

Background: Legionella pneumophila glucosyltransferase Lgt1 modifies elongation factor 1A and Hbs1 (Hsp70 subfamily B suppressor 1).
Results: In Saccharomyces cerevisiae deleted of endogenous eEF1A and Hbs1, Lgt1 inhibits growth by glucosylation of ectopically expressed eEF1A at serine 53.
Conclusion: Glucosylation of eEF1A but not of Hbs1 is essential for Lgt1-induced toxicity.
Significance: Yeast as a model to study the action of Legionella glucosyltransferases.
Keywords: Glycosylation, Glycosyltransferases, Host-pathogen Interactions, Microbiology, Protein Synthesis, Toxins, Yeast, Eukaryotic Elongation Factor eEF1A
Abstract
Legionella is a pathogenic Gram-negative bacterium that can multiply inside of eukaryotic cells. It translocates numerous bacterial effector proteins into target cells to transform host phagocytes into a niche for replication. One effector of Legionella pneumophila is the glucosyltransferase Lgt1, which modifies serine 53 in mammalian elongation factor 1A (eEF1A), resulting in inhibition of protein synthesis and cell death. Here, we demonstrate that similar to mammalian cells, Lgt1 was severely toxic when produced in yeast and effectively inhibited in vitro protein synthesis. Saccharomyces cerevisiae strains, which were deleted of endogenous eEF1A but harbored a mutant eEF1A not glucosylated by Lgt1, were resistant toward the bacterial effector. In contrast, deletion of Hbs1, which is also an in vitro substrate of the glucosyltransferase, did not influence the toxic effects of Lgt1. Serial mutagenesis in yeast showed that Phe54, Tyr56 and Trp58, located immediately downstream of serine 53 of eEF1A, are essential for the function of the elongation factor. Replacement of serine 53 by glutamic acid, mimicking phosphorylation, produced a non-functional eEF1A, which failed to support growth of S. cerevisiae. Our data indicate that Lgt1-induced lethal effect in yeast depends solely on eEF1A. The region of eEF1A encompassing serine 53 plays a critical role in functioning of the elongation factor.
Introduction
Legionella pneumophila is a Gram-negative bacterium, which causes severe pneumonia in humans known as Legionnaires' disease (1, 2). The pathogen is able to multiply inside eukaryotic cells, including free-living protozoa or mammalian cells (3). A type 4B secretion system, encoded by dot/icm gene clusters, translocates hundreds of Legionella proteins (effectors) into target cells and thereby changes the hostile intracellular environment of phagocytes into a niche for Legionella replication (4, 5).
Among L. pneumophila effectors are cytotoxic glucosyltransferases of the Lgt family (6). These enzymes use UDP-glucose as a cofactor and target eukaryotic substrates by covalent attachment of a glucosyl moiety (7). The crystal structure of Lgt1 has been solved, allowing the characterization of the Legionella enzyme as a GT-A glucosyltransferase structurally related to clostridial glucosylating toxins (8, 9). Known substrates of Lgts are eukaryotic elongation factor 1A (eEF1A)3 and eRF3 (eukaryotic release factor 3)-related protein Hbs1 (Hsp70 subfamily B suppressor 1) (10).
Lgt1 monoglucosylates eEF1A on serine 53 and yeast Hbs1 on serine 213 (10). These target serine residues are located within conserved regions, exhibiting significant homology between the two proteins. Recently, it has been shown that the preferred substrate for modification of eEF1A is its complex with charged tRNA and GTP (11). In relation to Hbs1, it is not known whether any cofactors are able to increase the level of its modification by Lgt1.
Glucosylation by Lgt1 parallels inhibition of protein synthesis both in vitro and in vivo and leads to death of target mammalian cells (12, 13). Thus far, it is not clear whether the lethal effect of Lgt1 is linked solely to modification of eEF1A or glucosylation of Hbs1 also contributes to toxic effects caused by the Legionella enzyme. Moreover, it was demonstrated recently that a short peptide encompassing region of the elongation factor from glycine 50 to valine 59 was effectively recognized and glucosylated by Lgt1 (10). This fact suggested that other not yet identified eukaryotic protein(s), possessing homologous sequences can be also substrate(s) of Legionella glucosyltransferases and participate in intoxication mechanisms. In contrast, natural target selection of a region encompassing serine 53 in eEF1A for glucosylation by a microbial effector toxin suggests importance of this area for biological functions of the elongation factor.
To address these issues, the budding yeast Saccharomyces cerevisiae was used as a model. This is appropriate because mammalian eEF1A, which is a target of L. pneumophila glucosyltransferase Lgt1, shares >80% of identical amino acid residues with the yeast analogs elongation factors Tef1 and Tef2. Here, we report that Lgt1 is toxic for yeast. This effect depends solely on yeast eEF1A, containing the glucose acceptor serine 53 but not on Hbs1. Moreover, we show that the narrow region of eEF1A, which is recognized by Lgt1, is characterized by several functionally essential amino acid residues.
