

Abstract
AIM: To investigate the mechanism of interleukin (IL)-6 secretion through blocking the IL-17A/IL-17A receptor (IL-17RA) signaling pathway with a short hairpin RNA (shRNA) in hepatic stellate cells (HSCs) in vitro.
METHODS: HSCs were derived from the livers of adult male Sprague-Dawley rats. IL-6 expression was evaluated using real-time quantitative polymerase chain reaction and enzyme linked immunosorbent assay. The phosphorylation activity of p38 mitogen activated protein kinases (MAPK) and extracellular regulated protein kinases (ERK) 1/2 upon induction by IL-17A and suppression by IL-17RA shRNA were examined using Western blotting.
RESULTS: IL-6 expression induced by IL-17A was significantly increased compared to control in HSCs (P < 0.01 in a dose-dependent manner). Suppression of IL-17RA using lentiviral-mediated shRNA inhibited IL-6 expression induced by IL-17A compared to group with only IL-17A treatment (1.44 ± 0.17 vs 4.07 ± 0.43, P < 0.01). IL-17A induced rapid phosphorylation of p38 MAPK and ERK1/2 after 5 min exposure, and showed the strongest levels of phosphorylation of p38 MAPK and ERK1/2 at 15 min in IL-17A-treated HSCs. IL-6 mRNA expression induced by IL-17A (100 ng/mL) for 3 h exposure was inhibited by preincubation with specific inhibitors of p38 MAPK (SB-203580) and ERK1/2 (PD-98059) compared to groups without inhibitors preincubation (1.67 ± 0.24, 2.01 ± 0.10 vs 4.08 ± 0.59, P < 0.01). Moreover, Lentiviral-mediated IL-17RA shRNA 1 inhibited IL-17A-induced IL-6 mRNA expression compared to random shRNA in HSCs (1.44 ± 0.17 vs 3.98 ± 0.68, P < 0.01). Lentiviral-mediated IL-17RA shRNA 1 inhibited phosphorylation of p38 MAPK and ERK1/2 induced by 15 min IL-17A (100 ng/mL) exposure.
CONCLUSION: Down-regulation of the IL-17RA receptor by shRNA decreased IL-6 expression induced by IL-17A via p38 MAPK and ERK1/2 phosphorylation in HSCs. Suppression of IL-17RA expression may be a strategy to reduce the inflammatory response induced by IL-17A in the liver.
Keywords: Interleukin 17A, Interleukin 6, Hepatic stellate cells, Liver fibrosis
INTRODUCTION
Hepatic stellate cells (HSCs), also known as fat-storing cells, are a major cell type involved in liver fibrosis. Activation and proliferation of HSCs are associated with liver injury[1]. Activated HSCs accumulate excess extracellular matrix and produce a variety of pro-inflammatory cytokines, including macrophage inflammatory protein-2, monocyte chemo-attractant protein-1, interleukin (IL)-6, IL-8, and transforming growth factor-β1 (TGF-β1)[2-3]. These pro-inflammatory cytokines can eventually lead to liver fibrosis.
IL-17A is the founding member of the IL-17 cytokine family, which now includes six major isoforms, IL-17A, -B, -C, -D, -E, and -F[4]. The original “IL-17” has been designated IL-17A and is considered a T cell (Th17 cells)-specific cytokine[5]. The IL-17A receptor (IL-17RA) is expressed on many cell types in the human body[6]. IL-17A exerts pro-inflammatory, pro-apoptotic, and pro-mitogenic effects via binding to IL-17RA[6,7].
In this study, we constructed a highly efficient lentiviral short hairpin RNA (shRNA) targeting IL-17RA to study the level of IL-6 secretion in the absence of IL-17RA in activated HSCs and the underlying mechanism(s). Our results showed that the silencing effect of IL-17RA shRNA eliminated IL-6 secretion in activated HSCs. We suggest that secretion of IL-6 involves the mitogen activated protein kinases (MAPK) signaling pathway through phosphorylation of p38 MAPK and extracellular regulated protein kinases (ERK) 1/2. Our finding may provide a novel interventional therapy against hepatic inflammatory responses and hepatic fibrosis.
MATERIALS AND METHODS
Reagents
Recombinant murine IL-17A was purchased from Peprothech (Princeton Business Park, NJ). Anti-IL-17R (sc-1902, dilution factor 1:200) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). PD-98509 (inhibitor of MEK1) and SB-203580 (inhibitor of p38 MAPK) were obtained from Sigma-Aldrich (Saint Louis, MO). The p38 MAPK antibody (dilution factor 1:200), phospho-p38 MAPK rabbit mAb (dilution factor 1:200), ERK1/2 MAPK rabbit mAb (dilution factor 1:250), and phospho-ERK1/2 MAPK rabbit mAb (dilution factor 1:250) were obtained from Cell Signaling Technology (Beverly, MA).
shRNA design and plasmid constructs
Candidate sequences targeting rat IL-17RA mRNA (GenBank Accession NM 001107883) were designed using the Dharmacon siDESIGN Center procedure. Two complementary single-strand oligonucleotides containing the target sequences were synthesized chemically and annealed. The double-stranded oligonucleotides were inserted between AgeI and EcoRI restriction sites in the pGCL-green fluorescent protein (GFP) small interfering RNA (siRNA) vector that contains a cytomegaoviyns-driven enhanced green fluorescent protein reporter gene. The ligated plasmid was transformed into Escherichia coli DH5α competent cells for pGCSIL/IL-17RA shRNA plasmid amplification.
shRNA lentivirus transduction
Plasmids containing the IL-17RA shRNA lentivirus were transfected into 293T cells using the ViraPower packaging mix (pGCSIL/IL-17RA shRNA plasmids, pHelper 1.0, and pHelper 2.0) and Lipofectamine 2000 (Invitrogen). After 48 h, the harvested viral supernatant was used to infect HSCs at a multiplicity of infection of 10 for 24 h. Cells with the GFP label were harvested after 48 h, and total RNA was extracted. The interference efficiency of IL-17RA shRNA was determined using real-time quantitative polymerase chain reaction (qPCR), and the most efficient silencing sequence of IL-17RA shRNA was selected for subsequent studies.
