

Abstract
Esophageal atresia (EA) with or without tracheoesophageal fistula (TEF) is the most common congenital anomaly of the esophagus. The improvement of survival observed over the previous two decades is multifactorial and largely attributable to advances in neonatal intensive care, neonatal anesthesia, ventilatory and nutritional support, antibiotics, early surgical intervention, surgical materials and techniques. Indeed, mortality is currently limited to those cases with coexisting severe life-threatening anomalies. The diagnosis of EA is most commonly made during the first 24 h of life but may occur either antenatally or may be delayed. The primary surgical correction for EA and TEF is the best option in the absence of severe malformations. There is no ideal replacement for the esophagus and the optimal surgical treatment for patients with long-gap EA is still controversial. The primary complications during the postoperative period are leak and stenosis of the anastomosis, gastro-esophageal reflux, esophageal dysmotility, fistula recurrence, respiratory disorders and deformities of the thoracic wall. Data regarding long-term outcomes and follow-ups are limited for patients following EA/TEF repair. The determination of the risk factors for the complicated evolution following EA/TEF repair may positively impact long-term prognoses. Much remains to be studied regarding this condition. This manuscript provides a literature review of the current knowledge regarding EA.
Keywords: Esophageal atresia, Tracheoesophageal fistula, Esophageal stenosis, Long-gap, Gastro-esophageal reflux
INTRODUCTION
Thomas Gibson first described esophageal atresia (EA) associated with tracheoesophageal fistula (TEF) in 1697. However, it was not until 1941 that Cameron Haight performed the first successful surgical repair of this anomaly following innumerable attempts by other surgeons[1-3]. EA with or without TEF remains the most common congenital anomaly of the esophagus. Although EA with or without TEF is a relative rare condition, this complex anomaly is still a challenging problem in pediatric surgery[1,2]. In developing countries, many infants that present with EA with TEF exhibit pneumonitis due to late referral, and these patients usually have a low birth weight.
The overall incidence of EA/TEF ranges from one in every 2500 to 4500 live births[1-9]. The vast majority of cases are sporadic, although the incidence is higher in twins. It has been reported that the relative risk for EA/TEF in twins was 2.56 higher than in singletons[6-8].
An improvement in survival has been observed over the most recent decades. This finding is likely multifactorial and largely attributable to advances in neonatal intensive care, neonatal anesthesia, ventilatory and nutritional support, antibiotics, surgical materials and techniques[3,5,9,10]. Indeed, mortality is currently limited to those cases with coexistent severe life-threatening anomalies. It should be noted that the care and treatment of EA is a measure of the surgical expertise and facilities available in a particular center or country. Despite an increased number of patients with severe associated anomalies, survival rates as high as 95% have been reported in centers offering the best neonatal care[11]. Today, practically all patients without concomitant severe malformation survive. Moreover, the high mortality of very low birth weight patients, patients with severe cardiac malformation and of infants with long-gap defects has significantly decreased. Because of this increase in survival, morbidity associated with EA/TEF repair has become an important issue during the follow-up of these children.
There are a limited number of reports concerning the long-term outcome of patients with EA. Relationships between esophageal dysmotility, gastroesophageal reflux, esophagitis and epithelial metaplastic changes, including esophageal cancer, should be studied further[3,5,8,9]. The present manuscript provides gastroenterologists with a literature review focused on EA with the aim of providing recent data regarding the embryology, diagnosis, therapeutic approaches, complications and outcomes of this very common congenital anomaly.
CLASSIFICATION
The classification of EA anomalies is determined by the location of the atresia and the presence of any associated fistula to the trachea. In this respect, five different variants have been clinically described. The first classification was published by Vogt in 1929 and was modified by Gross in 1953. Thus, two classifications are used today. The primary types of congenital EA are EA with distal TEF (85%, Vogt IIIb, Gross C), isolated EA without TEF (8%, Vogt II, Gross A), TEF without atresia or H-type TEF (4%, Gross E), EA with proximal TEF (3%, Vogt III, Gross B) and EA with proximal and distal TEF (< 1%, Vogt IIIa, Gross). Figure 1 illustrates the EA classification scheme. An understanding of these anatomical variants is important to aid in medical and surgical management[1,4,5,12,13].
Figure 1.
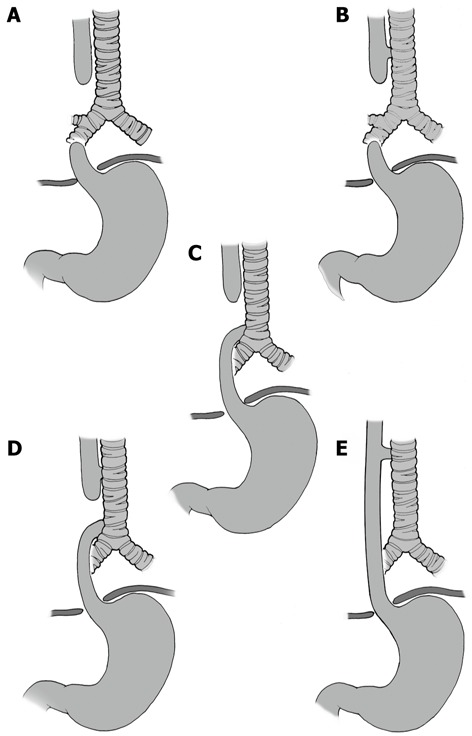
Classification of esophageal atresia/tracheoesophageal fistula. A: Esophageal atresia (EA) without tracheoesophageal fistula (TEF); B: Proximal TEF with distal EA; C: Distal TEF with proximal EA; D: Proximal and distal TEF; E: TEF without EA or “H”-type TEF.
EMBRYOLOGY
Successful treatment of esophageal anomalies requires knowledge of their embryological origin. Although significant improvements in clinical treatment have been made in recent years, our understanding of the etiology of these defects is still incomplete[14-17].
The primitive digestive tube (PDT) emerges from the primitive endoderm and subsequently gives rise to the esophagus and trachea. There are three primary theories that attempt to explain this phenomenon[6,14-19]. The first theory postulates that the evagination of a tracheal diverticulum begins with the PDT, which grows rapidly in the caudal direction, resulting in the separation of the trachea and esophagus (what remains of the PDT). In the context of this developmental mechanism, tracheoesophageal malformations result from tracheal growth failure[14]. Another theory suggests that the formation of a mesenchymal septum in the coronal plane of the PDT, separating the trachea ventrally and the esophagus dorsally from the distal to the proximal ends of PDT. A failure in this process would then result in a tracheoesophageal malformation[14]. In these two theories, the origin of EA is a cellular rearrangement of the remaining, distal PDT[14].
The third theory combines elements of the first two and suggests that rapid growth of the tracheal diverticulum occurs in concert with a mesenchymal septation of the PDT, separating the trachea from the esophagus. Unlike the previous theories, however, in this proposed mechanism, EA is believed to result from the loss of a portion of the previously formed tube due to regression toward the main part of the embryo[14].
Although syndromic cases of EA/TEF are rare, examining the specific genetic anomalies involved may provide valuable information regarding the abnormal developmental processes leading to EA/TEF. Many genes and genetic pathways have been implicated in the development of EA/TEF, but few have been shown to be involved in humans, animals, or both[15].
There are also molecular and morphogenetic factors related to EA, such as apoptosis, the Sox2, Shh, Gli-2, Gli-3, Pcsk5 and FOX genes and the transcription factors Nkx2.1 and Tbx4[4,5,15-19]. A failure in the expression of these genes or in the apoptotic programs that they regulate is responsible for EA. However, a whole understanding of these processes remains incomplete[4,15]. In addition, environmental factors have been suggested to increase the risk for the development of tracheoesophageal anomalies[15]. Further studies are required for a universally accepted explanation for the pathophysiology of EA/TEF.
DIAGNOSIS
The diagnosis of EA is most commonly made during the first 24 h of life but may be made either antenatally or may be delayed[1,5,7-9,20].
Ultrasound (US) scanning is currently a routine method used in prenatal care between the 16th and 20th week of gestation. The suspicion of EA is based on the presence of polyhydramnios and the absence of the gastric bubble, but these are non-specific criteria[7,21]. US features highly suggestive of EA/TEF are only observed in a small minority of fetuses with EA/TEF (< 10%) on prenatal scans. The combination of polyhydramnios and the absence of a stomach bubble in two previous reports of small patient series gave modest positive predictive values of 44% and 56%, respectively[7,21].