EXPERIMENTAL PROCEDURES
Strains, Vectors, and Culture Conditions
Cloning and recombinant protein production was performed in Escherichia coli DH10B and BL21(DE3) (Invitrogen). Genomic DNA from strain D273-10B was used as a source for the amplification of TRP1, HIS3, and LEU2 marker genes. MH272–3fα (ura3, leu2, his3, trp1, ade2) or the diploid MH272–3fα/a (ura3/ura3, leu2/leu2, his3/his3, trp1/trp1, ade2/ade2) (14) are the parental strains of the mutant S. cerevisiae employed in this study. (A summary of all strains is given in supplemental Table S1.) Plasmids for cloning and recombinant protein expression in E. coli are based on pUC19 (New England Biolabs, Frankfurt am Main, Germany), pBC KS(+), pBluescript KS(+) (Stratagene, Waldbronn, Germany), and pET28a (Novagen, Madison, WI). Yeast expression vectors were constructed in pRS313 (15), pESC-Ura (Stratagene), YEPLac195 (16), YCPLac444, and YEPLac555 (17). A summary of all plasmids is given in supplemental Table S2; primers employed for cloning are listed in supplemental Table S3.
Yeast strains were grown on rich medium containing glucose (YPD: 1% yeast extract, 2% peptone, 2% glucose) or on minimal medium (SD: 0.67% yeast nitrogen base without amino acids (Difco), 2% glucose; SGal: 0.67% yeast nitrogen base without amino acids, 2% galactose). SD and SGal media were supplemented with the appropriate supplements. Yeast transformations were performed by the lithium acetate method (18).
Purification of Lgt1, Hbs1, and Tef1
The coding sequence of glucosyltransferase Lgt1 was amplified from genomic DNA of the L. pneumophila Philadelphia-1 strain (19) and was cloned into the EcoRI/SacI restriction sites of pET28a. The resulting plasmid p558 (supplemental Table S2) encodes His-tagged Lgt1 and served as the template for the construction of mutated lgt1 versions. Lgt1 containing D246N (plasmid p562), D246N/W520A (p563), or D246N/W520A/N293A (p578) were prepared stepwise by QuikChange mutagenesis on p558 (Promega, Mannheim, Germany) using primers 183–184, 923–924, or 548–549 (supplemental Table S3), respectively. Yeast expression vectors were constructed by transferring wild type and mutated versions of lgt1 into pESC-Ura under the control of GAL promoter. The resulting pESC-Ura series plasmids p569, p590, p570, and p591 (supplemental Table S2) encode for wild type Lgt1, Lgt1-D246N, Lgt1-D246N/W520A, and Lgt1-D246N/W520A/N293A.
Purification of His-tagged Hbs1 from E. coli and His-tagged Tef1 (eEF1A) from S. cerevisiae was described previously (10, 11). Wild type and mutant versions of His-tagged Lgt1 were expressed from plasmids p558, p562, p563, and p578 (supplemental Table S2) in E. coli BL21(DE3). Induction was performed overnight with 0.2 mm isopropyl-β-d-thiogalactopyranoside at 22 °C. Proteins were subsequently purified via nickel-affinity chromatography using HisTrap columns (GE Healthcare) connected to an ÄKTA Purifier (GE Healthcare) according to the instructions of the manufacturer and stored in 10% glycerol/Tris-buffered saline (20 mm Tris-HCl, pH 7.4, 150 mm NaCl) at −20 °C.
Cloning and Mutagenesis of TEF1
TEF1 (yeast eEF1A) plus 500-bp regions up- and downstream of the orf was amplified using primers 518–492 (supplemental Table S3) and yeast genomic DNA as a template. The PCR product was cloned into the BamHI/SalI sites of YEPlac195, YCPlac444, and pRS313. The resulting plasmids are termed p561 (pYE-TEF1), p815 (pYC-TEF1), and p572 (pRS-TEF1) (supplemental Table S2). For mutagenesis experiments, a portion of TEF1, containing the coding sequence of the recognized by Lgt1 decapeptide Gly50–Val59 (10), was cut from p572 (pRS-TEF1) with SacI/ClaI and was ligated into pBC, resulting in plasmid p574 (supplemental Table S2). QuikChange mutagenesis of p574 was employed to generate the following substitutions in Tef1: G50A (p644), S53A (p575), S53E (p688), S53C (p686), S53K (p687), F54A (p579), K55A (p645), Y56A (p580), W58A (p581), and V59A (p668) (supplemental Table S2; for primers used, see supplemental Table S3). SacI/ClaI fragments from these pBC-based mutagenised plasmids were then transferred back to p572, substituting the wild type portion of the gene. The resulting pRS313-based yeast expression plasmids encode Tef1-G50A (p664), Tef1-S53A (p577), Tef1-S53E (p697), Tef1-S53C (p701), Tef1-S53K (p696), Tef1-F54A (p584), Tef1-K55A (p665), Tef1-Y56A (p585), Tef1-W58A (p586), and Tef1-V59A (p670) under the control of TEF1 promoter. To construct a S53A-mutated variant of p815 (pYC-TEF1), the BamHI/SalI fragment from p577 (pRS-TEF-S53A) was cloned into YCPLac444 to give rise p828 (pYC-TEF-S53A). Plasmids p815 and p828 were used in co-transformation studies (see “Results” and Fig. 5).
FIGURE 5.

