

Myosin II is an ATPase motor protein essential for many cellular functions including cell migration[1] and division.[2] In nonmuscle cells, myosin modulates protrusions at the leading edge and promotes retraction at the trailing edge during migration,[3] while during cytokinesis, myosin is required for contraction of the cleavage furrow.[4] For nonmuscle myosin, these varied functions are regulated by phosphorylation of the associated myosin regulatory light chain (mRLC) protein at Ser19, which activates the myosin complex to promote myosin assembly, contractility, and stress fiber formation.[5] Upon phosphorylation of the mRLC at both Thr18 and Ser19, these activities are further enhanced.[6 ] The dramatic effects of phosphorylation can also be recapitulated in vitro. Specifically, myosin and the proteolytic derivative heavy meromyosin (HMM),[7] which contains only one-third of the C-terminal myosin tail, exhibit low in vitro activities when associated with the nonphosphorylated mRLC. Phosphorylation of Ser19 amplifies actin-activated ATPase activities 10 – 1000-fold[8] and leads to myosin-mediated actin translocation.[9]
While myosin has been studied extensively, questions surrounding the dynamic interactions of the protein in live cells remain. Methods to study myosin and modulate activity include gene deletions or siRNA-mediated knockdown of gene expression,[3] mRLC kinase overexpression,[10] and the use of small molecule inhibitors.[11] While these methods have provided a wealth of information, they do not enable studies of the spatial dynamics of myosin regulation because localized activation cannot be achieved. Additionally, genetic approaches provide imprecise temporal control, preventing real-time studies of protein function. Thus, we sought to develop chemical tools to overcome these drawbacks by enabling direct and controlled myosin activation through the semisynthesis of a photoactivated mRLC. The light-mediated activation is achieved by the incorporation of a photolabile “caging group” onto the essential phosphate of pSer19 within the full-length mRLC. The caging group masks the phosphate functionality and renders the protein biologically inactive until irradiation, which releases the native phosphoprotein. By using light as the trigger, this strategy offers a kinase-independent method to activate myosin with precise spatial and temporal resolution and enables researchers to obtain real-time information about the downstream effects of myosin phosphorylation within a complex network.[12]
The 1-2-(nitrophenyl)ethyl (NPE) caging group has been employed for cellular applications because it is efficiently released, under biologically-compatible conditions, at 365 nm.[13] Peptides and proteins containing NPE-caged phosphorylated amino acids have been successfully exploited for the study of diverse systems.[14] Additionally, a general method for incorporating NPE-caged thiophosphoamino acids, which, upon irradiation, function similarly to the phosphorylated species but with greater phosphatase resistance, has been reported[15] and can be used for cellular studies of myosin.
Herein we report a chemical approach to investigate myosin function through the preparation of unnatural amino acid mutants of the mRLC. We present an efficient semisynthesis of full-length mRLC through expressed protein ligation for the site-specific incorporation of phosphorylation at Ser19 (pSer19) and Thr18 (pThr18) and the genesis of caged phosphoserine (cpSer) and caged thiophosphoserine (c(S)pSer) at position 19. Caging of pSer19 eliminates myosin and HMM activities, and irradiation releases the native phospho-mRLC to restore activity to nearly native phosphorylated levels (Figure 1). Microinjection of myosin exchanged with the caged protein into live cells and subsequent irradiation releases the phosphoprotein within cells. This tool is now poised to facilitate future investigations of the downstream effects of myosin activation.
Figure 1.

Installation of NPE-caged pSer19 into the mRLC is achieved by expressed protein ligation. The caging group masks the phosphate necessary for myosin activation until irradiation releases it to generate the native phosphoprotein and restore activity. Image was modified from Protein Data Bank file 1WDC.
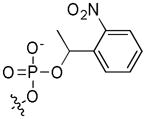
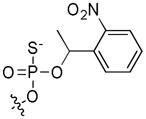
Semisynthesis of the mRLC was achieved through native chemical ligation (NCL)[16] between a synthetic peptide thioester corresponding to the N-terminal region of the mRLC (residues 1 – 23) and a recombinant protein fragment comprising the remaining C-terminal residues (residues 25 – 171) and a Met24Cys mutation (Scheme 1). To probe the effects of phosphorylation at discrete sites of the mRLC, the protein was synthesized with no phosphorylation (1) and with pSer19 (2), pThr18 (3), pThr18 pSer19 (4), cpSer19 (5), and c(S)pSer19 (6).