Dilatation of the blind fundus of the upper segment of the atresic esophagus, the “upper pouch sign”, may also be observed during fetal deglutition at approximately the 32nd gestational week[7,21]. Moreover, an upper pouch sign may not be detected even with specific examination[7].
The diagnostic criterion of EA through the use of magnetic resonance imaging is the non-visualization of the intra-thoracic portion of the esophagus. This imaging modality is complementary to ultrasound due to a high percentage of false positives when images are analyzed in an isolated fashion[21,22]. Even with advances in technological imaging, no ideal prenatal diagnostic method for EA exists.
In the delivery room, the primary sign of EA is the impossibility of the passage of an orogastric catheter beyond 11 or 12 centimeters[23]. In the nursery, the most important clinical signs are abundant salivation, episodes of cyanosis and suffocation during breastfeeding[1,4-7]. The confirmation of the diagnosis of EA should be made with a simple chest X-ray using air as contrast in the proximal pouch to avoid aspiration of contrast fluid. If a distal TEF is present, air in the stomach will be present on X-ray films and abdominal distension may be evident[24]. Figure 2 shows the plain X-rays of two neonates with different forms of EA.
Figure 2.
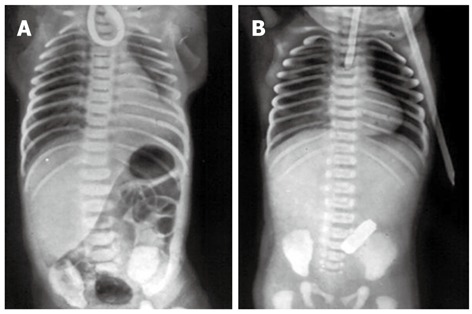
Plain X-rays of the chest and abdomen of two neonates with esophageal atresia. A: The non-progression of an orogastric catheter in the blind esophageal pouch and the presence of air in the stomach diagnose esophageal atresia with distal tracheoesophageal fistula; B: The radiopaque tube in the blind esophageal pouch and the absence of air in the stomach identify esophageal atresia without tracheoesophageal fistula.
Tracheobronchoscopy has been proposed as an imaging method to detect EA during the preoperative period[3,22,25]. This technique is used to determine the anatomy of the TEF with respect to the carina, to identify other airway anomalies and to occlude the TEF with a balloon, facilitating mechanical ventilation and avoiding both gastric distension and gastroesophageal reflux. In cases of presumed isolated EA, bronchoscopy also helps to rule out the presence of the less common proximal TEF[5,22].
PREOPERATIVE PERIOD
Once the diagnosis of EA has been established, the infant needs to be transferred to a regional pediatric surgical center with intensive care support facilities. The aim of the preoperative treatment of EA is to improve the general state of the newborn so that definitive surgery can be carried out under the best possible conditions[1,4,5].
Classification and risk factors
There are three primary classifications of preoperative risks regarding EA: the Waterston, Montreal and Spitz classifications[1,3,10,11,26,27].
According to Waterston, the risk factors to be considered are birth weight (BW), the presence or absence of pneumonia and complications from associated congenital anomalies. In this classification scheme, patients are categorized into group A (BW > 2500 g, with no other complications), group B (BW between 1800 g and 2500 g with no other complications or BW > 2500 g with moderate pneumonia/congenital anomaly) or group C (BW < 1800 g, with no other complications or BW > 2500 g with severe pneumonia/severe congenital anomaly)[1,3,11,27].
In the Montreal classification scheme for EA, factors such as dependence on mechanical ventilation (MV) and associated congenital anomalies are considered to be of high prognostic significance[1,11]. Patients are classified into group I (isolated major anomaly, isolated dependence on MV or the presence of non-significant anomalies) and group II (presence of severe congenital anomalies or dependence on MV associated with one major anomaly).
Spitz drafted the most recent classification method by associating BW and cardiac anomalies (CA) as risk factors for EA. In this classification scheme, patients are divided into group I (BW > 1500 g, without CA), group II (BW < 1500 g or the presence of CA) and group III (BW < 1500 g with CA)[11,21,26,27].
These classification systems serve as guides for the determination of the type of treatment for each case of EA. Some authors no longer consider BW to be a risk factor[26].
Associated congenital anomalies
What is perhaps of major clinical importance is the high frequency of anomalies associated with EA, with a frequency of over 50%, which may greatly impact both treatment and outcome. In addition to the high frequency of anomalies, their unequal distribution between patients is also important from a clinical perspective. Patients with isolated EA without TEF exhibit anomalies in as many as 65% of cases, while a much lower frequency is observed in patients with TEF without atresia (10%)[1,7,28].
The most common associated malformation occurs in the cardiovascular system (23% of cases), followed by musculoskeletal malformations (18%), anorectal and intestinal malformations (16%), genital-urinary malformations (15%), anomalies of the head and neck (10%), mediastinal anomalies (8%) and chromosomal anomalies (5.5%)[5]. Of the observed cardiac anomalies, the most common are ventricular septal defects and tetralogy of Fallot.
A concurrence of congenital anomalies unassociated with a genetic disturbance is referred to as VACTERL [vertebral, anal, cardiac, tracheal, esophageal, renal, and limb (pattern of congenital)] association, which is diagnosed if the patient with EA has 2 or more anomalies of the vertebral, anorectal, cardiac (excluding patent ductus arteriosus and patent foramen ovale), renal/genitourinary, or limb systems. Cardiac anomalies are the most common ones. This broad spectrum of anomalies suggests an alteration during the early stages of embryogenesis related to a deficiency in the regulation of the Shh gene[29]. The CHARGE (coloboma, heart, atresia choanal, retarded growth, genital hypoplasia, ear deformities) association may also include EA[7].
Life-threatening anomalies, including Potter’s Syndrome (bilateral renal agenesis, pulmonary hypoplasia, typical dysmorphic facies), cerebral hypoplasia and chromosomal anomalies, such as trisomy of chromosomes 13, 14 and 18, may be present. These severe conditions directly predict adverse outcomes[5]. Similarly, infants with totally uncorrectable major cardiac defects or with grade IV intraventricular hemorrhage should be considered for non-operative management[1].
Nasr et al[30] demonstrated in 2010 that normal clinical and radiologic examination predicts the absence of significant cardiac abnormalities on echocardiography in 100% of cases. Therefore, these authors conclude that routine pre-surgical echocardiography may not always be necessary, but should be reserved for infants with abnormal clinical and/or radiologic findings.
Vascular components are often overlooked in the investigation of anomalies associated with EA[31]. The presence of the right aortic arch in association with EA is most often discovered during the surgical intervention for EA correction. According to Babu et al[32], this condition occurs in 2.5% to 5% of cases of EA, with a greater frequency in males. The method of choice for detecting this anomaly is echocardiography, although this technique is not routinely employed during the investigation of EA. Following the discovery of this anomaly, a left thoracotomy should be performed to the correct the EA[32].
The persistence of the left superior vena cava (LSVC), which results from the persistence of the left inferior cardinal vein, is observed in nearly 10% of infants with EA. Depending on the trajectory of the LSVC, complications may occur, such as thrombosis of the coronary sinus and arrhythmia[31,33].
Antibiotic prophylaxis
Colonization by bacterial flora of the digestive tract in newborns with EA is related to the establishment of enteral nutrition. However, strains of Pseudomonas and Serratia have been isolated in the portion of the esophagus that is present in these infants. Antibiotic prophylaxis using amoxicillin and clavulanate is therefore indicated in such patients[1,3,34].
Neonatal care
Neonatal care includes stabilization of the infant’s respiratory status with avoidance of endotracheal intubation; suction tube drainage of the blind, proximal esophageal pouch; semi-prone positioning of the child to minimize the risk of gastroesophageal reflux; and aspiration via the occult fistula of the distal trachea[5]. Monitoring of vital signs and vascular access should also be performed as precautionary measures.
SURGICAL TREATMENT
The surgical treatment of EA is considered urgent but not an emergency, except in premature infants with respiratory distress[1,3].
Anesthetic care has focused on minimizing ventilation through the fistula, usually by placing the end of the endotracheal tube distal to the fistula, preventing gastric distension/perforation and/or ventilator compromise[1,7,25]. However, if the fistula is located at the level of the carina, distal placement of the endotracheal tube is impossible. Gastric distention can complicate the ventilation of patients with large TEFs. Gastric distension can also result in the aspiration of gastric contents or in elevation of the diaphragm, leading to decreased tidal volumes, decreased venous return, cardiovascular collapse and ultimately perforation. This results in tension pneumoperitoneum[1,7,25].