Isolation and culture of rat HSCs
Adult male Sprague-Dawley rats (body weight, 400-500 g) were used for HSC isolation as described previously[8]. The liver tissues were digested with collagenase IV (0.5 g/L) and deoxyribonuclease I (0.03 g/L) before fractionation on a discontinuous gradient of iodixanol. HSCs were harvested from the 11.5% medium interface, washed, and seeded in tissue culture plates. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, United States) with 10% fetal bovine serum (Sijiqing Bio. Co. Hangzhou, China), 100 U/mL penicillin, and 100 μg/mL streptomycin. Culture medium was changed every third day. All experiments were performed with cells from passage numbers 3-6.
Identification of primary HSCs and activated HSCs
The harvested primary HSCs were studied at days 1, 2, and 5 after isolation. Primary d1 HSCs showed intrinsic fluorescence of lipid droplet and desmin expression (Boster, Wuhan, China) under a fluorescence microscope. Nuclei cells were stained with DAPI. After the first subculture passage, activated HSC purity was assessed using α-smooth muscle actin (α-SMA, dilution factor 1:100; Boster) and immunocytochemical staining.
IL-6 secretion
Cells cultured in 6-well plates were exposed to different concentrations of IL-17A for 24 h. The amount of IL-6 in supernatants was determined using enzyme linked immunosorbent assay kits purchased from R and D Systems (Minneapolis, MN).
Western blotting analysis
Cells exposed to IL-17A in the presence or absence of inhibitors for the indicated time period were extracted with lysis buffer containing a phosphatase inhibitor cocktail and a protease inhibitor cocktail. The protein concentration was measured using the Bradford assay. Proteins from each sample were loaded equally and separated on a 6% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel. Proteins were electrophoretically transferred onto polyvinylidene fluoride membranes, which were then incubated with primary antibodies at 4 °C overnight. On the following day, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at 37 °C for 2 h and then signals were detected using chemiluminescence (ECL Plus, GE Healthcare). Glyceraldehyde-3-phosphate dehydrogenase [(GAPDH); dilution factor 1:2000, Santa Cruz Biotechnology] was used as a loading control.
RNA extraction and gene expression analysis
Real-time qPCR was used to examine the expression of IL-17RA and IL-6 in HSCs. Total RNAs were isolated from HSCs using the Trizol reagent following the manufacturer’s protocol and reverse-transcribed using a cDNA synthesis kit (Promega). SYBR Green detection was used and the values were normalized using GAPDH. Real-time qPCR was performed with a DNA Engine (ABi 7500) using SYBR GREENER qPCR UNIVERSAL. Primer sequences were: IL-17RA (forward) 5'-TGGCGGTTCTCCTTCAGTC-3' and IL-17RA (reverse) 5'-CGGTGTAGTCATCTTCATCTCC-3', IL-6 (forward) 5'-CGTTTCTACCTGGAGTTTGTG-3' and IL-6 (reverse) 5'-ATTAGGAGAGCATTGGAAGTTGG-3', and GAPDH (forward) 5'-TTCAACGGCACAGTCAAGG-3' and GAPDH (reverse) 5'-CTCAGCACCAGCATCACC-3'. Relative expression levels of each primer set were normalized to GAPDH expression.
Statistical analysis
Data were expressed as mean ± SD. The statistical significance of changes was determined using the t-test. P values < 0.05 were considered to indicate statistical significance.
RESULTS
IL-17RA shRNA lentiviral construction and transduction
The sequences of IL-17RA shRNAs 1, 2, 3, and 4 and random shRNA are shown in Table 1. The recombinant plasmid of pGCSIL/IL-17RA shRNA 1 was confirmed by sequence analysis (Figure 1A) and titers were approximately 1 × 109 TU/mL. The interference efficiency of IL-17RA shRNAs 1, 2, 3, and 4 are shown in Figure 1B. The relative expression of IL-17RA mRNA for IL-17RA shRNAs 1, 2, 3, and 4 were 0.253 ± 0.011, 0.643 ± 0.022, 0.673 ± 0.051, and 0.444 ± 0.043, respectively. IL-17RA shRNA 1 exhibited a significant silencing effect, with 74.7% interference efficiency. Figure 1C shows HSC morphology with successfully transduced GFP-labeled IL-17RA shRNA 1 using a fluorescence or light microscope (× 200) after 72 h.
Table 1.