Scheme 1.

Semisynthesis of the full-length mRLC. The C-terminal portion of the mRLC is expressed heterologously in E. coli. TEV proteolysis releases GST and reveals the N-terminal cysteine, which reacts in the NCL with the synthetic peptide thioester to generate the full-length mRLC.
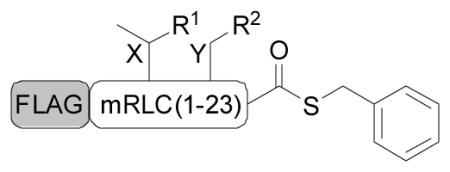
The peptide thioesters containing the phosphorylated or caged residues were made by solid phase peptide synthesis (Tables 1, S1), and the C-terminal portion of the protein was expressed in E. coli as a glutathione S-transferase (GST) fusion to enhance expression and aid purification. Following tobacco etch virus (TEV) proteolysis to expose the N-terminal cysteine required for ligation, the peptide and protein fragments were combined in the NCL reaction, which efficiently afforded milligram quantities of the full-length mRLC (Figure S1). N-terminal FLAG epitope and C-terminal His6 tags facilitated isolation of the product from unligated protein and excess peptide, respectively. The mass of the protein was then confirmed by MALDI analysis.
Table 1.
Peptide thioester derivatives used in the semisynthesis of mRLC.[a]
| |||
|---|---|---|---|
| FLAG-mRLC(1–23): DYKDDDDK-SSKKAKTKTTKKRPQRA XY NVFA
| |||
| Entry | Derivative | R1 | R2 |
| 1 | NonP | OH | OH |
| 2 | pSer19 | OH | OPO32− |
| 3 | pThr18 | OPO32− | OH |
| 4 | pThr18 pSer19 | OPO32− | OPO32− |
| 5 | cpSer19 | OH |
|
| 6 | c(S)pSer19 | OH |
|
Peptides were synthesized by Fmoc-based solid phase peptide synthesis as C-terminal thioesters.
We characterized the ability of the semisynthetic protein to regulate in vitro myosin activity and to enable myosin photoactivation. Semisynthetic mRLC was exchanged for the native mRLC in chicken gizzard smooth muscle HMM and myosin (Figure S2) and then tested in ATPase[17] and sliding filament assays.[18] We first focused on ATPase assays. Due to greater tractability in solution, HMM, rather than myosin, was used.[7] Similar to HMM with the native nonphosphorylated mRLC, the actin-activated ATPase activity of HMM exchanged with 1 was negligible (Figure 2a). HMM exchanged with 2 displayed activity similar to that of HMM phosphorylated by myosin light chain kinase (MLCK) (0.80 ± 0.07 and 0.98 ± 0.13 s−1, respectively). These experiments establish that the semisynthetic mRLC faithfully regulates HMM enzymatic activity. Additionally, the FLAG epitope and His6 tags do not influence function.
Figure 2.

Actin-activated ATPase activities of HMM. The values are the means ± SD of at least three trials. NonP, nonphosphorylated; P, phosphorylated by MLCK. a) ATPase activity of HMM with native mRLC (gray bars) and noncaged semisynthetic derivatives (black bars). b) ATPase activity of HMM with semisynthetic noncaged (black bars) and caged derivatives (open bars) before (−UV) and after (+UV) irradiation at 365 nm for 90 s.