The semi-prone position is the position of choice, with the right side elevated at 45º and the right arm placed over the head. Anesthesia is maintained during the surgical procedure with a volatile anesthetic agent. The patient is ventilated with positive-pressure MV and hydrated with a crystalloid solution[34].
The primary correction of EA and TEF is the best treatment option in the absence of severe malformations[35-38]. Standard right posterolateral extrapleural thoracotomy below the tip of the scapula is extremely useful and allows the for the repair of other complex anatomic variants[2]. If a right-sided aortic arch is observed on preoperative echocardiography, a left thoracotomy must be performed and the chest is entered through the fourth intercostal space. Care should be taken to avoid entry into the pleura. Extrapleural dissection proceeds posteriorly and superiorly to identify the azygos vein. Division of the azygos vein arch allows for full exposure of the posterior mediastinum. The TEF and vagus nerve are often encountered beneath the azygos arch. This procedure begins with the closure of the fistula. The TEF is divided near the trachea and sewn with fine non-absorbable sutures, which is followed by the correction of the EA. The upper atresic esophageal pouch is identified with downward tension on an oro-esophageal tube, and its dissection is facilitated by the placement of a traction suture at the end of the pouch. Blunt and sharp dissection can mobilize the proximal pouch to the level of the thoracic inlet. Esophageal continuity is accomplished using a single-layer, end-to-end anastomosis with monofilament absorbable sutures[5]. With respect to unstable patients, however, the procedure should be performed in steps[35].
There is limited evidence to support the use of a trans-anastomotic tube. The majority of surgeons do not routinely use an intercostal catheter if the repair is extrapleural. Alabbad et al[39] observed that a trans-anastomotic feeding tube may lead to a shorter total parenteral nutrition duration and decreased cholestasis. It was also demonstrated that central venous catheters tended to be removed earlier when trans-anastomotic tubes were used, decreasing the risk of future infection. Furthermore, hospital stays tended to be shorter[39]. However, this study provides only preliminary evidence of the benefits of trans-anastomotic tubes. Larger prospective studies will be required to conclusively demonstrate these benefits and to ensure that this technique does not increase anastomotic leaks.
No ideal replacement for the esophagus is available. Nonetheless, the substitute should function as similarly as possible to the original tissue[35-37]. The reconstruction of the esophagus using only its atresic portions is preferable to the use of any other material, even in cases of long-gap EA[40-46]. However, a number of authors do not agree with this procedure[47,48].
Long-gap EA still presents a challenge to pediatric surgeons. A long gap between the two ends of the esophagus in cases of EA has been defined as a gap longer than 3 cm or greater than the height of 2 vertebral bodies[49].
The historical treatment of EA has included a gastrostomy, the estimation of the extent of the gap between the proximal and distal esophagus and proximal pouch decompression. These measures allow time for potential lengthening of the esophagus with linear patient growth. Delayed surgical intervention is accomplished at approximately three months of age with attempts at achieving a primary anastomosis. Failed attempts at primary anastomosis may then be addressed with colonic interposition, reverse or antegrade gastric tube interposition, gastric transposition, or free jejunal graft interposition. Alternatively, these approaches have been attempted during the neonatal period as a primary repair[5,50-52].
The ideal surgical treatment for patients with long-gap EA has not been determined, and the topic is still very controversial. In fact, it remains somewhat difficult to determine which surgical procedure achieves the best results. The infant’s own functional esophagus is superior to any esophageal replacement. Familiarity with different techniques used to preserve this tissue is therefore important. A number of techniques are proposed for the treatment of long-gap EA. One is the dissection and mobilization of the distal esophageal stump using circular myotomy[41]. Some authors believe that this technique can be used without vascular impairment of the esophagus due to the abundant irrigation of the esophagus, and that it is preferable to surgery under tension[41]. Others believe that this technique may result in necrosis of the distal stump and consequent leaking of the anastomosis[47].
Suboptimal results with these strategies have led to the development of techniques that attempt to elongate the esophagus sufficiently to bridge the gap. This principle was the basis of the technique described by Foker et al[43]. The so-called Foker technique consists of the use of external traction sutures to elongate the esophageal portions and to approximate one stump to the other prior, thus completing the anastomosis. As the esophagus exhibits spontaneous growth during the first three months of life, the technique is performed following this period to ensure that the esophagus has the necessary thickness to support the traction. This procedure is extremely beneficial despite the requirement for a second thoracotomy, and its success is ensured by esophageal mobilization and the secondary anastomosis[43,44,53]. A complication that may occur with this technique is the undesired cutting of the esophagus by the sutures[43]. To avoid this outcome, a modification of the Foker technique has emerged, in which small tubes of silastic are attached to the terminal portions of the two esophageal stumps and the thoracic wall, where the tension is applied[43].
Nagaya et al[54] proposed a method for assessing the tension of the anastomosis by placing a metal clip in the trachea at the site of the TEF and assessing the distance between this clip and both the site of anastomosis and the esophagogastric junction. This method is based on the notion that the stretching of the esophagus is directly related to the tension of the anastomosis and that this information can predict possible postoperative complications. Stretching of up to 5 mm is considered well tolerated[54].
Tamburri et al[55] showed that Kimura’s technique[56] is a useful surgical option for a select group of patients with (1) a complex long-gap EA that requires a primary esophagostomy; or (2) any type of EA with the development of severe complications following a primary repair and that required a secondary esophagostomy. The operative procedure consists of an initial cutaneous esophagostomy, multistaged extrathoracic esophageal elongation (ETEE), and definitive esophageal anastomosis. The ETEE includes the mobilization and dissection of the esophagus up to the cricoid cartilage level, with the opening placed on the previously made superior clavicle surgical scar. Variable elongation of the esophagus is achieved following completion of the dissection. The esophagus is again exteriorized as an esophagostomy at a level of a few centimeters below the previous esophagostomy site[55,56].
More recently, Stringel et al[57] reported another technique used to repair long gap EAs without anastomosis. This technique consisted of suture approximation and subsequent endoscopic and fluoroscopic placement of string for guided dilatations[57].
In addition to techniques that employ the patient’s own esophagus, there are those that use pedicle grafts from the jejunum or colon, which are indicated when the motility of the esophagus does not allow adequate oral feeding and gastric transposition[3,36,47,48]. The creation of a jejunal graft involves a cross-sectional cut proximal to the ligament of Treitz, separating the first two mesenteric arteries from the peripheral arch. A second cut is made at the level of the third mesenteric artery. The distal portion of the proximal jejunum is removed and transferred to the thorax, leaving on the proximal portion of the jejunum. An anastomosis is then made between the esophageal stumps and the graft. The justification for this technique is that the peristalsis remains in the jejunal graft, but it does not have a tendency to elongate[48]. With respect to the colon pedicle technique for esophago colic anastomosis, the graft is chosen from the portion of the colon irrigated by the left colic artery and transferred to the thorax posterior to the mediastinum. The colon also needs to be attached to the muscles of the neck. This technique should only be employed after four months of life to ensure adequate vascularization of the colon[36,58]. The advantage of using the colon is the presence of long marginal arches, which allows for ample mobilization of extensions to the cervical region. Another advantage is the graft’s extreme resistance to gastric fluid due to the continuous production of mucus. The most feared complication is necrosis of the transposed loop[36,58].
A number of authors prefer gastric transposition[1,3,37]. This technique has the advantages of being simple, of using the vascularization of the stomach and of easily raising the stomach to the neck for the anastomosis. The primary disadvantages are respiratory distress and late-onset gastric leakage[37].
For thoracoscopic repair of EA, the patient is placed in a ¾ prone position and the trocars are inserted at the angle of the scapula in the right axillary region and between the vertebra and the angle of the scapula at the 7th to 8th right intercostal space. The azygos vein and TEF are linked, maintaining the separation between the portions of the esophagus until the mobilization of the proximal stump to avoid retraction of the distal stump toward the diaphragm[34,59]. A preferentially termino-terminal anastomosis is performed between the esophageal portions, beginning with the posterior wall of the esophagus[34]. Thoracoscopy is not indicated for the correction of long-gap EA, as this defect requires more invasive dissection and there may be difficulty with the anastomosis of the proximal and distal portions of the esophagus, leading to an increased surgical time, which does not justify the minimally invasive repair[34].
