

Background: γ-Secretase, which is composed of presenilin and three subunits, is an intramembrane-cleaving protease related to Alzheimer disease.
Results: Water accessibility of the catalytic pore of presenilin was decreased during assembly of active γ-secretase.
Conclusion: Binding of the subunits allosterically contributes to the formation of catalytic pore.
Significance: The γ-secretase subunits modulate the structure of presenilin.
Keywords: Alzheimer Disease, Amyloid, Intramembrane Proteolysis, Presenilin, Protein Folding, Secretases
Abstract
γ-Secretase is an intramembrane-cleaving protease related to the etiology of Alzheimer disease. γ-Secretase is a membrane protein complex composed of presenilin (PS) and three indispensable subunits: nicastrin, Aph-1, and Pen-2. PS functions as a protease subunit forming a hydrophilic catalytic pore structure within the lipid bilayer. However, it remains unclear how other subunits are involved in the pore formation. Here, we show that the hydrophilic pore adopted with an open conformation has already been formed by PS within the immature γ-secretase complex. The binding of the subunits induces the close proximity between transmembrane domains facing the catalytic pore. We propose a model in which the γ-secretase subunits restrict the arrangement of the transmembrane domains of PS during the formation of the functional structure of the catalytic pore.
Introduction
γ-Secretase is the responsible enzyme for generation of amyloid-β peptide (Aβ),3 which is the major component of senile plaques in the brains of patients with Alzheimer disease (1). Genetic studies revealed that numerous point mutations in presenilin (PSEN) genes are linked to familial Alzheimer disease. These mutations affect the γ-secretase activity in a way that increases the generation of Aβ ending at the 42nd residue (i.e. Aβ42), which is the most aggregable species and predominantly deposited in the brains of Alzheimer disease patients (2, 3). Genetic and chemical biology approaches revealed that PS, a nine membrane-spanning protein, is the catalytic subunit of the γ-secretase (1, 2). Thus, PS/γ-secretase is an attractive molecular target for the development of Alzheimer disease therapeutics. γ-Secretase belongs to the family of intramembrane-cleaving protease that cleaves transmembrane domains (TMDs) of a number of membrane proteins. Recent structural analyses revealed the atomic molecular structures of rhomboid and site-2 protease, which are the other members of the intramembrane-cleaving proteases. Both proteases harbor an intramembranous chamber in which the catalytic residues are located. This atypical structure enables water to have access to the catalytic center within the lipid bilayer (4). However, γ-secretase is a membrane protein complex composed of PS and three other subunits: nicastrin (Nct), Aph-1, and Pen-2 (5, 6). PS interacts with the Nct-Aph-1 subcomplex and Pen-2 in the endoplasmic reticulum. PS then undergoes endoproteolysis to generate N- and C-terminal fragments (NTF and CTF, respectively). Thus, very limited information regarding the structure of the γ-secretase is available so far as the active form of the enzyme is composed of five transmembrane protein subunits with 19 TMDs. To overcome this issue, we have employed chemical biology approaches, i.e. substituted cysteine accessibility method (SCAM) and chemical cross-linking experiments coupled with application of γ-secretase inhibitors (7–10). Using these approaches, we and others have shown that PS, a catalytic subunit of the γ-secretase, forms a hydrophilic structure within the lipid bilayer (7, 11). However, it still remains unknown whether the other γ-secretase subunits contribute to the formation of the catalytic pore structure in PS during the γ-secretase assembly. Here, we analyzed the structural differences of the pore of PS1 mutants that are unable to bind with the subunits and found that the binding of the subunits regulated the structure of the catalytic site.
EXPERIMENTAL PROCEDURES
Plasmid Construction, Cell Culture, Transfection, Retroviral Infection, and Baculoviral Infection
For expression in mammalian cells, cDNAs encoding human PS1 mutants were inserted into pMXs-puro (7–10, 12). cDNAs encoding mutant PS1 were generated by long PCR-based QuikChangeTM strategy (Stratagene). For the expression of γ-secretase subunits in Sf9 cells, PS1 mutants and Aph-1aL-Myc/His were inserted into pFastBac Dual (Invitrogen). Nct-V5/His and Pen-2 were inserted into pBlueBac4.5 as described (13, 14). Maintenance of embryonic fibroblast obtained from PS1/PS2 double knock-out mice (DKO cells), viral packaging in Plat-E cells, retroviral infection, and generation of stable infectant pools were performed as described previously (12, 15). γ-Secretase complex was reconstituted in Spodoptera frugiperda Sf9 insect cells as described previously (13, 14). Briefly, P3 virus stock for each subunit was simultaneously added to adherent culture of Sf9 cells in a 75-cm2 flask and collected 72 h later.
Antibodies and Immunochemical Analyses
Anti-G1L3 and PNT3 antibodies against GST-fused human PS1 loop or a synthetic peptide corresponding to the N-terminal 26 amino acids of human/mouse Pen-2, respectively, have been previously described (16, 17). Anti-PS1NT antibody was kindly provided by Dr. G. Thinakaran (The University of Chicago, Chicago, IL) (18). Other antibodies were purchased from Chemicon (anti-PS1 loop (MAB5232)), Covance (anti-Aph-1aL (O2C2)), or Sigma (anti-Nct (N1660)). Membrane fractionation, immunoblot analysis, and immunoprecipitation of CHAPSO-solubilized lysates were performed as described previously (7, 19, 20). 1% digitonin-solubilized cell lysates were analyzed by Blue Native-PAGE as per the manufacturer's instructions (10).
Photoaffinity Labeling
31C and 31C-Bpa were kindly provided by Drs. N. Umezawa and T. Higuchi (Nagoya City University, Nagoya, Japan) (21). pep.11 and pep.11-Bt (22) were purchased from Ito Life Sciences (Moriya, Japan) and BEX (Tokyo, Japan), respectively. Photoaffinity labeling was performed as described (12, 23, 24) with minor modifications. Briefly, membrane fractions were incubated with the indicated compounds and collected by centrifugation after UV irradiation. The pellets were solubilized by 1% SDS buffer, and streptavidin-Sepharose (GE Healthcare) was added to the supernatants to pull down the biotinylated proteins. Samples were then analyzed by immunoblotting.
