

Background: Human Nit2/ω-amidase is a putative tumor suppressor.
Results: Both the catalytic triad and loop 116–128 of hNit2 play an essential role in the enzyme-substrate binding and enzymatic activity.
Conclusion: The results of MD simulations are consistent with the kinetic analysis obtained with substrates α-ketoglutaramate and succinamate.
Significance: This work provides the basis for new areas of research into tumor glutamine metabolism and hyperammonemic diseases.
Keywords: Enzyme Kinetics, Homology Modeling, Molecular Dynamics, Site-specific Mutagenesis, Tumor Suppressor Gene, Human Nit2/ω-Amidase
Abstract
Human nitrilase-like protein 2 (hNit2) is a putative tumor suppressor, recently identified as ω-amidase. hNit2/ω-amidase plays a crucial metabolic role by catalyzing the hydrolysis of α-ketoglutaramate (the α-keto analog of glutamine) and α-ketosuccinamate (the α-keto analog of asparagine), yielding α-ketoglutarate and oxaloacetate, respectively. Transamination between glutamine and α-keto-γ-methiolbutyrate closes the methionine salvage pathway. Thus, hNit2/ω-amidase links sulfur metabolism to the tricarboxylic acid cycle. To elucidate the catalytic specificity of hNit2/ω-amidase, we performed molecular dynamics simulations on the wild type enzyme and its mutants to investigate enzyme-substrate interactions. Binding free energies were computed to characterize factors contributing to the substrate specificity. The predictions resulting from these computations were verified by kinetic analyses and mutational studies. The activity of hNit2/ω-amidase was determined with α-ketoglutaramate and succinamate as substrates. We constructed three catalytic triad mutants (E43A, K112A, and C153A) and a mutant with a loop 116–128 deletion to validate the role of key residues and the 116–128 loop region in substrate binding and turnover. The molecular dynamics simulations successfully verified the experimental trends in the binding specificity of hNit2/ω-amidase toward various substrates. Our findings have revealed novel structural insights into the binding of substrates to hNit2/ω-amidase. A catalytic triad and the loop residues 116–128 of hNit2 play an essential role in supporting the stability of the enzyme-substrate complex, resulting in the generation of the catalytic products. These observations are predicted to be of benefit in the design of new inhibitors or activators for research involving cancer and hyperammonemic diseases.
Introduction
A major route for the metabolism of glutamine in mammals is its hydrolysis to glutamate catalyzed by glutaminases K and L, followed by conversion of glutamate to α-ketoglutarate by transamination or by the glutamate dehydrogenase reaction. However, mammals also possess another pathway (the glutaminase II pathway) for the metabolism of glutamine. In the glutaminase II pathway, glutamine is transaminated to α-ketoglutaramate (KGM),3 followed by ω-amidase-catalyzed hydrolysis of KGM to α-ketoglutarate (1–12). Similarly, transamination of asparagine yields α-ketosuccinamate (KSM), followed by ω-amidase-catalyzed hydrolysis of KSM to oxaloacetate (1–3). The net reaction of both pathways is shown in supplemental Equation S1.
Although glutamine transamination is freely reversible, the reaction is drawn in the direction of glutamine utilization because KGM reversibly cyclizes to a lactam (2-hydroxy-5-oxoproline) (1, 4, 8) and is a substrate of ω-amidase. Under physiological conditions, ∼99.7% of KGM is in the lactam form, and only about 0.3% is in the open-chain (substrate) form (8). ω-Amidase has a high affinity for the open-chain form (8, 10, 11).
Nature makes use of the favorable direction of glutamine transamination to salvage α-keto acids arising through nonspecific transamination reactions and to close the last step of the methionine salvage pathway (6, 7) at the expense of glutamine. In the methionine salvage pathway, α-keto-γ-methiolbutyrate is formed from 5′-methylthioribose. α-keto-γ-methiolbutyrate is then converted to methionine by the glutaminase II pathway, thereby linking sulfur metabolism to the tricarboxylic acid cycle (11). KSM is unstable and can give rise to a plethora of aromatic heterocyclic compounds. An additional important role of ω-amidase, therefore, may be to remove potentially harmful KSM as oxaloacetate, which can then be metabolized through the tricarboxylic acid cycle (13).
The glutaminase II pathway is of clinical interest, particularly in regard to liver disease, certain inborn errors of metabolism, and cancer (14–21). A link between cancer biology and the glutaminase II pathway has recently been strengthened by the finding that nitrilase-like protein 2 (Nit2), which was previously shown to be a putative tumor suppressor (22), is identical to ω-amidase (10, 11, 23). On the basis of sequence analysis, Pace et al. reported that Nit2 belongs to the nitrilase (Nit) superfamily (24, 25). These authors have divided the nitrilase superfamily into 13 branches. Branch 10 in the Pace and Brenner (24) classification contains only two enzymes, both of which are found in mammalian tissues, designated Nit1 and Nit2 (26). All enzymes of the nitrilase family contain a highly conserved catalytic Glu-Lys-Cys triad in the active site (24, 27). Novo et al. (28) reported that mutation of any residue of this triad will result in loss of aliphatic amidase activity in Pseudomonas aeruginosa. The crystal structures of Caenorhabditis elegans NitFhit (nitrilase-fragile histidine fusion protein) (25), Saccharomyces cerevisiae Nit3 (29), and mouse Nit2 (mNit2) (26) have been determined. All of the Nit portions of these proteins possess an α-β-β-α sandwich structure, whereas the tertiary structure of human Nit2 (hNit2) has not yet been determined.
A number of fundamental techniques have been implemented to probe the microscopic interactions and to understand the mechanisms relating to the association between ligand and macromolecule/substrate and cognate enzyme (30). The following questions concerning hNit2/ω-amidase may be posited. How does the substrate interact with the enzyme to facilitate the catalytic reaction? When the substrate binds to hNit2/ω-amidase, how does the substrate induce a conformation change in the enzyme? What are the factors that determine the substrate specificity? To help answer these questions we have used molecular dynamics (MD) simulation, which has proved to be a useful tool for understanding the interaction between hNit2/ω-amidase and substrate and to predict structural features contributing to molecular recognition.