Cases of “H-type” TEF without EA (Type E) are unique in their presentation and repair[5]. Infants with this anomaly often present with a long-standing history of recurring aspiration and multiple episodes of pneumonia. The diagnosis is often made following a contrast esophagram or an upper gastrointestinal series by observing the fistula at the lower cervical esophagus or by bronchoscopy. The operative approach for these cases begins with a rigid bronchoscopy and attempts to cannulate the fistula with a small feeding tube or Fogarty catheter. This approach will often aid fistula identification at the time of the operation. The fistula is approached through a right lateral, oblique cervical incision with the neck extended. Dissection begins anterior to the sternocleidomastoid muscle with lateral reflection of the carotid sheath. Exposure of the esophagus continues into the upper mediastinum with identification of the fistula. Primary repair of the tracheal and esophageal portions of the fistula are performed with interrupted sutures. Separation of the adjacent suture lines is aided by interposition of mobilized strap muscles[5].
Some infants require respiratory support, especially those who are premature; those with associated cardiac anomalies; those in whom the diagnosis was initially missed; and in those with a splinted diaphragm secondary to gastric distension[1,7]. In infants requiring respiratory support, great care should be taken during intubation and in supervising ventilation. This challenging and potentially rapidly fatal situation appears to be best managed by an emergency ligation of the distal fistula with a transpleural approach. In the majority of cases, ligation of the fistula improves respiratory status. The thoracotomy is closed pending resolution of respiratory distress over the subsequent few days. Endoscopic placement of a Fogarty balloon catheter, while conceptually an elegant temporizing maneuver, requires a high degree of expertise and has not been proven to be a successful approach in most centers[1,7].
POST-OPERATIVE PERIOD
In most centers, the infant returns to the neonatal intensive care ventilated and with the neck flexed to reduce anastomotic tension. When the esophageal anastomosis has been performed under tension, the infant is electively paralyzed and mechanically ventilated for 5-7 d postoperatively. If the surgeon has inserted an transanastomotic feeding tube, feeding through the tube should progress slowly, usually beginning 48 h following the surgery[39]. When the infant can swallow saliva, oral feeding may be started. A routine contrast study appears unnecessary in many cases, but if there is any doubt regarding the integrity of the anastomosis, a water-soluble contrast study should be performed[1,7].
The primary complications during the postoperative period are leak and stenosis of the anastomosis, gastro-esophageal reflux (GER), esophageal dysmotility, fistula recurrence, scoliosis, deformities of the thoracic wall and respiratory disorders[1-5,7,37,47,60-65].
The outcome following EA/TEF repair is variable. Some patients have an uneventful postoperative period, while others experience several esophageal or respiratory complications that can significantly affect their health through adulthood and can predict the patient’s ability to develop adaptive behaviors[7,49,62-65].
Anastomotic leaks
Anastomotic leaks are considered minor or major. These leaks occur in 15%-20% of patients but are a significant disruption to recovery in less than a third of these cases[1-5,7,37,38]. Leaks result from the small, friable lower segment, ischemia of the esophageal ends, excess anastomotic tension, sepsis, poor suturing techniques, the type of suture, excessive mobilization of the distal pouch and increased gap length[66].
Minor leaks are spontaneously reabsorbed by the body, with the vast majority healing within a few days. These leaks are nonetheless associated with a greater incidence of subsequent stricture development. Alternatively, major leaks may cause tension pneumothorax and require the placement of a drain or an early thoracotomy. The thoracotomy is carried out with the intention of repairing the anastomosis. However, if there has been a complete disruption that precludes any attempt at re-anastomosis, the repair includes a cervical esophagostomy, the closure of the distal esophagus and a subsequent esophageal replacement. Antibiotics and continuous suctioning of the upper pouch may be instituted to reduce saliva egress from the esophagus. The institution of either parenteral nutrition or trans-anastomotic tube feeding is also recommended. A contrast study prior to oral feeding should be performed at the discretion of the surgeon[1-5,7,37].
A close association exists between anastomotic leakage and the tension of the anastomosis on the suture line[66]. Uchida et al[67] demonstrated the efficacy of postoperative elective ventilatory support for leakage protection in primary anastomoses of EAs.
Esophageal strictures
Anastomotic strictures are the most common cause of recurrent surgery in children with EA/TEF, and the incidence varies between 30%-40% of cases. The majority of these cases respond to one or two dilatations[1,2,66,68]. Risk factors that have been implicated in stricture formation include anastomotic tension, anastomotic leakage and GER. With meticulous handling of the esophageal ends, the preservation of the blood supply and careful inclusion of mucosa in each suture of the anastomosis, strictures can be kept to a minimum[1]. The occurrence of stenosis of the anastomosis is more common in patients with long-gap EA, which is thought to be due to the fact that the repair is under tension[7,48,59].
The definition of a stricture, however, is not universally accepted as a mild radiological narrowing on a contrast esophagram. Occasionally, this finding may not have any clinical relevance to the physician or to the patient, who may be able to swallow satisfactorily[2].
Treatment may require successive endoscopic dilatations under general anesthesia[1,40]. Most strictures respond to dilatation, but it is crucial that reflux is aggressively treated to diminish the impact of acid reflux on recurrent stricture formation[2]. There is disagreement as to whether dilatation should be performed in a prophylactic or therapeutic fashion. According to Koivusalo et al[69], therapeutic dilatation has the advantages of requiring fewer procedures, shorter hospitalization times and lower costs when compared to prophylactic dilatation.
GER
GER is extremely common among infants following EA repair and may affect between 40% and 65% of patients[2]. The presence of significant GER is generally believed to be due in part to an intrinsic deficiency in the motor function of the esophagus itself. However, it is likely that GER is exacerbated by the surgical repair and gastrostomy, causing an alteration of the anatomical gastro-esophageal junction and the angle of His[2]. GER is more common following tension anastomosis[1,37,70,71]. The symptoms include acute or chronic respiratory problems, regurgitation, recurring vomiting and growth failure[4]. The diagnosis is made primarily through esophagoscopy, a 24 h pH probe, intraluminal impedance or a contrast swallow[2,72].
The treatment of GER can be either clinical or surgical, the latter constituting approximately 28% of cases. The clinical treatment of GER includes dietary modification, adequate positioning of the infant and medication[5]. The most frequently recommended drug is omeprazole, the effective dose of which is 1.9 mg/kg to 2.5 mg/kg per day, until resolution of the GER and stenosis[54,73-75].
Surgical treatment consists of Nissen fundoplication, in which the closure of the esophageal hiatus occurs with the approximation of the diaphragm pillars. This is followed by the construction of anti-reflux valves using the gastric fundus to completely envelop the abdominal esophagus[1,5]. Unfortunately, however, a significant number of infants (> 40%) develop recurrent GER, which may be due in part to the inherent dysmotility of the esophagus[2].
Long-standing GER is associated with considerable morbidity, resulting in chronic esophageal inflammation and contributing to recurrent pulmonary infections and abnormalities of pulmonary function in a significant number of patients. Moreover, GER is associated with the development of columnar metaplasia that may undergo dysplastic changes, giving rise to esophageal adenocarcinoma through a metaplasia-dysplasia-carcinoma sequence[37,76-82].
It has been shown that symptoms of GER and histologic findings are poorly correlated. Therefore, long-term endoscopic and pH-metric follow-up of all patients following repair of an EA is warranted. Endoscopic follow-up is recommended for all patients following EA repair irrespective of symptoms. The endoscopic follow-up of children with completely normal esophageal biopsies can be discontinued at the age of 3 years. In patients with mild esophagitis, routine follow-up should be extended to at least 6 years of age[83-85]. Patients who have undergone anti-reflux surgery should also be followed long-term[85].
Esophageal dysmotility
Esophageal dysmotility is a very common long-term finding in children with EA/TEF and has been demonstrated in 75% to 100% of patients post-EA/TEF repair. Likewise, in patients who have had some form of esophageal replacement, dysmotility with symptoms such as aspiration, dysphagia, or food bolus obstruction is often reported[2]. Esophageal dysmotility can be caused by abnormal neural development of the esophagus or may result from complications of EA repair, resulting in deficient peristalsis and uncoordinated contractions of the distal portion of the reconstructed esophagus[37,86-91]. Esophageal emptying is therefore achieved primarily by gravity. Esophageal and gastric dysmotility can be considered causes of GER[1,37,86-89]. Scintigraphy and manometry exams are used to diagnose esophageal dysmotility[86,87,92].