Interleukin-17A receptor short hairpin RNA and random short hairpin RNA sequences
| Target sequence | Double strand DNA oligo sequence | |
| IL-17RA shRNA 1 | CAAGTCCAAGACCATCTTA | 5'-ccggcaCAAGTCCAAGACCATCTTAttcaagagaTAAGATGGTCTTGGACTTGtgtttttg-3' |
| 5'-aattcaaaaacaCAAGTCCAAGACCATCTTAtctcttgaaTAAGATGGTCTTGGACTTGtg-3' | ||
| IL-17RA shRNA 2 | CCAGCGATCCAATGTCACA | 5'-ccggcaCCAGCGATCCAATGTCACAttcaagagaTGTGACATTGGATCGCTGGtgtttttg-3' |
| 5'-aattcaaaaacaCCAGCGATCCAATGTCACAtctcttgaaTGTGACATTGGATCGCTGGtg-3' | ||
| IL-17RA shRNA 3 | CAGCAGCCATGAACATGAT | 5'-ccggcaCAGCAGCCATGAACATGATttcaagagaATCATGTTCATGGCTGCTGtgtttttg-3' |
| 5'-aattcaaaaacaCAGCAGCCATGAACATGATtctcttgaaATCATGTTCATGGCTGCTGtg-3' | ||
| IL-17RA shRNA 4 | GGAAGAAAGTGGAGTGGTA | 5'-ccgggaGGAAGAAAGTGGAGTGGTAttcaagagaTACCACTCCACTTTCTTCCtctttttg-3' |
| 5'-aattcaaaaagaGGAAGAAAGTGGAGTGGTAtctcttgaaTACCACTCCACTTTCTTCCtc-3' | ||
| Random shRNA | TTCTCCGAACGTGTCACGT | 5'-ccggTTCTCCGAACGTGTCACGTttcaagagaACGTGACACGTTCGGAGAAtttttg-3' |
| 5'-aattcaaaaaTTCTCCGAACGTGTCACGTtctcttgaaACGTGACACGTTCGGAGAA-3' |
shRNA: Short hairpin RNA; IL-17RA: Interleukin-17A receptor.
Figure 1.
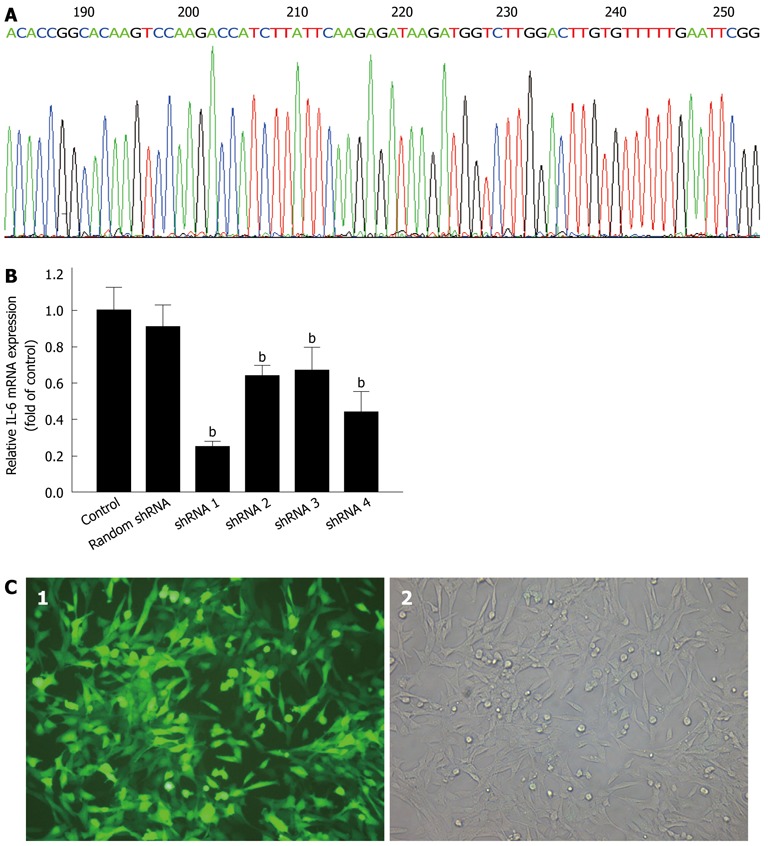
Short hairpin RNA design, interference efficiency assay, and lentivirus transduction. A: DNA sequence analysis of pGCSIL/Interleukin-17A receptor (IL-17RA) short hairpin RNA (shRNA) 1; B: Lentiviral-mediated IL-17RA shRNA 1, 2, 3, and 4 (shRNA 1, 2, 3, and 4) inhibited IL-17RA mRNA expression in hepatic stellate cells (HSCs). HSCs infected with lentivirus at a multiplicity of infection of 10 for 72 h, and IL-17RA mRNA expression was quantified by polymerase chain reaction. IL-17RA shRNA 1 was the most efficient silencing tool for IL-17RA. bP < 0.01 vs control; C: Cell morphology of HSCs under fluorescence (1) and light (2) microscope after 72 h transduction.
Isolation and culture of rat HSCs
Primary HSC culture after isolation on days 1, 2, and 5 are shown in Figure 2A. The cultured HSCs were assessed for intrinsic fluorescence of lipid droplet and desmin expression (Boster; Figure 2B). The purity of activated HSCs was confirmed using immunocytochemical staining for α-smooth muscle actin (Boster; Figure 2C); the purity reached > 98%.
Figure 2.