Rescue of toxic effects of Lgt1 in the S. cerevisiae (Δtef1Δtef2+pRS-TEF1-WT) strain by pYC-TEF1-S53A. A, growth of yeast strains in liquid SGal. S. cerevisiae (Δtef1Δtef2+pRS-TEF1-WT) co-transformed by pESC-Ura and pYC-TEF-WT (squares), by pESC-Ura and pYC-TEF1-S53A (diamonds), by pESC-lgt1 and pYC-TEF1-WT (triangles), or by pESC-lgt1 and pYC-TEF1-S53A (circles). B, spot-test assay of strains as in A on SD (left panel, glucose, Glc) or SGal (right panel, galactose, Gal). C, Lgt1 production in S. cerevisiae strains grown in SGal. S. cerevisiae (Δtef1Δtef2+pRS-TEF1-WT) co-transformed by pESC-Ura and pYC-TEF-WT (lane 1), by pESC-Ura and pYC-TEF1-S53A (lane 2), by pESC-lgt1 and pYC-TEF1-WT (lane 3), and by pESC-lgt1 and pYC-TEF1-S53A (lane 4). Western blots were probed with anti-Lgt1 serum (upper panel) or anti-eEF1A antibody (lower panel).
Generation of Δtef1 and Δtef2 Deletion Strains
tef1::TRP1 (Δtef1) was constructed by replacing nucleotides 363 to 967 within the TEF1 orf with the TRP1 marker gene. tef2::LEU2 (Δtef2) was constructed by replacing nucleotides 10 to 1368 within the TEF2 orf with the LEU2 marker gene. The deletion constructs were integrated into the genome of the diploid S. cerevisiae MH272a/α strain using standard yeast genetics methods (18). Disruptions were confirmed via PCR using genomic DNA of grown transformants. Due to the fact that the Δtef1Δtef2 double mutation is lethal (20), a diploid strain containing one copy of each tef1::TRP1 and tef2::LEU2 disrupted genes was transformed with p561 (pYE-TEF1) before been sporulated and dissected (Singer Instruments, Somerset, UK). After tetrad analysis, a haploid Δtef1Δtef2 strain rescued by pYE-TEF1 was selected (S. cerevisiae SC33) and was used for further 5-fluoroorotic acid (5-FOA) experiments. pYE-TEF1 was replaced with pRS313-based plasmids encoding for the different TEF1 mutants via plasmid shuffling using the 5-FOA method (21). The resulting yeast strains are summarized in supplemental Table S1. Description of Δtef1 and Δtef2 deletion procedure is given in the supplemental “Results.”
Generation of Δhbs1 Deletion Strains
hbs1::HIS3 (Δhbs1) was constructed by replacing nucleotides 436 to 1212 within the HBS1 orf with the HIS3 marker gene. The construct was transformed into the haploid S. cerevisiae MH272α strain. The hbs1::HIS3 deletion was confirmed by PCR and Western blotting with anti-Hbs1 serum, and the corresponding strain (S. cerevisiae SC31) was used in subsequent experiments.
Δtef1Δtef2Δhbs1 triple deletion strains were constructed in S. cerevisiae SC39 (Δtef1Δtef2+pRS-TEF1) or S. cerevisiae SC40 (Δtef1Δtef2+pRS-TEF1-S53A) (supplemental Table S1). To that end, HBS1 was replaced with the hbs1::kanMX4 deletion cassette from strain Y16000 (Euroscarf). Recombinant clones were selected on YPD supplemented with 0.4 mg/ml G418 (Invitrogen). The HBS1 deletion was confirmed by PCR and Western blotting with anti-Hbs1 serum and the resulting strains, termed S. cerevisiae SC200 (Δtef1Δtef2Δhbs1+pRS-TEF1) and S. cerevisiae SC201 (Δtef1Δtef2Δhbs1+pRS-TEF1-S53A), were used in subsequent experiments. Analysis of hbs1 deletion strains is presented on supplemental Figs. S3 and S4.
Yeast in Vitro Translation System
Yeast translation extracts were prepared as described (22). Transcription was performed using SP6 polymerase (22) and pSP-luc+ (Promega) as a template to generate firefly luciferase mRNA. Translation reactions were performed as described (22–24). To test for the ability of Lgt1 to inhibit translation in yeast system, purified His-tagged Lgt1 at 9, 90, or 280 nm final concentrations and 10 μm UDP-glucose as a Lgt1 co-substrate were added to the translational mixes lacking luciferase mRNA. The reactions were preincubated for 10 min at 20 °C. Translation was started after this preincubation step by adding luciferase mRNA and was allowed to proceed for 50 min at 20 °C. After that, 10 μl of the reaction mix was diluted into 100 μl of luciferase assay buffer (100 mm KH2PO4, 1 mm EDTA, 1 mm DTT, pH adjusted to 7.8 with KOH). The diluted reaction mixes (100 μl) were mixed with 100 μl of luciferase reagent (20 mm Tricine, 5 mm MgCl2, 0.1 mm EDTA, 3.3 mm DTT, 270 μm coenzyme A, 500 μm d-luciferin, 500 μm ATP), and luminescence was determined using a Lumat LB 9507 device (Berthold Technologies GmbH, Wildbad, Germany) (25). Luciferase activity obtained in the absence of Lgt1 was set to 100%.
