

Abstract
Chronic malaria severely affects the immune system and causes polyclonal B-cell activation, as evidenced by the presence of hypergammaglobulinemia, elevated levels of autoantibodies, loss of B-cell memory and the frequent occurrence of Burkitt’s lymphomas (BL) in children living in malaria endemic areas.
Previous studies have shown that the cysteine-rich interdomain region 1α (CIDR1α) of the Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) of the FCR3S1.2 strain, subsequently named CIDR1α, interacts with B cells partially through the binding to the B-cell receptor (BCR). This interaction leads to an activated phenotype, increased survival, and a low degree of proliferation. CIDR1α preferentially activates the memory B-cell compartment, therefore PfEMP1 is considered to act as a polyclonal B-cell activator and its role in memory maintenance has been suggested.
In this report, we extend the analysis of the PfEMP1–CIDR1α B-cell interaction and demonstrate that PfEMP1–CIDR1α increases the expression of TLR7 and TLR10 mRNA transcripts and sensitizes B cells to TLR9 signalling via the MyD88 adaptor molecule. Furthermore, despite its ability to bind to surface Igs, PfEMP1–CIDR1α-induced B-cell activation does not seem to proceed through the BCR, since it does not induce Lyn and/or phospho-tyrosine mediated signalling pathways. Rather PfEMP1–CIDR1α induces the phosphorylation of downstream kinases, such as ERK1/2, p38 and IKBα, in human B cells. These findings indicate that PfEMP1–CIDR1α induces a persistent activation of B cells, which in turn can contribute to the exhaustion and impairment of B-cell functions during chronic malaria infection.
Keywords: B cells, Toll-like receptors (TLRs), Malaria, Signal transduction
1. Introduction
Infection with Plasmodium falciparum is still a major health problem worldwide, causing about 225 million new malaria cases each year, according to the WHO malaria report 2010. Malaria severely affects the immune system, in particular the B-cell compartment, as indicated by the presence of hypergammaglobulinemia, elevated autoantibody titres, and the frequent occurrence of Burkitt’s lymphoma in children living in malaria holoendemic regions (Abele et al., 1965; Adu et al., 1982; McGregor et al., 1956; Greenwood and Vick, 1975; Banic et al., 1991; Bates and Bedu-Addo, 1997). The mechanisms leading to this B-cell disregulation are not fully understood.
A variety of malarial proteins that might affect B-cell functions are expressed at the surface of the parasitized red-blood cells (pRBCs). Attention has been focussed on the P. falciparum erythrocyte membrane protein 1 (PfEMP1) family, a highly polymorphic and modular family of proteins composed of Duffy binding-like (DBL) and cysteine-rich interdomain regions (CIDR) (Su et al., 1995; Chen et al., 2000; Flick et al., 2001). Previous studies have shown that the CIDR1α of PfEMP1 from the FCR3S1.2 strain binds to CD36, PECAM-1/CD31, and to the Fab- and Fc-fragments of immunoglobulins (Ig) from various classes (IgG, IgM) and different species (Chen et al., 1998; Donati et al., 2004). Furthermore, CIDR1α binds to and directly activates purified human B cells from non immune donors inducing activation, proliferation, increased survival and antibody secretion. These characteristics led to the definition of PfEMP1–CIDR1α as a polyclonal B-cell activator (Donati et al., 2004, 2006).
At present, little is known about the intracellular mechanisms triggered by the binding of PfEMP1–CIDR1α to B cells. Earlier characterization and comparison of the gene-expression profile induced by PfEMP1–CIDR1α and by anti-Ig activation of human B cells demonstrated a difference in the signatures imposed by these stimuli (Donati et al., 2006). The results suggested that the PfEMP1–CIDR1α-induced activation involves receptors other than Igs or concomitantly through Igs with additional receptors, which would lead to the activation of different signalling pathways (Donati et al., 2006).
The B-cell receptor (BCR) found on mature B cells is a multiprotein complex consisting of an antigen binding subunit, the membrane Ig (mIg), and a signalling subunit. The latter is a disulfide-linked heterodimer comprising the Igα and Igβ proteins, each containing a single immunoreceptor tyrosine-based activation motif (ITAM) within their cytoplasmic tail. Following BCR cross-linking, the Src-family protein tyrosine kinase (PTK) Lyn phosphorylates the ITAMs favouring the induction of several kinase cascades, such as mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), IkB kinase (IKK) complex, and phosphatidylinositol 3-kinase (PI3K)/Akt (Campbell, 1999; Dal Porto et al., 2004).