Deformities of the thoracic wall and scoliosis
Chest wall and spinal deformities can be very disfiguring for patients and may be inadvertently overlooked from a pediatric surgeon’s perspective, as these deformities may only become apparent later in life. These anomalies may then be referred to a different specialty, such as orthopedics or plastic surgery. Open thoracotomy can result in significant musculoskeletal morbidity if care is not taken to ensure proper muscle-sparing surgical techniques[93]. Associated vertebral anomalies can contribute to chest wall or spinal deformities by directly affecting the ribs and vertebral column[94,95]. A “winged” scapula secondary to neuromuscular injury to the latissimus dorsi muscle has been reported in 24% of patients undergoing a standard posterolateral thoracotomy for EA/TEF repair, with up to 21% of patients exhibiting scoliosis[2]. Scoliosis is more common in patients who have undergone more than one thoracotomy or division of portions of the serratus anterior and latissimus dorsi muscle groups or their nerve supplies[2]. Deformities of the thoracic wall are more common in patients who have undergone multiple thoracotomies but can be avoided or diminished with the use of thoracoscopy.
Respiratory disorders
The etiological factors involved in respiratory problems are thought to be retained secretions caused by tracheomalacia, aspiration related to impaired esophageal peristalsis and esophageal stricture and recurrence of TEF or GER. Primary respiratory complications, such as recurrent bronchitis, pneumonias, wheezing illnesses, daily coughing and bronchiectasis are common in patients with repaired EA, but become less frequent with time[96-102].
Tracheomalacia is due to a structural and functional weakness in the wall of the trachea, resulting in partial and occasionally complete obstruction of the tracheal lumen. This condition results from abnormal tracheal rings, deficiencies in cartilage, and an increase in length of the transverse muscle. Tracheomalacia is a consistent finding in most if not all children with TEF, but is reported to be clinically significant in only 10%-20% of patients and tends to improve with age[1,7,37]. Approximately half of symptomatic patients require surgical correction, the aortopexy. This repair is reserved for those with near-death episodes or recurrent pneumonia. Success rates of 35% to 88% have been reported[2].
Bronchoscopy is the gold standard for the diagnosis of tracheomalacia. The severity of tracheomalacia is based on a macroscopic estimation. It is considered severe when the anteroposterior collapse is ≥ 75% with cough or expiration, moderate when the collapse is 50% to 75%, and mild when the collapse is < 50%[96].
CONCLUSION
Improvements have been made in the treatment of EA over the years[44-49,55]. However, there is no ideal surgical procedure for this congenital anomaly. The search for simpler, less burdensome, more efficient methods for the treatment of EA is closely tied to the difficulty in obtaining sophisticated equipment and qualified personnel for the follow-up of these patients. Long-term outcomes and follow-up data are limited for post-EA/TEF repairs. GER affects these children at the five-year follow-up and gradually declines in children followed for over ten years. Additional long-term morbidities include recurrent respiratory infections, tracheomalacia, dysphagia and choking episodes. Each of these afflictions diminishes over long-term follow-up, but morbidity from esophageal dysmotility may be a life-long problem. Indeed, although EA primarily affects patients during the neonatal period, postoperative complications may lead to common gastroenterological disorders that require long-term follow-up by gastroenterologists. Therefore, determining the risk factors of the complicated evolution of a EA/TEF repair can positively impact the long-term prognosis of these patients, likely avoiding relevant gastroenterological symptoms during adulthood. Knowledge of these risk factors will allow the identification of patients who may benefit from a more intensive follow-up program. Much remains to be understood about EA. Meanwhile, in order to treat patients with individualized care, it is necessary for physicians to stay current regarding treatments for EA.
Footnotes
Supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Brazil; Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil; FAPEMIG: CBB-APQ-00075-09/CNPq 573646/2008-2; and Programa de Grupos de Excelência-FINEP, Brazil
Peer reviewers: Jianyuan Chai, Assistant Professor, PhD, MS, BS, Research (09-151), VA Long Beach Healthcare System, 5901 E. 7th St, Long Beach, CA 90822, United States; Piero Marco Fisichella, Assistant Professor of Surgery, Medical Director, MD, Swallowing Center, Loyola University Medical Center, Department of Surgery, Stritch School of Medicine, 2160 South First Avenue, Room 3226, Maywood, IL 60153, United States; Dr. Jeff Butterworth, MB, FRCP, Department of Gastroenterology, Shrewsbury and Telford Hospital NHS Trust, Mytton Oak Road, Shrewsbury, Shropshire SY3 8XQ, United Kingdom
S- Editor Cheng JX L- Editor A E- Editor Zhang DN
References
- 1.Spitz L. Oesophageal atresia. Orphanet J Rare Dis. 2007;2:24. doi: 10.1186/1750-1172-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortell AE, Azizkhan RG. Esophageal atresia repair with thoracotomy: the Cincinnati contemporary experience. Semin Pediatr Surg. 2009;18:12–19. doi: 10.1053/j.sempedsurg.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Spitz L. Esophageal atresia. Lessons I have learned in a 40-year experience. J Pediatr Surg. 2006;41:1635–1640. doi: 10.1016/j.jpedsurg.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Kovesi T, Rubin S. Long-term complications of congenital esophageal atresia and/or tracheoesophageal fistula. Chest. 2004;126:915–925. doi: 10.1378/chest.126.3.915. [DOI] [PubMed] [Google Scholar]
- 5.Grosfeld JL, Ladd AP. Anomalias congênitas. In: Silva ACS e, Pereira RM, Pinheiro PFM., editors. Cirurgia Pediátrica-Condutas clínicas e cirúrgicas. Rio de Janeiro: Guanabara Koogan; 2005. pp. 291–298. [Google Scholar]
- 6.Seo J, Kim do Y, Kim AR, Kim DY, Kim SC, Kim IK, Kim KS, Yoon CH, Pi SY. An 18-year experience of tracheoesophageal fistula and esophageal atresia. Korean J Pediatr. 2010;53:705–710. doi: 10.3345/kjp.2010.53.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holland AJ, Fitzgerald DA. Oesophageal atresia and tracheo-oesophageal fistula: current management strategies and complications. Paediatr Respir Rev. 2010;11:100–106; quiz 106-107. doi: 10.1016/j.prrv.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Spitz L. Esophageal atresia and tracheoesophageal malformations. In: Ashcraft KW, Holcomb GW, Murphy JP, editors. Pediatrics Surgery. Philadelphia: Saunders; 2005. pp. 352–370. [Google Scholar]
- 9.Nakayana DK. Congenital abnormalities of the esophagus. In: O’Neill Jr JA, Grosfeld JL, Foukalsrud EW, Coran AG, Caldanone AA, et al., editors. Principles of Pediatric Surgery. 2nd ed. St. Louis, MO: Mosby; 2003. pp. 385–394. [Google Scholar]
- 10.Goyal A, Jones MO, Couriel JM, Losty PD. Oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child Fetal Neonatal Ed. 2006;91:F381–F384. doi: 10.1136/adc.2005.086157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta DK, Sharma S. Esophageal atresia: the total care in a high-risk population. Semin Pediatr Surg. 2008;17:236–243. doi: 10.1053/j.sempedsurg.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Vogt EC. Congenital esophageal atresia. AJR Am J Roentgenol. 1929;22:463–465. [Google Scholar]