SCAM
Membrane fractions with final protein concentration at 0.3 mg/ml (DKO cells) or 0.1 mg/ml (Sf9 cells) were incubated with N-biotinaminoethyl methanethiosulfonate (MTSEA-biotin) (Toronto Research Chemicals, Toronto, Canada) for 10 min at 37 °C. MTSEA-biotin was dissolved in dimethyl sulfoxide (DMSO) at 10 mm and stored at −80 °C until use. Concentration of MTSEA-biotin was shown in each figure. The reaction was terminated by centrifugation. The subsequent procedure was done as described (7–10). Briefly, biotinylated PS1 was captured by streptavidin-Sepharose (GE Healthcare) and eluted by boiling in Laemmli sample buffer containing 2-mercaptoethanol. Eluate was separated and subjected to immunoblot analysis using anti-PS1 antibodies. To estimate the SCAM ratio (see below) in a quantitative manner, we confirmed that each concentration of MTSEA-biotin did not reach saturation of the reaction.
Cysteine-based Cross-linking
Cross-linking experiment using 1,2-ethanediyl bismethanethiosulfonate (M2M) (Toronto Research Chemicals) was performed as described previously (7–9). In copper-phenanthroline (Cu-Phe) cross-linking experiments, membrane fractions were dissolved in 17 mm Tris-HCl (pH 8.0) buffer containing 1 mm CaCl2, 100 mm NaCl, and CompleteTM protease inhibitor mixture (Roche Applied Science). After the addition of CuSO4 and 1,10-phenanthroline (final concentration at 3 and 15 mm, respectively), samples were incubated for 10 min at 4 °C. Reaction was terminated by the further addition of sample buffer containing 20 mm N-ethylmaleimide (NEM) and 10 mm EDTA and analyzed by immunoblot analysis.
RESULTS
Analysis of the Subunit Binding-defective PS1 Mutants
During the assembly of the γ-secretase, the subunits sequentially interact with PS to form an enzymatically active complex (5). Nct forms a subcomplex with Aph-1 and binds to the C terminus of PS1 (25, 26). Pen-2 directly associates at the WNF motif in TMD4 of PS to regulate the enzymatic activity (12, 27). We have previously found that PS1 mutant carrying alanine substitutions at the WNF motif (WNF mt) failed to interact with Pen-2. In this study, we also analyzed a PS1 mutant that lacks the C-terminal 12 amino acid residues (455st mt), the latter being the binding site for the Nct-Aph-1 subcomplex (26). We further designed a compound PS1 mutant in which both regions are mutated (WNF/455 mt) (Fig. 1A). Overexpression of wild-type (wt) PS1 in DKO cells resulted in the generation of PS fragments and recovered the levels of mature Nct and the accumulation of Pen-2, as described previously (Fig. 1B) (12). However, all PS1 mutants remained as a holoprotein and failed to restore the maturation of Nct. In addition, neither WNF nor WNF/455 mt rescued the expression of Pen-2. Immunoprecipitation analysis revealed that WNF and 455st mt failed to bind with Pen-2 and Nct-Aph-1aL, respectively, as described previously (Fig. 1C). Notably, WNF/455 mt was co-precipitated with neither of the subunits, indicating that the PS1 mutants lost the binding ability with specific subunits as designed. Formation of γ-secretase complex as a high molecular mass complex can be visualized by Blue Native-PAGE using 1% digitonin-solubilized cell lysates (Fig. 1D). In this analysis, γ-secretase complex was detected as an ∼440-kDa band as described previously (12). WNF mt was also migrated at ∼400–440 kDa as a doublet band, in which Nct and Aph-1aL were detected. However, consistent with the results of the immunoprecipitation experiment, no Pen-2 was detected, similarly to that in mock-transfected cells. The doublet band of this high molecular mass complex might represent a structural heterogeneity in conformation or binding mode to unidentified partner(s). In addition, an additional ∼120-kDa-band that reacted only with anti-PS1 antibody appeared. In fact, this 120-kDa band was detected in all PS1-expressing cell lysates including WNF/445 mt-expressing cells. Thus, the 120-kDa band might represent the PS1 holoprotein without binding to known subunits. Further analysis would be needed to clarify whether the 120-kDa PS1 holoprotein corresponds to the monomeric or dimeric forms of PS1 or PS1 complexed with unknown partner(s). In lysate of cells expressing 455st mt, PS1-Pen-2 complex was mainly detected as a 160-kDa band. However, the migration pattern of the 270-kDa band that reacted with anti-Nct as well as Aph-1aL antibodies was almost similar to that in mock-infected cells. This is consistent with the finding that C-terminal deletion mutation in PS1 diminished its binding with Nct-Aph-1 subcomplex.
FIGURE 1.

Biochemical analysis of the subunit binding-defective PS1 mutants. A, schematic representation of PS1 mutants used in this study. During the assembly of the γ-secretase, PS1 wild-type (wt) interacts with Nct-Aph-1 subcomplex and Pen-2 followed by endoproteolysis to generate PS1 NTF and CTF. WNF mt carries triple alanine substitutions at the Pen-2-binding WNF motif on TMD4 and interacts only with the Nct-Aph-1 subcomplex. 455st mt lacks the 12 amino acids of the most C-terminal region, which is the binding site of the Nct-Aph-1 subcomplex, and interacts only with Pen-2. WNF/455 mt harbors a compound mutation of WNF mt and 455st mt. B, immunoblot analysis of DKO cells stably expressing wt PS1 or PS1 mt (as indicated above the panel). FL, full-length; mNct, mature Nct; imNct, immature Nct. C, complex formation of PS1 mutants. Immunoprecipitated samples (IPed) with Mab5232 antibody (anti-PS1 loop) were analyzed by immunoblotting. D, Blue Native-PAGE analysis of cell lysates solubilized with 1% digitonin. Molecular mass and content of the complex were indicated at the left and right, respectively, of the panel. Antibodies used for detection were G1L3 (anti-PS1 CTF), N1660 (anti-Nct), O2C2 (anti-Aph-1aL), and PNT3 (anti-Pen-2).
To test whether the PS mutants formed an enzymatically active structure without the subunits, we took the photoaffinity labeling approach (10, 21). PS1 in an active γ-secretase complex harbors the catalytic and the initial substrate-binding sites, which are targeted by the transition state analog-type and the helical peptide-type γ-secretase inhibitors, respectively. 31C-Bpa, a transition state analog-type probe (28), was bound to wt PS1 NTF as well as CTF (Fig. 2), in agreement with previous studies (10, 21, 28). In contrast, neither wt PS1 holoprotein nor the subunit binding-incompetent PS1 mutants specifically bound to 31C-Bpa. A similar result was obtained using pep.11-Bt, which targeted exclusively PS1 NTF (10, 22). These results suggested that association of all subunits is required for the formation of both catalytic and initial substrate-binding sites in PS.