In addition to the specific immune response mediated by BCR engagement, Toll-like receptors (TLRs) are known to play a key role in B-cell interaction with pathogens (Bernasconi et al., 2002; Trinchieri and Sher, 2007). To date ten different TLRs have been identified in humans. TLRs are pattern recognition receptors (PRRs) that recognize a wide range of microbial motifs at the cell surface (TLRs 1, 2, 4, 5, 6 and 10) or within endosomes (TLRs 3, 7, 8 and 9), leading to the activation of innate immune responses (Takeda et al., 2003). All TLRs, except TLR3, use the downstream adaptor molecule myeloid differentiation factor 88 (MyD88), whereas TLR3 signals via the TIR domain-containing adaptor-inducing IFN-γ (TRIF); TLR4 recruits both (MyD88 and TRIF). Upon activation, MyD88 initiates signalling cascades that promote NF-kB and activator protein 1 (AP-1) activation leading to subsequent inflammatory responses (Akira and Takeda, 2004).
TLRs are highly expressed on antigen-presenting cells, such macrophages and dendritic cells (DCs), and promote their recruitment and maturation. Consequently, TLRs indirectly control T-and B-cell responses (Reis e Sousa, 2006; Bourke et al., 2002). On human B cells, TLRs (TLR2, 6, 7, 9 and 10) are expressed almost exclusively in the memory subset (Ruprecht and Lanzavecchia, 2006; Lanzavecchia and Sallusto, 2007). Although their relevance in the formation of Ab responses remains controversial, a role for TLRs in regulating human B-cell immunity is widely accepted (Ruprecht and Lanzavecchia, 2006; Lanzavecchia and Sallusto, 2007; Bernasconi et al., 2003; Poeck et al., 2003; Hornung et al., 2002; Bekeredjian-Ding et al., 2005).
Recent findings in a murine model point to the occurrence of a possible synergy between TLRs and other immune mechanisms in the host responsiveness to different antigens (Trinchieri and Sher, 2007). Noteworthy, the internalization of the BCR–antigen complex induces re-localization of TLR9 from endosomes to the same autophagosome-like compartments as the antigen-bound internalized BCR, which leads to a synergistic signalling through NF-kB and MAPKs phosphorylation (Chaturvedi et al., 2008).
To further understand the mechanisms involved in the PfEMP1–CIDR1α induced B-cell activation we studied the effect of PfEMP1–CIDR1α on TLRs, their expression and TLRs-related signalling pathways. Our results provide evidence that the PfEMP1–CIDR1α-mediated B-cell interaction results in an increased expression of TLR7 and TLR10 mRNA, and an enhanced TLR9-driven signalling via MyD88. The signalling induced by PfEMP1–CIDR1α does not seem to involve the BCR structure, since Lyn and other major BCR-triggered phosphorylation pathways are not activated. On the other hand, PfEMP1–CIDR1α triggers the phosphorylation of two members of the MAPK family: ERK1/2 and p38MAPK, and also of the inhibitor of kBα (IkBα). The results suggest that PfEMP1–CIDR1α-induced B-cell activation is exerted through these downstream kinases and may involve recruitment of nuclear factor-kB (NF-kB).
Taken together, PfEMP1–CIDR1α– induced B-cell activation seems to involve at least two distinct signalling pathways. The results of these multiple stimulation outcomes may play an important role in the aberrant B-cell activation that is characteristic of malaria infections.
2. Materials and methods
2.1. Production of recombinant Ags
PfEMP1–CIDR1α, from the cloned strain FCR3S1.2var1, was cloned in the pGEX-4T plasmid (Amersham Biosciences, Uppsala, Sweden) and expressed in Escherichia coli (BL21) as previously described (Chen et al., 2000). The PfEMP1–CIDR1α-GST fusion protein, referred to as PfEMP1–CIDR1α, was expressed and purified according to the manufacturer’s instructions. GST produced by the empty vector was used as control and is referred to as GST. The purity was determined by SDS-PAGE and Western blot, as described (Chen et al., 1998).
2.2. Cell isolation and cell cultures
Buffy coats from peripheral venous blood of healthy individuals who had not been previously exposed to malaria were obtained from the blood bank of the Karolinska Hospital. Mononuclear cells were isolated by centrifugation over Lymphoprep (Nycomed Pharma, Zurich, Switzerland). CD19+ B cells were purified by positive selection using an AutoMACS sorter (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instruction. In all the experiments more than 94% of recovered cells were CD19 positive as revealed by FACS analysis.
Purified B cells were resuspended in RPMI 1640 supplemented with 10% foetal calf serum (FCS) (GIBCO, Invitrogen Life Technologies, Carlsbad, CA, USA), 100 U/mL of penicillin and 2 mM glutamine, plated into 24-well plates (2 × 106 cells/well) in a final volume of 1 mL and cultured for up to 16 h at 37 °C in 5% CO2, in either medium alone or medium containing anti-Ig F(ab′)2 (Jackson ImunoResearch Laboratories), anti-human CD40 mAb S2C6 (Mabtech, Stockholm, Sweden), phosphorothioate-backbone modified CpG ODN 2006 (CpG) (Invitrogen), Imiquimod-R837 (Invivogen, San Diego, CA, USA), GST or PfEMP1–CIDR1α at final concentrations of 10 μg/mL, 1 μg/mL, 2.5 μg/mL, 1 μg/mL, 50 μg/mL and 100 μg/mL, respectively.
