

Background: Elucidation of molecular mechanisms controlling vascular calcification is critical for chronic kidney disease patients.
Results: High phosphate induced Klf4 expression in SMCs. Klf4 knockdown attenuated high phosphate-induced SMC phenotypic switching into osteogenic cells.
Conclusion: Results suggest that Klf4 contributes to high phosphate-induced conversion of SMCs into osteogenic cells.
Significance: Control of Klf4 might be a novel therapeutic target for vascular calcification.
Keywords: Atherosclerosis, Calcification, Krüppel-like Factor, Transcription Factors, Vascular Biology, Vascular Smooth Muscle Cells
Abstract
Hyperphosphatemia in chronic kidney disease is highly associated with vascular calcification. Previous studies have shown that high phosphate-induced phenotypic switching of vascular smooth muscle cells (SMCs) into osteogenic cells plays an important role in the calcification process. In the present study, we determined whether Krüppel-like factor 4 (Klf4) and phosphorylated Elk-1, transcriptional repressors of SMC differentiation marker genes activated by intimal atherogenic stimuli, contributed to this process. Rat aortic SMCs were cultured in the medium with normal (0.9 mmol/liter) or high (4.5 mmol/liter) phosphate concentration. Results showed that high phosphate concentration induced SMC calcification. Moreover, high phosphate decreased expression of SMC differentiation marker genes including smooth muscle α-actin and SM22α, whereas it increased expression of osteogenic genes, such as Runx2 and osteopontin. High phosphate also induced Klf4 expression, although it did not phosphorylate Elk-1. In response to high phosphate, Klf4 selectively bound to the promoter regions of SMC differentiation marker genes. Of importance, siRNA-mediated knockdown of Klf4 blunted high phosphate-induced suppression of SMC differentiation marker genes, as well as increases in expression of osteogenic genes and calcium deposition. Klf4 was also induced markedly in the calcified aorta of adenine-induced uremic rats. Results provide novel evidence that Klf4 mediates high phosphate-induced conversion of SMCs into osteogenic cells.
Introduction
Vascular calcification is highly prevalent in patients with chronic kidney disease and is associated with increased risk of cardiovascular disease and mortality (1, 2). In atherosclerotic lesions of these patients, calcium deposits are localized mainly in the medial layer of arteries, and this phenotype is known as Mönckeberg's medial sclerosis (3, 4). Hyperphosphatemia in chronic kidney disease is a key factor promoting vascular medial calcification (4). In addition to a passive process involving spontaneous calcium phosphate precipitation in necrotic tissue, multiple recent studies have shown that high phosphate concentration induces phenotypic conversion of vascular smooth muscle cells (SMCs)2 into osteogenic cells, thereby enhancing medial calcification in the vessels (4–6). In response to high phosphate stimulus, SMCs decrease expression of SMC differentiation marker genes including smooth muscle (SM) α-actin and SM22α, and increase expression of osteogenic genes such as Runx2, osteopontin, osteocalcin, and alkaline phosphatase (ALP). Elucidation of the molecular mechanisms controlling phenotypic conversion from differentiated SMCs into osteogenic cells is likely to provide important insights toward novel therapeutic approach of vascular calcification.
Molecular mechanisms controlling the differentiated state of SMCs have been progressively clarified in the last two decades (7, 8). Most SMC differentiation maker genes contain common cis-elements including multiple CC(A/T-rich)6GG (CArG) elements and a transforming growth factor-β control element (TCE) in their promoter-enhancer regions. The binding factor of CArG elements is serum response factor (SRF), which regulates expression of SMC differentiation marker genes by cooperating with its co-activator, myocardin or its co-repressor, phosphorylated Elk-1 (7–11). Krüppel-like factor 4 (Klf4) is a binding factor of TCE, and it potently represses SMC differentiation marker genes (12, 13). Down-regulation of SMC differentiation marker genes by platelet-derived growth factor-BB (PDGF-BB) and oxidized phospholipids, both of which contribute to the development of intimal atherosclerosis, has been shown to be mediated through these cis-elements and trans-binding factors (9, 11–15). Indeed, results of our previous studies and others showed that PDGF-BB and oxidized phospholipids, respectively, caused phosphorylation of Elk-1 via activation of the MEK-ERK1/2 pathway, and phosphorylated Elk-1 displaced myocardin from SRF, resulting in the transcriptional repression of SMC differentiation marker genes (9, 11, 14). These factors also induced Klf4 expression, and siRNA-induced knockdown of Klf4 partially attenuated PDGF-BB or oxidized phospholipid-induced suppression of SMC differentiation marker genes in cultured SMCs (13–15). In addition, we demonstrated that conditional knock-out of the Klf4 gene in mice delayed repression of SMC differentiation markers but accelerated neointimal formation following carotid injury in vivo (16). As such, studies thus far have indicated that Klf4 and phosphorylated Elk-1 play critical roles in phenotypic switching of SMCs in response to intimal atherogenic stimuli.