FIGURE 2.
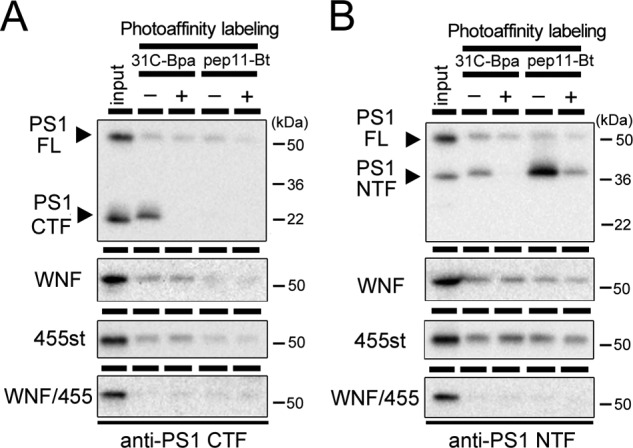
Photoaffinity labeling experiment of PS1 mutants. Microsome fractions of DKO cells stably expressing PS1 mutant were incubated with 31C-Bpa or pep.11-Bt in the presence (+) or absence (−) of parent compound 31C or pep.11, respectively. After 1 h of UV irradiation, samples were lysed with 1% SDS/PBS and incubated with streptavidin-Sepharose beads. Eluates were analyzed by immunoblotting using G1L3 (anti-PS1 CTF) (A) and PS1NT (anti-PS1 NTF) (B). The loaded amount of input was 6.5% relative to labeled fraction. Note that only PS1 fragments (NTF and CTF), but not PS1 holoprotein, were specifically labeled by both probes. FL, full-length.
Effects of the Subunit Binding on the Water Accessibility to the Catalytic Pore of PS1
The results of the photoaffinity labeling experiments prompted us to examine the structural change of the catalytic pore in PS1 mutants. The catalytic pore structure within the membrane was assessed by thiol-based chemistry in SCAM (7, 8). Cys-less PS1 (PS1/Cys(−))-based subunit-binding mutants (WNF, 455st, WNF/455) also failed to form an enzymatically active high molecular mass complex (data not shown). We then constructed mutant PS1/Cys(−) having a single cysteine residue at the residues facing the catalytic pore in PS1 CTF (i.e. L383C, I387C, L435C, S438C, L443C, and Y446C) and examined the water accessibility of the substituted Cys residues by specific labeling using MTSEA-biotin (Fig. 3D). We also analyzed the residues located at the hydrophilic loop (H351C, A398C) as controls. All cysteine mutants were specifically biotinylated. Intriguingly, in addition to PS1 CTF that represents the enzymatically active form, PS1 holoprotein was also labeled by MTSEA-biotin. This suggested that a pore-like structure has already been formed with the holoprotein form of PS1, consistent with the previous finding that PS holoprotein functions as a Ca2+ leak channel in endoplasmic reticulum (29). Moreover, the amount of biotinylated holoprotein was larger than that of CTF bound to MTSEA-biotin. Different biotinylation levels in holoprotein and fragment were also observed in L250C, which directly faces the pore on the NTF (Fig. 3B). We routinely detected some degradation bands just under the holoproteins (i.e. 40–45-kDa bands) in PS DKO cell lysates due to overexpression, as shown in Fig. 1B. These bands were also labeled by MTSEA-biotin. Furthermore, we tested the water accessibility of Cys-419, which is located in a hydrophobic environment and was never biotinylated in wt PS1 CTF. However, no labeling was observed in C419C holoprotein nor CTF (Fig. 3C). To compare the labeling efficiency in the holoprotein and the fragment forms of PS1, we defined the SCAM ratio as the biotinylated PS1 levels divided by the total PS1 levels in the input fraction (Fig. 4). The SCAM ratio of L383C holoprotein was 0.92 ± 0.43. In contrast, the SCAM ratio of L383C CTF was 0.13 ± 0.04 (n = 3, mean ± S.E., p < 0.05 by Student's t test), suggesting that the water accessibility of Leu-383 was significantly decreased in CTF. In addition, similar trends were detected in all mutants except for H351C, in which cysteine is located at the cytoplasmic hydrophilic loop 6. Notably, the SCAM ratio of A398C was also decreased in CTF, implicating the structural change of the loop region connected with TMD7 and TMD8. These results suggested that the microenvironment of the residues facing the catalytic pore as well as Ala-398 was altered in the fragment form of PS1.
FIGURE 3.

SCAM analysis of PS1 mutants. A, schematic representation of residues in PS1 analyzed in this study. The letters in white circles indicate the residues that are substituted to cysteine. Gray stars denote the catalytic center aspartates. In this preparation, MTSEA-biotin had access to Cys residues from both the extracellular and the intracellular sides (arrows). B–D, SCAM analysis of PS1 mts carrying Cys substitutions at NTF (B) or CTF (C and D). Minus and Plus denote samples incubated without or with MTSEA-biotin, respectively. Final concentration of MTSEA-biotin was 6.6 μm for L250C and S438C and 3.3 μm for other Cys mutants. Biotinylated proteins were precipitated by streptavidin beads, and the entire eluate was analyzed by immunoblotting. The loaded amount of input was 6.5% relative to labeled fraction. C, labeling to C419C mt by MTSEA-biotin at 0, 10, and 100 μm as denoted above the lanes. PS1 was detected by immunoblotting using PS1NT (anti-PS1 NTF) (B) and G1L3 (anti-PS1 CTF) (C and D). FL, full-length.
FIGURE 4.

Quantification of SCAM analysis on PS1 mutants. Quantification of SCAM ratio by densitometric analysis of each band as shown in Fig. 3 was performed. SCAM ratio was calculated as the ratio of each PS1 polypeptide in MTSEA-biotin (+) to that in the input. *, p < 0.05; **, p < 0.01 (Student's t test, n = 3–8, mean ± S.E. when compared with that of NTF or CTF (white bar)); #, p < 0.05; ##, p < 0.01 (Student's t test, n = 3–8, mean ± S.E., sample pairs indicated by lines). FL, full-length; n.s., not significant.