2.3. Proliferation assays
To assess cellular proliferation, purified B cells were plated into round-bottomed 96-well plates (5 × 104 cells/well) in a final volume of 200 μL. Cultures were seeded in triplicate in the presence of the same concentration of stimulants as above. A mixture of Phorbol myristate acetate (PMA) (5 ng/mL) plus iono-mycin (500 ng/mL) (Sigma–Aldrich, Taufkirchen, Germany) and Pansorbin (1:4000) (Calbiochem, San Diego, CA, USA) was used as positive control. To understand the effects of coincubation with both stimuli, 1 μg/mL Imiquimod-R837 (Invivogen) was added to PfEMP1–CIDR1α containing cultures at the initiation of the culture and after 12 h of incubation.
Cultures were incubated for 72 h and pulsed with 1 μCi of [3H]thymidine (Amersham Biosciences) during the last 12 h of the incubation period. Cells were harvested onto a fiberglass filter, and [3H]thymidine incorporation was determined by liquid scintillation counting. Results are expressed as the reactivity index (RI), calculated as follows: RI = counts per minute in stimulated cultures versus counts per minute in control cultures. The standard deviations (SD) of the triplicate cultures ranged between 10 and 15% of the mean.
2.4. Confocal microscopy
Purified CD19+ B cells attached to poly-lysine coated chamber slides (Bio-Coat, Horsham, PA, USA) were treated with anti-Ig F(ab′)2 (10 μg/mL), GST (50 μg/mL), PfEMP1–CIDR1α (100 μg/mL) at 37 °C from 2 to 60 min according to pilot (time-course) experiments carried out to determine the best stimulation time point for each protein kinase under consideration. Some experiments involved stimulation with a combination of anti-Ig F(ab′)2 and PfEMP1–CIDR1α. After incubation cells were fixed with 3.7% paraformaldehyde for 20 min at 25°, permeabilized with 0.2% TX-100 in PBS-BSA, blocked with 10% donkey serum for 20 min at 25°, and further incubated overnight at 4° with rabbit antibodies specific for phospho-Lyn, phospho-tyrosine, phospho-JNK, phospho-Akt, Phospho-PKC and total-IKK (Cell Signalling Technology, Beverly, MA, USA), phospho-ERK and phospho-p38 (R&D Systems, Minneapolis, MN, USA), conjugated phospho-PLCγ PE (Upstate, Billerica, MA, USA), and mouse antibody specific for phospho-IkBα (Santacruz Biotechnology, Santa Cruz, CA, USA). Thereafter, the Alexa-fluor-conjugated goat antibody (Invitrogen) specific for rabbit Ig and the Alexa-fluor-conjugated goat antibody (Invitrogen) specific for mouse Ig were added in the dark and kept for 45 min at 25°. The coverslips were mounted in Prolong Antifade (Invitrogen) and examined by fluorescence microscopy using a Zeiss 510 Meta confocal microscope (Carl Zeiss, Jena Germany).
2.5. Reverse transcriptase PCR (RT-PCR) and real-time PCR
Total RNA was extracted from the B-cell cultures at 3 h and 15 h using Qiagen’s QIamp RNA Blood minikit (Qiagen, Hilden, Germany) and reverse-transcribed using the Amersham T-Primed First-Strand Kit (Amersham Biosciences) according to the manufacturer’s instructions.
Four microliters of the cDNA (diluted 1:10) were used in the final 10 μl Real-time PCR reaction volume; each sample was measured in triplicate. Real-time PCR was performed using predesigned gene assays for TLR7 (ID Hs00152971 m1), TLR9 (ID Hs00152973 m1) and TLR10 (ID Hs00999403 m1) (Applied Biosystems, Carlsbad, CA, USA). The GAPDH gene was used as endogenous control and the relative amounts of mRNA were normalized to GAPDH RNA levels within each sample using an ABI PRISM 7500 sequence detector (Applied Biosystems) and Taqman reagents. Data are expressed as the fold increase between stimulated and unstimulated cells (value normalized to the unit: 1).
2.6. Immunoblot analysis
For analysis of MyD88 protein expression, B cells stimulated for 16 h were lysed in RIPA buffer (0.1% SDS, 0.1% sodium deoxy-cholate, 1% NP-40; 250 mM NaCl, 50 mM TrisHCl, pH 7.5). Lysates were subjected to 10% SDS gel separation, whereafter the proteins were transferred to a nitrocellulose membrane and analyzed by immunoblotting. Equal loading of the cell lysate was reassured by the determination of the protein concentration by the BCA assay (Pierce Biotechnology, Rockford, IL, USA), using BSA as standard. Blots were probed with anti-MyD88 antibody (1:1000) (Stressgen, Ann Arbor, MI, USA) and HRP-conjugated secondary Ab (Dako-Cytomation, Glostrup, Denmark), followed by ECL reagent (ECL) detection (Amersham Biosciences).