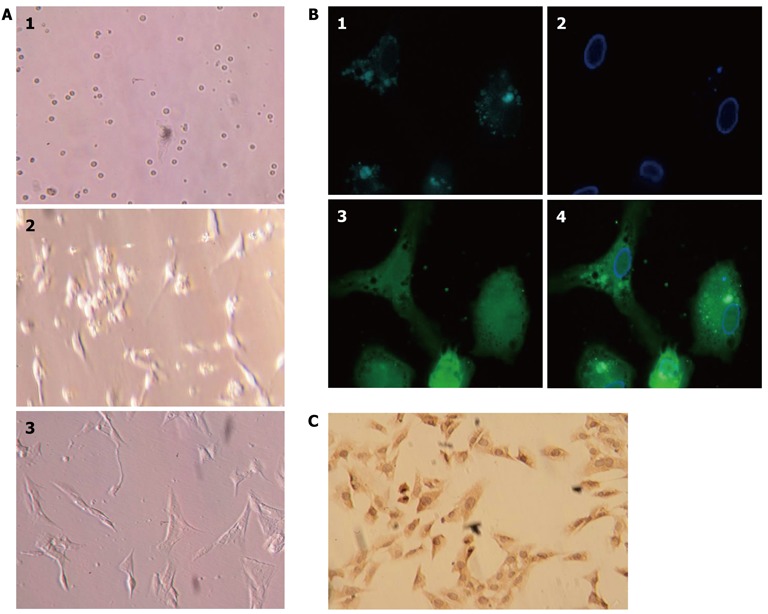
Isolation, culture, and identification of primary rat hepatic stellate cells. A: Primary rat hepatic stellate cells (HSCs) on days 1, 2, and 5 after isolation and plated in culture; B: HSCs isolated from Sprague-Dawley rats showed characteristic multiple lipid droplets with a rapid fade of the blue-green autofluorescence at 328 nm. 1: Lipid droplets; 2: Cell nucleus; 3: Cell body. 4: Merged images; C: HSC culture after the first passage and stained with anti-α-smooth muscle actin antibody.
IL-17A induces IL-6 expression
HSCs were incubated with increasing concentrations of IL-17A for 24 h, and the levels of IL-6 secretion in the supernatant were measured by ELISA. As shown in Figure 3A, IL-17A-induced IL-6 secretion increased in a dose-dependent manner. The expression level of IL-6 mRNA by IL-17A was quantified using real-time qPCR (Figure 3B). A rapid increase in IL-17A-induced IL-6 mRNA expression reached a maximum level at 3 h of IL-17A exposure. However, the level of IL-6 mRNA expression decreased gradually after 3 h exposure.
Figure 3.
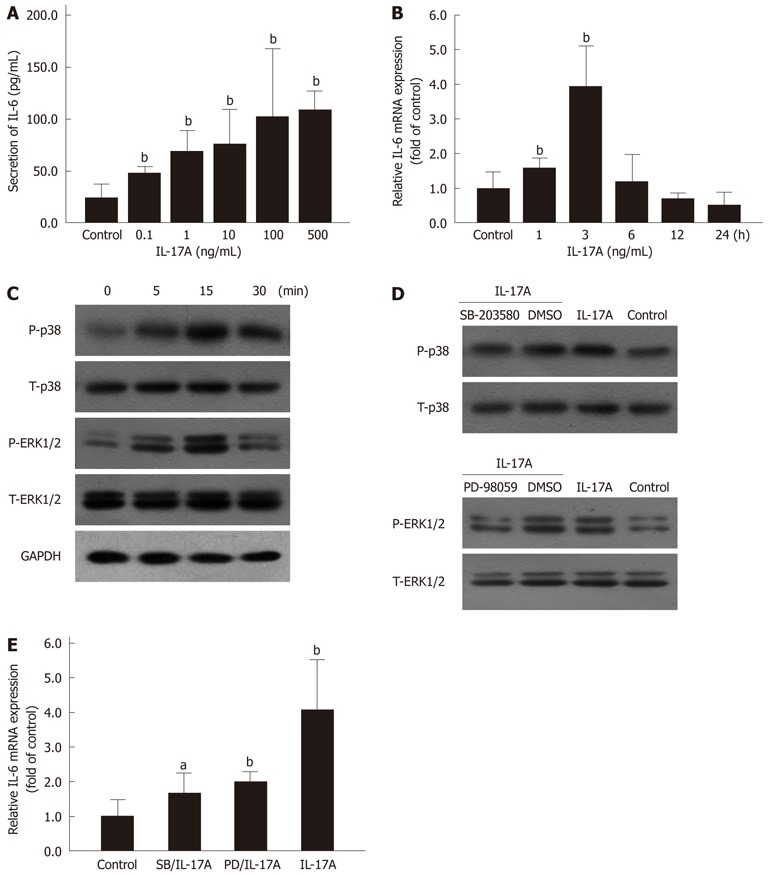
Mitogen activated protein kinases pathway involved in interleukin 17A induced interleukin 6 expression. A: Secretion of interleukin (IL)-6 in hepatic stellate cells (HSCs) induced by interleukin 17A (IL-17A) was determined using enzyme-linked immunosorbent assay. bP < 0.01 vs control; B: IL-6 mRNA expression in HSCs induced by IL-17A (100 ng/mL) was measured by polymerase chain reaction. bP < 0.01 vs control; C: Phosphorylation of p38 MAPK and ERK1/2 induced by IL-17A (100 ng/mL) in HSCs was detected using Western blotting; D: Phosphorylation of p38 MAPK and ERK1/2 induced by IL-17A (100 ng/mL) for 15 min was blocked by preincubation with MAPKs inhibitors, SB-203580 (1 μmol/L in DMSO, 30 min) and PD-98059 (10 μmol/L in DMSO, 1 h); E: IL-6 mRNA expression induced by IL-17A (100 ng/mL) for 3 h exposure was inhibited by preincubation with SB-203580 (SB) and PD-98059 (PD) for the same time period as above. aP < 0.05, bP < 0.01 vs control. T: Total; P: Phosphorylation; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; MAPK: Mitogen activated protein kinases; ERK: Extracellular regulated protein kinases.