Next we analyzed the effects of the subunit binding-incompetent mutations on the water accessibility of the residues facing the pore. The SCAM ratio of L383C in the subunit-binding mutants was almost comparable with that of wt PS1 holoprotein (i.e. WNF, 1.25 ± 0.15; 455st, 1.85 ± 0.33; WNF/455, 1.57 ± 0.25, n = 3–6) (Fig. 4), suggesting that the water accessibility of Leu-383 in holoprotein form was almost the same as that in the subunit-binding mutants. Similar results were obtained in all single cysteine PS1 mutants except for H351C. These results suggested that the complex assembly caused reduction in water accessibility around the residues facing the catalytic pore. Notably, the SCAM ratio of Y446C/WNF mt (0.27 ± 0.16) was lower than that of 455st mt (0.72 ± 0.11) or WNF/455 mt (0.71 ± 0.10). However, the SCAM ratio of Y446C CTF was much lower (0.06 ± 0.03), raising the possibility that the water accessibility in Tyr-446 was differently affected by binding of Nct-Aph-1 or Pen-2. In addition, the SCAM ratio of L250C/WNF mt (0.56 ± 0.09) as well as of L250C/455st mt (0.73 ± 0.23) was significantly lower than that of L250C/WNF/455 mt (1.78 ± 0.40), suggesting that binding of either of the subunits decreased the water accessibility of L250C. No subunit-binding mutant carrying C419C mutation was labeled by MTSEA-biotin, indicating that the subunit-binding mutations did not alter the general topology and conformation of PS1 (Fig. 3C). To examine whether increased water accessibility reflected the misfolded conformation of PS1 polypeptides, we conducted SCAM analysis after heating the cell lysates to induce misfolding. However, preincubation at 50 or 70 °C lowered the labeling efficiency of mutant PS1 holoproteins (i.e. I387C WNF/455 and L435C WNF/455) (Fig. 5A), indicating that increased water accessibility was not correlated with the misfolded conformation of PS1.
FIGURE 5.
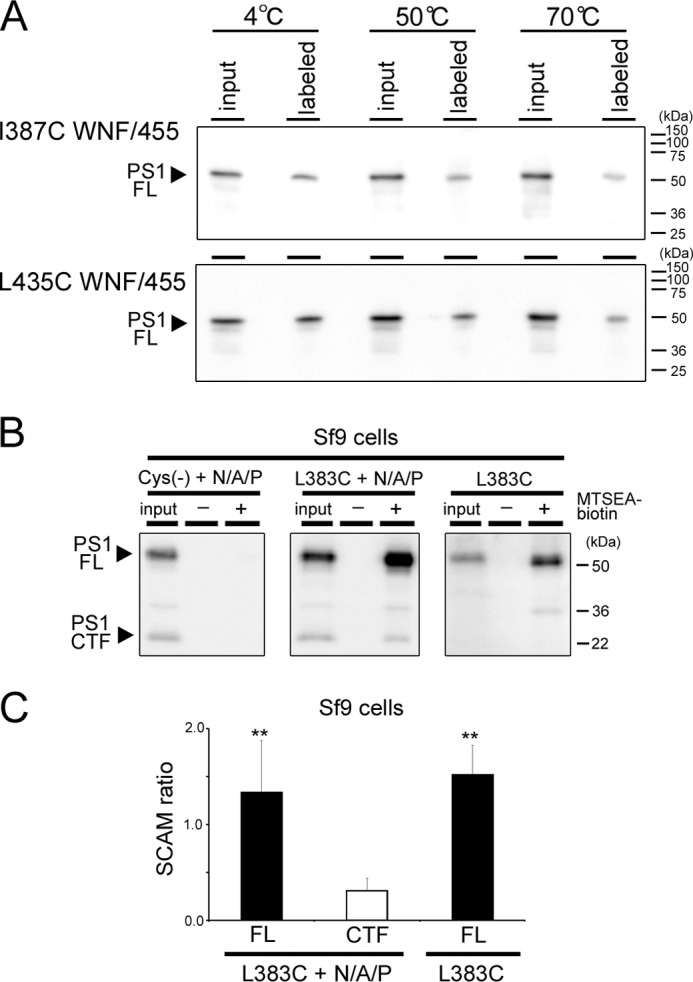
SCAM analysis of misfolded PS1 and recombinant PS1. A, temperature-dependent changes in labeling efficiency. Membrane fractions were incubated at the indicated temperatures for 10 min before SCAM analysis using MTSEA-biotin (3.3 μm, 10 min at 37 °C). G1L3 (anti-PS1 CTF) was used to immunodetection. FL, full-length. B, SCAM analysis of reconstituted γ-secretase in Sf9 cells. + N/A/P indicates co-infection of Nct-V5/His, Aph-1aL-Myc/His, and Pen-2. Final concentration of MTSEA-biotin was 3.3 μm. The loaded amount of input was 6.5% relative to the labeled fraction. C, quantification of the SCAM ratio in B. PS1 was detected by immunoblotting using G1L3 (anti-PS1 CTF) *, p < 0.05; **, p < 0.01 (Student's t test, n = 3–8, mean ± S.E. when compared with that of CTF (white bar)).
Finally, to test whether the changes in the SCAM ratio were caused by the allosteric effect of WNF or 455st mutation, we analyzed the recombinant PS1 proteins expressed in Sf9 cells (13, 14). We previously reported that Sf9 cells do not have detectable levels of endogenous γ-secretase (13). Co-expression of human PS1, Nct, Aph-1, and Pen-2 in Sf9 cells resulted in the reconstitution of functional γ-secretase complex, whereas single infection of PS1 did not show any γ-secretase activity (13). PS1 Cys(−) expressed with other subunits was endoproteolyzed, but neither PS1 holoprotein nor CTF was labeled by MTSEA-biotin (Fig. 5B). In contrast, both holoprotein and CTF of L383C expressed with other subunits were biotinylated (Fig. 5B). The SCAM ratio of PS1 CTF was lower (0.31 ± 0.05) than that of the holoprotein (1.34 ± 0.22) in a similar fashion to that observed in DKO cells (Fig. 5C). Notably, the SCAM ratio of L383C infected without other subunits was retained at a high level (1.52 ± 0.43) comparable with that of holoprotein in L383C with other subunits. This result supports our view that the change of the water accessibility was induced by the binding of other subunits, but not by the introduction of the subunit binding-defective mutations. Collectively, these data suggest that the hydrophilic pore structure in PS1 has already been formed without binding of the subunits and that the water accessibility of the pore is reduced by the complex assembly.