2.7. Statistical analysis
Paired two-tailed Student’s t test was performed. A value of p < 0.05 was considered significant.
3. Results
3.1. PfEMP1–CIDR1α stimulation affects TLRs mRNA expression in human B cells
Given the well known fact that TLR signalling affects B-cell activation (Ruprecht and Lanzavecchia, 2006; Lanzavecchia and Sallusto, 2007) and that the innate immune response plays an important role in host–pathogen interactions, we hereby investigated the effect of the PfEMP1–CIDR1α on TLRs expression on B cells isolated from non-malaria exposed donors.
This study focuses on the three TLRs, which have been demonstrated to be involved in human B-cell activation: TLR7, TLR9 which reside in intracellular compartment, and TLR10, expressed on the cell surface. The possibility of a positive control such as LPS stimulation is ruled out since human B cells do not express TLR4. Considering the possible differences in the kinetics of the expression of TLRs, measurements were done after 3 and 15 h of stimulation.
PfEMP1–CIDR1α stimulation led to a significant (p < 0.05) increase in TLR7 mRNA expression in B cells stimulated for 3 h as compared to the control GST. At 15 h of stimulation, TLR7 mRNA expression levels reached a 4.5 fold increase as compared to the GST control, and the other stimuli (p < 0.005) (Fig. 1A and D). No similar effects were seen after stimulation with anti-CD40, anti-Ig and/or CpG stimulation (Fig. 1A).
Fig. 1.

Regulation of the expression of TLRs mRNA in human B cells upon differential stimulation. B cells (2 × 106/mL in 24-well plates) were stimulated with anti-CD40, anti-Ig F(ab′)2, anti-Ig F(ab′)2 + anti-CD40, CpG 2006, GST and PfEMP1–CIDR1α for 3 h and 15 h. Total RNA was prepared, converted to cDNA, and analyzed for expression of (A) TLR7, TLR9 (not shown), and (B) TLR10 by real-time PCR as outlined in Section 2. GAPDH expression was used to standardize all results. Expression is shown as the fold increase versus unstimulated control (value normalized to the unit: 1) of the relative transcripts amounts for each TLR under consideration. The means ± SD of 5 independent experiments are shown (**p < 0.005; *p < 0.05). The average fold increase of TLR7, TLR9 and TLR10 transcripts following GST versus medium and PfEMP1–CIDR1α versus GST stimulation for 15 h in human B cells is shown (C). The up-regulation of TLR7 and TLR10 mRNA following PfEMP1–CIDR1α stimulation (15 h) is dose-dependent (D).
Neither PfEMP1–CIDR1α nor CD40, anti-Ig and/or CpG stimulation induced a significant increase in TLR9 transcripts at any time point tested (data not shown).
For the TLR10 mRNA transcript expression, negligible levels were observed after 3 h of incubation with all stimuli. However, after 15 h PfEMP1–CIDR1α induced a 4-fold increase as compared to the GST control (Fig. 1B and C). In the same conditions anti-CD40 but not anti-Ig triggered a slight up-regulation of TLR10. The positive control for TLR10 activation, CpG, led to a 12 fold up-regulation (Fig. 1B and C).
The increase in the expression of TLR7 and TLR10 induced by PfEMP1–CIDR1α was dose dependent with an optimal concentration of around 20 μg/mL and most prominent at 15 h of incubation (Fig. 1D).
Considering the up-regulation of TLR7 induced by Imiquimod (Tomai et al., 2000), we investigated the combined effect of PfEMP1–CIDR1α and the TLR7 ligand Imiquimod, on B-cell proliferation and TLR7 mRNA expression. To this end Imiquimod was added to PfEMP1–CIDR1α cultures at different time points (0, 12 h). Imiquimod induced by itself a stronger proliferation than the one induced by PfEMP1–CIDR1α (3 fold difference). No proliferation was observed in cultures containing both stimuli irrespective of the time of addition of Imiquimod (Fig. 2A).
Fig. 2.