Spot-test Assay
The toxicity of Lgt1 in different yeast strains was analyzed on SD or SGal plates. To that end, 5-fold serial dilutions of cultures were spotted onto agar plates with the required supplements and either glucose or galactose as indicated on the corresponding figures. Plates were incubated for the times and at temperatures indicated in the figure legends. For liquid culture experiments, 3 ml of SGal medium in plastic tubes (catalog no. 62.515.006, Sarstedt) was inoculated with yeast cells to a starting A600 of 0.1. Cultures were then incubated at 30 °C for up to 48 h under vigorous shaking.
Production of Polyclonal Antibodies
Monospecific sera against Lgt1 and Hbs1 were produced by injecting 10 μg of purified recombinant proteins into mice intraperitoneally three times with 4-day intervals. Sera were collected 5 days after the last injection. Monospecific anti-Tef1 serum was produced by GeneScript (Piscataway, NJ) in rabbits by injecting synthesized Tef1-derived peptide (31CGGIDKRTIEKFEK44) conjugated to keyhole limpet hemocyanin via the first cysteine residue of the peptides.
Electrophoresis and Western Blotting
SDS-PAGE and Western blotting were performed according to published protocols (26, 27). Preparation of yeast cell extracts was accomplished by NaOH/trichloroacetic acid procedure or by glass bead disruption (28).
Glucosyltransferase Assay
Glucosylation was performed with 167 pmol of recombinant His-tagged wild type or mutated Lgt1 and 67 pmol of GST-tagged target decapeptide in a total volume of 20 μl (10). The standard reaction was allowed to proceed at 37 °C for up to 60 min in 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm MnCl2, and 10 μm UDP-[14C]glucose (American Radiolabeled Chemicals, St. Louis, MO). The reaction was stopped at various time points by the addition of SDS sample buffer and incubation at 95 °C for 5 min. Subsequently, samples were subjected to SDS-PAGE and autoradiography. Radiolabeled bands were analyzed by PhosphorImager and quantified with ImageQuant (version 5.2, GE Healthcare).
RESULTS
Toxicity of Lgt1 in Wild Type S. cerevisiae Background
First, we studied whether expression of lgt1 in the cytosol of S. cerevisiae produces toxic effects. To this end, we generated mutated variants of the glucosyltransferase with gradually decreased enzymatic activity. Such Lgt1 mutations included the single replacement D246N, the double substitution D246N/W520A and the triple mutant D246N/W520A/N293A. Aspartate 246 represents an important element of the DxD motif and has been shown to participate in orienting the distal part of glucose moiety of the co-factor UDP-glucose. Tryptophan 520 represents a critical residue of a flexible C-terminal loop, which is suggested to fix the co-substrate into the correct position for catalysis. Finally, asparagine 293 appears to be involved in the guidance and/or binding of the protein substrate eEF1A (8, 9). The proteins with described mutations were initially purified from recombinant E. coli cultures and tested in the 14C-glucosylation assay. As shown in Fig. 1A, substitution of D246N resulted in a drastic decrease of glucosylation activity of Lgt1. The double D246N/W520A mutant demonstrated further diminished enzymatic activity, whereas the triple mutation produced a protein with catalytic activity below the detection limit of the method.
FIGURE 1.

Toxic effects of Lgt1 in wild type yeast. A, 14C-glucosylation of GST-tagged target decapeptide 50GKGSFKYAWV59 of eEF1A by wild type Lgt1 (circles), Lgt1 with D246N substitution (diamonds), Lgt1 with D246N/W520A substitutions (squares), and Lgt1 with D246N/W520A/N293A substitutions (triangles). The amount of the 14C-glucosylated eEF1A-derived peptide at indicated time periods is given. Left and right panels represent graphs with different Y-scales to accommodate different glucosylation activities of wild type Lgt1 and its mutated forms. Reaction with the wild type Lgt1 is not shown on the right panel. B, spot-test assay of Lgt1-accomplished yeast toxicity. S. cerevisiae MH272α was transformed with the vector pESC-Ura or pESC-Ura-based plasmids, coding for different variants of Lgt1 (wild type and mutated), titrated 5-fold and spotted onto supplemented SD agar (with glucose, Glc) or SGal agar (with galactose, Gal). Variants of proteins, coded by the plasmids, are indicated on the left. C, Western blotting analysis of Lgt1 production in yeast, transformed with vector pESC-Ura or pESC-Ura-based plasmids, coding for different variants of Lgt1 (wild type and mutated). S. cerevisiae were cultivated in liquid SD. (i.e. with glucose, lanes 1–5) or SGal (i.e. with galactose, lanes 6–10). Lanes 1 and 6, pESC-Ura; lanes 2 and 7, lgt1-WT; lanes 3 and 8, lgt1-D246N; lanes 4 and 9, lgt1-D246N/W520A; lanes 5 and 10, lgt1-D246N/W520A/N293A. Nitrocellulose membranes were probed with anti-Lgt1 serum overnight at 4 °C and anti-mouse horseradish peroxidase conjugate for 1 h at 22 °C. The position of reacting protein bands is marked by an asterisk.