A homology modeling method was used to construct the structure of hNit2. We constructed an hNit2-substrate complex by docking and MD simulations to search for the potential substrate binding site. The substrate binding free energy for hNit2 was computed to characterize the substrate specificity for hNit2. The simulation results are consistent with the findings of the enzyme kinetic experiments reported herein; the enzyme was purified to homogeneity from recombinant wild type (WT) and mutated hNit2 genes. In addition, we carried out some inhibitor studies. Because glycylglycine was previously reported to be a good competitive inhibitor of rat liver ω-amidase (3), we used this dipeptide for comparative inhibitor studies with hNit2/ω-amidase. The enzymatic activity of hNit2/ω-amidase was measured in the presence of different substrate concentrations or various inhibitor concentrations. The kinetic parameters Vmax, Km, and Ki were obtained by global fitting of the experimental data.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Kinetic Analysis
The hNit2 gene was amplified with pFLAG-Nit2 as described previously (22). It was subcloned into a pQE70 plasmid and transformed in Escherichia coli M15. The catalytic triad mutant clones (E43A, K112A, and C153A) were prepared with the QuikChange II site-directed mutagenesis kit (Stratagene), followed by use of the pQE-70/M15 expression system to generate each desired mutant. The construction of the recombinant hNit2 Δ346–384 clone and purification of hNit2 Δ116–128 protein are described in the supplemental Materials and Methods. The mutagenic oligonucleotides are listed in supplemental Table S1. The amino acid sequences of WT hNit2 and Δ116–128 mutant are listed in supplemental Table S2. A single colony of WT and mutated M15[pQE70-hNit2] was inoculated into 25 ml of Luria-Bertani broth containing 100 μg/ml ampicillin and 25 μg/ml kanamycin and was incubated overnight at 37 °C. Protein overexpression was induced with 2 mm isopropyl 1-thio-β-d-galactopyranoside and maintained for 6 h; the overexpressed protein in the crude protein fraction obtained from the periplasmic fraction was further purified. We chose KGM, SM, and 3-PPN for assay of the enzymatic activity of hNit2/ω-amidase (11, 23, 31). The proteins were then subjected to kinetic analysis. Protein concentrations were measured by the Bradford procedure using bovine serum albumin as a standard. The apparent kinetic parameters were obtained by nonlinear regression analysis using the SigmaPlot enzyme kinetic module 1.3 program. The experimental data were best fitted to a competitive inhibition equation,
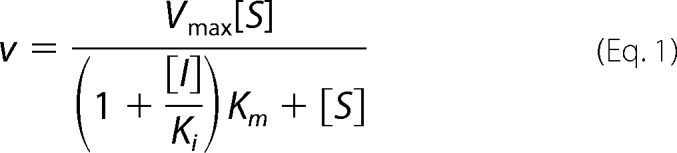 |
where [S] and [I] are the substrate and inhibitor concentration, respectively, and Ki is the inhibition constant for the enzyme-inhibitor complex. Detailed construction, purification, and kinetic analysis methods are described in the supplemental Materials and Methods and supplemental Figs. S1–S5).
Molecular Modeling of the hNit2 Structure
A homology modeling method (32) was used to construct the tertiary structure of hNit2. First, the BLAST program was used to search for template proteins from the Protein Data Bank. From a comparison of these template proteins, a structure that has the highest sequence identity with the lowest E-value was chosen as the template. Next, a sequence alignment of the template and the hNit2 sequences by the ClustalW program was performed, followed by a structure-based sequence alignment using the Profile-3D program (33). Based on the sequence alignment, the MODELLER program (32, 34–36) was used to build the model, followed by energy minimization. All molecular visualizations and graphics were carried out using the VMD program (37). Structural similarities were estimated from the root mean square deviation (RMSD).
Docking and Molecular Dynamics Simulation
To identify substrate binding modes in the catalytic cavity of hNit2, we performed docking by the Autodock Vina program (38). This approach permits an evaluation of scoring function during the docking process, so that as many conformations as possible can be detected. The minimum scoring value indicates the most likely conformation. Based on these results, the complex structure of hNit2 docked with substrate was selected as a starting structure for the MD simulation. The initial substrate structures were generated by the InsightII (Molecular Simulations Inc.) package, and the charges were calculated using the GAMESS (40) program. All MD simulations were performed by using the NAMD program (41) and the CHARMM27 force field (42). Each form of hNit2 structure was minimized and then solvated with TIP3P (43) water molecules in a 80 × 80 × 80-Å3 cubic box with 150 mm KCl ionic strength. Energy minimization and 40-ns MD were applied to equilibrate the system. A periodic boundary condition and particle mesh Ewald algorithm (44) were applied, and the non-bonding cut-off was set at 14 Å. The simulation was carried out under an NPT ensemble at 300 K and 1 atm.
Binding Free Energy Calculation by the Molecular Mechanics Poisson-Boltzmann Surface Area Method
The binding affinity between hNit2 and substrates (i.e. KGM, KSM, and SM) was computed by using the molecular mechanics Poisson-Boltzmann surface area (45) method. The binding free energy ΔGbind of substrate to enzyme is calculated according to Equation 2.
If ΔGX represents the free energy of species X, it can be calculated from Equation 3.
EMM is the molecular mechanical energy obtained from the equation, EMM = EInternal + EvdW + Eelec, where EInternal, EvdW, and Eelec are the internal energy of the molecule, the non-bonded interaction energy from the van der Waals interaction, and electrostatic interaction, respectively. The solvation free energy (ΔGsolv) represents the polar free energy (ΔGpolar) and the nonpolar free energy (ΔGnonpolar) of solvation; the polar part is calculated by the APBS program (46), and the nonpolar part is calculated from the equation,
In Equation 4, ΔA is the solvent-accessible surface area change of the molecule (47), the parameter β is 0.92 kcal/mol, and γ is 0.00542 kcal/mol Å2 (45). For the solute entropic contribution to the system (TΔS), ΔS is calculated by normal mode analysis with the CHARMM (48) program, and T is temperature in Kelvin.
Circular Dichroism (CD) Spectral Analysis
CD spectra were recorded by means of a JASCO J-810 spectropolarimeter equipped with a 0.1-cm quartz cuvette. All reported spectra are the average of 10 scans. The percentage of protein secondary structure was analyzed by accessing the DichroWeb Web site (39).
Fluorescence Spectroscopy
All intrinsic fluorescence measurements of WT hNit2 and each mutated hNit2 protein were carried out on a PerkinElmer LS508 luminescence spectrometer with an excitation wavelength of 280 nm. The excitation and emission slits were set at 5.0 and 10.0 nm, respectively.
RESULTS
Purification and Confirmation of Expression of hNit2 and Its Mutants
The various proteins were overexpressed in the M15[pREP4] system for the production of the hNit2-His6 fusion protein (hereinafter referred to simply as hNit2) and then were subjected to 12% SDS-PAGE followed by staining with Coomassie Brilliant Blue. A single band for each form of hNit2 (WT and single-point mutants) with a mass of about 32 kDa and that for the Δ116–128 mutant with a mass of 30 kDa are shown in Fig. 1A. Purified WT hNit2 or Δ116–128 protein was subjected to 12% SDS-PAGE, and the protein band was excised and trypsinized. LC/MS/MS spectral analysis showed a highly significant probability that the protein overexpressed in E. coli is indeed hNit2 and hNit2 Δ116–128 mutant (see supplemental Fig. S6).
FIGURE 1.

Gel electrophoresis and enzyme kinetic analysis. A, 12% SDS-PAGE analysis of recombinant hNit2 and mutants eluted with 200 mm imidazole through a nickel-nitrilotriacetic acid column, respectively. An aliquot (20 μg) of each purified protein was applied to individual lanes. B and C, enzyme kinetic analysis of hNit2/ω-amidase activity. The graphs are shown with the Lineweaver-Burk plot. B, α-ketoglutarate formation in the presence of varying concentrations of KGM and 1 μg of enzyme. The reactions were carried out at 37 °C in the presence of 100 mm Tris-HCl buffer, pH 8.5, for 3 min. The concentration of KGM (1.5–20 μm) is that of the open-chain form (note that the interconversion between open-chain (substrate) and lactam structures is very rapid at pH 8.5 (8) and, at the concentration of enzyme used (1 μg in a 20-μl assay mixture), is not limiting for enzyme activity). C, succinyl hydroxamate formation in the presence of 100 mm hydroxylamine, 1 μg of enzyme, and varying concentrations of SM (20–120 mm) in 50 μl of reaction mixture. The reactions were carried out for 3 min at 37 °C in the presence of 100 mm potassium phosphate buffer, pH 7.2. Each assay was carried out in the presence of 0 mm (●), 10 mm (○), 20 mm (▾), or 30 mm (▿) glycylglycine. Results are shown as the mean ± S.E. of three repeats. The solid lines are the best fit by the competitive inhibition equation. D, enzymatic activity of WT hNit2/ω-amidase and various mutants. The assay mixture (50 μl) contained 100 mm SM, 100 mm hydroxylamine, and 0.5 μg of protein in 100 mm potassium phosphate buffer, pH 7.2. Each mixture was incubated at 37 °C for 3 min, followed by measurement of succinyl hydroxamate. Error bars, S.E.