IL-17A induces activation of MAPKs
In various cells, the MAPK family has been shown to play an important role in regulating gene expression in response to inflammatory mediators[7]. To investigate whether IL-17A induced activation of p38 MAPK and ERK1/2 in HSCs, phosphorylation of p38 MAPK and ERK1/2 in HSCs were evaluated after IL-17A stimulation. IL-17A induced rapid phosphorylation of p38 MAPK and ERK1/2 after 5 min, and showed the strongest levels of phosphorylation of p38 MAPK and ERK1/2 at 15 min (Figure 3C). However, IL-17A did not affect the expression of total p38 MAPK or ERK1/2. This result indicated that IL-17A enhanced p38 MAPK and ERK1/2 phosphorylation in HSCs.
MAPK inhibitors suppress IL-6 expression
To further assess the phosphorylation of p38 and ERK1/2 induced by IL-17A, two specific inhibitors, SB-203580 (p38 MAPK) and PD-98059 (ERK1/2), were used. As shown in Figure 3D, SB-203580 inhibited p38 MAPK phosphorylation and PD-98059 blocked phosphorylation of ERK1/2 induced by IL-17A in HSCs. SB-203580 and PD-98059 were used to further evaluate the roles of p38 MAPK and ERK1/2 in IL-6 mRNA expression induced by IL-17A in HSCs. Both inhibitors reduced IL-17A-induced IL-6 mRNA expression significantly (Figure 3E).
shRNA suppresses IL-17RA and IL-6 expression
Western blotting and real-time qPCR were performed to study the silencing effect of lentiviral-mediated shRNA on IL-17RA-induced IL-6 expression in HSCs. The protein levels of IL-17RA in shRNA 1-treated HSCs were reduced, compared with random shRNA and control (Figure 4A). HSCs treated with IL-17A alone or IL-17A with random shRNA showed increased IL-6 mRNA expression, whereas, IL-6 mRNA expression in HSCs pretreated with IL-17RA shRNA 1 was decreased significantly (Figure 4B).
Figure 4.
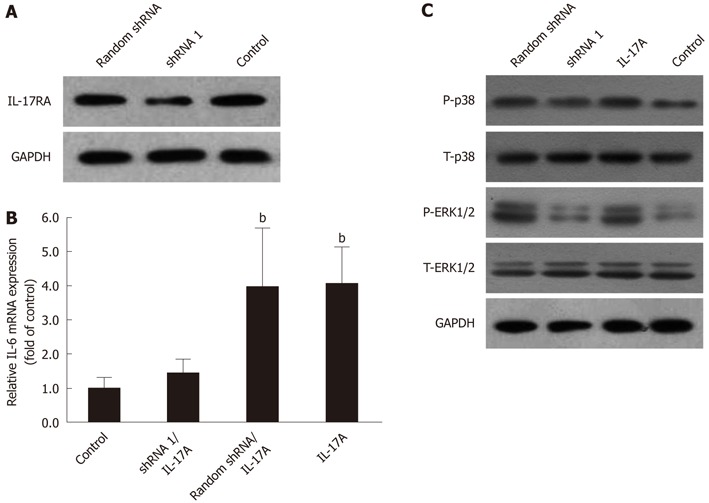
Lentiviral-mediated interleukin 17A receptor short hairpin RNA arrested interleukin 6 expression, partly through suppressing phosphorylation of p38 mitogen activated protein kinases and extracellular regulated protein kinases 1/2. A: Lentiviral-mediated interleukin (IL)-17A receptor short hairpin RNA (shRNA) 1 [at multiplicity of infection (MOI) = 10 for 72 h] inhibited the expression of IL-17RA in hepatic stellate cells (HSCs); B: Lentiviral-mediated IL-17RA shRNA 1 inhibited IL-17A-induced IL-6 mRNA expression in HSCs. HSCs were infected with IL-17RA shRNA 1 or random shRNA lentivirus at MOI = 10 for 72 h, and followed by 3 h exposure to IL-17A (100 ng/mL). bP < 0.01 vs control; C: Lentiviral-mediated IL-17RA shRNA 1 inhibited phosphorylation of p38 MAPK and ERK1/2. HSCs were transduced with IL-17RA shRNA 1 or random shRNA lentiviral at MOI = 10 for 72 h, and followed by 15 min exposure to IL-17A (100 ng/mL). T: Total, P: Phosphorylation; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
shRNA suppresses phosphorylation of p38 and ERK1/2
IL-17A induced IL-6 expression via p38 MAPK and ERK1/2 phosphorylation. In Figure 4C, we show that HSCs treated with IL-17A transduced with shRNA 1 exhibited reduced protein levels of phosphorylation of p38 MAPK and ERK1/2, compared with IL-17A alone and IL-17A transduced with random shRNA group. Phosphorylation of p38 MAPK and ERK1/2 was blocked in the group treated with lentiviral-mediated IL-17RA shRNA 1, whereas there was almost no change in the IL-17A alone or IL-17A transduced with random shRNA groups.