Next, coding sequences of the wild type and mutated Lgt1 variants were cloned into a galactose-inducible yeast expression vector and were transferred into wild type S. cerevisiae. Wild type Lgt1 was severely toxic and resulted in cell death upon induction in the presence of galactose (Fig. 1B). Yeast cells demonstrated a slow growth even under glucose-repressing conditions, possibly due to “promoter leakage.” Similar to wild type Lgt1, the enzyme with the D246N substitution also inhibited S. cerevisiae plated on galactose-containing medium, whereas growth on glucose-supplemented agar was restored to control level. Decreasing enzymatic activity in the double D246N/W520A and the triple D246N/W520A/N293A mutants paralleled with a diminished toxic phenotype (Fig. 1B). However, even the triple Lgt1 mutant exhibited reduced growth on galactose-containing medium as compared with control yeast transformed with the empty pESC-Ura vector (compare the upper and lower rows on Fig. 1B).
Next, we tested Lgt1 production by Western blotting using anti-Lgt1 serum. As expected, no protein production was observed when yeast strains were cultivated in the presence of glucose (Fig. 1C, lanes 1–5). Wild type Lgt1 and Lgt1-D246N were also not detected when the respective strains were cultivated in the presence of galactose (Fig. 1C, lanes 7 and 8). Low toxicity of the double and triple mutants allowed production of Lgt1 detected by Western blotting (Fig. 1C, lanes 9 and 10).
Using a reticulocyte-based translation system, we recently showed that Lgt1 effectively blocks mammalian protein synthesis in vitro (12, 13). Similar results were obtained, when translation extracts from S. cerevisiae were programmed with mRNA coding for firefly luciferase. In the presence of UDP-glucose, increasing concentrations of purified Lgt1 resulted in a dose-dependent inhibition of luciferase mRNA translation, observed already at 9 nm Lgt1 (Fig. 2A, bar 2). Inhibition of translation was paralleled by glucosylation of Tef1/2 (the homologs of elongation factor 1A in yeast) as shown in 14C-glucosylation assay (Fig. 2B).
FIGURE 2.
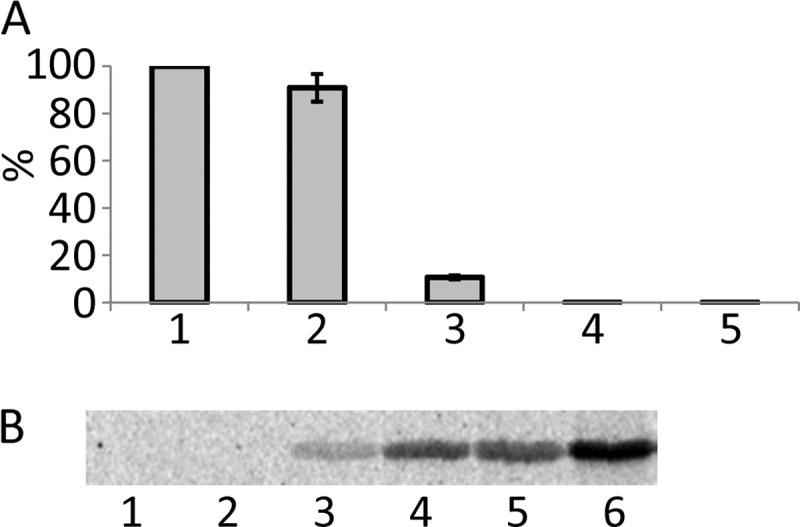
Influence of Lgt1 upon protein synthesis in S. cerevisiae translation extracts. A, synthesis of luciferase in yeast translation extracts in the absence (bars 1 and 5) or presence of 9 nm (bar 2), 90 nm (bar 3), and 280 nm (bar 4) Lgt1. Bar 5, experiment without luciferase-coding mRNA (i.e. no translation). Each bar represents means of two experiments ± S.D. Data are shown as percentage of maximal translation without added Lgt1. B, modification of eEF1A in yeast translational extracts by Lgt1. The reaction conditions were exactly the same as for in vitro translation but with 10 μm UDP-[14C]glucose instead of unlabeled UDP-glucose. Subsequently, the reaction mix was subjected to SDS-PAGE and autoradiography. Lanes 1 and 2, 9 nm Lgt1; lanes 3 and 4, 90 nm Lgt1; lanes 5 and 6, 280 nm Lgt1. Lanes 1, 3, and 5, incubation time of 10 min; lane 2, 4, and 6, incubation time of 60 min.
Engineering of S. cerevisiae Strains with Mutations in Elongation Factor 1A Gene
Because Lgt1 has been reported to use eEF1A as a protein substrate both in vitro and in vivo through the recognition of a 10-amino acid peptide (10), we wanted to know whether amino acid residues comprising this recognition sequence were essential for eEF1A function. Therefore, we engineered S. cerevisiae strains by disrupting both chromosomal copies of eEF1A (TEF1 and TEF2). The lethal phenotype of the Δtef1Δtef2 mutation was rescued by plasmid encoded TEF1 or mutated versions of it (see “Experimental Procedures” and supplemental “Results”).
As shown in our previous investigations with mammalian eEF1A, substitution of the glucose-accepting serine 53 of the elongation factor by an alanine residue resulted in a variant protein, which is not glucosylated by Lgt1 in vitro (12). Moreover, F54A, Y56A, and W58A substitutions severely affected glucosylation efficiency of mutated eEF1A-derived peptides, whereas G50A, K51A, G52A/K55A, and V59A produced only minor effects (10).