- 13.Gross RE. The surgery of infancy and chilhood. Philadelphia: WB Saunders; 1953. [Google Scholar]
- 14.Ioannides AS, Copp AJ. Embryology of oesophageal atresia. Semin Pediatr Surg. 2009;18:2–11. doi: 10.1053/j.sempedsurg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felix JF, de Jong EM, Torfs CP, de Klein A, Rottier RJ, Tibboel D. Genetic and environmental factors in the etiology of esophageal atresia and/or tracheoesophageal fistula: an overview of the current concepts. Birth Defects Res A Clin Mol Teratol. 2009;85:747–754. doi: 10.1002/bdra.20592. [DOI] [PubMed] [Google Scholar]
- 16.de Jong EM, Felix JF, de Klein A, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: “mind the gap”. Curr Gastroenterol Rep. 2010;12:215–222. doi: 10.1007/s11894-010-0108-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunner HG, van Bokhoven H. Genetic players in esophageal atresia and tracheoesophageal fistula. Curr Opin Genet Dev. 2005;15:341–347. doi: 10.1016/j.gde.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Gohary Y, Gittes GK, Tovar JA. Congenital anomalies of the esophagus. Semin Pediatr Surg. 2010;19:186–193. doi: 10.1053/j.sempedsurg.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 20.de Jong EM, de Haan MA, Gischler SJ, Hop W, Cohen-Overbeek TE, Bax NM, de Klein A, Tibboel D, Grijseels EW. Pre- and postnatal diagnosis and outcome of fetuses and neonates with esophageal atresia and tracheoesophageal fistula. Prenat Diagn. 2010;30:274–279. doi: 10.1002/pd.2466. [DOI] [PubMed] [Google Scholar]
- 21.Houben CH, Curry JI. Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula. Prenat Diagn. 2008;28:667–675. doi: 10.1002/pd.1938. [DOI] [PubMed] [Google Scholar]
- 22.Atzori P, Iacobelli BD, Bottero S, Spirydakis J, Laviani R, Trucchi A, Braguglia A, Bagolan P. Preoperative tracheobronchoscopy in newborns with esophageal atresia: does it matter? J Pediatr Surg. 2006;41:1054–1057. doi: 10.1016/j.jpedsurg.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 23.Lahdes-Vasama TT, Sihvonen R, Iber T. Perforation of the upper and lower segments of atretic esophagus (type C) secondary to nasogastric tube insertion. Pediatr Surg Int. 2009;25:537–538. doi: 10.1007/s00383-009-2377-z. [DOI] [PubMed] [Google Scholar]
- 24.McDuffie LA, Wakeman D, Warner BW. Diagnosis of esophageal atresia with tracheoesophageal fistula: is there a need for gastrointestinal contrast? J Pediatr. 2010;156:852. doi: 10.1016/j.jpeds.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 25.Alabbad SI, Shaw K, Puligandla PS, Carranza R, Bernard C, Laberge JM. The pitfalls of endotracheal intubation beyond the fistula in babies with type C esophageal atresia. Semin Pediatr Surg. 2009;18:116–118. doi: 10.1053/j.sempedsurg.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Sugito K, Koshinaga T, Hoshino M, Inoue M, Goto H, Ikeda T, Hagiwara N. Study of 24 cases with congenital esophageal atresia: what are the risk factors? Pediatr Int. 2006;48:616–621. doi: 10.1111/j.1442-200X.2006.02288.x. [DOI] [PubMed] [Google Scholar]
- 27.Yagyu M, Gitter H, Richter B, Booss D. Esophageal atresia in Bremen, Germany--evaluation of preoperative risk classification in esophageal atresia. J Pediatr Surg. 2000;35:584–587. doi: 10.1053/jpsu.2000.0350584. [DOI] [PubMed] [Google Scholar]
- 28.Eghbalian F, Monsef A, Mousavi-Bahar SH. Urinary tract and other associated anomalies in newborns with esophageal atresia. Urol J. 2009;6:123–126. [PubMed] [Google Scholar]
- 29.Keckler SJ, St Peter SD, Valusek PA, Tsao K, Snyder CL, Holcomb GW, Ostlie DJ. VACTERL anomalies in patients with esophageal atresia: an updated delineation of the spectrum and review of the literature. Pediatr Surg Int. 2007;23:309–313. doi: 10.1007/s00383-007-1891-0. [DOI] [PubMed] [Google Scholar]
- 30.Nasr A, McNamara PJ, Mertens L, Levin D, James A, Holtby H, Langer JC. Is routine preoperative 2-dimensional echocardiography necessary for infants with esophageal atresia, omphalocele, or anorectal malformations? J Pediatr Surg. 2010;45:876–879. doi: 10.1016/j.jpedsurg.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Mowery N, Billmire DF, Schamberger M, Szotek P, West KW, Rescorla FJ, Scherer LR, Engum S, Rouse T, Grosfeld JL. Incidence of persistent left superior vena cava in esophageal atresia. J Pediatr Surg. 2006;41:484–486. doi: 10.1016/j.jpedsurg.2005.10.094. [DOI] [PubMed] [Google Scholar]
- 32.Babu R, Pierro A, Spitz L, Drake DP, Kiely EM. The management of oesophageal atresia in neonates with right-sided aortic arch. J Pediatr Surg. 2000;35:56–58. doi: 10.1016/s0022-3468(00)80013-5. [DOI] [PubMed] [Google Scholar]
- 33.Snider AR, Serwer GA, Ritter SB. Abnormal Vascular Connections and Structures. In: Echocardiografy in pediatric heart disease, et al., editors. 2 ed. St. Louis: Mosby; 1997. pp. 452–496. [Google Scholar]
- 34.Krosnar S, Baxter A. Thoracoscopic repair of esophageal atresia with tracheoesophageal fistula: anesthetic and intensive care management of a series of eight neonates. Paediatr Anaesth. 2005;15:541–546. doi: 10.1111/j.1460-9592.2005.01594.x. [DOI] [PubMed] [Google Scholar]
- 35.Seitz G, Warmann SW, Schaefer J, Poets CF, Fuchs J. Primary repair of esophageal atresia in extremely low birth weight infants: a single-center experience and review of the literature. Biol Neonate. 2006;90:247–251. doi: 10.1159/000094037. [DOI] [PubMed] [Google Scholar]
- 36.Hamza AF. Colonic replacement in cases of esophageal atresia. Semin Pediatr Surg. 2009;18:40–43. doi: 10.1053/j.sempedsurg.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Rintala RJ, Sistonen S, Pakarinen MP. Outcome of esophageal atresia beyond childhood. Semin Pediatr Surg. 2009;18:50–56. doi: 10.1053/j.sempedsurg.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Sharma AK, Shekhawat NS, Agrawal LD, Chaturvedi V, Kothari SK, Goel D. Esophageal atresia and tracheoesophageal fistula: a review of 25 years’ experience. Pediatr Surg Int. 2000;16:478–482. doi: 10.1007/s003830000393. [DOI] [PubMed] [Google Scholar]
- 39.Alabbad SI, Ryckman J, Puligandla PS, Shaw K, Nguyen LT, Laberge JM. Use of transanastomotic feeding tubes during esophageal atresia repair. J Pediatr Surg. 2009;44:902–905. doi: 10.1016/j.jpedsurg.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 40.Pane A, Foschia F, Caldaro T, De Angelis P, Torroni F, Federici G, Servedio D, Dall’Oglio L. Endoscopy. 2008;40 Suppl 2:E254–E255. doi: 10.1055/s-2008-1077650. [DOI] [PubMed] [Google Scholar]
- 41.Farkash U, Lazar L, Erez I, Gutermacher M, Freud E. The distal pouch in esophageal atresia -- to dissect or not to dissect, that is the question. Eur J Pediatr Surg. 2002;12:19–23. doi: 10.1055/s-2002-25091. [DOI] [PubMed] [Google Scholar]
- 42.Séguier-Lipszyc E, Bonnard A, Aizenfisz S, Enezian G, Maintenant J, Aigrain Y, de Lagausie P. The management of long gap esophageal atresia. J Pediatr Surg. 2005;40:1542–1546. doi: 10.1016/j.jpedsurg.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Hadidi AT, Hosie S, Waag KL. Long gap esophageal atresia: lengthening technique and primary anastomosis. J Pediatr Surg. 2007;42:1659–1662. doi: 10.1016/j.jpedsurg.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 44.Foker JE, Kendall Krosch TC, Catton K, Munro F, Khan KM. Long-gap esophageal atresia treated by growth induction: the biological potential and early follow-up results. Semin Pediatr Surg. 2009;18:23–29. doi: 10.1053/j.sempedsurg.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Hagberg S, Rubenson A, Sillén U, Werkmäster K. Management of long-gap esophagus: experience with end-to-end anastomosis under maximal tension. Prog Pediatr Surg. 1986;19:88–92. doi: 10.1007/978-3-642-70777-3_8. [DOI] [PubMed] [Google Scholar]