DISCUSSION
IL-17A is a pro-inflammatory cytokine secreted by a subset of T helper cells, named Th17 cells. IL-17A signals exert miscellaneous effects through binding to the IL-17A receptor, which is expressed on a variety of cells and tissues[9,10]. The IL-17A pathway plays important roles in the human inflammatory and autoimmune diseases and tumor progression[11,12]. Clinical studies have shown that high levels of IL-17A and other cytokines related to the IL-17A pathway are seen in sera or tissues of patients with diseases such as psoriasis, multiple sclerosis, systemic sclerosis, ankylosing spondylitis, and juvenile idiopathic arthritis[13-17]. The IL-17RA is found on hepatocytes, Kupffer cells, HSCs, biliary epithelial cells, and sinusoidal endothelial cells[18]. Thus, IL-17A function in liver diseases has attracted much attention. Previous studies indicated that IL-17A is elevated in various liver diseases, including liver autoimmunity and inflammatory diseases, alcoholic liver disease (ALD), and hepatocellular carcinoma[19]. Studies based on acute hepatic injury (AHI) and hepatitis C virus (HCV) showed that patients with AHI and HCV have significantly higher serum IL-17A levels than controls[20-22]. Furthermore, percentages of circulating Th17 cells and Th17-associated cytokines, including IL-17A, IL-6, and IL-23, were increased markedly in peripheral blood and in liver tissues of chronic hepatitis B patients compared with controls[23,24]. Recent studies indicate that patients with ALD had significantly higher plasma levels of IL-17A compared with healthy subjects. Moreover, the number of liver Th17 cells was correlated positively with hepatocellular damage and the severity of the disease in a cohort study of patients with ALD[25].
HSCs are the major source of extracellular matrix in liver fibrosis[26] and participate in modulating liver inflammation during liver fibrogenesis[27]. IL-17RA is expressed on HSCs, and thus blockade of IL-17A/IL-17RA signal transduction in HSCs may be a strategy to interfere with hepatic inflammatory processes in chronic liver diseases. In our study, we sought to investigate the secretion of IL-6, one of the major cytokines in various liver injuries[28-30], stimulated by IL-17A in HSCs. Our data show that IL-17A induces a large amount of IL-6 secretion (68.96 ± 8.30 pg/mL) even at a low concentration of IL-17A treatment (1 ng/mL). With increasing concentrations of IL-17A, the secretion of IL-6 also increased. Surprisingly, a higher concentration of IL-17A (500 ng/mL) induced only moderate expression of IL-6 (109.16 ± 6.91 pg/mL). This may suggest that there is saturation of binding between IL-17A and IL-17RA at a particular concentration or that there may be a negative feedback response on IL-6 expression on IL-17A induction. Our data further show that the IL-6 mRNA expression detected in real-time qPCR reached a maximum level after 3 h exposure to IL-17A and declined thereafter. These results are based on data obtained from the expression of IL-6 protein in HSCs supernatants and indicate that IL-17A induces IL-6 expression in vitro. We made a lentivirus-mediated IL-17RA shRNA construct to repress endogenous IL-17RA in HSCs. As a result, we found that IL-6 mRNA expression was notably reduced. Our results further suggest that the MAPK pathway in IL-6 expression is induced by IL-17A in HSCs. We showed that the phosphorylation of p38 MAPK and ERK1/2 is increased after 15 min exposure to IL-17A, and lentivirus-mediated IL-17RA shRNA decreased the phosphorylation of p38 MAPK and ERK1/2. Moreover, use of specific inhibitors further revealed the importance of the MAPK pathway in IL-17A-induced IL-6 mRNA expression in HSCs. The imidazole compound SB-203580, a specific inhibitor of p38 MAPK, caused a significant decrease in IL-17A-induced IL-6 mRNA expression. Additionally, PD-98059, a specific inhibitor of MEK1 that is the directly upstream protein kinase of ERK1/2, promotes marked suppression of IL-17A induced IL-6 mRNA expression. Thus, we conclude that the p38 MAPK and ERK1/2 pathways are involved in IL-6 mRNA expression induced by IL-17A in HSCs. Hot et al[31] indicated that IL-17A stimulated IL-6 secretion by inducing activation of all three MAPKs (ERK, p38, JNK). Inhibition of IL-17RA expression via siRNA lead to near complete abrogation of IL-6 expression mediated by IL-17A.
IL-6 is an important cytokine in regulating different inflammatory responses in liver diseases and is a marker in the diagnosis of symptoms of liver cirrhosis[32-34]. Previous studies have described that IL-6, in conjunction with TGF-β, promotes Th17 cell differentiation and drives IL-17A production[35,36]. On the other hand, IL-17A can also stimulate IL-6 expression[37] and IL-6 is a key downstream target gene for IL-17A in non-immune cells, such as fibroblasts[38]. IL-17A triggers a positive feedback loop of IL-6 signaling and forms the “IL-6 amplifier”[37,38]. IL-17A plays a pivotal role in immune and non-immune tissues[37]. The IL-6 amplifier exerts its effects through activation and phosphorylation of both NF-κB and STAT3 proteins. It is also known that HSCs are involved in extracellular matrix degradation and maintenance, imbalances in which lead to liver fibrosis, as a result of unmatched levels of metalloproteinases, inhibitors of fibrillary collagen, and tissue inhibitors of metalloproteinase-1 and -2[2]. Thus, it is important to examine the role of the IL-17A/IL-6 pathway in HSCs. Zhao et al[39] indicated that IL-17A induced IL-6 expression via the MAPK signaling pathway in hepatocytes, which, in turn, may further stimulate Th17 cells and forms a positive feedback loop. They concluded that Th17 cells and the IL-17A signaling pathway played an important role in targets for AIH. Yan et al[40] demonstrated that acute Con A-induced liver injury was reduced in mice treated with an adenovirus vector encoding a soluble IL-17R immunoglobulin (Ig)G fusion (AdIL-17R:Fc) that neutralized the interaction between IL-17A and IL-17RA. This supported the key role of IL-17A/IL-17RA signaling in Con A-induced hepatitis. Blockade of the IL-17A/IL-17RA signaling pathway may provide a novel therapeutic target in human autoimmune-related hepatitis. Genovese et al[41] also reported that IL-17A neutralization can achieve positive results in rheumatoid arthritis.