hNit2/ω-Amidase-catalyzed Hydrolysis of KGM and Hydroxaminolysis of SM
We chose KGM, SM, and 3-PPN for a detailed study of the substrate specificity of highly purified recombinant hNit2. Homogeneous (as judged by the criterion of SDS-PAGE) hNit2 exhibited considerable ω-amidase activity. The highest specific activity among the compounds studied as substrates (at saturating substrate concentration) was exhibited by SM, followed by KGM. The preparation exhibited no detectable catalytic activity toward 3-PPN at pH 8.0 as assessed by a sensitive ammonia assay. The activities toward KGM and SM were also determined at different concentrations of substrates and inhibitor glycylglycine, and the kinetic parameters, Vmax, Km, and Ki, were obtained by global and best fitting of the experimental data, as shown in Fig. 1, B and C.
Kinetic analyses showed Vmax and Km values for KGM to be 30.3 ± 1.7 μmol/min/mg and 0.0027 ± 0.0004 mm, respectively, and showed the corresponding values for SM to be 169 ± 6 μmol/min/mg and 10.9 ± 2.4 mm, respectively (Table 1). The calculation for the Km exhibited toward KGM takes into account the finding that only 0.3% of KGM is in the open-chain (substrate) form, whereas 99.7% is in a lactam (inactive) form (8). Moreover, interconversion between the two forms is OH−-catalyzed (8). The pH of the buffer (pH 8.5) and the amount of enzyme present in the assay mixture were such that the rate of ring opening was not rate-limiting. Evidently, under the assay conditions employed, hNit2 shows relatively higher affinity for KGM but higher catalytic activity with SM.
TABLE 1.
Kinetic data exhibited by hNit2/ω-amidase and mouse ω-amidase toward various substrates
All of the kinetic data for hNit2/ω-amidase were determined in triplicate at 37 °C and are reported as means ± S.E.
| Substrate | Vmax | kcat | Km | kcat/Km |
|---|---|---|---|---|
| μmol/min/mg | s−1 | mm | s−1 m−1 | |
| hNit2/ω-amidase | ||||
| KGM (pH 8.5) | 30.3 ± 1.7 | 16.1 ± 0.9 | 0.0027 ± 0.0004 | 6.1 × 106 |
| SM (pH 7.2) | 169 ± 6 | 89.7 ± 3.4 | 10.9 ± 2.4 | 8.3 × 103 |
| Mouse ω-amidasea | ||||
| KGM (pH 8.5) | 32.0 ± 4.0 | 17.5 ± 2.2 | 0.195 ± 0.023 (∼0.0006)b | 89.7 × 103 (∼29.7 × 106)b |
| KSM (pH 7.2) | 2.1 ± 0.1 | 1.15 ± 0.05 | 0.003 ± 0.001 | 385 × 103 |
| SM (pH 7.2) | 3.6 ± 0.5 | 2.0 ± 0.3 | 0.23 ± 0.03 | 8.7 × 103 |
When the assay with either KGM or SM was conducted in the presence of varying amounts of glycylglycine, competitive inhibitor kinetics were noted (Fig. 1, B and C). The Ki values exhibited toward glycylglycine in the presence of KGM and SM were found to be 5.6 ± 0.7 and 6.1 ± 1.2 mm, respectively. These values are similar to that reported previously (5 mm) for rat liver ω-amidase when KGM was used as substrate (3).
Kinetic Analyses of hNit2 Mutants
Three catalytic triad mutated hNit2/ω-amidase proteins, namely E43A, K112A, and C153A, did not exhibit any detectable ω-amidase activity when enzymatic assays were performed using 100 mm succinamate as substrate, compared with the activity of the WT hNit2; the Δ116–128 mutant exhibited less than 3% of the activity exhibited by the WT enzyme (Fig. 1D).
hNit2 Model
A multiple sequence alignment of hNit2 with the sequences of representative proteins of the nitrilase superfamily, including examples from mice, yeast, C. elegans, Pyrococcus horikoshii ot3, and Agrobacterium sp. was constructed, showing that hNit2 (residues 1–276) shares a high degree of sequence homology with other family members (Fig. 2). The highest sequence identity is 89% between hNit2 and mNit2. Remarkably, a structure-based sequence alignment revealed that hNit2 and mNit2 are highly similar with a Z-score value of 49.5. Accordingly, the crystal structure of mNit2 (Protein Data Bank entry 2w1v) (26) was taken as the template to construct the tertiary structure of the dimeric and monomeric forms of hNit2 (Fig. 3, A and B). The monomeric structure of the hNit2 was optimized and followed by MD simulation to maximally satisfy the spatial restraints. Note, however, that the C terminus (residues 261–276) of the monomeric hNit2 structure changes to a random coil after MD simulation (Fig. 3B). The evidence is due to the lack of a stabilizing β-sheet formed from the dimeric interface interaction. The surface electrostatic potential representation of hNit2 is shown in Fig. 3C.
FIGURE 2.

Sequence alignment of hNit2 and selected homologous nitrilase superfamily proteins. NP_064587 is the hNit2 sequence from the NCBI. Other sequences were obtained from the Protein Data Bank. Identical residues in aligned sequences are highlighted; the catalytic triad residues are boxed.
FIGURE 3.

Structures of hNit2/ω-amidase. Shown are dimeric (A) and monomeric (B) hNit2 structures. In B, the catalytic triad (Glu43, Lys112, and Cys153) is shown in a stick representation and is color-coded according to atom type. α-Helix, β-sheet, and coil regions are shown in red, yellow, and gray, respectively. The loop in a closed state B (green) in the conformation of hNit2 shown here is based on the known crystal structure of mNit2. The loop in an open state A (purple) is based on the crystal structure of the Nit portion of C. elegans NitFhit fusion protein. The loop in a semiopen state C (orange) is derived from a 40-ns MD simulation. C, surface electrostatic potential representation of the monomeric hNit2. Positive, negative, and neutral charges are colored blue, red, and white, respectively.
Structural alignment of the hNit2 model with the crystal structure of mNit2 shows an RMSD value of 0.94 Å. Structurally, the hNit2 monomer is composed of seven α helices, 12 anti-parallel β strands, and one loop (residues 116–128); the catalytic site is located in the deep cleft covered with the loop 116–128, consisting of a β-turn-β-structure (Fig. 3B). A comparison of the structures shows that this loop 116–128 is in a closed conformation for the mNit2 (Protein Data Bank entry 2w1v) (26) and in an open conformation for the Nit portion of C. elegans NitFhit (Protein Data Bank entry 1ems) (25). An open conformation for the loop 116–128 is required to allow substrate access to the hNit2 catalytic pocket. Therefore, we constructed an open conformation for hNit2 based on the loop conformation of the C. elegans NitFhit. The loop conformation changes from an initial open state A (purple) to a semiopen state C (orange) after a 40-ns MD equilibration (Fig. 3B). Our analysis demonstrated that the loop 116–128 undergoes a liplike motion after MD simulation. The loop acts as a gate. Opening of the gate results in an enzymatically active form of hNit2.