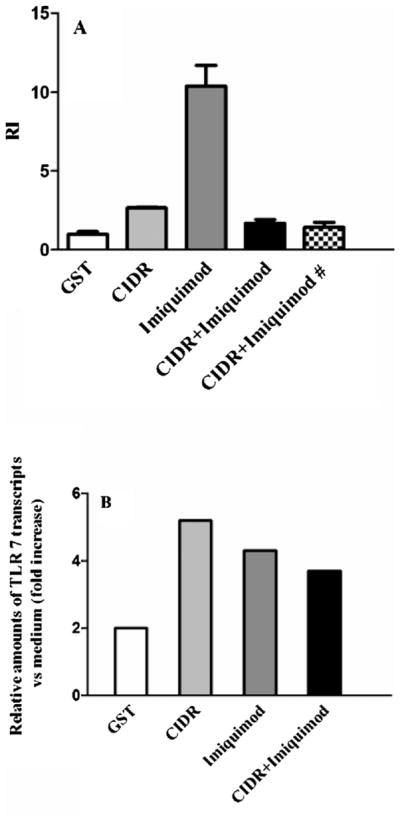
Effects of Imiquimod alone, or coincubated with PfEMP1–CIDR1α, on B cell proliferation and TLRs mRNA expression. (A) Proliferative response of B cells to GST, PfEMP1–CIDR1α, Imiquimod, PfEMP1–CIDR1α + Imiquimod, PfEMP1–CIDR1α + Imiquimod # (# added after 12 h); values represent mean ± standard error of the RI of 5 independent experiments. (*p < 0.05) (B) Relative amounts of TLR7 transcripts versus unstimulated control (value normalized to the unit: 1), after incubation of B cells with GST, PfEMP1–CIDR1α, Imiquimod, and PfEMP1–CIDR1α + Imiquimod for 15 h. The experiment shown is representative of two yielding similar results.
Interestingly, the levels of TLR7 mRNA expression were similar in PfEMP1–CIDR1α and Imiquimod containing cultures. Despite the lack of proliferation on cultures containing both stimuli, there were no significant differences in the mRNA expression. Thus, the inhibitory effect of PfEMP1–CIDR1α on Imiquimod-induced proliferation does not seem to correlate with the level of TLR7 expression (Fig. 2B).
3.2. Effect of PfEMP1–CIDR1α on TLRs signalling
B-cell responses to DNA-containing antigens involve BCR-driven recruitment of TLR9 to the autophagosomes resulting in hyperactivation of MAPKs (Chaturvedi et al., 2008). Thus, it became of interest to analyse the effect of CpG on PfEMP1–CIDR1α-induced expression of TLR7 and 9 and the MyD88 adapter protein, the latter indicative of innate TLR-mediated signalling (Fig. 3A). A combination of CpG and PfEMP1–CIDR1α induced a synergistic effect on TLR9 and TLR10 mRNA expression (Fig. 3A), and on the MyD88 adaptor protein (Fig. 3B).
Fig. 3.

CpG 2006 coincubation with PfEMP1–CIDR1α on TLR9 and TLR10 mRNA expression and MyD88 protein level. (A) Relative amounts of TLR9 and TLR10 transcripts versus unstimulated control (value normalized to the unit: 1), after incubation of B cells with GST, PfEMP1–CIDR1α, CpG 2006 and PfEMP1–CIDR1α + CpG 2006 for 15 h. The experiment shown is representative of two yielding similar results. (B) Expression of MyD88 was evaluated by Western blotting after 16 h of culture following stimulation with GST, PfEMP1–CIDR1α, Imiquimod, PfEMP1–CIDR1α + Imiquimod, CpG2006 and PfEMP1–CIDR1α + CpG2006. 12 μg of proteins was loaded on each lane of the gel and actin was used as loading control (not shown).
In contrast to the inhibitory effect observed by PfEMP1–CIDR1α on Imiquimod-induced proliferation, PfEMP1–CIDR1α did not affect the Imiquimod-induced MyD88 expression (Fig. 3B). These findings imply that the synergistic effect of PfEMP1–CIDR1α and CpG-induced activation may involve signalling through the MyD88 adaptor molecule.
3.3. CIDR1α induces MAPKs phosphorylation independently of BCR-signalling activation
Despite the ability of PfEMP1–CIDR1α to bind Igs, our previous data suggest that CIDR1α signalling proceeds via other receptor(s) than Igs, or involves engagement of additional receptors (Donati et al., 2006). To further clarify the role of BCR engagement in PfEMP1–CIDR1α-induced B-cell responses we determined the phosphorylation pattern of different protein kinases, known to be involved in BCR signalling, by confocal microscopy. To this end the analysis was done on human B-cells treated with F(ab′)2 anti-Ig and PfEMP1–CIDR1α.
In contrast to the robust phosphorylation of Lyn already seen after 2 min upon anti-Ig stimulation, PfEMP1–CIDR1α did not trigger the phosphorylation of Lyn, the first kinase involved in the BCR-signalling pathway (Fig. 4A). Similar results were obtained when measuring the phosphorylation of Syk at 5 min following PfEMP1–CIDR1α treatment (data not shown). Moreover, PfEMP1–CIDR1α did not induce measurable tyrosine-phosphorylation after 3 min of treatment as compared to anti-Ig stimulation (Fig. 4B). Interestingly, we found that upon PfEMP1–CIDR1α stimulation, two MAPKs: ERK1/2 and p38MAPK, were phosphorylated (Fig. 4C and D). The stimulation times chosen were 10 min and 60 min, respectively. Evaluation of the other main kinase cascades which are activated upon BCR triggering with anti-Ig showed that PfEMP1–CIDR1α and anti-Ig stimulation gave rise to the phosphorylation of a member of the IkB family, IkBα (after 45 min) (Fig. 4E), whereas JNK (60 min), PI3K/Akt (30 min), PKC (30 min) and PLCγ2 (10 min) were not activated (data not shown).