Effects of the Subunit Binding to the Proximity between Transmembrane Domains of PS1
Decreased water accessibility of the residues within the pore would be caused by the steric hindrance by the subunits or the conformational change of TMDs involved in the formation of the pore. To test the latter possibility, we conducted disulfide cross-linking experiments to detect the proximity between TMDs using two different reagents. M2M is a sulfhydryl-to-sulfhydryl MTS cross-linking reagent with spacer arms of 5.2 Å long (7, 30). Copper and 1,10-phenanthroline (Cu-Phe) acts as a redox catalyst to oxidize free sulfhydryls and form a disulfide bond in cysteines that can collide (31). We chose a cysteine pair at Leu-250 and Leu-435 located at the PS1 TMD6 and PALP motif, respectively, within the catalytic pore (Fig. 6A) (7, 8). Using M2M, the cross-linked NTF and CTF were observed in cell lysates expressing L250C/L435C, as described previously (Fig. 6B) (8). Moreover, Cu-Phe treatment resulted in an appearance of the NTF/CTF heterodimer, which was abolished by pretreatment with NEM. In fact, a small amount of NTF/CTF heterodimer, which was abolished by NEM pretreatment, was observed in the lysate without any cross-linking reagent. The mobilities of cross-linked products were slightly distinct, presumably due to difference in the structural flexibility of NTF/CTF heterodimer by M2M and Cu-Phe treatments. In contrast, no cross-linked product was observed in cell lysates expressing the single cysteine mutant (i.e. L250C and L435C) (Fig. 6C), indicating that Leu-250 and Leu-435 are located in a close proximity within the pore. Next we tested the effect of the subunit-binding mutations. Notably, no PS1 mutant was cross-linked by Cu-Phe treatment, whereas these mutants were cross-linked by M2M (Fig. 6D). Finally, to examine the effect of the endoproteolysis of PS1 on the proximity of these residues, we tested the M292D mutation, which is known to block endoproteolysis without affecting the complex formation and the enzymatic activity of the γ-secretase (32). Again, a cross-linked product of L250C/L435C with the M292D mutation was observed by both Cu-Phe and M2M treatment similarly to that in PS1/Cys(−) (Fig. 6E). This result suggested that the formation of an enzymatically active complex, but not the endoproteolysis, is the prerequisite to maintain the close proximity of Leu-250 and Leu-435 within the pore. These data suggest that the hydrophilic pore structure of PS1 has already been formed prior to binding of any subunits and that the subunit binding is correlated with the reduction of the water accessibility as well as the formation of the catalytic structure of the pore.
FIGURE 6.

Cross-linking analysis of PS1 mutants. A, schematic representation of Leu-250 and Leu-435 in PS1. The letters in white circles with thicker line indicate the residues that are substituted to cysteine. Gray stars denote the catalytic center aspartates. In this preparation, MTSEA-biotin had access to Cys residues from both the extracellular and the intracellular sides. B, chemical cross-linking of PS1/Cys(−) mutant carrying L250C/L435C mutations. Microsome fractions of DKO cells stably expressing PS1 mt were treated with Cu-Phe or M2M at 4 °C and analyzed by immunoblotting. The lower panel shows the result using samples pretreated with NEM (NEM pretreat). Black arrowheads and arrows denote PS1 holoprotein and fragments, respectively. White arrowheads indicate cross-linked NTF/CTF heterodimer. NEM pretreatment significantly reduced the amount of cross-linked products. FL, full-length. C, cross-linking experiment using PS1/Cys(−) with single cysteine substitution. D, cross-linking experiment of WNF, 455st, and WNF/455 mt carrying L250C/L435C substitutions. The lower panels show the results using NEM-pretreated lysates. Note that cross-linked PS1 mutants were detected only in M2M-treated samples in these mutants. E, chemical cross-linking of M292D mutant carrying L250C/L435C. PS1 was detected by immunoblotting with PS1NT (anti-PS1 NTF) (B) and G1L3 (anti-PS1 CTF) (B–E).
DISCUSSION
In this study, we analyzed the effects of the γ-secretase subunits on the structure of PS1 using chemical biology approaches. SCAM analysis revealed that water-accessible pore structure has already been formed by PS1 alone, the latter being an enzymatically inactive form (Fig. 3). Importantly, an increased SCAM ratio was observed in both the subunit binding-defective mutant PS1 as well as recombinant PS1 holoprotein singly expressed without other subunits. These results suggest that the binding of the subunits renders the pore structure narrower to form an enzymatically active catalytic site within the pore. Supporting this notion, Fluorescence-lifetime imaging microscopic analysis revealed that the interaction with different isoforms of Aph-1 or mutant Pen-2 affected the distance between PS1 NTF and CTF (33, 34). To date, several different functions of the γ-secretase subunits have been suggested: activity modulation, substrate recognition, trafficking, and stabilization of the enzyme (35). Here, our data suggest that in addition to these known functions, the subunits harbor a “chaperone”-like function acting on the arrangement of TMDs of PS during the assembly of the complex. Several ion or protein channels form a membrane protein complex containing auxiliary subunits. In SecYEG complex, a translocon in bacteria, SecY is a pore-forming subunit (36). SecE, which partly participates in the pore structure, is required for the stabilization of SecY. SecG is not essential for protein translocation, but stimulates preprotein translocation. X-ray crystallographic analysis revealed that the transmembrane segment of SecE embraces the SecY subunit as a clamp (37). Moreover, the function of voltage-dependent K+ channels is regulated by voltage-gated potassium channel-interacting protein (KChIP) subunits (20, 38). Structural study indicated that the binding of voltage-gated potassium channel-interacting protein affected the gating kinetics as well as the stability of the pore-forming α-subunit of the channel (39). These data suggest that the auxiliary subunits harbor chaperone-like activity to affect the structure of the water-filled cavity within the membrane of the functional subunit in these channel structures. Importantly, we found that some residues in PS1 were differently affected by the binding of distinct subunits, whereas the hydrophilicity of residues around the pore was similarly affected in all mutants tested. The biotinylation of Y446C was significantly decreased only in the WNF mutant PS1, which can interact with Nct-Aph-1 (Fig. 4E). This result is consistent with the finding that Nct-Aph-1 interacts with PS1 CTF (25, 26, 40). Intriguingly, it has been shown that a point mutation at TMD9 rendered PS1 enzymatically active without binding to Nct (41). These results support our notion that the structure of TMD9 was affected by binding of the Nct-Aph-1 subcomplex. In addition, we found that the water accessibility of L250C was slightly decreased either in WNF or 455st mt when compared with that in the compound mt PS1. However, the SCAM ratios of these mutants were higher than that in L250C NTF. This suggests that the binding of Pen-2 directly to the TMD4 within PS1 NTF (12) affected the structure of TMD6 in NTF. In accordance with these results, it has been shown that recombinant Pen-2 activated the PS1 holoprotein in a proteoliposome preparation (42), suggesting that Pen-2 directly modulates the structure of the catalytic structure of PS1. In contrast, no direct interaction between NTF and Nct-Aph-1 has so far been reported. Thus, the reduction of the SCAM ratio of L250C NTF in the WNF mt background would reflect the steric hindrance induced by structural changes within CTF. Nevertheless, these data support the notion that the pore-like structure around the catalytic center formed by PS1 TMDs is closely regulated by the γ-secretase subunits (Fig. 7).