Structural Comparisons of hNit2/ω-Amidase Mutants with the WT Protein
To elucidate the role of each mutated residue and the deleted loop 116–128, it is essential that the mutant hNit2 structures do not undergo major structural changes as a result of the mutation. Importantly, CD spectral analysis indicates that the secondary structures of the WT hNit2 and the Δ116–128 mutant are similar; the percentages of α-helix, β-strand, and random loop domains in the WT hNit2 are about 26, 34, and 40%, respectively, whereas the percentages for the Δ116–128 mutant are 26, 33, and 41%, respectively, as shown in supplemental Fig. S7. The CD spectra of the three catalytic triad mutants and the Δ116–128 mutant did not show any gross protein misfolding compared with that of the WT hNit2/ω-amidase protein (also see supplemental Fig. S7). Moreover, fluorescence spectra show that the emission maxima at 335 nm of WT hNit2 and each of the mutants are not altered, as shown in supplemental Fig. S8. Taken together, both the CD and fluorescence spectral data confirmed that there are no significant secondary structural changes associated with the hNit2 mutants relative to the WT protein.
Analysis of Conformational Changes at the Catalytic Active Site of hNit2
The catalytic triad (Glu43, Lys112, and Cys153) of hNit2 is located in the proximal pocket (Fig. 3B). The spatial positioning of the catalytic triad residues and the lid region 116–128 are important factors in the activity of hNit2. To examine the effect of mutation on the conformation of the pocket, the distances between the triad residues were calculated, and the results are shown in Fig. 4A and supplemental Fig. S9. These distances are important determinants in the stabilization of the enzyme-substrate complex during the catalytic reaction. For hNit2 and the Δ116–128 mutant, the average distances of OE1 (Glu43)–NZ (Lys112), OE2 (Glu43)–S (Cys153), and NZ (Lys112)–S (Cys153) are relatively short, and the hydrogen bond OE1 (Glu43)–NZ (Lys112) is preserved during 90 ns of simulation. However, for the E43A and K112A mutants, the average distances of CB (Ala43)–NZ (Lys112) and CB (Ala112)–S (Cys153) are increased dramatically, resulting in a loss of hydrogen bonding, an enlargement of the catalytic pocket, and a decrease in the substrate binding affinity. In the key step of the ω-amidase enzymatic reaction, Cys153 plays a crucial role as a nucleophile by attacking the carbonyl carbon of the substrate amide group, a process promoted by Lys112. In the case of the C153A mutant, there are no apparent distance changes among the active site triad residues, but the nucleophile role of Cys153 has been removed with total loss of enzyme activity. The spatial positioning of the catalytic triad in hNit2 changes little in the presence of water molecules, but in the mutants there is a large change in the positioning. The size of the catalytic pocket was also estimated from the number of water molecules required to fill the pocket along with the MD simulation for hNit2 and its mutants (Fig. 4B and supplemental Fig. S10). For the E43A, K112A, and Δ116–128 mutants, the pocket size is much bigger than that of WT hNit2 and the C153A mutant. Structural analyses showed that these mutants (E43A, K112A, and Δ116–128) enlarge the size of the pocket, thereby decreasing the substrate binding affinity with concomitant loss of activity. Kinetic analysis indicated that these are inactivating mutations. The RMSD of the backbone atoms for each mutant was calculated (supplemental Fig. S11). The result shows that the core structure, lacking the loop 116–128, N terminus (residues 1–4), and C terminus (residues 245–260), remained intact during MD simulations.
FIGURE 4.
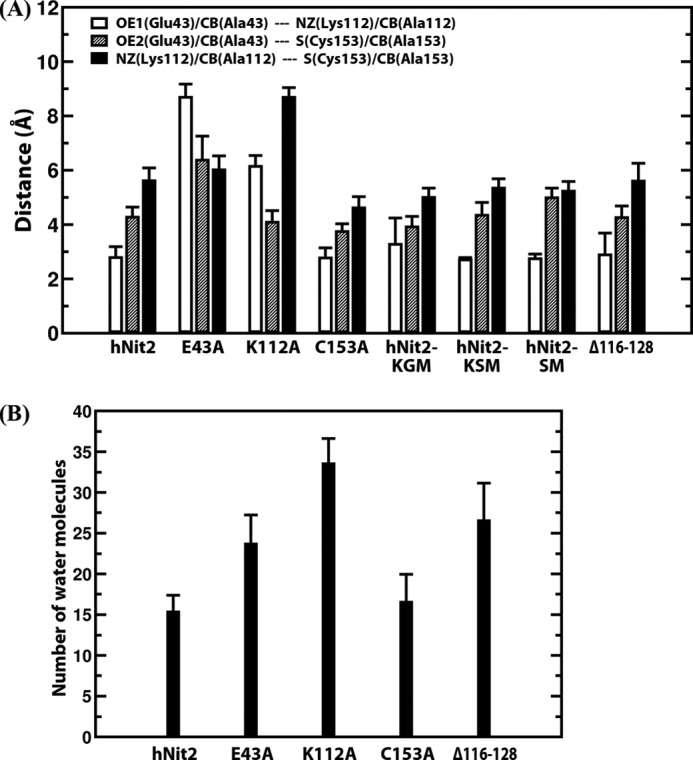
Analyses of the catalytic triad cavity. A, average distances of OE1 (Glu43)–NZ (Lys112), OE2 (Glu43)–S (Cys153), and NZ (Lys112)–S (Cys153) for the hNit2, hNit2-substrate complexes, and Δ116–128; the average distances of CB (Ala43)–NZ (Lys112), CB (Ala43)–S (Cys153), and NZ (Lys112)–S (Cys153) for E43A; the average distances of OE1 (Glu43)–CB (Ala112), OE1 (Glu43)–S (Cys153), and CB (Ala112)–S (Cys153) for K112A; the average distances of OE1 (Glu43)–NZ (Lys112), OE1 (Glu43)–CB (Ala153), and NZ (Lys112)–CB (Ala153) for C153A. B, the number of water molecules estimated to fill the active site cavity of WT hNit2 and mutants. In each case, the data were calculated from MD simulations for WT hNit2, mutants, and substrate-bound proteins, respectively. Error bars, S.E.