Despite the importance of high phosphate-induced repression of SMC differentiation marker genes during the vascular calcification process (4, 6), underlying mechanisms have not been fully determined yet. Elevated concentrations of extracellular phosphate have been shown to activate multiple signaling pathways and transcription factors including the caspase-dependent apoptosis pathway (17), mitochondrial reactive oxygen species and nuclear factor-κB pathway (18), and Runx2 (6, 19, 20) in part via the uptake of phosphate by the type III sodium-dependent phosphate co-transporter, Pit-1 (19), in SMCs. Of these, Runx2 is a factor capable of repressing SMC differentiation marker genes. It has been shown to be expressed in the medial cells of the calcified arteries in humans and in a rat model of chronic renal failure (21) and also to be induced by high phosphate concentration in cultured SMCs (6, 19). Recently, Tanaka et al. (22) showed that overexpression of Runx2 decreased expression of SMC differentiation marker genes, such as SM α-actin and SM22α, by inhibiting the binding of myocardin to the SRF-CArG elements. Moreover, Speer et al. (20) showed that Runx2, but not loss of myocardin, was required for high phosphate-induced vascular calcification. These results suggest a critical role of Runx2 in high phosphate-induced SMC phenotypic switching into osteogenic cells. However, it remains unknown whether transcription factors required for repression of SMC differentiation marker genes by intimal atherogenic stimuli also play important roles for medial calcification. Therefore, the aims of the present studies were to determine whether Klf4 and phosphorylated Elk-1 contribute to high phosphate-induced SMC phenotypic switching into osteogenic cells, and if so, to determine their relationship with Runx2.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The SM α-actin promoter-luciferase (−2.6 kb/+2.8 kb) construct and its mutants were described previously (23). The SM α-actin promoter-luciferase (−2.6 kb/+2.8 kb) construct containing combinatorial mutations of two CArG elements and TCE was generated by QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). The human osteopontin promoter-luciferase (−1500/+87 bp and −970/+87 bp) plasmids were constructed as described (24). The mouse osteopontin promoter (−2287/+132 bp) was amplified by PCR and subcloned into KpnI/NheI sites of pGL3-basic vector (Promega) to make the mouse osteopontin promoter-luciferase (−2287/+132 bp) construct. An expression plasmid for Myc-tagged Klf4 was described previously (14). An expression plasmid for FLAG-tagged Runx2 was constructed by inserting mouse Runx2 cDNA into p3xFLAG-CMV-7.1 vector (Sigma).
Cell Culture
Rat aortic SMCs were cultured as described previously (11, 14). One day after plating at 10,000 cells/cm2, SMCs were started to be incubated in DMEM (glucose: 4.5 g/liter)/10% fetal bovine serum (Invitrogen) with normal (0.9 mmol/liter) or high (4.5 mmol/liter) phosphate concentration. Serum-containing DMEM was used throughout the studies because high phosphate-induced SMC calcification was not evident in serum-free DMEM. Medium was changed every 2 days. Cells were harvested for subsequent analyses. To determine the calcification status, von Kossa staining was performed after 10-day incubation with normal or high phosphate medium. In addition, SMCs were decalcified with 0.6 mol/liter HCl, and calcium content in the supernatant was determined by the o-cresolphthalein complexone method (17). A quantitative index of apoptosis was determined by a cell-death detection ELISAplus kit (Roche Applied Science) (17). For siRNA experiments, siRNA for Klf4 and Runx2 (Stealth RNAi; Invitrogen) or a scramble sequence was transfected with Oligofectoamine (Invitrogen) before starting incubation with normal or high phosphate medium.
Real-time RT-PCR
Total RNA was used for real-time RT-PCR. Primer and probe sequences for SM α-actin, SM22α, Klf4, and 18 S rRNA were described previously (13, 23, 25). Primer sequences for Runx2, osteopontin, ALP, Klf2, and Klf5 were as follows: Runx2-F, 5′-CCACCTCTGACTTCTGCCTC-3′; Runx2-R, 5′-GACTGGCGGGTGTAAGTGAA-3′; osteopontin-F, 5′-CCGATGAGGCTATCAAGGTC-3′; osteopontin-R, 5′-ACTGCTCCAGGCTGTGTGTT-3′; ALP-F, 5′-TCCGTGGGTCGGATTCCT-3′; ALP-R, 5′-GCCGGCCCAAGAGAGAAA-3′; Klf2-F, 5′-ACCAAGAGTTCGCACCTAAA-3′; Klf2-R, 5′-GTGGCACTGGAAGGGCCTGT-3′; Klf5-F, 5′-TGGTTGCACAAAAGTTTATAC-3′; Klf5-R, 5′-GGCTTGGCGCCCGTGTGCTTCC-3′.
Western Blotting
Western blotting was performed as described previously (14). Antibodies used were as follows: ERK1/2 (Cell Signaling Technology), phospho-ERK1/2 (E10, Cell Signaling Technology), Elk-1 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Elk-1 (Cell Signaling Technology), Klf4 (14), SRF (Santa Cruz Biotechnology), vimentin (V9; Dako), GAPDH (6C5; Chemicon International), and FLAG (M2; Sigma).
Quantitative Chromatin Immunoprecipitation (ChIP) Assays
Quantitative ChIP assays were performed using antibodies for Klf4, SRF, and Runx2 (Santa Cruz Biotechnology) as described previously (25). Real-time PCR was performed to amplify the promoter region of the SM α-actin and SM22α genes, which contain CArG elements and a TCE or the intron 1 region of the SM22α gene (9, 25).
Luciferase Assays
Rat aortic SMCs were transfected with plasmids using Superfect (Qiagen) (23). The total amount of DNA per well was kept constant by adding the corresponding amount of expression vector without a cDNA insert. Luciferase activity was measured and normalized by cellular protein concentrations.
Mammalian Two-hybrid Assays
Mammalian two-hybrid assay was performed according to the manufacturer's instructions (Promega) (25). Expression plasmids of fusion proteins for GAL4-SRF and VP16-Klf4 were constructed, and expression was confirmed by Western blotting. Expression plasmids for GAL4 fusion protein, VP16 fusion protein, and Runx2 were co-transfected into NIH/3T3 cells, and luciferase activity was measured.