In addition to Ser19, the mRLC can also be phosphorylated at Thr18.[19] Previous studies of Thr18 phosphorylation alone have relied on a Ser19Ala mutation because Ser19 is normally phosphorylated first.[20] Moreover, mRLC diphosphorylation has been observed in vitro and in cells, but complete in vitro phosphorylation requires high concentrations of MLCK.[19] Our semisynthetic approach provides convenient access to homogenously phosphorylated proteins, allowing the effects of defined phosphorylation to be examined without the need for additional mutations. ATPase assays of HMM exchanged with 3 showed that phosphorylation of Thr18 moderately increases activity to 0.18 ± 0.03 s−1, whereas phosphorylation at both Thr18 and Ser19 (4) generates even greater activity (1.16 ± 0.11 s−1) than pSer19 alone (Figure 2a). These trends are consistent with previous studies on the effects of kinase-mediated Thr18 and Thr18 Ser19 phosphorylation.[20]
Next, we investigated the ability to photoactivate the protein. We used RP-HPLC analysis to examine the kinetics of NPE removal after irradiation of the caged peptide at 365 nm (Figure S3). Nearly maximal release of the free phosphopeptide (70%) was achieved after irradiation for 90 s, a dosage previously shown to be compatible for cellular studies.[14b] Western blot analysis of the full-length caged proteins (5 and 6) with an anti-pSer19 mRLC antibody confirmed that the phospho- and thiophosphoproteins were generated upon irradiation (Figure S4).
After exchange of caged mRLCs 5 and 6 into HMM, actin-activated ATPase assays demonstrated that the activity of the caged proteins was low and mimicked that of nonphosphorylated mRLC 1 (Figure 2b). However, irradiation at 365 nm for 90 s increased activity about 20-fold to levels near that of HMM exchanged with semisynthetic pSer19 mRLC 2. Importantly, the caged proteins completely suppress HMM ATPase activity, indicating that the NPE group is sufficient to maintain the inhibited state of the protein. The activities following uncaging (0.48 ± 0.04 and 0.43 ± 0.05 s−1 for 5 and 6, respectively) are consistent with restoration of about 60% activity compared to that of HMM with 2 and lie within the range expected based on the HPLC analysis. Thus, irradiation enables direct control over release of the phosphorylated mRLC and, correspondingly, over HMM activation.
To further characterize the system, we performed sliding filament assays, which assess the force-generating ability of myosin. In this assay, we measure the velocities of fluorescently-labeled actin filaments propelled by myosin bound to a nitrocellulose-coated glass coverslip. Myosin was used in these assays because it produced more consistent filament movement than HMM. Nonphosphorylated myosin and myosin exchanged with 1 did not move the actin filaments, but both MLCK-phosphorylated myosin and myosin exchanged with 2 led to significant movement with velocities around 0.9 μm s−1 (Figure 3a). Each phosphorylated semisynthetic derivative generated filament movement at velocities between 0.7 and 1.0 μm s−1. A one-way ANOVA followed by Tukey’s post-hoc test indicated that the differences among myosin exchanged with 2, 3, and 4 are statistically significant, with all comparisons yielding p < 0.0001 (Figure 3a). These results are consistent with a previous study in which myosin with a pThr18 Ser19Ala mutant mRLC generated slightly lower filament velocities than pSer19 and pThr18 pSer19 derivatives.[20b] However, our results indicate differences between phosphorylation at Ser19 and diphosphorylation (pThr18 pSer19), which have not been previously reported.
Figure 3.

In vitro myosin sliding filament assays. a) The mean velocities ± SD of at least 45 actin filaments during incubation with native myosin (gray bars) and myosin exchanged with noncaged semisynthetic (black bars) and caged semisynthetic (open bars) mRLCs. NMO, no motility observed; NonP, nonphosphorylated; P, phosphorylated by MLCK. b) Actin filament paths from a representative field before (−UV) and after (+UV) 90 s irradiation of myosin exchanged with cpSer19 mRLC 5.