- 46.Boyle EM, Irwin ED, Foker JE. Primary repair of ultra-long-gap esophageal atresia: results without a lengthening procedure. Ann Thorac Surg. 1994;57:576–579. doi: 10.1016/0003-4975(94)90548-7. [DOI] [PubMed] [Google Scholar]
- 47.Bagolan P, Iacobelli Bd B, De Angelis P, di Abriola GF, Laviani R, Trucchi A, Orzalesi M, Dall’Oglio L. Long gap esophageal atresia and esophageal replacement: moving toward a separation? J Pediatr Surg. 2004;39:1084–1090. doi: 10.1016/j.jpedsurg.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 48.Bax KM. Jejunum for bridging long-gap esophageal atresia. Semin Pediatr Surg. 2009;18:34–39. doi: 10.1053/j.sempedsurg.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Castilloux J, Noble AJ, Faure C. Risk factors for short- and long-term morbidity in children with esophageal atresia. J Pediatr. 2010;156:755–760. doi: 10.1016/j.jpeds.2009.11.038. [DOI] [PubMed] [Google Scholar]
- 50.Sri Paran T, Decaluwe D, Corbally M, Puri P. Long-term results of delayed primary anastomosis for pure oesophageal atresia: a 27-year follow up. Pediatr Surg Int. 2007;23:647–651. doi: 10.1007/s00383-007-1925-7. [DOI] [PubMed] [Google Scholar]
- 51.Konkin DE, O’hali WA, Webber EM, Blair GK. Outcomes in esophageal atresia and tracheoesophageal fistula. J Pediatr Surg. 2003;38:1726–1729. doi: 10.1016/j.jpedsurg.2003.08.039. [DOI] [PubMed] [Google Scholar]
- 52.Engum SA, Grosfeld JL, West KW, Rescorla FJ, Scherer LR. Analysis of morbidity and mortality in 227 cases of esophageal atresia and/or tracheoesophageal fistula over two decades. Arch Surg. 1995;130:502–508; discussion 508-509. doi: 10.1001/archsurg.1995.01430050052008. [DOI] [PubMed] [Google Scholar]
- 53.Lopes MF, Reis A, Coutinho S, Pires A. Very long gap esophageal atresia successfully treated by esophageal lengthening using external traction sutures. J Pediatr Surg. 2004;39:1286–1287. doi: 10.1016/j.jpedsurg.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 54.Nagaya M, Kato J, Niimi N, Tanaka S, Iio K. Proposal of a novel method to evaluate anastomotic tension in esophageal atresia with a distal tracheoesophageal fistula. Pediatr Surg Int. 2005;21:780–785. doi: 10.1007/s00383-005-1540-4. [DOI] [PubMed] [Google Scholar]
- 55.Tamburri N, Laje P, Boglione M, Martinez-Ferro M. Extrathoracic esophageal elongation (Kimura’s technique): a feasible option for the treatment of patients with complex esophageal atresia. J Pediatr Surg. 2009;44:2420–2425. doi: 10.1016/j.jpedsurg.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 56.Takamizawa S, Nishijima E, Tsugawa C, Muraji T, Satoh S, Tatekawa Y, Kimura K. Multistaged esophageal elongation technique for long gap esophageal atresia: experience with 7 cases at a single institution. J Pediatr Surg. 2005;40:781–784. doi: 10.1016/j.jpedsurg.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 57.Stringel G, Lawrence C, McBride W. Repair of long gap esophageal atresia without anastomosis. J Pediatr Surg. 2010;45:872–875. doi: 10.1016/j.jpedsurg.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Burgos L, Barrena S, Andrés AM, Martínez L, Hernández F, Olivares P, Lassaletta L, Tovar JA. Colonic interposition for esophageal replacement in children remains a good choice: 33-year median follow-up of 65 patients. J Pediatr Surg. 2010;45:341–345. doi: 10.1016/j.jpedsurg.2009.10.065. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen T, Zainabadi K, Bui T, Emil S, Gelfand D, Nguyen N. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula: lessons learned. J Laparoendosc Adv Surg Tech A. 2006;16:174–178. doi: 10.1089/lap.2006.16.174. [DOI] [PubMed] [Google Scholar]
- 60.Yang CF, Soong WJ, Jeng MJ, Chen SJ, Lee YS, Tsao PC, Hwang B, Wei CF, Chin TW, Liu C. Esophageal atresia with tracheoesophageal fistula: ten years of experience in an institute. J Chin Med Assoc. 2006;69:317–321. doi: 10.1016/S1726-4901(09)70265-5. [DOI] [PubMed] [Google Scholar]
- 61.Chetcuti P, Phelan PD. Gastrointestinal morbidity and growth after repair of oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child. 1993;68:163–166. doi: 10.1136/adc.68.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deurloo JA, Ekkelkamp S, Hartman EE, Sprangers MA, Aronson DC. Quality of life in adult survivors of correction of esophageal atresia. Arch Surg. 2005;140:976–980. doi: 10.1001/archsurg.140.10.976. [DOI] [PubMed] [Google Scholar]
- 63.Deurloo JA, Ekkelkamp S, Bartelsman JF, Ten Kate FJ, Schoorl M, Heij HA, Aronson DC. Gastroesophageal reflux: prevalence in adults older than 28 years after correction of esophageal atresia. Ann Surg. 2003;238:686–689. doi: 10.1097/01.sla.0000094303.07910.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koivusalo A, Pakarinen M, Vanamo K, Lindahl H, Rintala RJ. Health-related quality of life in adults after repair of congenital diaphragmatic defects--a questionnaire study. J Pediatr Surg. 2005;40:1376–1381. doi: 10.1016/j.jpedsurg.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 65.Tomaselli V, Volpi ML, Dell’Agnola CA, Bini M, Rossi A, Indriolo A. Long-term evaluation of esophageal function in patients treated at birth for esophageal atresia. Pediatr Surg Int. 2003;19:40–43. doi: 10.1007/s00383-002-0887-z. [DOI] [PubMed] [Google Scholar]
- 66.Upadhyaya VD, Gangopadhyaya AN, Gupta DK, Sharma SP, Kumar V, Pandey A, Upadhyaya AD. Prognosis of congenital tracheoesophageal fistula with esophageal atresia on the basis of gap length. Pediatr Surg Int. 2007;23:767–771. doi: 10.1007/s00383-007-1964-0. [DOI] [PubMed] [Google Scholar]
- 67.Uchida K, Inoue M, Otake K, Okita Y, Morimoto Y, Araki T, Miki C, Kusunoki M. Efficacy of postoperative elective ventilatory support for leakage protection in primary anastomosis of congenital esophageal atresia. Pediatr Surg Int. 2006;22:496–499. doi: 10.1007/s00383-006-1700-1. [DOI] [PubMed] [Google Scholar]
- 68.Chittmittrapap S, Spitz L, Kiely EM, Brereton RJ. Anastomotic stricture following repair of esophageal atresia. J Pediatr Surg. 1990;25:508–511. doi: 10.1016/0022-3468(90)90561-m. [DOI] [PubMed] [Google Scholar]
- 69.Koivusalo A, Turunen P, Rintala RJ, van der Zee DC, Lindahl H, Bax NM. Is routine dilatation after repair of esophageal atresia with distal fistula better than dilatation when symptoms arise? Comparison of results of two European pediatric surgical centers. J Pediatr Surg. 2004;39:1643–1647. doi: 10.1016/j.jpedsurg.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 70.Taylor AC, Breen KJ, Auldist A, Catto-Smith A, Clarnette T, Crameri J, Taylor R, Nagarajah S, Brady J, Stokes K. Gastroesophageal reflux and related pathology in adults who were born with esophageal atresia: a long-term follow-up study. Clin Gastroenterol Hepatol. 2007;5:702–706. doi: 10.1016/j.cgh.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 71.Krug E, Bergmeijer JH, Dees J, de Krijger R, Mooi WJ, Hazebroek FW. Gastroesophageal reflux and Barrett’s esophagus in adults born with esophageal atresia. Am J Gastroenterol. 1999;94:2825–2828. doi: 10.1111/j.1572-0241.1999.1423_c.x. [DOI] [PubMed] [Google Scholar]
- 72.Fröhlich T, Otto S, Weber P, Pilic D, Schmidt-Choudhury A, Wenzl TG, Köhler H. Combined esophageal multichannel intraluminal impedance and pH monitoring after repair of esophageal atresia. J Pediatr Gastroenterol Nutr. 2008;47:443–449. doi: 10.1097/MPG.0b013e3181638ca2. [DOI] [PubMed] [Google Scholar]