Based on this information, we developed a panel of pRS313-based plasmids, containing as an insert TEF1-WT, TEF1-G50A, TEF1-S53A, TEF1-S53E, TEF1-S53C, TEF1-S53K, TEF1-F54A, TEF1-K55A, TEF1-Y56A, TEF1-W58A, and TEF1-V59A under the control of the TEF1 promoter. These constructs were transformed into S. cerevisiae (Δtef1Δtef2+pYE-TEF1). After cultivation of yeast on 5-FOA-supplemented medium, causing elimination of URA3-containing pYE-TEF1, plates were inspected for growth of the resulting strains. As expected, S. cerevisiae, containing pRS313 vector without the TEF1 insert, failed to grow (Fig. 3A), whereas the strain with the wild type TEF1 produced numerous colonies on the plate. Surprisingly, yeast strains containing eEF1A with S53A, S53C, and S53K mutations were viable, whereas substitution of S53E was lethal for S. cerevisiae. Remarkably, analysis of the growth phenotype in the other mutated strains displayed critical importance for functional activity of eEF1A amino acid residues Tyr56 and Trp58. Substitution of these amino acids by alanine residues precluded growth of yeast. In addition, a strain with F54A substitution demonstrated growth on agar plates under favorable conditions but displayed a growth-limiting phenotype under conditions of cold and heat stress (Fig. 3B). By contrast, alanine substitutions of Gly50, Lys55, and Val59 in Tef1 failed to produce growth defects under the cultivation conditions tested.
FIGURE 3.

Growth phenotypes of yeast strains expressing wild type (WT) and mutated TEF1. A, Δtef1Δtef2 yeast strain expressing TEF1 on a URA3-containing plasmid p561 (pYE-TEF1) was transformed with different variants of TEF1 cloned into pRS313. Strains harboring both plasmids were cultivated on 5-FOA-containing plates for 1 week at 30 °C. Only strains that had lost the URA3-containing plasmid and in addition harbored a functional variant of TEF1 as an insert in pRS313 vector were able to form colonies. Constructions resulting in non-functional Tef1 are shown in boldface type. The experiment was repeated twice with identical results. Representative plates are shown. B, spot-test assay of growth phenotypes of selected TEF1 mutants grown on YPD at 30, 10, or 40 °C and in the presence of 0.9 m NaCl. TEF1 mutants, encoded on pRS313-based plasmids, are shown on the left.
Toxicity of Lgt1 in S. cerevisiae Tef1 Mutants
As demonstrated in the above experiments, yeast expressing exclusively TEF1-S53A was viable. Therefore, we were able to test the toxic activity of Lgt1 in this yeast mutant, in which eEF1A could not be modified by Lgt1. To this end, S. cerevisiae (Δtef1Δtef2+pRS-TEF1) and (Δtef1Δtef2+pRS-TEF1-S53A) strains were transformed with galactose-inducible lgt1-containing plasmids. In line with our previous findings with wild type S. cerevisiae, transformation of lgt1 into yeast strain (Δtef1Δtef2+pRS-TEF1) produced a lethal effect on galactose-containing medium and a partial defect on glucose-containing agar. lgt1-D246A/W520A was less toxic when expressed in this yeast variant (Fig. 4A). In contrast, S. cerevisiae (Δtef1Δtef2+pRS-TEF1-S53A) was protected against the toxic activity of Lgt1 and demonstrated an improved survival rate (Fig. 4A). Western blotting analysis with anti-Lgt1 serum demonstrated that good growth of S. cerevisiae (Δtef1Δtef2+pRS-TEF1-S53A) was accompanied by high level of Lgt1 production, whereas even undetectable amounts of the wild type glucosyltransferase efficiently killed yeast strain (Δtef1Δtef2+pRS-TEF1) (Fig. 4B, compare lanes 2 and 6).
FIGURE 4.

Toxic effect of Lgt1 in S. cerevisiae variants expressing wild type and mutated TEF1 genes. A, spot-test assay of growth phenotypes of S. cerevisiae containing plasmid-born wild type Tef1 and Tef1-S53A as the only eEF1A present in yeast cells. Yeast variants were transformed with pESC-Ura or pESC-Ura-based plasmids coding for wild type lgt1 and lgt1-D246A/W520A. Strains were analyzed as described under “Experimental Procedures” on SD (left panel, glucose, Glc) and SGal (right panel, galactose, Gal). The transformed variants of lgt1 and types of Tef1 in recipient yeast strains are indicated on the left. B, Western blotting, demonstrating Lgt1 production in S. cerevisiae strains transformed by pESC-Ura-based plasmids coding for wild type lgt1 and lgt1-D246A/W520A. Yeast cultures were grown in SD liquid medium (i.e. with glucose, lanes 1, 3, 5, and 7) or SGal (i.e. with galactose, lanes 2, 4, 6, and 8). Lanes 1 and 2, lgt1-WT in S. cerevisiae expressing wild type TEF1; lanes 3 and 4, lgt1-D246A/W520A in S. cerevisiae expressing wild type TEF1; lanes 5 and 6, lgt1 in S. cerevisiae expressing TEF1-S53A; lanes 7 and 8, lgt1-D246A/W520A in S. cerevisiae expressing Tef1-S53A. Western blotting was probed with anti-Lgt1 serum. The position of the glucosyltransferase is labeled with an asterisk. C, In vitro glucosylation of yeast extract prepared from S. cerevisiae containing Tef1-S53A and transformed with wild type lgt1. After cultivation in SGal, yeast cells were collected, disrupted by glass beads, and tested for Lgt1-dependent glucosylation in the 14C-glucosylation assay without (lane 1) and with (lane 2) addition of purified His-tagged Tef1. Coomassie-stained purified His-tagged Tef1 (lane 3) and molecular mass markers (lane 4) are shown. Molecular masses of used markers are shown on the right. The position of a protein band representing glucosylated Tef1 is labeled with an asterisk.