Adenine-induced Uremic Rats
Animal protocols were approved by Keio University Animal Care and Use Committee. Seven-week-old male Sprague-Dawley rats were fed a standard diet (CE2: 1.06% calcium and 0.99% phosphorus; CLEA, Tokyo) alone or a diet containing 0.75% adenine (Sigma) for 4 weeks to induce uremia (21). After adenine withdrawal, all animals were maintained on the standard diet for additional 4 weeks and sacrificed. Frozen sections of the thoracic aorta were subjected to von Kossa staining or immunohistochemistry for SM α-actin and Klf4, as described previously (25, 26).
Statistical Analyses
Data are presented as mean ± S.E. Statistical analyses were performed by one-way ANOVA with a post hoc Fisher protected least significant difference test. p values < 0.05 were considered significant.
RESULTS
High Phosphate Concentration Decreased SMC Differentiation Marker Genes, but Increased Osteogenic Genes in Cultured SMCs
The initial aim of the present studies was to determine whether our cultured rat aortic SMCs had an ability to convert into osteogenic cells by high phosphate concentration, as described previously by other laboratories (4–6, 17–20). Rat aortic SMCs were cultured in normal (0.9 mmol/liter) or high (4.5 mmol/liter) phosphate medium for 10 days, and the calcification status was examined by von Kossa staining. As shown in Fig. 1A, incubation with high phosphate medium markedly increased calcification of cultured SMCs. Expression of SMC differentiation marker genes and osteogenic genes was then examined by real-time RT-PCR. High phosphate medium significantly decreased expression of SM α-actin and SM22α by 38–57% (Fig. 1, B and C). In contrast, expression of Runx2, osteopontin, and ALP was increased by high phosphate concentration (Fig. 1, D–F). Indeed, incubation of SMCs with high phosphate medium for 8 days increased Runx2, osteopontin, and ALP mRNA expression by 1.7-fold, 2.4-fold, and 2.0-fold, respectively. In addition, high phosphate induced calcium deposition and apoptosis in SMCs (supplemental Fig. I), both of which are known to be increased during the calcification process (4–6, 17–20). These results indicate that incubation with high phosphate concentration decreased expression of SMC differentiation marker genes, whereas it increased expression of osteogenic genes and thereby induced calcification in our cultured SMCs.
FIGURE 1.

High phosphate concentration induced calcification of SMCs. A, rat aortic SMCs were cultured with normal (left) or high (right) phosphate concentration for 10 days. Representative pictures of von Kossa staining are shown (n = 3). Top, appearance of 60-mm dishes. Bottom, magnification x100. B–F, SMCs were incubated with normal (NP) or high (HP) phosphate concentration for 2, 4, 6, and 8 days. Expression of SM α-actin (B), SM22α (C), Runx2 (D), osteopontin (E), and ALP (F) mRNA was determined by real-time RT-PCR. Values represent the means ± S.E. *, p < 0.05 compared with SMCs with normal phosphate medium (n = 4).
High Phosphate Concentration Induced Klf4 Expression, but It Did Not Phosphorylate Elk-1
Results of our previous studies and others showed that suppression of SMC differentiation marker genes by either PDGF-BB or oxidized phospholipids was mediated in part by Klf4 induction and phosphorylation of Elk-1 (9, 11, 13–15). We examined whether the decrease in SMC differentiation marker gene expression by high phosphate concentration also utilized these mechanisms. First, expression of Klf4 mRNA was measured by real-time RT-PCR. Results showed that incubation of SMCs with high phosphate concentration for 2 days induced significantly Klf4 mRNA expression by 2.2-fold (Fig. 2A). After 4-, 6-, and 8-day incubation with high phosphate medium, there was a trend that Klf4 mRNA level was still higher than incubation with normal phosphate medium. Klf4 induction was also detectable at the protein level (Fig. 2B). Indeed, high phosphate incubation for 2 days markedly induced Klf4 expression in a nuclear fraction, but not in a cytosolic fraction of cultured SMCs. Expression levels of Klf2 and Klf5 were also examined because these Klf family members have been shown to be expressed in vascular SMCs (27). Expression of Klf2 and Klf5 was, however, unaltered by high phosphate concentration (Fig. 2, C and D), indicating that Klf4 induction was selective. On the other hand, high phosphate concentration did not induce phosphorylation of Elk-1 at any time points examined in cultured SMCs (Fig. 2E), although treatment of SMCs with 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC) at 10 μg/ml for 6 h (14), a positive control, obviously induced phosphorylation of Elk-1. In addition, high phosphate medium only slightly phosphorylated ERK1/2, an upstream signaling factor of Elk-1. These results suggest that suppression of SMC differentiation marker genes by high phosphate concentration utilizes a Klf4-dependent, but a phosphorylated Elk-1 independent mechanism.
FIGURE 2.