FIGURE 7.

Proposed model for the formation of the catalytic pore of PS1. A, PS1 TMDs are shown as columns with numbers. Without any subunits, PS forms a relatively open pore structure within the membrane. B, upon the binding of subunits, the catalytic structure is activated by the structural changes in PS TMDs, and the PALP motif moves to the proximity to the catalytic center.
Structural regulation of the disordered catalytic site in zymogen is an essential activation mechanism in several proteases such as trypsin, caspase, and calpain (43–45). Our cross-linking data suggest that the distance between Leu-250 and Leu-435 of PS1, which are located in proximity to the catalytic aspartates (7, 8), is altered by the assembly leading to the activation of the enzyme. Leu-250 on TMD6 is a highly conserved residue beyond species and functions as a subsite of the catalytic structure. Helical alignment of TMD6 suggests that Leu-250 locates on the same interface as Ala-246 and Asp-257 on the pore-facing α-helix. Leu-435 is a part of PALP motif in TMD9, which also participates in the formation of the catalytic site (46, 47). Intriguingly, NMR analysis revealed that TMD9 is a highly flexible domain in PS1 CTF (48). Thus, we propose a model in which PALP motif at the cytoplasmic side of TMD9 moves up to be located in proximity with the catalytic aspartates by the binding of PS1 to the subunits (Fig. 7). This structural arrangement of the catalytic site is highly reminiscent of the movement of the plug structure in SecY by SecE interaction (37). Intriguingly, classical proteases are activated by cleavage of their prodomain after acquiring the functionally active conformation. In this regard, it is tempting to speculate that the hydrophilic loop region of PS could represent an autoinhibitory domain of the γ-secretase (49, 50). However, endoproteolytic cleavage at the loop is dispensable for the activation of PS (32), supporting our notion that the allosteric changes of the structure of the catalytic pore by the subunit binding are critical for the acquisition of the proteolytic activity. Recently, crystallographic structural analysis of the first GxGD-type membrane protease FlaK has been reported (51). Notably, the catalytic aspartates sit in distant locations within the open cavity. Considering the predicted catalytic mechanism of the aspartic protease, these aspartates should locate in proximity to each residue in an enzymatically active conformation. We compared the location of the residues in FlaK structure with that corresponding to the analyzed residues in our study (Fig. 8). Essentially, almost all tested residues seemed to face a single cavity where the catalytic aspartates reside. Thus, our results suggested that the conformation with the open cavity could be a common inactive structure of GxGD-type protease. Structural analysis of the active form of FlaK with transition state analog-type inhibitor would provide further structural information. In conclusion, we propose the first detailed structural rearrangement in TMDs of PS1 induced by binding of the γ-secretase subunits during the assembly of the γ-secretase complex. This result also raised the possible chaperone-like function of the subunits during the assembly as well as the activation of γ-secretase complex. Further structural analyses (i.e. x-ray crystallography, NMR, molecular dynamics simulation) would pave the way to deeper understanding of the functions of the subunits as well as the structural changes in PS1 in the activation process of the γ-secretase complex.
FIGURE 8.

Modified FlaK structural model replaced with the corresponding residues of PS1. A, sequence alignment of Homo sapiens PS1 (hPS1) and Methanococcus maripaludis C6 FlaK around the catalytic and PALP motifs by ClustalW. The numbers of colored residues in PS1 and FlaK are shown above and below sequences, respectively. Asterisks, colons, and periods denote identical, conserved, and semiconserved residues in ClustalW. B, modified structural model of FlaK (Protein Data Bank (PDB) 3S0X) viewed from the cytosol (46). Colored residues were replaced with the corresponding amino acid residues in PS1 sequence (A) using PyMOL. The highlighted residues are shown as spheres, and other residues are shown as gray ribbons and sticks. C, closer view surface presentation around the catalytic site of the model.
Acknowledgments
We are grateful to Drs. N. Umezawa and T. Higuchi (Nagoya City University) and Dr. G. Thinakaran (The University of Chicago) for valuable reagents and to our current and previous laboratory members for helpful discussions and technical assistance.
This work was supported in part by grants-in-aid for young scientists (S) (to T. T.) from the Japan Society for the Promotion of Science (JSPS), by the Targeted Proteins Research Program of the Japan Science and Technology Corporation (JST) (to T. T. and T. I.), by Core Research for Evolutional Science and Technology of JST (to T. T. and T. I.), and by the Strategic Research Program for Brain Sciences “Analysis of the molecular basis of brain aging and degeneration due to failure in metabolic homeostasis and environmental stress” by the Ministry of Education, Culture, Sports, Science and Technology, Japan.
- Aβ
- amyloid-β peptide
- CTF
- carboxyl-terminal fragment
- NTF
- amino-terminal fragment
- DKO
- PS1/PS2 double knockout fibroblasts
- Nct
- nicastrin
- mt
- mutant
- MTSEA
- N-biotinaminoethyl methanethiosulfonate
- PS
- presenilin
- SCAM
- substituted cysteine accessibility method
- TMD
- transmembrane domain
- NEM
- N-ethylmaleimide
- M2M
- 1,2-ethanediyl bismethanethiosulfonate
- Cu-Phe
- copper-phenanthroline
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate.