Fig. 4.

PfEMP1–CIDR1α does not induce Lyn and Tyrosine phosphorylation but triggers the phosphorylation of ERK1/2, p38 and IkBα. (A) CD19+ B cells were either untreated or stimulated for 2 min with either anti-Ig F(ab′)2 10 μg/mL or PfEMP1–CIDR1α 100 μg/mL and GST 50 μg/mL as control. In each case the cells were fixed, permeabilized, and stained with primary rabbit antibody specific for phospho-Lyn while the secondary goat anti-rabbit antibody was labelled with Alexa-488. (B–D) The same as in (A), except for the stimulation times: 3, 10 and 60 min, respectively, and the specific primary rabbit antibody for phospho-tyrosine used. (E) CD19+ B cells were stimulated as above for 30 min and stained with specific primary mouse antibody for phospho-IkBα. The secondary goat anti-mouse antibody was labelled with Alexa-488. Representative confocal images of Alexa-488 are shown; approximately 200 cells were analyzed from three independent experiments for each kinase. A and B present a doubled magnification compared with C, D and E.
In an attempt to control for any inhibitory effect that PfEMP1–CIDR1α might exert on BCR-signalling pathways, we supplemented the B-cell cultures with PfEMP1–CIDR1α in the presence of anti-Ig and using the stimulation times mentioned above. The coincubation did not exert any inhibitory effect on the BCR signalling pathways, since Lyn, Syk and MAPKs phosphorylation was not reduced (data not shown).
4. Discussion
We have previously shown based on competition assays that the CIDR1α domain of PfEMP1 does bind to B cells and that the interaction may involve binding to the BCR, as demonstrated by competition assays (Donati et al., 2004). However, analysis of the differential gene-expression profiles induced by PfEMP1–CIDR1α and anti-Ig stimulation suggested that different receptors and signalling pathways maybe involved (Donati et al., 2006). In addition PfEMP1–CIDR1α preferentially interacts with memory-B cells (Donati et al., 2006), known to express the intracellular TLR7, TLR9 and the surface expressed TLR10 (Hornung et al., 2002).
In this study we further investigated the interaction of PfEMP1–CIDR1α with particular emphasis on the expression of TLR7, 9 and 10 and signalling. Our data show that PfEMP1–CIDR1α induced TLR7 and TLR10 expression and that this expression is distinct from that seen after BCR-engagement, supporting the notion that PfEMP1–CIDR1α binds to other receptors in addition to the BCR.
The differential cross-talk between TLRs and PfEMP1–CIDR1α-induced signalling is intriguing but at the same time poorly understood. On one hand PfEMP1–CIDR1α did not induce TLR9 transcripts but had a strong synergistic effect on its expression when added together with CpG. On the other hand we could not demonstrate a correlation between proliferation and levels of TLR7 mRNA expression. The latter might be due to the fact that mRNA expression and proliferation do not always go hand in hand and/or that PfEMP1–CIDR1α may compete with TLR7 ligand Imiquimod. The mechanisms responsible for the conflicting results we obtained in the experiments involving PfEMP1–CIDR1α and Imiquimod or CpG remain unclear. The synergistic effect of PfEMP1–CIDR1α and CpG on TLR9 expression is intriguing in view of the proposed connection between TLR9 and BCR during B-cell stimulation. The BCR is known to be an important molecule in the internalization and processing of antigens that have bound to the receptor. Thus, one likely mechanism whereby TLR ligands are delivered to the intracellular TLRs in B cells might depend on the BCR, the latter functioning as an antigen-scavanger receptor (Chaturvedi and Pierce, 2009). Such mechanisms have recently been reported by Chaturvedi et al. (2008) who showed that crosslinking of BCR through DNA-containing antigens initiates signalling resulting in the recruitment of TLR9 to the autophagosomes, where the antigen is delivered. In support of the interaction between BCR and TLRs, several investigators have shown upon synergistic effects between BCR and TLR7 and 9 in response to DNA- and RNA-containing antigens and that these interactions lead to the development of autoimmune disorders (Leadbetter et al., 2002; Viglianti et al., 2003; Christensen et al., 2005; Pisitkun et al., 2006; Lau et al., 2005; Deane et al., 2007).
During chronic malaria, the B-cell compartment is stressed. It could be assumed that the binding of PfEMP1–CIDR1α to the BCR can lead to its endocytic internalization, bringing CIDR1α in close proximity of the endoplasmic compartment where TLR7 and 9 are located. This could allow the intracellular TLRs to interact with the BCR who will present ligands involved in triggering TLRs-related signalling pathways.