Consistent with previous reports in other cells, our data indicate that IL-17A induces IL-6 expression in HSCs. Suppression of IL-17RA with lentivirus-mediated IL-17RA shRNA inhibited IL-6 expression and blocked IL-17A-triggered positive-feedback loop of IL-6 signaling. Additionally, phosphorylation of p38 MAPK and ERK1/2, induced by IL-17A, was blocked in the presence of IL-17RA shRNA. Negative regulation of MAPK signaling using two specific inhibitors (SB-203580 and PD-98059) for p38 MAPK and ERK1/2, respectively, significantly attenuated IL-6 mRNA expression. These results indicated that blocking IL-17RA expression may be an alternative strategy to reduce the inflammatory response induced by IL-17A in the liver. Further, this study allows us to further understand the function of IL-17RA in HSCs. We would like to examine more potential target substrates that may be affected by the IL-17A/IL-17RA signaling pathway in the future and to further examine the underlying mechanism of HSCs in liver fibrosis.
ACKNOWLEDGMENTS
The study protocol and animal care were approved by the Animal Care and Use Committee of Wenzhou Medical College.
COMMENTS
Background
Liver fibrosis results from chronic damage to the liver in accompany with repeated chronic liver inflammation and evenly evolves into liver cirrhosis. Following chronic injury, Hepatic stellate cells (HSCs) activate, migrate and accumulate at the sites of tissue repair, acquiring contractile, proinflammatory, and fibrogenic properties. Activated HSCs secrete large amounts of extracellular matrix (ECM) and regulate ECM degradation. HSCs are also involved in modulating liver inflammation in liver fibrogenesis. But the mechanism involved in the secretion of interleukin (IL) 6 (which is a crucial cytokine in regulating different inflammatory responses in liver diseases and is a marker to diagnose symptoms of liver cirrhosis) mediated by IL-17 stimulation in HSCs is not understood totally.
Research frontiers
IL-17A is a pro-inflammatory cytokine secreted by a new subset T help cells named Th17 cells. IL-17A signals exert miscellaneous effects through binding to the IL-17A receptor (IL-17RA) which is expressed in a variety of cells and tissues. In recently years, Clinical studies have shown that high levels of IL-17A and other cytokines related to IL-17A pathway exhibit various liver diseases including liver autoimmunity and inflammatory diseases, viral hepatitis, alcoholic liver disease and hepatocellular carcinoma. The research hotspot is how to reduce liver injury by modify liver inflammatory response, and then inhibit or postpone liver fibrosis and cirrhosis.
Innovations and breakthroughs
The present study indicated that IL-17A induces IL-6 secretion via p38 MAPK and ERK1/2 pathway by binding to IL-17RA in HSCs. Suppression of IL-17RA using lentiviral-mediated IL-17RA shRNA inhibits IL-6 secretion.
Applications
The study results suggest that IL-17 could stimulate IL-6 secretion via p38 MARK and ERK1/2 pathway in HSCs. Downregulation of IL-17RA receptor by Lentiviral vector-mediated IL-17RA shRNA decreases IL-6 expression induced by IL-17A via p38 MAPK and ERK1/2 Phosphorylation in HSCs. Suppression of IL-17RA expression may be an optional strategy to impair the inflammation response induced by IL-17A in the liver. This suggests that modulating IL-17RA expression might be a strategy to interfere with hepatic inflammatory processes in various liver diseases.
Terminology
HSCs are the main ECM-producing cells in the injured liver. In the normal liver, HSCs reside in the space of Disse and are the major storage sites of vitamin A. Following chronic injury, HSCs activate or transdifferentiate into myofibroblast-like cells, acquiring contractile, proinflammatory, and fibrogenic properties. Activated HSCs migrate and accumulate at the sites of tissue repair, secreting large amounts of ECM and regulating ECM degradation. If the hepatic injury persists, then eventually the liver regeneration fails, and hepatocytes are substituted with abundant ECM, including fibrillar collagen. As fibrotic liver diseases advance, disease progression from collagen bands to bridging fibrosis to frank cirrhosis occurs.
Peer review
The manuscript is addressing clearly the objective of the study. The results sustain the conclusion obtained and the experiments are performed properly. The novelty is enough to be an interesting paper.