Simulation of Substrate Binding to hNit2
The structures of three substrates KGM, KSM, and SM, and the partial charges at pH 7.2 for each atom are shown in Fig. 5. A 40-ns MD simulation was performed for each hNit2-substrate complex. The distances between the triad residues for each complex and the RMSD of the backbone atoms were calculated to ascertain whether the hNit2-substrate complexes are stable (Fig. 6A and supplemental Figs. S9 and S11). The findings showed that, without including the loop 116–128 and the N-terminal and C-terminal regions, the RMSD of the main structure is around 1.0 Å, which means that the conformation of hNit2 remains stable in the hNit2-substrate complexes. The loop 116–128, however, undergoes a significant change when substrate is bound, with the value of the RMSD larger than 1.0 Å. How does the substrate binding induce the conformational change of hNit2? The difference of the root mean square fluctuation for the equivalent residues between hNit2 and hNit2-substrate complex was calculated (Fig. 6B). Remarkably, the flexibility of the loop 116–128 is dramatic in the hNit2-substrate complex, but this loop becomes much less flexible in hNit2, as a result of a salt bridge interaction between Asp117 and Arg157 stabilizing the loop conformation. In the hNit2-substrate complex, the substrate occupies the catalytic cavity and breaks the salt bridge interaction, leading to a higher freedom of the loop 116–128, whereas residues His187, Tyr154, and Arg157 interact directly with substrate. The effect of mutation on the motion of the loop 116–128 was simulated. The conformation of the loop became much wider than the open state in the K112A mutant, but for the E43A and C153A mutants, the loop remained in a state similar to that exhibited by hNit2.
FIGURE 5.
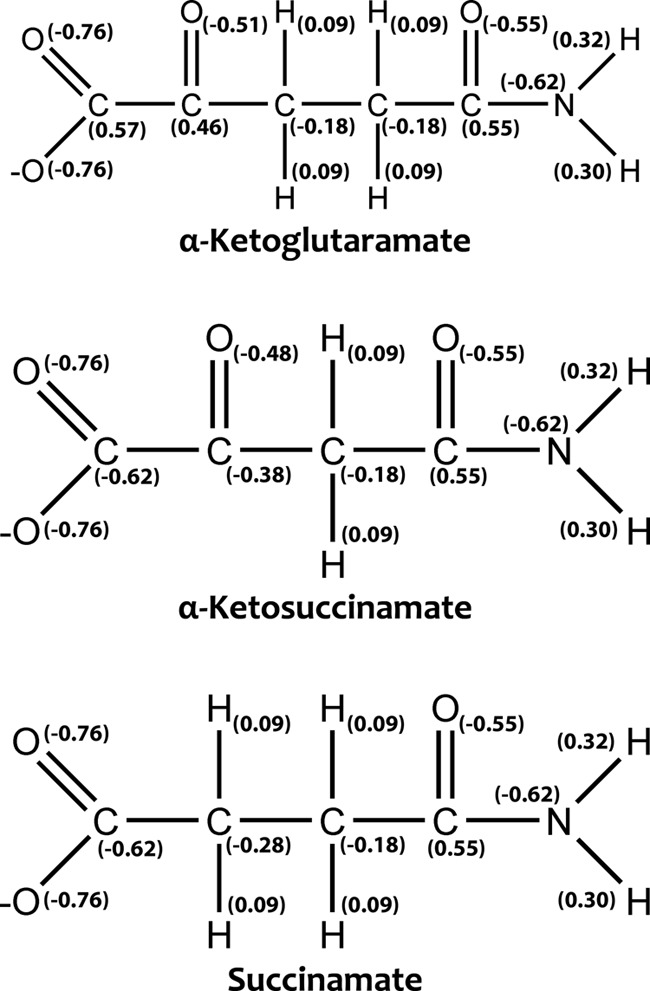
Structures and partial charges of three substrates: KGM (open-chain form), KSM, and SM.
FIGURE 6.

Structural analysis of the hNit2, hNit2-substrate complexes, and Δ116–128 mutant. A, RMSD of the hNit2 backbone atoms (residues 1–260) (white bar) and that of a structure lacking the loop (residues 116–128), N terminus (residue 1–4), and C terminus (residues 245–260) (black bar). The C terminus (residues 261–276) of the hNit2 monomer is a random coil and is not calculated. B, profiles of the difference of root mean square fluctuation for each residue of the hNit2-substrate complexes. The substrates are KGM (a), KSM (b), and SM (c). The reference structure is hNit2 in which substrate is not present in the active site. Error bars, S.E.
Remarkably, in hNit2, the simulation showed that one water molecule is juxtaposed between Glu43 and Lys112 and forms hydrogen bonds with both residues at 53 ns of MD time (supplemental Fig. S9a). A similar result was observed with the hNit2-KGM complex during 4.5–13.5-ns MD time, the water molecule disrupting the hydrogen bond OE1 (Glu43)–NZ (Lys112), leading to an increased distance between Glu43 and Lys112. Once the water molecule has departed from this region, the side chains of both Glu43 and Lys112 return to their initial positions (supplemental Fig. S9e). Indeed, both the substrate-free and substrate-bound forms of hNit2 display a highly spatial conservation of the catalytic triad during the MD time course. In future work, it will be important to determine whether the hydrogen bonds between the water molecules and triad residues participate in the catalytic mechanism and stabilization of the transition state.
Simulated docking of three substrates (KGM, KSM, and SM) to E43A, K112A, C153A, and Δ116–128 mutants was also performed. However, these simulations were difficult to perform, because, unlike the case with hNit2, these substrates reside only fleetingly in the active site of the hNit2 mutants compared with residence time in the wild type enzyme. Therefore, the substrate binding free energy cannot be calculated for these mutants of hNit2. The simulation results are consistent with the mutational experiments, which revealed that these mutations severely decrease the affinity of hNit2 for the substrate. 3-PPN was found to bind very poorly in the catalytic pocket of the wild type hNit2, and, as a result, the free energy of binding of this compound to hNit2 was also not calculated.
Comparison of the hNit2 active site structure with three binding substrates (KGM, KSM, and SM) and two non-binding substrates (Gln and Asn) is shown in Fig. 7A. (The binding mode of KSM is based on literature values for Km and Vmax (3 μm and 2.1 μmol/min/mg, respectively, at pH 7.2 and 30 °C) published previously for mNit2/ω-amidase (10).) The active site cavity is formed by residues Glu43, Lys112, Phe116, Asp117, Asp119, Glu128, Cys153, Tyr154, Arg157, Thr183, His187, and Tyr216. KGM forms hydrogen bonds with Lys112, Cys153, Glu128, Arg157, and His187 (Fig. 7B). A water molecule is interposed between Glu43 and KGM and leads to hydrogen bond formation between Glu43 and Cys153. The amide group of KGM directly interacts with Glu128, which replaces the role of Glu43, and forms a hydrogen bond with KGM. KSM is an analog of KGM that is shorter than KGM by one methylene group. The amide group of KSM is oriented toward Glu43 and forms a hydrogen bond with OE2 (Glu43). The carboxyl group of KSM is stabilized by Arg157 and Thr183 through formation of hydrogen bonds (Fig. 7C). For SM, the binding mode is similar to that of KGM, except that the lack of an α-keto group results in the absence of hydrogen bonding to Arg157 (Fig. 7D). As a result, the binding affinity to hNit2 for SM is weaker than that for KGM. In addition, the average distances between the Cys153 sulfur and the carbon atom of the amide groups of KGM, KSM, and SM are 3.95, 3.65, and 4.22 Å, respectively; the distance trend is a contributing factor to the binding affinity.
FIGURE 7.

Binding modes of three substrates and two non-substrates in the hNit2 catalytic cavity. A, structural alignment of five hNit2 complexes. Substrates are KGM (B), KSM (C), and SM (D). Non-substrates are Gln (E) and Asn (F). The carbon (green), oxygen (red), nitrogen (blue), and polar hydrogen (white) atoms of the substrates are shown in a ball-and-stick representation. Hydrogen bonding is shown in orange, and distances are indicated by a number in Å. A water molecule (W) is shown in a ball-and-stick representation and color-coded by atom type. These structures were taken from the last 20-ns MD trajectories.