REFERENCES
- 1. Holtzman D. M., Morris J. C., Goate A. M. (2011) Alzheimer disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tomita T. (2009) Secretase inhibitors and modulators for Alzheimer disease treatment. Expert Rev. Neurother 9, 661–679 [DOI] [PubMed] [Google Scholar]
- 3. Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y. (1994) Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13, 45–53 [DOI] [PubMed] [Google Scholar]
- 4. Erez E., Fass D., Bibi E. (2009) How intramembrane proteases bury hydrolytic reactions in the membrane. Nature 459, 371–378 [DOI] [PubMed] [Google Scholar]
- 5. Takasugi N., Tomita T., Hayashi I., Tsuruoka M., Niimura M., Takahashi Y., Thinakaran G., Iwatsubo T. (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441 [DOI] [PubMed] [Google Scholar]
- 6. Edbauer D., Winkler E., Regula J. T., Pesold B., Steiner H., Haass C. (2003) Reconstitution of γ-secretase activity. Nat. Cell Biol. 5, 486–488 [DOI] [PubMed] [Google Scholar]
- 7. Sato C., Morohashi Y., Tomita T., Iwatsubo T. (2006) Structure of the catalytic pore of γ-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 26, 12081–12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato C., Takagi S., Tomita T., Iwatsubo T. (2008) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci. 28, 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takagi S., Tominaga A., Sato C., Tomita T., Iwatsubo T. (2010) Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the γ-secretase. J. Neurosci. 30, 15943–15950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watanabe N., Takagi S., Tominaga A., Tomita T., Iwatsubo T. (2010) Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of γ-secretase. J. Biol. Chem. 285, 19738–19746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tolia A., Chávez-Gutiérrez L., De Strooper B. (2006) Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the γ-secretase complex. J. Biol. Chem. 281, 27633–27642 [DOI] [PubMed] [Google Scholar]
- 12. Watanabe N., Tomita T., Sato C., Kitamura T., Morohashi Y., Iwatsubo T. (2005) Pen-2 is incorporated into the γ-secretase complex through binding to transmembrane domain 4 of presenilin 1. J. Biol. Chem. 280, 41967–41975 [DOI] [PubMed] [Google Scholar]
- 13. Hayashi I., Urano Y., Fukuda R., Isoo N., Kodama T., Hamakubo T., Tomita T., Iwatsubo T. (2004) Selective reconstitution and recovery of functional γ-secretase complex on budded baculovirus particles. J Biol Chem 279, 38040–38046 [DOI] [PubMed] [Google Scholar]
- 14. Ogura T., Mio K., Hayashi I., Miyashita H., Fukuda R., Kopan R., Kodama T., Hamakubo T., Iwatsubo T., Iwastubo T., Tomita T., Sato C. (2006) Three-dimensional structure of the γ-secretase complex. Biochem. Biophys. Res. Commun. 343, 525–534 [DOI] [PubMed] [Google Scholar]
- 15. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 16. Tomita T., Takikawa R., Koyama A., Morohashi Y., Takasugi N., Saido T. C., Maruyama K., Iwatsubo T. (1999) C terminus of presenilin is required for overproduction of amyloidogenic Aβ42 through stabilization and endoproteolysis of presenilin. J. Neurosci. 19, 10627–10634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Isoo N., Sato C., Miyashita H., Shinohara M., Takasugi N., Morohashi Y., Tsuji S., Tomita T., Iwatsubo T. (2007) Aβ42 overproduction associated with structural changes in the catalytic pore of γ-secretase: common effects of Pen-2 N-terminal elongation and fenofibrate. J. Biol. Chem. 282, 12388–12396 [DOI] [PubMed] [Google Scholar]
- 18. Leem J. Y., Vijayan S., Han P., Cai D., Machura M., Lopes K. O., Veselits M. L., Xu H., Thinakaran G. (2002) Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J. Biol. Chem. 277, 19236–19240 [DOI] [PubMed] [Google Scholar]
- 19. Tomita T., Maruyama K., Saido T. C., Kume H., Shinozaki K., Tokuhiro S., Capell A., Walter J., Grünberg J., Haass C., Iwatsubo T., Obata K. (1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid-β protein ending at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. U.S.A. 94, 2025–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morohashi Y., Hatano N., Ohya S., Takikawa R., Watabiki T., Takasugi N., Imaizumi Y., Tomita T., Iwatsubo T. (2002) Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem. 277, 14965–14975 [DOI] [PubMed] [Google Scholar]
- 21. Imamura Y., Watanabe N., Umezawa N., Iwatsubo T., Kato N., Tomita T., Higuchi T. (2009) Inhibition of γ-secretase activity by helical β-peptide foldamers. J. Am. Chem. Soc. 131, 7353–7359 [DOI] [PubMed] [Google Scholar]
- 22. Das C., Berezovska O., Diehl T. S., Genet C., Buldyrev I., Tsai J. Y., Hyman B. T., Wolfe M. S. (2003) Designed helical peptides inhibit an intramembrane protease. J. Am. Chem. Soc. 125, 11794–11795 [DOI] [PubMed] [Google Scholar]
- 23. Morohashi Y., Kan T., Tominari Y., Fuwa H., Okamura Y., Watanabe N., Sato C., Natsugari H., Fukuyama T., Iwatsubo T., Tomita T. (2006) C-terminal fragment of presenilin is the molecular target of a dipeptidic γ-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem. 281, 14670–14676 [DOI] [PubMed] [Google Scholar]
- 24. Ohki Y., Higo T., Uemura K., Shimada N., Osawa S., Berezovska O., Yokoshima S., Fukuyama T., Tomita T., Iwatsubo T. (2011) Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaether C., Capell A., Edbauer D., Winkler E., Novak B., Steiner H., Haass C. (2004) The presenilin C terminus is required for ER retention, nicastrin binding, and γ-secretase activity. EMBO J. 23, 4738–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bergman A., Laudon H., Winblad B., Lundkvist J., Näslund J. (2004) The extreme C terminus of presenilin 1 is essential for γ-secretase complex assembly and activity. J. Biol. Chem. 279, 45564–45572 [DOI] [PubMed] [Google Scholar]
- 27. Kim S. H., Sisodia S. S. (2005) Evidence that the “NF” motif in transmembrane domain 4 of presenilin 1 is critical for binding with PEN-2. J. Biol. Chem. 280, 41953–41966 [DOI] [PubMed] [Google Scholar]
