs. These discrepancies might be attributable to the difference in cell types, but the greatest difference between these previous studies and ours is the concentration of mGluR agonists (100 μm DHPG in Topolnik et al., 400 μm t-ACPD in Chavis et al. and 5 μm DHPG in our study). In our preliminary experiments, calcium spikes were frequently observed in recorded neurons even in the presence of 10 μm DHPG, precluding precise measurement of calcium currents. Furthermore, considering the strong synaptic stimulation required to obtain a similar facilitation ratio to that at 5 μm DHPG (Fig. 1D: 120–150% facilitation), the use of 20-fold higher concentrations of DHPG might have over-activated mGluRs than in physiological conditions and/or produced side effects such as activation of other receptors than mGluRs. Therefore, we believe that the modulation of calcium currents by physiological activation of mGluR5 is mediated by CICR, but not by the modification of the L-VDCC itself.
Functional coupling between mGluR5, InsP3Rs and L-VDCCs
The most straightforward explanation for the InsP3R-activation mechanism is the synergistic action of InsP3 and calcium (Bezprozvanny et al. 1991; Finch et al. 1991). It has been reported that either InsP3 or calcium alone activates InsP3Rs only partially, and full activation of InsP3Rs requires both of them. The activation of mGluR5 is known to result in InsP3 production through the Gq/11-PLCβ pathway. Furthermore, the degradation of elevated InsP3 after mGluR activation is supposed to occur within 1 s (Nakamura et al. 1999), which is consistent with the short duration of the facilitation (shorter than 10 s) observed in our study (Fig. 1D). These reports support the idea that calcium and InsP3 derived from L-VDCC and mGluR5 activation, respectively, work synergistically on InsP3Rs to trigger large CICR. The disappearance of the calcium-current facilitation in the presence of the L-VDCC blocker (Fig. 5C) suggests a specific coupling between mGluR5, InsP3Rs and L-VDCCs, and the coimmunoprecipitation of mGluR5 with L-VDCC and Homer directly confirmed physical interaction between these proteins (Fig. 5D). In neurons, Homer/Vesl-family scaffold proteins form a large protein complex by bridging many proteins, including mGluR5, InsP3Rs and TRPC channels (Brakeman et al. 1997; Kato et al. 1998; Tu et al. 1998; Yuan et al. 2003). Our results suggest the existence of an even larger protein complex containing L-VDCCs as a member. We propose that this protein complex enables fine control of calcium dynamics through the close coupling between L-VDCCs, mGluR5, CICR machinery and CICR-coupled ion channels (Fig. 7).
Figure 7. The model for the mGluR5–InsP3R–L-VDCC complex.
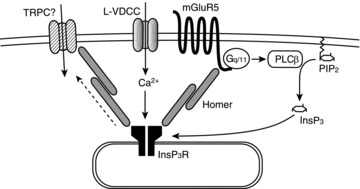
mGluR5, InsP3Rs and L-VDCCs constitute a large protein complex, which is tethered to the scaffold protein Homer. This protein complex enables the efficient functional coupling between each component. Activation of mGluR5 generates the second messenger InsP3, which acts on InsP3Rs synergistically with calcium entering through L-VDCCs, leading to the amplification of calcium signalling.
Another novel finding in our study is the requirement of coincident activation of mGluR5 and L-VDCCs not only for the induction of CICR but also for the generation of the large inward current, and the latter is, to our knowledge, the first demonstration in the literature of this research field. There is a report on the group I mGluR-dependent activation of presumptive TRPC channels (Gee et al. 2003). In contrast to our results, this report demonstrates a tonic inward current through TRPC channels during DHPG application without evoked depolarization of the recorded cells, even though the inward current is also dependent on Ca2+ influx through VDCCs. There are also differences in the results at the molecular level between the report and our study. In the previous study, mGluR1 is also associated with the inward current in addition to mGluR5, and TRPC channels are activated even in the presence of GDPβ-S. These inconsistencies might be derived from the differences in the intracellular Ca2+ concentrations and/or molecular compositions between cultured slices in the previous study and acute slices in ours. In more physiological conditions, where the basal intracellular Ca2+ concentration is kept lower, we believe that coincident activation of mGluR5 and L-VDCCs would be an important factor for the generation of the inward current. It is well known that the coincidence detection of presynaptic and postsynaptic activities realized by NMDARs is a critical molecular mechanism for learning and memory (Lisman, 1989). Cooperative generation of the large inward current by simultaneous activation of mGluRs and L-VDCCs might also work as an additional mechanism for the detection of coincidence between presynaptic glutamate release and postsynaptic depolarization. In future studies, it would be interesting to test whether this inward current is confined around the presynaptically active synapses using Ca2+ imaging.