With both caged proteins 5 and 6, negligible filament movement was observed before irradiation (Figures 3a, 3b, S5). In contrast, irradiation of myosin prior to the assay generated significant filament movement with velocities comparable to those observed with MLCK-phosphorylated myosin or myosin exchanged with 2. Although about 60% of the HMM ATPase activity was achieved after uncaging, the sliding filament velocities were fully restored following irradiation. Studies have shown that while steady-state ATPase activities increase proportionally with the degree of phosphorylation,[21] sliding filament velocities reach a maximal value even in the presence of nonphosphorylated myosin.[22]
The in vitro studies establish that caging of pSer19 provides effective photochemical control over myosin activity. Finally, in order to test these chemical tools in live cells, we microinjected the caged mRLC into COS7 cells and investigated uncaging in situ. Initially, the caged thiophosphorylated mRLC 6 was used to minimize potential complicating effects from cellular phosphatases, eliminating the need for nonspecific phosphatase inhibitors. Because incorporation of the injected mRLC into endogenous myosin complexes was slow, gizzard smooth muscle myosin exchanged with the caged protein was prepared in vitro and microinjected. Following irradiation of the injected cells, the cells were fixed, stained with an anti-pSer19 mRLC antibody, and analyzed by immunofluorescence microscopy. The signal from the anti-pSer19 mRLC antibody was significantly higher following uncaging compared to injected cells that had not been irradiated (Figures 4, S6). In addition, irradiation of only a portion of a fixed cell enabled spatial control over release of activated myosin (Figure S7). These studies indicate that the thiophosphorylated protein can be readily and reproducibly generated within a cellular system and represent the foundation for future investigations of myosin within living cells.
Figure 4.

Cells injected with myosin exchanged with 6 and Texas Red-labeled dextran marker before (Caged) or after (Uncaged) irradiation. The cells were fixed and stained with an antibody specific for pSer19 mRLC. Scale bar: 10 μm.
In summary, the semisynthetic approach provides convenient access to milligram quantities of various phosphorylated and caged phosphorylated mRLC derivatives to facilitate studies of individual sites of phosphorylation. This general method can be readily adapted to incorporate other unnatural elements into the N-terminal domain of the mRLC. Additionally, the caged protein enables precise photocontrol over HMM and myosin activity. Uncaging efficiently furnishes the phospho- and thiophosphoproteins that appropriately regulate HMM and myosin activity. The in vitro characterization of the semisynthetic protein and the cellular uncaging experiments provide the basis for subsequent studies of myosin in a cellular context. For instance, this system could be used to further address effects of myosin on stress fiber and focal adhesion formation. Offering the unique ability to activate myosin with exact spatial and temporal resolution, this approach promises to help unravel the complex role of the protein within the cell.
Supplementary Material
Footnotes
We thank Dr. Andreas Aemissegger for synthesis of the caged amino acids. This research was supported by the NIH Cell Migration Consortium (GM064346). B.N.G. was supported by the NIGMS Biotechnology Training Grant, and B.D.H. was supported by an AHA Fellowship.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Brenda N. Goguen, Departments of Chemistry and Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA), Fax: (+)1 617-452-2799
Dr. Brenton D. Hoffman, Department of Microbiology, University of Virginia, Charlottesville, VA 22908 (USA)
Dr. James R. Sellers, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892 (USA)
Prof. Martin A. Schwartz, Department of Microbiology, University of Virginia, Charlottesville, VA 22908 (USA)
Prof. Barbara Imperiali, Email: imper@mit.edu, Departments of Chemistry and Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA), Fax: (+)1 617-452-2799, Homepage: http://web.mit.edu/imperiali