- 28. Micchelli C. A., Esler W. P., Kimberly W. T., Jack C., Berezovska O., Kornilova A., Hyman B. T., Perrimon N., Wolfe M. S. (2003) γ-Secretase/presenilin inhibitors for Alzheimer disease phenocopy Notch mutations in Drosophila. FASEB J. 17, 79–81 [DOI] [PubMed] [Google Scholar]
- 29. Tu H., Nelson O., Bezprozvanny A., Wang Z., Lee S. F., Hao Y. H., Serneels L., De Strooper B., Yu G., Bezprozvanny I. (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer disease-linked mutations. Cell 126, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loo T. W., Clarke D. M. (2001) Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J. Biol. Chem. 276, 36877–36880 [DOI] [PubMed] [Google Scholar]
- 31. Klco J. M., Lassere T. B., Baranski T. J. (2003) C5a receptor oligomerization. I. Disulfide trapping reveals oligomers and potential contact surfaces in a G protein-coupled receptor. J. Biol. Chem. 278, 35345–35353 [DOI] [PubMed] [Google Scholar]
- 32. Steiner H., Romig H., Pesold B., Philipp U., Baader M., Citron M., Loetscher H., Jacobsen H., Haass C. (1999) Amyloidogenic function of the Alzheimer disease-associated presenilin 1 in the absence of endoproteolysis. Biochemistry 38, 14600–14605 [DOI] [PubMed] [Google Scholar]
- 33. Serneels L., Van Biervliet J., Craessaerts K., Dejaegere T., Horré K., Van Houtvin T., Esselmann H., Paul S., Schäfer M. K., Berezovska O., Hyman B. T., Sprangers B., Sciot R., Moons L., Jucker M., Yang Z., May P. C., Karran E., Wiltfang J., D'Hooge R., De Strooper B. (2009) γ-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer disease. Science 324, 639–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uemura K., Lill C. M., Li X., Peters J. A., Ivanov A., Fan Z., DeStrooper B., Bacskai B. J., Hyman B. T., Berezovska O. (2009) Allosteric modulation of PS1/γ-secretase conformation correlates with amyloid-β(42/40) ratio. PLoS One 4, e7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Strooper B., Annaert W. (2010) Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu. Rev. Cell Dev. Biol. 26, 235–260 [DOI] [PubMed] [Google Scholar]
- 36. du Plessis D. J., Nouwen N., Driessen A. J. (2011) The Sec translocase. Biochim. Biophys. Acta 1808, 851–865 [DOI] [PubMed] [Google Scholar]
- 37. Van den Berg B., Clemons W. M., Jr., Collinson I., Modis Y., Hartmann E., Harrison S. C., Rapoport T. A. (2004) X-ray structure of a protein-conducting channel. Nature 427, 36–44 [DOI] [PubMed] [Google Scholar]
- 38. Wang K. (2008) Modulation by clamping: Kv4 and KChIP interactions. Neurochem. Res. 33, 1964–1969 [DOI] [PubMed] [Google Scholar]
- 39. Wang H., Yan Y., Liu Q., Huang Y., Shen Y., Chen L., Chen Y., Yang Q., Hao Q., Wang K., Chai J. (2007) Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat. Neurosci. 10, 32–39 [DOI] [PubMed] [Google Scholar]
- 40. Steiner H., Winkler E., Haass C. (2008) Chemical cross-linking provides a model of the γ-secretase complex subunit architecture and evidence for close proximity of the C-terminal fragment of presenilin with APH-1. J. Biol. Chem. 283, 34677–34686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Futai E., Yagishita S., Ishiura S. (2009) Nicastrin is dispensable for γ-secretase protease activity in the presence of specific presenilin mutations. J. Biol. Chem. 284, 13013–13022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahn K., Shelton C. C., Tian Y., Zhang X., Gilchrist M. L., Sisodia S. S., Li Y. M. (2010) Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. U.S.A. 107, 21435–21440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 Å resolution. II. Crystallographic refinement, refined crystal structure, and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 44. Riedl S. J., Fuentes-Prior P., Renatus M., Kairies N., Krapp S., Huber R., Salvesen G. S., Bode W. (2001) Structural basis for the activation of human procaspase-7. Proc. Natl. Acad. Sci. U.S.A. 98, 14790–14795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hosfield C. M., Elce J. S., Davies P. L., Jia Z. (1999) Crystal structure of calpain reveals the structural basis for Ca2+-dependent protease activity and a novel mode of enzyme activation. EMBO J. 18, 6880–6889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomita T., Watabiki T., Takikawa R., Morohashi Y., Takasugi N., Kopan R., De Strooper B., Iwatsubo T. (2001) The first proline of PALP motif at the C terminus of presenilins is obligatory for stabilization, complex formation, and γ-secretase activities of presenilins. J. Biol. Chem. 276, 33273–33281 [DOI] [PubMed] [Google Scholar]
- 47. Wang J., Beher D., Nyborg A. C., Shearman M. S., Golde T. E., Goate A. (2006) C-terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation. J. Neurochem. 96, 218–227 [DOI] [PubMed] [Google Scholar]
- 48. Sobhanifar S., Schneider B., Löhr F., Gottstein D., Ikeya T., Mlynarczyk K., Pulawski W., Ghoshdastider U., Kolinski M., Filipek S., Güntert P., Bernhard F., Dötsch V. (2010) Structural investigation of the C-terminal catalytic fragment of presenilin 1. Proc. Natl. Acad. Sci. U.S.A. 107, 9644–9649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Knappenberger K. S., Tian G., Ye X., Sobotka-Briner C., Ghanekar S. V., Greenberg B. D., Scott C. W. (2004) Mechanism of γ-secretase cleavage activation: is γ-secretase regulated through autoinhibition involving the presenilin-1 exon 9 loop? Biochemistry 43, 6208–6218 [DOI] [PubMed] [Google Scholar]
- 50. Fukumori A., Fluhrer R., Steiner H., Haass C. (2010) Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of γ-secretase-mediated intramembrane proteolysis. J. Neurosci. 30, 7853–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu J., Xue Y., Lee S., Ha Y. (2011) The crystal structure of GXGD membrane protease FlaK. Nature 475, 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]