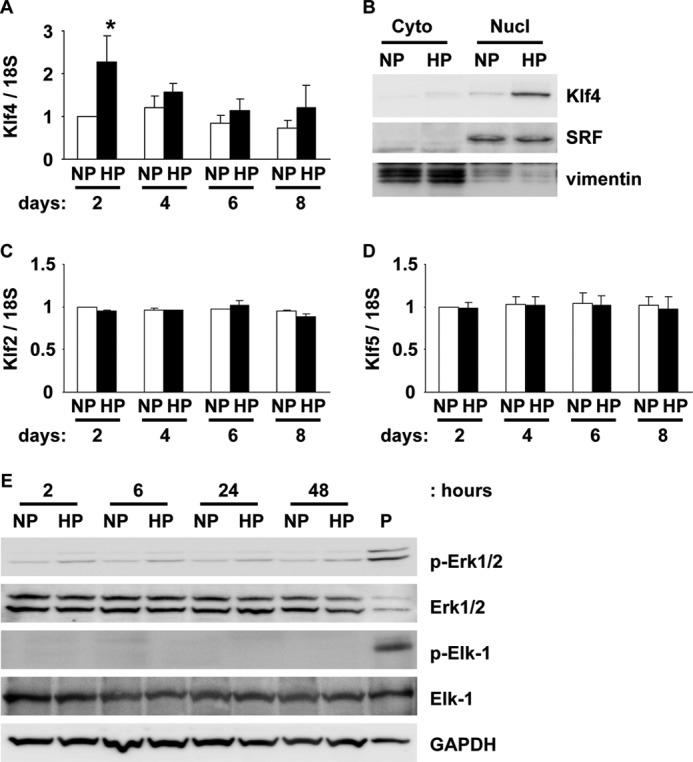
High phosphate induced Klf4 expression in cultured SMCs. SMCs were incubated with normal (NP) or high (HP) phosphate concentration for indicated times. Expression of Klf4 (A), Klf2 (C), and Klf5 (D) mRNA was determined by real-time RT-PCR. Values represent the means ± S.E. *, p < 0.05 compared with SMCs with normal phosphate medium (n = 4). B, expression of Klf4, SRF (nuclear protein), and vimentin (cytosolic protein) in SMCs with normal or high phosphate medium for 2 days was examined by Western blotting (n = 3). Cyto, cytosolic fraction; Nucl, nuclear fraction. E, expression of ERK1/2, phosphorylated ERK1/2 (p-ERK1/2), Elk-1, phosphorylated Elk-1 (p-Elk-1), and GAPDH was examined by Western blotting (n = 4). P, POVPC treatment at 10 μg/ml for 6 h.
Klf4 Was Induced in the Calcified Aorta of Uremic Rats
To determine whether Klf4 was involved in the calcification of the arteries in vivo, Klf4 expression was examined in the thoracic aorta of adenine-induced uremic rats. These rats are a model of uremia, which exhibits all key features of chronic renal failure, including elevated serum creatinine, hypocalcemia, hyperphosphatemia, reduced serum 1α,25-dihydroxyvitamin D, as well as arterial medial calcification (21). By feeding a diet containing 0.75% adenine for 4 weeks and by maintaining with a normal diet for additional 4 weeks subsequently, rats exhibited circumferential calcification in the media of thoracic arteries (Fig. 3, B and D). In contrast, rats received a control diet showed no evidence of calcification (Fig. 3, A and C). Immunohistochemical staining showed a dramatic decrease in SM α-actin expression in adenine-induced uremic rats compared with control rats (Fig. 3, E and F). The decrease in SM α-actin expression in the calcified arteries was consistent with the results of previous studies showing that expression of SM α-actin and SM22α was extensively suppressed in matrix GLA protein-null mice whose arteries calcify spontaneously (6). Notably, Klf4 expression was markedly induced in medial SMCs of adenine-induced uremic rats (Fig. 3H and supplemental Fig. IIA). Indeed, > 70% of medial cells were positive for Klf4, whereas Klf4-positive medial cells were undetectable in control rats (Fig. 3G). Klf4 mRNA expression was also induced in the aorta of adenine-induced uremic rats (supplemental Fig. IIB). These results suggest that adenine-induced uremic rats are a suitable model investigating SMC phenotypic switching into osteogenic cells in vivo, and that Klf4 plays a role in the medial calcification in these uremic rats.
FIGURE 3.

Klf4 was induced in the aorta of adenine-induced uremic rats. The calcification status was examined by von Kossa staining in the thoracic aorta of adenine-induced uremic rats (B and D) and controls (A and C). Expression of SM α-actin (E and F) and Klf4 (G and H) was examined by immunohistochemistry in the thoracic aorta of adenine-induced uremic rats (F and H) and controls (E and G). Representative pictures are shown from 4 mice analyzed per group. Bars: 1 mm (A and B); 50 μm (C–H). Inset in H is a magnified image from white lined square. Black and white arrowheads indicate Klf4-positive and Klf4-negative cells, respectively.
High Phosphate-induced Phenotypic Switching of SMCs Was in Part Mediated by Klf4
We determined whether Klf4 mediates high phosphate-induced suppression of SMC differentiation marker genes. Binding of Klf4 to the promoter region of SMC differentiation marker genes was first examined by quantitative ChIP assays. Results showed that high phosphate concentration in SMCs induced significantly Klf4 binding to the promoter region of the SM α-actin and SM22α genes, which contain two CArG elements and a TCE (Fig. 4, A and B). In contrast, Klf4 binding was undetectable at the intron 1 region of the SM22α gene, which lacks CArG elements and TCE, in SMCs incubated with normal or high phosphate medium (Fig. 4C). These results clearly indicate that Klf4 was selectively recruited to the CArGs/TCE-containing promoter regions of SMC differentiation marker genes by high phosphate concentration. We also examined whether the binding of SRF and Runx2 to these regions was altered by high phosphate concentration. The binding of SRF to the promoter regions of SMC differentiation marker genes was decreased by high phosphate, whereas Runx2 binding was induced in cultured SMCs (Fig. 4). These results were consistent with previous studies showing that overexpression of Klf4 decreased SRF binding to CArG elements within the SM α-actin promoter in SMCs (13) and that Runx2 was able to bind to SRF directly (22). Next, effects of Klf4 knockdown on high phosphate-induced phenotypic switching of SMCs were examined. siRNA specific for Klf4 was transfected into cultured SMCs, and SMCs were then incubated with normal or high phosphate medium for 2 or 6 days. By transfecting Klf4 siRNA, high phosphate-induced expression of Klf4 after 2-day incubation was blunted in cultured SMCs (Fig. 5A). Expression of SM α-actin, osteopontin, and ALP was determined after 6-day incubation with normal or high phosphate medium. As shown in Fig. 5C, high phosphate-induced decrease in SM α-actin expression was attenuated by knockdown of Klf4 in cultured SMCs. Of interest, high phosphate-induced increases in expression of osteogenic genes including osteopontin and ALP, but not Runx2, were blunted in Klf4 siRNA-transfected SMCs (Fig. 5, D–F). Moreover, knockdown of Klf4 attenuated high phosphate-induced calcium deposition in SMCs (Fig. 5G). Attenuation in high phosphate-induced decrease in SM α-actin expression, high phosphate-induced increases in osteopontin and ALP expression, and high phosphate-induced increase in calcium deposition was also seen by Runx2 siRNA (Fig. 5, B–G), although effects of Klf4 siRNA and Runx2 siRNA were not additive. Taken together, results suggest that Klf4 contributes to high phosphate-induced phenotypic switching of SMCs into osteogenic cells.