Our calculations suggest that the docked substrate induces a structural change in hNit2 to enhance the nucleophilicity of the Cys153 sulfhydryl. Lys112 forms a hydrogen bond with the carbonyl oxygen of the substrate amide, stabilizing the hNit2-substrate complex. The average distance between the ϵ-amino nitrogen (NZ) of Lys112 and the carbonyl oxygen (O) of the substrate amide for the three substrates, KGM, KSM, and SM, is ∼2.70 Å. The ϵ-amino group of Lys112 is a general base and participates in hydrogen bonding during the catalytic reaction. One oxygen (OE1) of the γ-carboxyl group of Glu43 is close enough to the ϵ-amino group (NZ) of Lys112 to form a salt bridge. For the KSM complex, the other carboxyl oxygen (OE2) of Glu43 stabilizes substrate binding via a hydrogen bond with the amide group nitrogen of the substrate. These hydrogen bonds are very important for the stabilization of the hNit2-substrate complex for the catalytic reaction. However, for the KGM and SM complexes, Glu128 plays a role similar to that of Glu43, forming a hydrogen bond with the substrate to enhance the binding affinity. Thus, the orientation of the amide group of the three substrates studied here depends on interactions with Glu43 and Glu128. The deprotonated Glu43 may act as a base by removing a proton from Cys153, thereby increasing its nucleophilicity.
In nature, separate enzymes are used to hydrolyze glutamine (glutaminase K and L), asparagine (asparaginase), and KGM/KSM (ω-amidase) despite the fact that the substrates are of similar size. However, there may be overlap in specificities between some asparaginases and glutaminases. Meister et al. (2) noted that partially purified rat liver ω-amidase has no activity with glutamine or asparagine, a finding that we have verified with purified hNit2/ω-amidase. To determine the mechanism for this remarkable selectivity, we investigated the simulated docking of glutamine and asparagine to the active site of hNit2. Docking scores for glutamine and asparagine binding to hNit2 were compared with those for KGM, KSM, and SM (Table 2). The results show that docking scores for both glutamine and asparagine are better than those for the three substrates. However, the molecular orientations of glutamine and asparagine within the active site pocket are not conducive to catalysis. The amide groups of glutamine and asparagine are directed away from the catalytic triad and oriented toward Asp117 and Tyr216 to form hydrogen bonds with these residues (Fig. 7, E and F). For this reason, hNit2 cannot effectively hydrolyze glutamine and asparagine.
TABLE 2.
Average energy contributions to the binding free energies for hNit2/ω-amidase and the substrate amides (KGM, KSM, and SM) and nonsubstrate amides (Gln and Asn)
Energy units are expressed as kcal/mol. Errors are given as S.E. The Ka units are expressed as m−1. The association constant (Ka) is 10−ΔGband/2.303RT, where R is the gas constant and T is the temperature in Kelvin. R2 is the correlation coefficient.
| Docking scorea | Docking orientationb | ΔEelec | ΔEvdW | ΔGpolar | ΔGnonpolar | −TΔS | ΔGbind | Ka | |
|---|---|---|---|---|---|---|---|---|---|
| Substrate amide | |||||||||
| KGM | −5.1 | Proper | −14.4 ± 0.2 | −9.6 ± 0.1 | −2.6 ± 0.2 | −10.9 ± 0.1 | 20.1 ± 0.6 | −17.3 ± 0.2 | 4.3 × 1012 |
| KSM | −4.9 | Proper | −56.2 ± 0.1 | −6.1 ± 0.1 | 37.5 ± 0.1 | −12.6 ± 0.1 | 16.7 ± 0.5 | −20.6 ± 0.2 | 1.1 × 1015 |
| SM | −4.1 | Proper | −22.3 ± 0.2 | −8.6 ± 0.1 | 4.8 ± 0.2 | −10.1 ± 0.1 | 19.3 ± 0.5 | −16.9 ± 0.2 | 2.1 × 1012 |
| R2 = 0.98 | R2 = 0.98 | R2 = 0.98 | R2 = 0.90 | R2 = 0.96 | R2 = 0.98 | ||||
| Nonsubstrate amidec | |||||||||
| Gln | −4.8 | Improper | |||||||
| Asn | −4.8 | Improper |
a The minimum value for the docking score was calculated by means of the AutoDock Vina program (42).
b If the amide group (-C(O)NH2) orients toward the catalytic triad, the orientation is referred to as proper; if not, the orientation is referred to as improper.
c Gln and Asn are included in the table for comparison of docking scores with those of substrates.
Free Energy of Substrate Binding to hNit2
Because the binding free energy between hNit2 and substrate is an important determinant for substrate specificity, we have computed the binding free energy of hNit2 with three substrates, separately. In addition, the binding free energy and correlation of individual energy terms with the experimental affinities were analyzed in order to better understand the mechanism(s) driving the formation of the hNit2-substrate complex. Our computed energy components to the binding free energies are listed in Table 2. We found that the SM and KSM substrates display the lowest and highest binding affinities toward hNit2, with binding free energies of −16.9 and −20.6 kcal/mol, respectively. Comparison of the KSM and SM structures revealed that the replacement of a methylene (-CH2-) by a keto group (-C(O)-) adjacent to the terminal carboxylate enhances substrate binding affinity, thus leading to a tighter binding of KSM relative to SM. KGM has one more extended carbon (methylene) relative to KSM. The binding free energy for KGM is 3.3 kcal/mol weaker than that for KSM but is 0.4 kcal/mol stronger than that for SM. Two energy components were considered as an aid to identifying a potential substrate candidate, namely the non-bonded interaction energy and the solvation free energy. In our present calculations, the non-bonded interaction energy (ΔEelec + ΔEvdW) has a high correlation coefficient R2 value of 0.98, but the nonpolar solvation free energy has a lower correlation coefficient R2 value of 0.90. Therefore, our results suggest that the non-bonded interaction is the dominant contribution to stabilization of the hNit2-substrate complex.
Upon binding of substrate, the hNit2-substrate complex is stabilized through a balance between the favorable entropy change of binding due to hydrophobic interactions and the unfavorable entropy change of binding due to loss of conformation freedom. The compensation of ΔEelec and ΔGpolar results in a major contribution to the binding affinity. For example, it costs more for KSM in electrostatic energy to bind to hNit2 than KGM and SM, but the desolvation penalty is large for KSM to offset the Coulombic interaction energy. Our analysis of binding components shows that the non-bonded van der Waals (ΔEvdW) and nonpolar solvation free energies (ΔGnonpolar) also drive binding for the three substrates.
DISCUSSION
In this study, we have employed both mutational analysis in the catalytic triad and Δ116–128 mutants and MD simulations to validate the enzymatic activity of hNit2/ω-amidase and substrate binding affinity. The point mutations in the loop 116–128, in theory, can be of many combinations. Therefore, to avoid this complexity, we have constructed a mutant in which the whole of the lid region (i.e. 116–128) is experimentally deleted in hNit2. The structural intactness of the Δ116–128 and three catalytic triad mutants was examined by CD and fluorescence analyses. The results show that there are no significant structural differences among the mutants and WT hNit2. In accord with the results from the enzymatic analysis of the Δ116–128 mutant and the MD simulations, these studies show that the loop 116–128 plays an essential role in supporting the stability of the enzyme-substrate complex, resulting in the generation of the catalytic products.