References
- 1.Lauffenburger DA, Horwitz AF. Cell. 1996;84:359. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 2.De Lozanne A, Spudich JA. Science. 1987;236:1086. doi: 10.1126/science.3576222. [DOI] [PubMed] [Google Scholar]
- 3.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Nat Rev Mol Cell Biol. 2009;10:778. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komatsu S, Yano T, Shibata M, Tuft RA, Ikebe M. J Biol Chem. 2000;275:34512. doi: 10.1074/jbc.M003019200. [DOI] [PubMed] [Google Scholar]
- 5.a) Chrzanowska-Wodnicka M, Burridge K. J Cell Biol. 1996;133:1403. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Watanabe T, Hosoya H, Yonemura S. Mol Biol Cell. 2007;18:605. doi: 10.1091/mbc.E06-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komatsu S, Ikebe M. J Cell Biol. 2004;165:243. doi: 10.1083/jcb.200309056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikebe M, Hartshorne DJ. Biochemistry. 1985;24:2380. doi: 10.1021/bi00330a038. [DOI] [PubMed] [Google Scholar]
- 8.a) Sellers JR, Pato MD, Adelstein RS. J Biol Chem. 1981;256:13137. [PubMed] [Google Scholar]; b) Sellers JR. J Biol Chem. 1985;260:15815. [PubMed] [Google Scholar]
- 9.Tan JL, Ravid S, Spudich JA. Annu Rev Biochem. 1992;61:721. doi: 10.1146/annurev.bi.61.070192.003445. [DOI] [PubMed] [Google Scholar]
- 10.a) Murata-Hori M, Fukuta Y, Ueda K, Iwasaki T, Hosoya H. Oncogene. 2001;20:8175. doi: 10.1038/sj.onc.1205055. [DOI] [PubMed] [Google Scholar]; b) Hecht G, et al. Am J Physiol. 1996;271:C1678. doi: 10.1152/ajpcell.1996.271.5.C1678. [DOI] [PubMed] [Google Scholar]
- 11.a) Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Science. 2003;299:1743. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]; b) Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. J Biol Chem. 1987;262:7796. [PubMed] [Google Scholar]; c) Uehata M, et al. Nature. 1997;389:990. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]; d) Chartier L, Rankin LL, Allen RE, Kato Y, Fusetani N, Karaki H, Watabe S, Hartshorne DJ. Cell Motil Cytoskeleton. 1991;18:26. doi: 10.1002/cm.970180104. [DOI] [PubMed] [Google Scholar]
- 12.Rothman DM, Shults MD, Imperiali B. Trends Cell Biol. 2005;15:502. doi: 10.1016/j.tcb.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Ellis-Davies GC. Nat Methods. 2007;4:619. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Vazquez ME, Nitz M, Stehn J, Yaffe MB, Imperiali B. J Am Chem Soc. 2003;125:10150. doi: 10.1021/ja0351847. [DOI] [PubMed] [Google Scholar]; b) Nguyen A, Rothman DM, Stehn J, Imperiali B, Yaffe MB. Nat Biotechnol. 2004;22:993. doi: 10.1038/nbt997. [DOI] [PubMed] [Google Scholar]; c) Hahn ME, Muir TW. Angew Chem. 2004;116:5924. doi: 10.1002/anie.200461141. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2004;43:5800. [Google Scholar]; d) Humphrey D, Rajfur Z, Vazquez ME, Scheswohl D, Schaller MD, Jacobson K, Imperiali B. J Biol Chem. 2005;280:22091. doi: 10.1074/jbc.M502726200. [DOI] [PubMed] [Google Scholar]; e) Vogel EM, Imperiali B. Protein Sci. 2007;16:550. doi: 10.1110/ps.062549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aemissegger A, Carrigan CN, Imperiali B. Tetrahedron. 2007;63:6185. doi: 10.1016/j.tet.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 17.Trybus KM. Methods. 2000;22:327. doi: 10.1006/meth.2000.1085. [DOI] [PubMed] [Google Scholar]
- 18.Sellers JR. Curr Protoc Cell Biol. 2001;Chapter 13(Unit 13):2. doi: 10.1002/0471143030.cb1302s00. [DOI] [PubMed] [Google Scholar]
- 19.Ikebe M, Hartshorne DJ, Elzinga M. J Biol Chem. 1986;261:36. [PubMed] [Google Scholar]
- 20.a) Kamisoyama H, Araki Y, Ikebe M. Biochemistry. 1994;33:840. doi: 10.1021/bi00169a027. [DOI] [PubMed] [Google Scholar]; b) Bresnick AR, Wolff-Long VL, Baumann O, Pollard TD. Biochemistry. 1995;34:12576. doi: 10.1021/bi00039a012. [DOI] [PubMed] [Google Scholar]
- 21.Ellison PA, Sellers JR, Cremo CR. J Biol Chem. 2000;275:15142. doi: 10.1074/jbc.275.20.15142. [DOI] [PubMed] [Google Scholar]
- 22.Cuda G, Pate E, Cooke R, Sellers JR. Biophys J. 1997;72:1767. doi: 10.1016/S0006-3495(97)78823-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.