FIGURE 4.
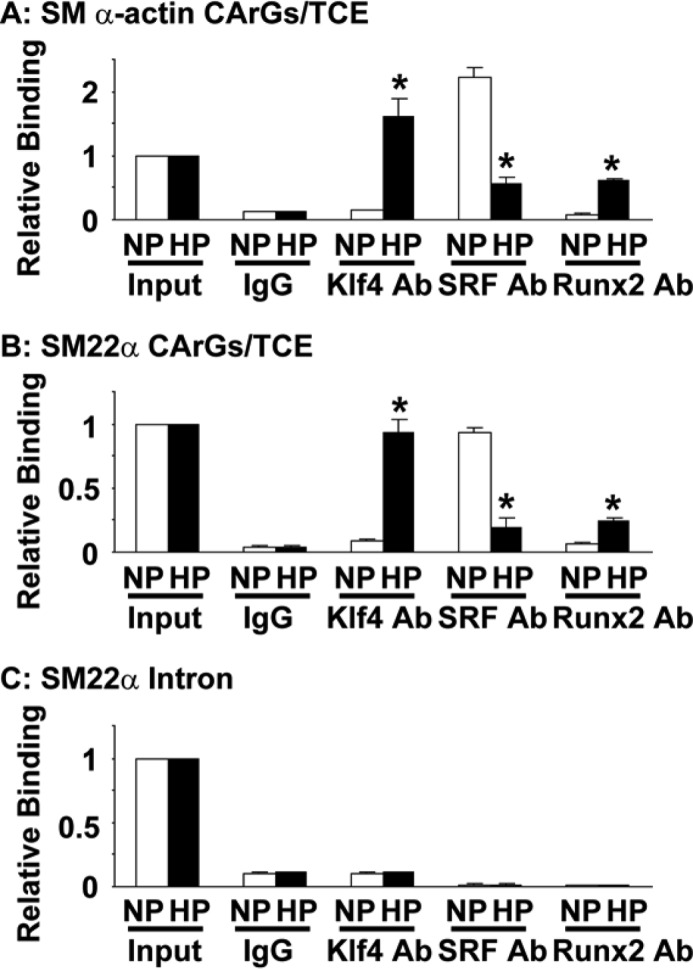
High phosphate concentration induced selectively Klf4 binding to the SMC promoters in SMCs. SMCs were cultured with normal (NP) or high (HP) phosphate concentration for 2 days, and the association of Klf4, SRF, and Runx2 with the CArGs/TCE-containing promoter regions of the SM α-actin gene (A) and the SM22α gene (B), or the intron 1 region of the SM22α gene (C) was determined by ChIP assays. Values represent the means ± S.E. *, p < 0.05 compared with SMCs with normal phosphate medium (n = 3).
FIGURE 5.

Knockdown of Klf4 attenuated high phosphate-induced phenotypic switching of SMCs. A, cultured SMCs were transfected with siRNA specific for Klf4 (siKlf4) or a scramble sequence (siScr) and incubated with normal (NP) or high (HP) phosphate concentration for 2 days. Expression of Klf4 and SRF in the nuclear fraction of SMCs was examined by Western blotting (n = 3). B, SMCs were transfected with siRNA for Runx2 (siRunx2) or siScr, followed by transfection of FLAG-Runx2 expression plasmid or control plasmid (Ct). Expression of Runx2 (anti-FLAG antibody) and GAPDH was examined by Western blotting (n = 3). C–G, cultured SMCs were transfected with siKlf4, siRunx2, and/or siScr and incubated with normal (NP) or high (HP) phosphate concentration for 6 days. Expression of SM α-actin (C), Runx2 (D), osteopontin (E), and ALP (F) mRNA was determined by real-time RT-PCR. Calcium deposition was measured and normalized by cellular protein content (G). Values represent the means ± S.E. *, p < 0.05 compared with SMCs with normal phosphate medium (n = 3).