Moreover, we have shown that PfEMP1–CIDR1α stimulation by itself might induce a hyperresponse and activation of MAPKs. Despite its capacity to bind Igs, PfEMP1–CIDR1α does not stimulate upstream kinases involved in BCR signalling in human B cells, whereas it does activate ERK1/2, p38 and IkBα through unknown signalling pathways. The failure of PfEMP1–CIDR1α to activate the BCR could be due either to a low affinity for the BCR, or to a more complex and wider interaction with other surface receptors. In support of this notion is the fact that PfEMP1–CIDR1α binds equally to Fab (κ or λ) and to Fc fragments of human Igs (Donati et al., 2004), which might stimulate specific intracellular signalling cascades as suggested by the observed activation of ERK1/2 and p38 MAPK. The phosphorylation of these MAPKs is consistent with the observation that PfEMP1–CIDR1α treatment increases B-cell survival (Donati et al., 2006), likely by suppressing proapoptotic signals. The finding of IKBα phosphorylation provides further support of this concept, since its proteosomal degradation leads to NF-kB translocation to the nucleus, which in turn triggers the expression of Bcl-2 and other anti-apoptotic family members (Aggarwal, 2000; Grossman et al., 2000).
Moreover, coincubation in vitro with CpG increases MyD88 levels. To further support the model published by Chaturvedi, a recent paper, examining B cells from children with tonsillar hyperplasia and interaction with the IgD-binding species Moraxella, demonstrates the existence of IgD-mediated endocytosis of whole bacteria with clustering of IgD and TLR9 on the vacuolar membrane-containing bacteria with an enhancement of the signalling (Jendholm et al., 2009). This is possibly achieved on one hand, via MAPKs and NF-kB activation due to PfEMP1–CIDR1α itself, and/or on the other, via MyD88 enhanced signalling, linked to the interaction of PfEMP1–CIDR1α with TLRs ligands.
As far as TLR10 mRNA expression in concerned, the demonstration of a significant increase in TLR10 transcripts induced by CpG and/or CpG plus PfEMP1–CIDR1α stimulation cannot relate to the hypothesized mechanism for relocalization of endosomic TLRs, since TLR10 is located on the cellular surface (Hasan et al., 2005). Rather, the enhancement of TLR10 expression in B cells induced by polyclonal activators, i.e., SAC and CpG DNA, is likely related to the B-cell progression from a resting to an activated status, as suggested by others (Bourke et al., 2002). TLR10 expression is restricted to B- and plasmacytoid dendritic cells, and because MyD88 adaptor molecule is involved in TLR10 signalling (Hasan et al., 2005) a possible involvement of TLR10 in response to specific pathogens, such as malaria P. falciparum, cannot be excluded.
Our findings are of great interest, because the continuous interaction between CIDR1α and B cells during chronic malaria might result in a decreased threshold for B-cell activation, causing persistent stimulation.
Based on our data it could be postulated that PfEMP1–CIDR1α, as a polyclonal activator, is able to trigger the phosphorylation of some kinases involved in B-cell homeostasis. This might increase the activation status of B cells in a general, non-specific fashion, resulting in sustained hyper-activation that leads to impaired immune responses. As is usually observed in homeostatic systems, the magnitude of the response is more related to the difference seen between the induced activation and the basal conditions, more than to the absolute levels of stimulation. Therefore, a persistent exposure to PfEMP1–CIDR1α during active and repeated and protracted malaria infections may affect both the polyclonal and antigen-specific responses.
In conclusion, our findings indicate that the PfEMP1–CIDR1α-induced activation is not related to the BCR rather goes through TLR signalling. The results suggest the capacity of the parasite to avert the innate and adaptive immune responses. This persistent activation of the immune system may result in B-cell dysfunction and “exhaustion” during malaria infection, that adds to the parasite’s survival.
Acknowledgments
This work was supported by grants to from the Swedish Agency for Research Development with Developing Countries (SIDA, SAREC), the Swedish Research Council (VR) as well as grants within the EviMalaR and BioMalPar European Networks of Excellence (LSMP-CT-2004-503578) and from the Priority 1 “Life Sciences, Genomics and Biotechnology for Health” in the 6th Framework Programme.