To confirm that Lgt1 was not only produced but was fully functional in S. cerevisiae with plasmid-born Tef1-S53A, we prepared extracts of yeast cells and tested them in the 14C-glucosylation assay. As shown in Fig. 4C, extracts obtained from S. cerevisiae (Δtef1Δtef2+pRS-TEF1-S53A) displayed no endogenous glucosylation of eEF1A in line with the fact that the major substrate Tef1 did not contain the acceptor serine 53 in this strain. However, addition of purified wild type Tef1 to the reaction mix resulted in 14C-glucosylation of the substrate.
Next, we investigated whether expression of TEF1-S53A would rescue the lethal effect of lgt1 expression in the strain (Δtef1Δtef2+pRS-TEF1). To that end, we transformed S. cerevisiae (Δtef1Δtef2+pRS-TEF1) with pYC-TEF1-S53A, or as control with pYC-TEF1. Then, these strains were transformed with the plasmid, containing wild type lgt1 under the control of galactose promoter or, as a control, with pESC-Ura vector. All four engineered S. cerevisiae strains were tested for growth on galactose-containing liquid and solid media.
As illustrated on Fig. 5, additional copies of Tef1-S53A reduced the lethal phenotype in comparison with the strain expressing an additional copy of wild type Tef1. The rescued phenotype was observed even despite high production of Lgt1, detected in Western blotting with anti-Lgt1 serum (Fig. 5C). Semiquantitative investigation of Lgt1-positive yeast extract in Western blotting suggested that single S. cerevisiae cell may contain up to 160,000 molecules of the glucosyltransferase (supplemental Fig. S1).
Toxicity of Lgt1 in HBS1-deleted S. cerevisiae
The experiments described above indicated that glucosylation of eEF1A causes a toxic effect in the S. cerevisiae model. However, to clarify whether modification of the other Lgt1 substrate, protein Hbs1, also influences yeast cell growth, we generated Δhbs1 S. cerevisiae strains by disrupting the gene either with the HIS3 marker or with geneticin antibiotic cassette. Next, we transformed the wild type or mutated lgt1 genes into the engineered S. cerevisiae strains and compared the toxic effects observed in Δhbs1 strains with that of the yeast strains containing an intact copy of chromosomal HBS1.
Expression of galactose-inducible lgt1-D246N resulted in a lethal phenotype in the wild type as well as in the Δhbs1 strain on galactose but not on glucose-containing medium (Fig. 6). Lgt1-D246N/W520A, the enzymatically less active glucosyltransferase mutant, produced moderate toxic effects in S. cerevisiae also irrespective of the presence or absence of HBS1. These results indicate that deletion of HBS1 in a wild type yeast background, i.e. in S. cerevisiae containing intact chromosomal TEF1/TEF2 genes, did not influence toxic effects caused by Lgt1.
FIGURE 6.

Toxicity of Lgt1 in wild type and Δhbs1 strains. Wild type and Δhbs1 S. cerevisiae, containing pESC-Ura or lgt1-D246N or lgt1-D246N/W520A within pESC-Ura-based plasmids, were analyzed on SD (left panel, glucose, Glc) or SGal (right panel, galactose, Gal). Description of strains is indicated on the left.
Next, we studied the impact of Hbs1 upon Lgt1-caused toxicity in S. cerevisiae with plasmid-born Tef1-S53A or Tef1-WT as sole elongation factors, i.e. in the yeast strains (Δtef1Δtef2+pRS-TEF1) and (Δtef1Δtef2+pRS-TEF1-S53A). As shown in Fig. 7, deletion of HBS1 did not alter the growth phenotypes of and Lgt1 production by the corresponding Hbs1-engineered S. cerevisiae. Strains without HBS1 but containing wild type TEF1 on a plasmid were efficiently killed by Lgt1-WT. In contrast, HBS1-deleted strains containing Tef1-S53A were protected against lethal effect of the highly produced wild type glucosyltransferase (Fig. 7C, lane 4) similar to that of HBS1-containing S. cerevisiae (Fig. 4B, lane 6). Interestingly, the S. cerevisiae strain lacking Hbs1 and containing non-modifiable eEF1A (i.e. lacking all known catalytic substrates for glucosyltransferases of L. pneumophila), still demonstrated a moderate growth-limiting phenotype even in the presence of triple-mutated Lgt1-D246N/W520A/N293A (Fig. 7B, lgt1-mut, lower row).
FIGURE 7.