Klf4 and Runx2 Independently Regulated SMC Phenotypic Switching into Osteogenic Cells
Results of previous studies showed that overexpression of Runx2 decreased expression of SMC differentiation marker genes by being recruited to the promoter region of SMC differentiation marker genes and competing SRF binding with myocardin (22). In addition, results of our previous studies showed that Klf4 repressed SMC differentiation marker genes through multiple mechanisms including direct binding to the TCE within the promoter region of SMC differentiation marker genes, interaction with SRF, and repression of myocardin expression (12, 13). Although siRNA-induced knockdown of Klf4 and Runx2 did not exhibit additive effects on SMC calcification (Fig. 5), we examined whether Klf4 and Runx2, both of which were induced by high phosphate concentration in SMCs, acted additively or synergistically to repress SMC differentiation marker genes by co-transfection of these expression plasmids and SM α-actin promoter-luciferase construct into cultured aortic SMCs. Results showed that Klf4 repressed SM α-actin mRNA expression (supplemental Fig. IIIA), as well as the transcriptional activity of the SM α-actin gene more potently than Runx2 and that effects of Klf4 and Runx2 were neither additive nor synergistic (Fig. 6A). Mutational analyses of the SM α-actin promoter-luciferase construct showed that repressive effect of Klf4 was dependent on both CArG elements and the TCE, whereas Runx2 repressed the transcriptional activity through CArG elements but not the TCE (Fig. 6B). CArG element-dependent repression of SMC differentiation marker genes by Runx2 was consistent with previous results showing that Runx2 repressed SMC differentiation marker genes via the inhibition of myocardin (22). Moreover, suppression of the SM α-actin promoter activity by Klf4 was mediated through CArG elements and the TCE because combinatorial mutations of these cis-elements abolished Klf4 effect. Next, we tested the effect of Runx2 on the interaction between Klf4 and SRF. Results of mammalian two-hybrid assays showed that Klf4 interacted with SRF, and this interaction was hindered by Runx2 in NIH/3T3 cells (Fig. 6C). These results suggest that, although both Klf4 and Runx2 were induced by high phosphate concentration in SMCs, repressive effects of these factors on SMC differentiation marker genes were not cooperative.
FIGURE 6.

Transcriptional regulation of the SM α-actin gene and the osteopontin gene by Klf4 and Runx2 was not interdependent. Expression plasmids for Klf4 and Runx2 (0, 30, 100, or 300 ng) were co-transfected with wild-type (A) and CArGs/TCE-mutant (B) of the SM α-actin promoter-luciferase construct (900 ng), human osteopontin promoter-luciferase (hOPN −1500/+87 or hOPN −970/+87) construct, mouse osteopontin promoter-luciferase (mOPN −2287/+132) construct or pGL3-basic plasmid (900 ng) (D) into rat aortic SMCs. Luciferase activities were measured and normalized to protein content. An arbitrary value of 1 was assigned to the activity of cells transfected with the wild-type SM α-actin promoter-luciferase construct (A and B) or hOPN (−1500/+87) construct and empty expression plasmids. C, NIH/3T3 cells were co-transfected with pG5Luc reporter plasmid, expression plasmids for fusion proteins of GAL4-SRF and VP16-Klf4, and Runx2 expression plasmid. Luciferase activity was measured and normalized to protein content. An arbitrary value of 1 was assigned to the activity of cells transfected with pG5Luc reporter plasmid and empty vectors for GAL4 and VP16. Values represent the means ± S.E. (n = 3). RLU, relative luciferase units.
Finally, we examined effects of Klf4 and Runx2 on the transcriptional activity of the osteopontin gene in SMCs. Both factors increased osteopontin mRNA expression in SMCs (supplemental Fig. IIIB). The binding site for Runx2 has been identified at −138/−132 bp and −136/−130 bp within human and mouse osteopontin promoters, respectively (28). We found two consensus Klf4 binding sites, 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′, at −1306/−1300 bp and −1140/−1134 bp within human osteopontin promoter. Two consensus Klf4 binding sites were also found at −2178/−2172 bp and −2090/−2084 bp within mouse osteopontin promoter. We made the human osteopontin promoter-luciferase (−1500/+87) construct, the human osteopontin promoter-luciferase (−970/+87) construct, which lacked two Klf4 binding sites, and the mouse osteopontin promoter-luciferase (−2287/+132) construct and tested their responsiveness to Klf4 and Runx2 in SMCs (Fig. 6D). Results showed that Klf4 increased the transcriptional activity of human osteopontin promoter-luciferase (−1500/+87) construct and mouse osteopontin promoter-luciferase (−2287/+132) construct, but not human osteopontin promoter-luciferase (−970/+87) construct, whereas Runx2 increased the activity of these constructs. Effects of Klf4 and Runx2 on the transcriptional activity of the osteopontin gene were not synergistic. These results suggest that Klf4 induces the transcription of the osteopontin gene at least in part through the region containing two consensus Klf4 binding sites and that this mechanism is independent of the action of Runx2.
DISCUSSION
Results of the present studies provide evidence that high phosphate-induced phenotypic switching of SMCs is mediated in part by Klf4. Indeed, incubation of SMCs with high phosphate concentration decreased expression of SMC differentiation marker genes such as SM α-actin and SM22α, whereas it increased expression of osteogenic genes including Runx2, osteopontin, and ALP. Phenotypic conversion of SMCs into osteogenic cells was accompanied by the induction of Klf4 both at mRNA and protein levels, but not by phosphorylation of Elk-1. Klf4 was also induced in the media of calcified aorta in adenine-induced uremic rats, where SM α-actin expression was suppressed markedly. Moreover, we demonstrated that high phosphate concentration induced Klf4 binding at the promoter regions of SMC differentiation marker genes and that siRNA-mediated knockdown of Klf4 blunted high phosphate-induced phenotypic switching of SMCs into osteogenic cells. Finally, we showed that effects of Klf4 and Runx2 on high phosphate-induced SMC calcification, as well as on the transcription of the SM α-actin gene or the osteopontin gene were not synergistic, although both factors were induced by high phosphate concentration. These results suggest the critical role of Klf4 for SMC phenotypic switching, not only in intimal atherosclerosis as demonstrated by previous studies (16), but also in a model of vascular medial calcification.