Facilitation of VDCC-dependent LTP by mGluR5 activation
Compared with the well-known role of group I mGluRs in long-term synaptic depression (Oliet et al. 1997; Watabe et al. 2002; Lüscher & Huber, 2010), it has been controversial whether they also contribute to LTP induction (Kullmann et al. 1992; Bashir et al. 1993; Manzoni et al. 1994; Lu et al. 1997; Francesconi et al. 2004). This controversy has been attributed to either the poor specificity of mGluR antagonists or the differences in LTP-inducing protocols, but the exact causes have been still unclear. Here, we propose a previously unknown mechanism by which group I mGluRs can enhance LTP: upregulation of VDCC–CICR coupling. Using subtype-specific mGluR KO mice, we have shown that mGluR5 activation facilitates VDCC-dependent LTP. Because this form of LTP is independent of NMDARs (Kato et al. 2009), it is unlikely that the metaplastic modulation through NMDAR upregulation (Aniksztejn et al. 1992) is associated with this LTP facilitation. Rather, it is reasonable to assume that mGluR5 increases calcium currents, resulting in the LTP facilitation, although we cannot exclude other mechanisms including additional change in intracellular signalling pathways (Miura et al. 2002; Bortolotto et al. 2005). Considering the involvement of L-VDCCs in both synaptic plasticity (Grover & Teyler, 1990; Aniksztejn & Ben-Ari, 1991; Kato et al. 2009) and spatial memory (Borroni et al. 2000; Moosmang et al. 2005), mGluR5 may contribute to memory formation through this novel mechanism.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (to A.M.W., A.A. and T.M.), Comprehensive Center of Education and Research for Chemical Biology of the Diseases (Global COE Program) and Strategic Research Program for Brain Sciences (to T.M.) from Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by the grants from The Ichiro Kanehara Foundation (to A.M.W.) and from the Uehara Memorial Foundation (to T.M.).
Glossary
- AMPAR
AMPA receptor
- 2-APB
2-aminoethoxydiphenyl borate
- CICR
calcium-induced calcium release
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CPA
cyclopiazonic acid
- d-APV
d-(–)-2-amino-5-phosphonovaleric acid
- DHPG
dihydroxyphenylglycine
- InsP3R
InsP3 receptor
- KO
knockout
- LTP
long-term potentiation
- L-VDCC
L-type VDCC
- mGluR
metabotropic glutamate receptor
- NMDAR
NMDA receptor
- PKA
protein kinase A
- PKC
protein kinase C
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- t-ACPD
(±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid
- TRPC
transient receptor potential canonical
- VDCC
voltage-dependent Ca2+ channel
- WT
wild-type
Author contributions
The experiments were designed by H.K.K., A.M.W. and T.M. and performed in the laboratory of T.M., except for the coimmunoprecipitation experiment, which was performed in the laboratory of A.A. The data were collected and analysed by H.K.K and H.K. Mutant mice were maintained and provided by A.A. The manuscript was written by H.K.K. and T.M., with the assistance of A.M.W., H.K. and A.A. All authors have read and approved the final version.
Author's present address
A. M. Watabe: Jikei University School of Medicine, Tokyo, 105-8461, Japan.
Supplementary material
Supplementary Figure S1
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Otani S, Ben-Ari Y. Quisqualate metabotropic receptors modulate NMDA currents and facilitate induction of long-term potentiation through protein kinase C. Eur J Neurosci. 1992;4:500–505. doi: 10.1111/j.1460-9568.1992.tb00900.x. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci. 2000;20:9272–9276. doi: 10.1523/JNEUROSCI.20-24-09272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolotto ZA, Collett VJ, Conquet F, Jia Z, van der Putten H, Collingridge GL. The regulation of hippocampal LTP by the molecular switch, a form of metaplasticity, requires mGlu5 receptors. Neuropharmacology. 2005;49(S1):13–25. doi: 10.1016/j.neuropharm.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, Worley PF. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–288. doi: 10.1038/386284a0. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Church PJ, Stanley EF. Single L-type calcium channel conductance with physiological levels of calcium in chick ciliary ganglion neurons. J Physiol. 1996;496:59–68. doi: 10.1113/jphysiol.1996.sp021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Flockerzi V, Oeken HJ, Hofmann F, Pelzer D, Cavalié A, Trautwein W. Purified dihydropyridine-binding site from skeletal muscle t-tubules is a functional calcium channel. Nature. 1986;323:66–68. doi: 10.1038/323066a0. [DOI] [PubMed] [Google Scholar]
- Francesconi W, Cammalleri M, Sanna PP. The metabotropic glutamate receptor 5 is necessary for late-phase long-term potentiation in the hippocampal CA1 region. Brain Res. 2004;1022:12–18. doi: 10.1016/j.brainres.2004.06.060. [DOI] [PubMed] [Google Scholar]