Hersh (8) previously showed that the γ-ethyl and γ-methyl esters of α-ketoglutarate are excellent substrates of rat liver ω-amidase. He also showed that the propyl, ethyl, and methyl monoesters of glutarate are substrates, as are the ethyl and methyl monoesters of succinate (8). Finally, he also showed that various aromatic γ-esters of glutarate are substrates of the rat liver enzyme (9). The glutarate and succinate monoesters were found to be less effective substrates than the γ-ethyl and γ-methyl esters of α-ketoglutarate (8, 9). In later work, Jaisson et al. (10) showed that the γ-ethyl ester of α-ketoglutarate is also an excellent substrate of the mouse enzyme. In summary, the “preferred” substrates for mammalian ω-amidases thus far characterized conform to the general structure, −O2CC(O)(CH2)nC(O)(NH2 or OR), where n = 1 or 2 and OR is part of an aliphatic or aromatic ester. The -C(O)- grouping in the α-position may be replaced with a -CH2- grouping but less effectively. However, the -C(O)- grouping cannot be replaced with a -CH(NH2)- grouping, as in glutamine and asparagine.
In the present work, the Km values reported from the experimental work for KGM and SM with hNit2/ω-amidase are in good agreement with the binding affinities calculated from the MD simulation. Furthermore, the binding affinities from our MD simulation for three substrates (KGM, KSM, and SM) show an excellent correlation with the Km values previously published for mNit2/ω-amidase (10), and the trend is in the order KSM > KGM > SM (Table 1). A higher binding affinity leads not only to a higher Ka (association constant) but also to a lower Km. Our results show that the Km for KGM is lower than that for SM. The apparent Km of KGM (not determined at pH 7.2) at pH 8.5 for mNit2/ω-amidase was reported to be 0.195 mm (10). However, the fact that the open-chain (substrate) form only represents 0.3% of the total KGM in solution (9) indicates that the true Km exhibited by the mouse enzyme toward KGM is ∼0.6 μm.
Catalytic Efficiency
Enzymatically active hNit2 is a homodimer (11). The exact Mr of the hNit2 monomer used in the present study is calculated to be ∼31,914 based on its translated recombinant sequence and the presence of a His6 tag in pQE70. Assuming that each monomer of dimeric hNit2 is catalytically competent and independent of the other subunit, then under substrate saturating conditions, the turnover numbers per monomer for KGM (pH 8.5) and SM (pH 7.2) are 16.1 ± 0.9 and 89.7 ± 3.4 s−1, respectively at 37 °C. The catalytic efficiency (turnover number/Km) is, however, 3 orders of magnitude greater for KGM (6.1 × 106 s−1 m−1) than for SM (8.3 × 103 s−1 m−1). These values may be compared with those reported previously by Jaisson et al. (10) for KGM, KSM, and SM as substrates of mNit2/ω-amidase. Catalytic efficiency values for KGM (pH 8.5), KSM (pH 7.2), and SM (pH 7.2) at 30 °C reported by Jaisson et al. (10) are ∼29.7 × 106 (corrected for the open-chain form), 385 × 103, and 8.7 × 103 s−1 m−1, respectively. mNit2/ω-amidase has a high affinity for KSM but a relatively low turnover rate with this substrate. It should be noted that it is not possible to obtain a meaningful Km value for KGM at pH 7.2 because at this pH, the rate of ring opening is rate-limiting for the amount of enzyme required for kinetic analysis (8). Moreover, the hydroxylamine used in the present kinetic study with SM as substrate is a much better nucleophile than is water. This is exemplified by the much higher Vmax reported in the present work for SM (∼169 μmol/min/mg, pH 7.2, 37 °C, hydroxylamine as attacking nucleophile) than that reported by Jaisson et al. (10) for SM (3.6 μmol/min/mg, pH 7.2, 30 °C, water as attacking nucleophile). It is clear from the present work that hNit2/ω-amidase catalytic efficiency values are in the order KGM > SM.
Although the enzyme is classified as a member of the nitrilase superfamily, hNit2/ω-amidase was not expected to have nitrilase-like activity. Indeed when 3-PPN was evaluated as a substrate, no ammonia production could be detected in the assay system in this study. The cysteine residue in the active site is not positioned for nucleophilic attack on the carbon of the nitrile moiety.
Conclusion
In the present studies, MD simulations have successfully verified the experimental trends in the binding affinities of hNit2/ω-amidase to various substrates. Extension of this work may suggest new strategies for engineering hNit2 to modulate and/or extend its substrate specificity. It will be important to determine whether the putative tumor suppressor properties of hNit2 are related to the ω-amidase activity or whether hNit2 is a bifunctional protein with separate ω-amidase and tumor suppressor domains. Our new findings should provide an impetus for more detailed clinical studies of KGM and stimulate more interest in understanding the biological roles of hNit2/ω-amidase. Moreover, in future studies, it will be important to understand the biochemical mechanisms leading to elevated KGM in hyperammonemic diseases (see Refs. 14–16). We suggest that further MD simulation studies may aid in the design of new inhibitors or activators that will be useful in research involving cancer and hyperammonemic diseases.
Supplementary Material
Acknowledgments
We thank Dr. Chi-Yuan Chou for assistance in CD and fluorescence spectral analysis. We are grateful to the National Center for High-Performance Computing of Taiwan for computer time and facilities and the Proteomics Research Center of the National Yang-Ming University for the performance of the LC/MS/MS analysis.
This work was supported in part by the National Science Council of Taiwan Grants NSC-99-2113-M-010-002-MY3 and NSC 99-2311-B-010-001 (to S. Y. S. and C. H. C.), National Yang-Ming University International Collaboration Grant 98A-D-N309 (to C. H. C.), and the Aim for the Top University Plan at the National Yang-Ming University supported by the Ministry of Education, Taiwan.

This article contains supplemental Equation S1, Materials and Methods, and Figs. S1–S11.
- KGM
- α-ketoglutaramate
- KSM
- α-ketosuccinamate
- SM
- succinamate
- hNit2 and mNit2
- human and mouse Nit2/ω-amidase, respectively
- MD
- molecular dynamics
- 3-PPN
- 3-phenylpropionitrile
- RMSD
- root mean square deviation
- Δ116–128
- residues 116–128 deleted from hNit2.