Unlike either skeletal or cardiac muscle cells that are terminally differentiated, SMCs retain more extensive plasticity to undergo modulation of their phenotype in response to changes in local environmental cues (7, 8). Although the major role of differentiated SMCs is contraction, they are able to change their phenotype into a wide spectrum of SMCs including proliferating SMCs, migrating SMCs, and SMCs producing extracellular matrix proteins. Previous studies have shown that Klf4 plays important roles in multiple scenes of phenotypic switching of SMCs (13–16, 29). Indeed, Klf4 decreased expression of SMC differentiation marker genes such as SM α-actin, SM-myosin heavy chain, and SM22α in response to PDGF-BB or oxidized phospholipids (13–15). We also showed that conditional deletion of the Klf4 gene in mice transiently delayed injury-induced suppression of SMC differentiation markers and enhanced neointimal formation after vascular injury (16). Suppression of SMC proliferation by Klf4 has been shown to be mediated through increased expression of p21WAF1/Cip1, a cell cycle inhibitor, by cooperating with p53 (16). Moreover, results of our recent studies showed that Klf4 mediated oxidized phospholipid-induced migration as well as production of type VIII collagen in SMCs (29). In the present studies, we have added a novel role of Klf4 for phenotypic switching of SMCs during the calcification process. As such, the preceding studies and our present studies strongly support a model wherein Klf4 serves as a point of convergence in mediating effects of local environmental cues on differentiated SMCs changing into a wide range of SMC phenotypes.
In contrast to the increase in Klf4 expression, incubation of SMCs with high phosphate concentration for 2, 6, 24, or 48 h did not induce phosphorylation of Elk-1. Instead of inducing Elk-1 phosphorylation, high phosphate induced expression of Runx2, a transcription factor shown to be able to repress SMC differentiation marker genes (22). Of interest, molecular mechanisms of repression of SMC differentiation marker genes by Runx2 and phosphorylated Elk-1 are quite similar. Tanaka et al. (22) showed that Runx2 repressed the transcriptional activity of SMC differentiation marker genes by inhibiting myocardin to bind with SRF. They demonstrated that the Runt domain of Runx2 was able to bind to the MCM1-Agamous-Deficiens-SRF (MADS) domain of SRF (22). This mechanism was confirmed and strengthened by results of our present studies showing that repressive effect of Runx2 was abolished by the mutation of CArG elements in the promoter region of the SM α-actin gene. On the other hand, results of previous studies showed that phosphorylated Elk-1 also displaced myocardin from the MADS domain of SRF at the CArG element-containing promoter region of SMC differentiation marker genes (9–11). It is highly likely that Runx2 and phosphorylated Elk-1 utilize a common mechanism to repress the transcription of SMC differentiation marker genes. It is of interest to determine whether Runx2 and phosphorylated Elk-1 are able to bind to the MADS domain of SRF simultaneously, or binding of these proteins to SRF is exclusive mutually.
Results of our present studies showed that Klf4 regulated the transcription of osteogenic genes in SMCs. Indeed, siRNA-mediated knockdown of Klf4 attenuated high phosphate-induced increases in expression of osteopontin and ALP in cultured SMCs. We also showed that Klf4 increased the transcriptional activity of the human and mouse osteopontin promoters containing two consensus Klf4 binding sites. Although the −1500/−970 bp region, which contains two Klf4 binding sites within human osteopontin promoter is relatively far from the transcriptional start site compared with the Runx2 binding site at −138/−132 bp, this region has been shown to be responsive to PPARγ ligand in THP-1 monocytic leukemia cells (24). It is interesting to determine whether other osteogenic genes also contain multiple Klf4 binding sites in their promoters.
In summary, we provide novel evidence that Klf4 contributes to high phosphate-induced phenotypic switching of SMCs in vitro as well as in an animal model of chronic renal failure. Results suggest that Klf4 has dual roles during the vascular calcification process: not only decreasing expression of SMC differentiation marker genes, but also inducing osteogenic genes. Further studies are needed to analyze the role of Klf4 for vascular calcification in loss-of-function animal models with chronic renal failure.
Supplementary Material
This study was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to M. H.), a grant for Research on Measures for Intractable Diseases from Japanese Ministry of Health Labour and Welfare (to M. H.), Takeda Science Foundation (to T. Y.), and Keio Gijuku Academic Development Fund (to T. Y.).

This article contains supplemental Figs. I–III.
- SMC
- smooth muscle cell
- Klf4
- Krüppel-like factor 4
- SM
- smooth muscle
- ALP
- alkaline phosphatase
- CArG
- CC(A/T-rich)6GG
- TCE
- transforming growth factor-β control element
- SRF
- serum response factor
- PDGF-BB
- platelet-derived growth factor-BB
- POVPC
- 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine.