- 73.Lüthold SC, Rochat MK, Bähler P. Disagreement between symptom-reflux association analysis parameters in pediatric gastroesophageal reflux disease investigation. World J Gastroenterol. 2010;16:2401–2406. doi: 10.3748/wjg.v16.i19.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ozin Y, Dagli U, Kuran S, Sahin B. Manometric findings in patients with isolated distal gastroesophageal reflux. World J Gastroenterol. 2009;15:5461–5464. doi: 10.3748/wjg.15.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Biervliet S, Van Winckel M, Robberecht E, Kerremans I. High-dose omeprazole in esophagitis with stenosis after surgical treatment of esophageal atresia. J Pediatr Surg. 2001;36:1416–1418. doi: 10.1053/jpsu.2001.26388. [DOI] [PubMed] [Google Scholar]
- 76.Deurloo JA, Ekkelkamp S, Taminiau JA, Kneepkens CM, ten Kate FW, Bartelsman JF, Legemate DA, Aronson DC. Esophagitis and Barrett esophagus after correction of esophageal atresia. J Pediatr Surg. 2005;40:1227–1231. doi: 10.1016/j.jpedsurg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 77.Deurloo JA, van Lanschot JJ, Drillenburg P, Aronson DC. Esophageal squamous cell carcinoma 38 years after primary repair of esophageal atresia. J Pediatr Surg. 2001;36:629–630. doi: 10.1053/jpsu.2001.22305. [DOI] [PubMed] [Google Scholar]
- 78.Alfaro L, Bermas H, Fenoglio M, Parker R, Janik JS. Are patients who have had a tracheoesophageal fistula repair during infancy at risk for esophageal adenocarcinoma during adulthood? J Pediatr Surg. 2005;40:719–720. doi: 10.1016/j.jpedsurg.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 79.Pultrum BB, Bijleveld CM, de Langen ZJ, Plukker JT. Development of an adenocarcinoma of the esophagus 22 years after primary repair of a congenital atresia. J Pediatr Surg. 2005;40:e1–e4. doi: 10.1016/j.jpedsurg.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Z, Huang Y, Su P, Wang D, Wang L. Experience in treating congenital esophageal atresia in China. J Pediatr Surg. 2010;45:2009–2014. doi: 10.1016/j.jpedsurg.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 81.Lambert R, Hainaut P. The multidisciplinary management of gastrointestinal cancer. Epidemiology of oesophagogastric cancer. Best Pract Res Clin Gastroenterol. 2007;21:921–945. doi: 10.1016/j.bpg.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 82.Schuchert MJ, Luketich JD. Barrett’s esophagus-emerging concepts and controversies. J Surg Oncol. 2007;95:185–189. doi: 10.1002/jso.20635. [DOI] [PubMed] [Google Scholar]
- 83.Schalamon J, Lindahl H, Saarikoski H, Rintala RJ. Endoscopic follow-up in esophageal atresia-for how long is it necessary? J Pediatr Surg. 2003;38:702–704. doi: 10.1016/jpsu.2003.50187. [DOI] [PubMed] [Google Scholar]
- 84.Fass R. Erosive esophagitis and nonerosive reflux disease (NERD): comparison of epidemiologic, physiologic, and therapeutic characteristics. J Clin Gastroenterol. 2007;41:131–137. doi: 10.1097/01.mcg.0000225631.07039.6d. [DOI] [PubMed] [Google Scholar]
- 85.Koivusalo A, Pakarinen MP, Rintala RJ. The cumulative incidence of significant gastrooesophageal reflux in patients with oesophageal atresia with a distal fistula--a systematic clinical, pH-metric, and endoscopic follow-up study. J Pediatr Surg. 2007;42:370–374. doi: 10.1016/j.jpedsurg.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 86.Romeo G, Zuccarello B, Proietto F, Romeo C. Disorders of the esophageal motor activity in atresia of the esophagus. J Pediatr Surg. 1987;22:120–124. doi: 10.1016/s0022-3468(87)80425-6. [DOI] [PubMed] [Google Scholar]
- 87.Romeo C, Bonanno N, Baldari S, Centorrino A, Scalfari G, Antonuccio P, Centonze A, Gentile C. Gastric motility disorders in patients operated on for esophageal atresia and tracheoesophageal fistula: long-term evaluation. J Pediatr Surg. 2000;35:740–744. doi: 10.1053/jpsu.2000.6048. [DOI] [PubMed] [Google Scholar]
- 88.Boleken M, Demirbilek S, Kirimiloglu H, Kanmaz T, Yucesan S, Celbis O, Uzun I. Reduced neuronal innervation in the distal end of the proximal esophageal atretic segment in cases of esophageal atresia with distal tracheoesophageal fistula. World J Surg. 2007;31:1512–1517. doi: 10.1007/s00268-007-9070-y. [DOI] [PubMed] [Google Scholar]
- 89.Li K, Zheng S, Xiao X, Wang Q, Zhou Y, Chen L. The structural characteristics and expression of neuropeptides in the esophagus of patients with congenital esophageal atresia and tracheoesophageal fistula. J Pediatr Surg. 2007;42:1433–1438. doi: 10.1016/j.jpedsurg.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 90.Sistonen SJ, Koivusalo A, Nieminen U, Lindahl H, Lohi J, Kero M, Kärkkäinen PA, Färkkilä MA, Sarna S, Rintala RJ, et al. Esophageal morbidity and function in adults with repaired esophageal atresia with tracheoesophageal fistula: a population-based long-term follow-up. Ann Surg. 2010;251:1167–1173. doi: 10.1097/SLA.0b013e3181c9b613. [DOI] [PubMed] [Google Scholar]
- 91.Tovar JA, Diez Pardo JA, Murcia J, Prieto G, Molina M, Polanco I. Ambulatory 24-hour manometric and pH metric evidence of permanent impairment of clearance capacity in patients with esophageal atresia. J Pediatr Surg. 1995;30:1224–1231. doi: 10.1016/0022-3468(95)90029-2. [DOI] [PubMed] [Google Scholar]
- 92.Dutta HK, Grover VP, Dwivedi SN, Bhatnagar V. Manometric evaluation of postoperative patients of esophageal atresia and tracheo-esophageal fistula. Eur J Pediatr Surg. 2001;11:371–376. doi: 10.1055/s-2001-19718. [DOI] [PubMed] [Google Scholar]
- 93.Jaureguizar E, Vazquez J, Murcia J, Diez Pardo JA. Morbid musculoskeletal sequelae of thoracotomy for tracheoesophageal fistula. J Pediatr Surg. 1985;20:511–514. doi: 10.1016/s0022-3468(85)80477-2. [DOI] [PubMed] [Google Scholar]
- 94.Chetcuti P, Dickens DR, Phelan PD. Spinal deformity in patients born with oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child. 1989;64:1427–1430. doi: 10.1136/adc.64.10.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chetcuti P, Myers NA, Phelan PD, Beasley SW, Dickens DR. Chest wall deformity in patients with repaired esophageal atresia. J Pediatr Surg. 1989;24:244–247. doi: 10.1016/s0022-3468(89)80003-x. [DOI] [PubMed] [Google Scholar]
- 96.Malmström K, Lohi J, Lindahl H, Pelkonen A, Kajosaari M, Sarna S, Malmberg LP, Mäkelä MJ. Longitudinal follow-up of bronchial inflammation, respiratory symptoms, and pulmonary function in adolescents after repair of esophageal atresia with tracheoesophageal fistula. J Pediatr. 2008;153:396–401. doi: 10.1016/j.jpeds.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 97.Robertson DF, Mobaireek K, Davis GM, Coates AL. Late pulmonary function following repair of tracheoesophageal fistula or esophageal atresia. Pediatr Pulmonol. 1995;20:21–26. doi: 10.1002/ppul.1950200105. [DOI] [PubMed] [Google Scholar]
- 98.Chetcuti P, Myers NA, Phelan PD, Beasley SW. Adults who survived repair of congenital oesophageal atresia and tracheo-oesophageal fistula. BMJ. 1988;297:344–346. doi: 10.1136/bmj.297.6644.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chetcuti P, Phelan PD. Respiratory morbidity after repair of oesophageal atresia and tracheo-oesophageal fistula. Arch Dis Child. 1993;68:167–170. doi: 10.1136/adc.68.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Biller JA, Allen JL, Schuster SR, Treves ST, Winter HS. Long-term evaluation of esophageal and pulmonary function in patients with repaired esophageal atresia and tracheoesophageal fistula. Dig Dis Sci. 1987;32:985–990. doi: 10.1007/BF01297188. [DOI] [PubMed] [Google Scholar]
- 101.Velanovich V. Gastroesophageal reflux disease and the airway-essentials for the surgeon. World J Gastrointest Surg. 2009;1:8–10. doi: 10.4240/wjgs.v1.i1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bresci G, Sacco R. Pulmonary or otolaryngologic extraesophageal manifestations in patients with gastroesophageal reflux disease. World J Gastrointest Endosc. 2010;2:47–49. doi: 10.4253/wjge.v2.i2.47. [DOI] [PMC free article] [PubMed] [Google Scholar]