- Freichel M, Vennekens R, Olausson J, Stolz S, Philipp SE, Weissgerber P, Flockerzi V. Functional role of TRPC proteins in native systems: implications from knockout and knock-down studies. J Physiol. 2005;567:59–66. doi: 10.1113/jphysiol.2005.092999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner DM, Grey RD. Membrane junctions in Xenopus eggs: their distribution suggests a role in calcium regulation. J Cell Biol. 1983;96:1159–1163. doi: 10.1083/jcb.96.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee CE, Benquet F, Gerber U. Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol. 2003;546:655–664. doi: 10.1113/jphysiol.2002.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Johnston D. Noradrenaline andβ-adrenoceptor agonists increase activity of voltage-dependent calcium channels in hippocampal neurons. Nature. 1987;327:620–622. doi: 10.1038/327620a0. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Xie M, Han J, Hümmer A, Melnik-Martinez KVR, Moreno RL, Mark MD. Targeting mechanisms of high voltage-activated Ca2+ channels. J Bioenerg Biomembr. 2003;35:621–637. doi: 10.1023/b:jobb.0000008027.19384.c0. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Kato A, Ozawa F, Saitoh Y, Fukazawa Y, Sugiyama H, Inokuchi K. Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J Biol Chem. 1998;273:23969–23975. doi: 10.1074/jbc.273.37.23969. [DOI] [PubMed] [Google Scholar]
- Kato HK, Watabe AM, Manabe T. Non-Hebbian synaptic plasticity induced by repetitive postsynaptic action potentials. J Neurosci. 2009;29:11153–11160. doi: 10.1523/JNEUROSCI.5881-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Mignery GA, Zhu MX, Muallem S. The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol Cell. 1999;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron. 1992;9:1175–1183. doi: 10.1016/0896-6273(92)90075-o. [DOI] [PubMed] [Google Scholar]
- Lahouratate P, Guibert J, Camelin JC, Bertrand I. Specific inhibition of cardiac and skeletal muscle sarcoplasmic reticulum Ca2+ pumps by H-89. Biochem Pharmacol. 1997;54:991–998. doi: 10.1016/s0006-2952(97)00320-1. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb process underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Weisskopf MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Lux HD. Glutamate selectively increases the high-threshold Ca2+ channel current in sensory and hippocampal neurons. Brain Res. 1992;580:341–344. doi: 10.1016/0006-8993(92)90965-c. [DOI] [PubMed] [Google Scholar]
- Miura M, Watanabe M, Offermanns S, Simon MI, Kano M. Group I metabotropic glutamate receptorsignaling via Gαq/Gα11 secures the induction of long-term potentiation in the hippocampal area CA1. J Neurosci. 2002;22:8379–8390. doi: 10.1523/JNEUROSCI.22-19-08379.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- Nagasaki K, Kasai M. Channel selectivity and gating specificity of calcium-induced calcium release channel in isolated sarcoplasmic reticulum. J Biochem. 1984;96:1769–1775. doi: 10.1093/oxfordjournals.jbchem.a135009. [DOI] [PubMed] [Google Scholar]
- Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Nakajo K, Chen L, Okumura Y. Cross-coupling between voltage-dependent Ca2+ channels and ryanodine receptors in developing ascidian muscle blastomeres. J Physiol. 1999;515:695–710. doi: 10.1111/j.1469-7793.1999.695ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara J-G, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara J-G, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Riedel G. Function of metabotropic glutamate receptors in learning and memory. Trends Neurosci. 1996;19:219–224. doi: 10.1016/0166-2236(96)20012-8. [DOI] [PubMed] [Google Scholar]
- Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer RJ, Schwindt PC, Crill WE. Metabotropic glutamate receptor-mediated suppression of L-type calcium current in acutely isolated neocortical neurons. J Neurophysiol. 1992;68:833–842. doi: 10.1152/jn.1992.68.3.833. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Challiss RAJ, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends Neurosci. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Chamberland S, Pelletier J-G, Ran I, Lacaille J-C. Activity-dependent compartmentalized regulation of dendritic Ca2+signaling in hippocampal interneurons. J Neurosci. 2009;29:4658–4663. doi: 10.1523/JNEUROSCI.0493-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O’Dell TJ. Post synaptic induction and presynaptic expression of group 1 mGluR-dependent LTD in the hippocampal CA1 region. J Neurophysiol. 2002;87:1395–1403. doi: 10.1152/jn.00723.2001. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Xu S-Z, Zeng F, Boulay G, Grimm C, Harteneck C, Beech DJ. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol. 2005;145:405–414. doi: 10.1038/sj.bjp.0706197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.