Footnotes
Supported by The Zhejiang Extremely Key Subject of Surgery; and The Wenzhou Key Laboratory Project in Surgery
Peer reviewers: Sabine Mihm, Professor, Department of Gastroenterology, Georg-August-University, Robert-Koch-Str.40, Göttingen D-37099, Germany; Juan-Ramón Larrubia, PhD, Gastroenterology Unit and Liver Research Unit, Guadalajara University Hospital, Donante de Sangre s/n, 19002 Guadalajara, Spain
S- Editor Cheng JX L- Editor A E- Editor Zheng XM
References
- 1.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 3.Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416. doi: 10.1055/s-2001-17554. [DOI] [PubMed] [Google Scholar]
- 4.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 5.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 6.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 7.Patel DN, King CA, Bailey SR, Holt JW, Venkatachalam K, Agrawal A, Valente AJ, Chandrasekar B. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-kappaB and C/EBPbeta activation. J Biol Chem. 2007;282:27229–27238. doi: 10.1074/jbc.M703250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiskirchen R, Gressner AM. Isolation and culture of hepatic stellate cells. Methods Mol Med. 2005;117:99–113. doi: 10.1385/1-59259-940-0:099. [DOI] [PubMed] [Google Scholar]
- 9.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 10.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 12.Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 13.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, Takabayashi K, Iwamoto I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000;43:2455–2463. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74:304–305. doi: 10.1016/j.jbspin.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal S, Misra R, Aggarwal A. Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J Rheumatol. 2008;35:515–519. [PubMed] [Google Scholar]
- 18.Lafdil F, Miller AM, Ki SH, Gao B. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol. 2010;7:250–254. doi: 10.1038/cmi.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye C, Li WY, Zheng MH, Chen YP. T-helper 17 cell: A distinctive cell in liver diseases. Hepatol Res. 2011;41:22–29. doi: 10.1111/j.1872-034X.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- 20.Yasumi Y, Takikawa Y, Endo R, Suzuki K. Interleukin-17 as a new marker of severity of acute hepatic injury. Hepatol Res. 2007;37:248–254. doi: 10.1111/j.1872-034X.2007.00040.x. [DOI] [PubMed] [Google Scholar]
- 21.Lan RY, Salunga TL, Tsuneyama K, Lian ZX, Yang GX, Hsu W, Moritoki Y, Ansari AA, Kemper C, Price J, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol. 2009;157:261–270. doi: 10.1111/j.1365-2249.2009.03947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge J, Wang K, Meng QH, Qi ZX, Meng FL, Fan YC. Implication of Th17 and Th1 cells in patients with chronic active hepatitis B. J Clin Immunol. 2010;30:60–67. doi: 10.1007/s10875-009-9328-2. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, Fu JL, Shi F, Shi M, Wang HF, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 2010;51:81–91. doi: 10.1002/hep.23273. [DOI] [PubMed] [Google Scholar]
- 25.Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 26.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 27.Marra F. Hepatic stellate cells and the regulation of liver inflammation. J Hepatol. 1999;31:1120–1130. doi: 10.1016/s0168-8278(99)80327-4. [DOI] [PubMed] [Google Scholar]
- 28.Choi I, Kang HS, Yang Y, Pyun KH. IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin Exp Immunol. 1994;95:530–535. doi: 10.1111/j.1365-2249.1994.tb07031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deviere J, Content J, Denys C, Vandenbussche P, Schandene L, Wybran J, Dupont E. High interleukin-6 serum levels and increased production by leucocytes in alcoholic liver cirrhosis. Correlation with IgA serum levels and lymphokines production. Clin Exp Immunol. 1989;77:221–225. [PMC free article] [PubMed] [Google Scholar]
- 30.Mas E, Danjoux M, Garcia V, Carpentier S, Ségui B, Levade T. IL-6 deficiency attenuates murine diet-induced non-alcoholic steatohepatitis. PLoS One. 2009;4:e7929. doi: 10.1371/journal.pone.0007929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hot A, Zrioual S, Toh ML, Lenief V, Miossec P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis. 2011;70:341–348. doi: 10.1136/ard.2010.132233. [DOI] [PubMed] [Google Scholar]
- 32.Malaguarnera M, Di Fazio I, Romeo MA, Restuccia S, Laurino A, Trovato BA. Elevation of interleukin 6 levels in patients with chronic hepatitis due to hepatitis C virus. J Gastroenterol. 1997;32:211–215. doi: 10.1007/BF02936370. [DOI] [PubMed] [Google Scholar]
- 33.Giannitrapani L, Cervello M, Soresi M, Notarbartolo M, La Rosa M, Virruso L, D’Alessandro N, Montalto G. Circulating IL-6 and sIL-6R in patients with hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:46–52. doi: 10.1111/j.1749-6632.2002.tb04093.x. [DOI] [PubMed] [Google Scholar]
- 34.Genesca J, Gonzalez A, Segura R, Catalan R, Marti R, Varela E, Cadelina G, Martinez M, Lopez-Talavera JC, Esteban R, et al. Interleukin-6, nitric oxide, and the clinical and hemodynamic alterations of patients with liver cirrhosis. Am J Gastroenterol. 1999;94:169–177. doi: 10.1111/j.1572-0241.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- 35.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 36.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 37.Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 38.Hirano T. Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:717–730. doi: 10.2183/pjab.86.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, Qiu D, Wei J, Liu Y, Shen L, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One. 2011;6:e18909. doi: 10.1371/journal.pone.0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan S, Wang L, Liu N, Wang Y, Chu Y. Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol Cell Biol. 2012;90:421–428. doi: 10.1038/icb.2011.59. [DOI] [PubMed] [Google Scholar]
- 41.Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929–939. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]