REFERENCES
- 1. Meister A. (1953) Preparation and enzymatic reactions of the keto analogues of glutamine and asparagines. J. Biol. Chem. 200, 571–589 [PubMed] [Google Scholar]
- 2. Meister A., Levintow L., Greenfield R. E., Abendschein P. A. (1955) Hydrolysis and transfer reactions catalyzed by ω-amidase. J. Biol. Chem. 215, 441–460 [PubMed] [Google Scholar]
- 3. Cooper A. J. (1977) Asparagine transaminase from rat liver. J. Biol. Chem. 252, 2032–2038 [PubMed] [Google Scholar]
- 4. Meister A., Otani T. T. (1957) ω-Amide and ω-amino acid derivatives of α-ketoglutaric and oxalacetic acids. J. Biol. Chem. 224, 137–148 [PubMed] [Google Scholar]
- 5. Cooper A. J., Meister A. (1974) Isolation and properties of a new glutamine transaminase from rat kidney. J. Biol. Chem. 249, 2554–2561 [PubMed] [Google Scholar]
- 6. Cooper A. J., Meister A. (1981) Comparative studies of glutamine transaminases from rat tissues. Comp. Biochem. Physiol. 69B, 137–145 [Google Scholar]
- 7. Cooper A. J. (2004) The role of glutamine transaminase K (GTK) in sulfur and α-keto acid metabolism in the brain and possible bioactivation of neurotoxicants. Neurochem. Int. 44, 557–577 [DOI] [PubMed] [Google Scholar]
- 8. Hersh L. B. (1971) Rat liver ω-amidase. Purification and properties. Biochemistry 10, 2884–2891 [DOI] [PubMed] [Google Scholar]
- 9. Hersh L. B. (1972) Rat liver ω-amidase. Kinetic evidence for an acyl-enzyme intermediate. Biochemistry 11, 2251–2256 [DOI] [PubMed] [Google Scholar]
- 10. Jaisson S., Veiga-da-Cunha M., Van Schaftingen E. (2009) Molecular identification of ω-amidase, the enzyme that is functionally coupled with glutamine transaminases, as the putative tumor suppressor Nit2. Biochimie 91, 1066–1071 [DOI] [PubMed] [Google Scholar]
- 11. Krasnikov B. F., Chien C. H., Nostramo R., Pinto J. T., Nieves E., Callaway M., Sun J., Huebner K., Cooper A. J. (2009) Identification of the putative tumor suppressor Nit2 as ω-amidase, an enzyme metabolically linked to glutamine and asparagine transamination. Biochimie 91, 1072–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duffy T. E., Cooper A. J., Meister A. (1974) Identification of α-ketoglutaramate in rat liver, kidney, and brain. Relationship to glutamine transaminase and ω-amidase activities. J. Biol. Chem. 249, 7603–7606 [PubMed] [Google Scholar]
- 13. Cooper A. J., Raps S. P., Meister A. (1987) Fluorometric determination of α-ketosuccinamic acid in rat tissues. Anal. Biochem. 167, 312–320 [DOI] [PubMed] [Google Scholar]
- 14. Vergara F., Duffy T. E., Plum F. (1973) α-Ketoglutaramate, a neurotoxic agent in hepatic coma. Trans. Assoc. Am. Physicians 86, 255–263 [PubMed] [Google Scholar]
- 15. Vergara F., Plum F., Duffy T. E. (1974) α-Ketoglutaramate. Increased concentrations in the cerebrospinal fluid of patients in hepatic coma. Science 183, 81–83 [DOI] [PubMed] [Google Scholar]
- 16. Kuhara T., Inoue Y., Ohse M., Krasnikov B. F., Cooper A. J. (2011) Urinary 2-hydroxy-5-oxoproline, the lactam form of α-ketoglutaramate, is markedly increased in urea cycle disorders. Anal. Bioanal. Chem. 400, 1843–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bertino J. R. (1979) Nutrients, vitamins, and minerals as therapy. Cancer 43, 2137–2142 [DOI] [PubMed] [Google Scholar]
- 18. Szeliga M., Obara-Michlewska M. (2009) Glutamine in neoplastic cells. Focus on the expression and roles of glutaminases. Neurochem. Int. 55, 71–75 [DOI] [PubMed] [Google Scholar]
- 19. Wang J. B., Erickson J. W., Fuji R., Ramachandran S., Gao P., Dinavahi R., Wilson K. F., Ambrosio A. L., Dias S. M., Dang C. V., Cerione R. A. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Erickson J. W., Cerione R. A. (2010) Glutaminase. A hot spot for regulation of cancer cell metabolism? Oncotarget 1, 734–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levine A. J., Puzio-Kuter A. M. (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344 [DOI] [PubMed] [Google Scholar]
- 22. Lin C. H., Chung M. Y., Chen W. B., Chien C. H. (2007) Growth inhibitory effect of the human Nit2 gene and its allelic imbalance in cancers. FEBS J. 274, 2946–2956 [DOI] [PubMed] [Google Scholar]
- 23. Krasnikov B. F., Nostramo R., Pinto J. T., Cooper A. J. (2009) Assay and purification of ω-amidase/Nit2, a ubiquitously expressed putative tumor suppressor, that catalyzes the deamidation of the α-keto acid analogues of glutamine and asparagine. Anal. Biochem. 391, 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pace H. C., Brenner C. (2001) The nitrilase superfamily. Classification, structure, and function. Genome Biol. 2, REVIEWS0001–REVIEWS0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pace H. C., Hodawadekar S. C., Draganescu A., Huang J., Bieganowski P., Pekarsky Y., Croce C. M., Brenner C. (2000) Crystal structure of the worm NitFhit Rosetta Stone protein reveals a Nit tetramer binding two Fhit dimmers. Curr. Biol. 10, 907–917 [DOI] [PubMed] [Google Scholar]
- 26. Barglow K. T., Saikatendu K. S., Bracey M. H., Huey R., Morris G. M., Olson A. J., Stevens R. C., Cravatt B. F. (2008) Functional proteomic and structural insights into molecular recognition in the nitrilase family enzymes. Biochemistry 47, 13514–13523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brenner C. (2002) Catalysis in the nitrilase superfamily. Curr. Opin. Struct. Biol. 12, 775–782 [DOI] [PubMed] [Google Scholar]
- 28. Novo C., Farnaud S., Tata R., Clemente A., Brown P. R. (2002) Support for a three-dimensional structure predicting a Cys-Glu-Lys catalytic triad for Pseudomonas aeruginosa amidase comes from site-directed mutagenesis and mutations altering substrate specificity. Biochem. J. 365, 731–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kumaran D., Eswaramoorthy S., Gerchman S. E., Kycia H., Studier F. W., Swaminathan S. (2003) Crystal structure of a putative CN hydrolase from yeast. Proteins 52, 283–291 [DOI] [PubMed] [Google Scholar]
- 30. Simonson T., Archontis G., Karplus M. (2002) Free energy simulations come of age. Protein-ligand recognition. Acc. Chem. Res. 35, 430–437 [DOI] [PubMed] [Google Scholar]
- 31. Bolleter W. T., Bushman C. J., Tidwell P. W. (1961) Spectrophotometric determination of ammonia as indophenol. Anal. Chem. 33, 592–594 [Google Scholar]
- 32. Sali A., Blundell T. L. (1993) Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 33. Lüthy R., Bowie J. U., Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 34. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 35. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 37. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 38. Trott O., Olson A. J. (2010) AutoDock Vina. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy. Methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt M. W., Baldridge K. K., Boatz J. A., Elbert S. T., Gordon M. S., Jensen J. H., Koseki S., Matsunaga N., Nguyen K. A., Su S., Windus T. L., Dupuis M., Montgomery J. A. (1993) General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 [Google Scholar]
- 41. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. MacKerell A. D., Jr., Bashford D., Bellott M., Dunbrack R. L., Jr., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., III, Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics Studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 43. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 44. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald. An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 45. Sitkoff D., Sharp K. A., Honig B. (1994) Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98, 1978–1988 [Google Scholar]
- 46. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanner M. F., Olson A. J., Spehner J. C. (1996) Reduced surface. An efficient way to compute molecular surfaces. Biopolymers 38, 305–320 [DOI] [PubMed] [Google Scholar]
- 48. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) Charmm. A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.