REFERENCES
- 1. Tonelli M., Wiebe N., Culleton B., House A., Rabbat C., Fok M., McAlister F., Garg A. X. (2006) Chronic kidney disease and mortality risk: a systematic review. J. Am. Soc. Nephrol. 17, 2034–2047 [DOI] [PubMed] [Google Scholar]
- 2. London G. M., Guérin A. P., Marchais S. J., Métivier F., Pannier B., Adda H. (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 18, 1731–1740 [DOI] [PubMed] [Google Scholar]
- 3. Moe S. M., O'Neill K. D., Duan D., Ahmed S., Chen N. X., Leapman S. B., Fineberg N., Kopecky K. (2002) Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 61, 638–647 [DOI] [PubMed] [Google Scholar]
- 4. Shanahan C. M., Crouthamel M. H., Kapustin A., Giachelli C. M. (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ. Res. 109, 697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shioi A., Nishizawa Y., Jono S., Koyama H., Hosoi M., Morii H. (1995) β-Glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 15, 2003–2009 [DOI] [PubMed] [Google Scholar]
- 6. Steitz S. A., Speer M. Y., Curinga G., Yang H. Y., Haynes P., Aebersold R., Schinke T., Karsenty G., Giachelli C. M. (2001) Smooth muscle cell phenotypic transition associated with calcification: up-regulation of Cbfa1 and down-regulation of smooth muscle lineage markers. Circ. Res. 89, 1147–1154 [DOI] [PubMed] [Google Scholar]
- 7. Miano J. M. (2003) Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell Cardiol. 35, 577–593 [DOI] [PubMed] [Google Scholar]
- 8. Yoshida T., Owens G. K. (2005) Molecular determinants of vascular smooth muscle cell diversity. Circ. Res. 96, 280–291 [DOI] [PubMed] [Google Scholar]
- 9. Wang Z., Wang D. Z., Hockemeyer D., McAnally J., Nordheim A., Olson E. N. (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428, 185–189 [DOI] [PubMed] [Google Scholar]
- 10. Zhou J., Hu G., Herring B. P. (2005) Smooth muscle-specific genes are differentially sensitive to inhibition by Elk-1. Mol. Cell. Biol. 25, 9874–9885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshida T., Gan Q., Shang Y., Owens G. K. (2007) Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am. J. Physiol. Cell Physiol. 292, C886–895 [DOI] [PubMed] [Google Scholar]
- 12. Adam P. J., Regan C. P., Hautmann M. B., Owens G. K. (2000) Positive- and negative-acting Krüppel-like transcription factors bind a transforming growth factor β control element required for expression of the smooth muscle cell differentiation marker SM22α in vivo. J. Biol. Chem. 275, 37798–37806 [DOI] [PubMed] [Google Scholar]
- 13. Liu Y., Sinha S., McDonald O. G., Shang Y., Hoofnagle M. H., Owens G. K. (2005) Krüppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 280, 9719–9727 [DOI] [PubMed] [Google Scholar]
- 14. Yoshida T., Gan Q., Owens G. K. (2008) Krüppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am. J. Physiol. Cell Physiol. 295, C1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pidkovka N. A., Cherepanova O. A., Yoshida T., Alexander M. R., Deaton R. A., Thomas J. A., Leitinger N., Owens G. K. (2007) Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ. Res. 101, 792–801 [DOI] [PubMed] [Google Scholar]
- 16. Yoshida T., Kaestner K. H., Owens G. K. (2008) Conditional deletion of Krüppel-like factor 4 delays down-regulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 102, 1548–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Son B. K., Kozaki K., Iijima K., Eto M., Nakano T., Akishita M., Ouchi Y. (2007) Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 556, 1–8 [DOI] [PubMed] [Google Scholar]
- 18. Zhao M. M., Xu M. J., Cai Y., Zhao G., Guan Y., Kong W., Tang C., Wang X. (2011) Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 79, 1071–1079 [DOI] [PubMed] [Google Scholar]
- 19. Li X., Yang H. Y., Giachelli C. M. (2006) Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 98, 905–912 [DOI] [PubMed] [Google Scholar]
- 20. Speer M. Y., Li X., Hiremath P. G., Giachelli C. M. (2010) Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell Biochem. 110, 935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neven E., Dauwe S., De Broe M. E., D'Haese P. C., Persy V. (2007) Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. 72, 574–581 [DOI] [PubMed] [Google Scholar]
- 22. Tanaka T., Sato H., Doi H., Yoshida C. A., Shimizu T., Matsui H., Yamazaki M., Akiyama H., Kawai-Kowase K., Iso T., Komori T., Arai M., Kurabayashi M. (2008) Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells. Mol. Cell. Biol. 28, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yoshida T., Sinha S., Dandré F., Wamhoff B. R., Hoofnagle M. H., Kremer B. E., Wang D. Z., Olson E. N., Owens G. K. (2003) Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 92, 856–864 [DOI] [PubMed] [Google Scholar]
- 24. Oyama Y., Akuzawa N., Nagai R., Kurabayashi M. (2002) PPARγ ligand inhibits osteopontin gene expression through interference with binding of nuclear factors to A/T-rich sequence in THP-1 cells. Circ. Res. 90, 348–355 [DOI] [PubMed] [Google Scholar]
- 25. Shang Y., Yoshida T., Amendt B. A., Martin J. F., Owens G. K. (2008) Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J. Cell Biol. 181, 461–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoshida T., Gan Q., Franke A. S., Ho R., Zhang J., Chen Y. E., Hayashi M., Majesky M. W., Somlyo A. V., Owens G. K. (2010) Smooth and cardiac muscle-selective knock-out of Krüppel-like factor 4 causes postnatal death and growth retardation. J. Biol. Chem. 285, 21175–21184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki T., Aizawa K., Matsumura T., Nagai R. (2005) Vascular implications of the Krüppel-like family of transcription factors. Arterioscler. Thromb. Vasc. Biol. 25, 1135–1141 [DOI] [PubMed] [Google Scholar]
- 28. Sato M., Morii E., Komori T., Kawahata H., Sugimoto M., Terai K., Shimizu H., Yasui T., Ogihara H., Yasui N., Ochi T., Kitamura Y., Ito Y., Nomura S. (1998) Transcriptional regulation of osteopontin gene in vivo by PEBP2αA/CBFA1 and ETS1 in the skeletal tissues. Oncogene 17, 1517–1525 [DOI] [PubMed] [Google Scholar]
- 29. Cherepanova O. A., Pidkovka N. A., Sarmento O. F., Yoshida T., Gan Q., Adiguzel E., Bendeck M. P., Berliner J., Leitinger N., Owens G. K. (2009) Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ. Res. 104, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.