References
- Abele DC, Tobie JE, Hill GJ, Contacos PG, Evans CB. Alterations in serum proteins and 19S antibody production during the course of induced malaria infections in man. Am J Trop Med Hyg. 1965;14:191–197. doi: 10.4269/ajtmh.1965.14.191. [DOI] [PubMed] [Google Scholar]
- Adu D, Williams DG, Quakyi IA, Voller A, Anim-Addo Y, Bruce-Tagoe AA, Johnson GD, Holborow EJ. Anti-ssDNA and antinuclear antibodies in human malaria. Clin Exp Immunol. 1982;49:310–316. [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB. Apoptosis and nuclear factor-kappa B: a tale of association and dissociation. Biochem Pharmacol. 2000;60:1033–1039. doi: 10.1016/s0006-2952(00)00393-2. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Banic DM, Viana-Martins FS, De Souza JM, Peixoto TD, Daniel-Ribeiro C. Polyclonal B-lymphocyte stimulation in human malaria and its association with ongoing parasitemia. Am J Trop Med Hyg. 1991;44:571–577. doi: 10.4269/ajtmh.1991.44.571. [DOI] [PubMed] [Google Scholar]
- Bates I, Bedu-Addo G. Chronic malaria and splenic lymphoma: clues to understanding lymphoma evolution. Leukemia. 1997;11:2162–2167. doi: 10.1038/sj.leu.2400878. [DOI] [PubMed] [Google Scholar]
- Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, Hartmann G. Plasmacytoid dendritic cells control TLR7 sensitivity of naïve B cells via Type I IFN. J Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naïve B cells and constitutive expression in memory B cells. Blood. 2003;101:4500–4504. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- Bernasconi N, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;289:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- Bourke E, Bosisio D, Golay J, Polentarutti N, Mantovani A. The Toll-like repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2002;102:956–963. doi: 10.1182/blood-2002-11-3355. [DOI] [PubMed] [Google Scholar]
- Campbell KS. Signal transduction from the B cell antigen-receptor. Curr Opin Immunol. 1999;11:256–264. doi: 10.1016/s0952-7915(99)80042-9. [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic. 2009;10:621–628. doi: 10.1111/j.1600-0854.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Barragan A, Fernandez V, Sundstrom A, Schichtherle M, Sahlen A, Carlson J, Datta S, Wahlgren M. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum. J Exp Med. 1998;187:15–23. doi: 10.1084/jem.187.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Heddini A, Barragan A, Fernandez V, Pearce SF, Wahlgren M. The semiconserved head structure of Plasmodium falciparum erythrocyte membrane protein 1 mediates binding to multiple independent host receptors. J Exp Med. 2000;192:1–10. doi: 10.1084/jem.192.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulo L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Porto JM, Gauld SB, Merrel KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling. Mol Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Deane JA, Pisitkun P, Barret RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati D, Mok B, Chene A, Xu H, Thangarajh M, Glas R, Chen Q, Wahlgren M, Bejarano MT. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J Immunol. 2006;177:3035–3044. doi: 10.4049/jimmunol.177.5.3035. [DOI] [PubMed] [Google Scholar]
- Donati D, Zhang LP, Chen Q, Chene A, Flick K, Nystrom M, Wahlgren M, Bejarano MT. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect Immun. 2004;72:5412–5418. doi: 10.1128/IAI.72.9.5412-5418.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K, Scholander C, Chen Q, Fernandez V, Pouvelle B, Gysin J, Wahlgren M. Role of nonimmune IgG bound to PfEMP1 in placental malaria. Science. 2001;293:2098–2100. doi: 10.1126/science.1062891. [DOI] [PubMed] [Google Scholar]
- Greenwood BM, Vick RM. Evidence for a malaria mitogen in human malaria. Nature. 1975;257:592–594. doi: 10.1038/257592a0. [DOI] [PubMed] [Google Scholar]
- Grossman M, O’Reilly LA, Gugasyan R, Strasse A, Adams JM, Gerondakis S. The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J. 2000;19:6351–6360. doi: 10.1093/emboj/19.23.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebeque S, Trinchieri G, Bates EEM. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174:2942–2950. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of Toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- Jendholm J, Mörgelin M, Perez Vidakovics MLA, Carlsson M, Leffler H, Cardell LO, Riesbeck K. Superantigen- and TLR-dependent activation of tonsillar B cells after receptor-mediated endocytosis. J Immunol. 2009;182:4713–4720. doi: 10.4049/jimmunol.0803032. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A, Sallusto F. Toll-like receptors and innate immunity in B-cell activation and antibody responses. Curr Opin Immunol. 2007;19:268–274. doi: 10.1016/j.coi.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rikfin IR, Hohlbaum AM, Baudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- McGregor IA, Giles HM, Walter JH, Davies AH. Effects of heavy and repeated malaria infections of Gambian infants and children. Br Med J. 1956;2:686–691. doi: 10.1136/bmj.2.4994.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood. 2003;103:3058–3064. doi: 10.1182/blood-2003-08-2972. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- Ruprecht CR, Lanzavecchia A. Toll-like receptor as a third signal required for activation of human naïve B cells. Eur J Immunol. 2006;36:810–816. doi: 10.1002/eji.200535744. [DOI] [PubMed] [Google Scholar]
- Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tomai MA, Imbertson LM, Stanczak TL, Tygrett LT, Waldschmidt TJ. The immune response modifiers imiquimod and R-848 are potent activators of B lymphocytes. Cell Immunol. 2000;203:55–65. doi: 10.1006/cimm.2000.1673. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]