Toxicity of Lgt1 in Δhbs1, expressing exclusively plasmid-born TEF1-WT or TEF1-S53A. Growth of S. cerevisiae (Δtef1Δtef2Δhbs1+pRS-TEF1-S53A) harboring pESC-Ura, pESC-Ura-coded lgt1, or pESC-Ura-coded lgt1-D246N/W520A/N293A was compared with growth of the corresponding strain expressing wild type HBS1. A, growth of engineered yeast strains in liquid SGal. Δtef1Δtef2Δhbs1+pRS-TEF1+lgt1 (closed triangles), Δtef1Δtef2Δhbs1+pRS-TEF1-S53A+pESC-Ura (circles), Δtef1Δtef2+pRS-TEF1-S53A+lgt1 (squares) and Δtef1Δtef2Δhbs1+pRS-TEF1-S53A+lgt1 (open triangles). B, growth phenotypes of yeast strains on SD and SGal plates. A description of the strains is indicated on the left. lgt1-mut indicates lgt1-D246N/W520A/N293A. A strain expressing firefly luciferase (lucifer) served as an additional control. C, expression of lgt1 in SGal by engineered S. cerevisiae strains: Δtef1Δtef2Δhbs1+pRS-TEF1+lgt1 (lane 1), Δtef1Δtef2Δhbs1+pRS-TEF1+lgt1-D246N/W520A/N293A (lane 2), Δtef1Δtef2Δhbs1+pRS-TEF1-S53A+pESC-Ura (lane 3), Δtef1Δtef2Δhbs1+pRS-TEF1-S53A+lgt1 (lane 4), and Δtef1Δtef2Δhbs1+pRS-TEF1-S53A+lgt1-D246N/W520A/N293A (lane 5). Western blots were probed with anti-Lgt1 serum (upper panel) or anti-eEF1A antibody (lower panel).
DISCUSSION
As a first step toward elucidation of S. cerevisiae as a model to study the toxic activity of Legionella glucosylating enzymes, we checked the effects of Lgt1 in in vitro translation using yeast extract. Addition of purified glucosyltransferase to S. cerevisiae translational extract inhibited protein synthesis in a dose-dependent manner. These results were similar to those obtained with mammalian reticulocyte lysates recently (12, 13). Also similar to experiments with mammalian cells, expression of lgt1 in the cytosol of S. cerevisiae resulted in severe toxic effects. A slight inhibitory growth phenotype was observed already in the presence of glucose in the medium (i.e. under non-inducing conditions) precluding overexpression of the recombinant protein. Induction of lgt1 gene with galactose completely stopped growth of yeast. Such inhibition was observed despite very low synthesis of the toxic protein because Lgt1 was not detected by Western blotting with the monospecific anti-Lgt1 serum. Transformation of yeast with mutated lgt1 genes produced toxic effects proportional to glucosylation activity of the proteins. These results demonstrated that S. cerevisiae is an adequate model to study Lgt activity in intact cells.
As shown in our previous studies, substitution of serine 53 by alanine turned elongation factor into a protein not modifiable in vitro by L. pneumophila glucosyltransferases Lgt (12, 13). In the current study, we engineered S. cerevisiae strains, which contained such non-modifiable Tef1-S53A as the only elongation factor present. In contrast to S. cerevisiae with modifiable wild type Tef1, Tef1-S53A-containing yeast cells were protected toward toxic action of Lgt1. The positive impact of Tef1-S53A upon survival of S. cerevisiae is even more evident when the high level of Lgt1 production in the Tef1-S53A-possessing yeast strains is compared with the very low amount of toxic protein that produced lethal effect in yeast with wild type eEF1A. These results demonstrated that eEF1A is a major target responsible for toxic effect after its glucosylation by Lgt1.
Another substrate of L. pneumophila glucosyltransferases, Hbs1 (Hsp70 subfamily B suppressor 1 (29)), was shown to be modified by in vitro glucosylation. For Hbs1, the target amino acid residue was identified as serine 314 (Hbs1-like protein of human origin, GenBankTM accession no. AK292656) or the corresponding serine 213 of Hbs1 in S. cerevisiae (10).
Both substrates of Lgt, eEF1A and Hbs1, represent important players in translational processes in eukaryotes. eEF1A is necessary for active translation and represents an essential protein. In contrast, disruption of HBS1 does not cause an obvious phenotype, but the protein has been shown to play an important role during situations of translational stalls due to inhibitory structures (e.g. strong hairpin loops), premature or lacking stop codons, and other defects of mRNA or deleterious mutations in rRNA (30, 31). Thus far, a role of Hbs1 in toxic effects of Lgt1 was not clarified. To address this issue, we deleted the HBS1 gene in wild type S. cerevisiae or in S. cerevisiae containing Tef1-S53A. However, when such deletion strains were transformed with the lgt1 gene, toxic effects produced by Lgt1 did not differ considerably from that of control background strains, containing Hbs1. Thus, these findings indicate that modification of Hbs1 is not essential for toxicity.
Of interest is our observation that the triple mutant of Lgt1, possessing undetectable level of glucosyltransferase activity, still demonstrated a noticeable growth-limiting effect toward S. cerevisiae (TEF1-S53A, Δhbs1) variant. Such an effect cannot be explained simply by nonspecific action of overproduced foreign protein upon cell metabolism because the control yeast strain, transformed with the similarly highly expressed luciferase gene-containing plasmid (Fig. 7B) or hyper-producing fragments of Lgt1 (data not shown), did not demonstrate a decrease in growth. One possible explanation for the mechanism of the observed phenomenon can be non-productive interaction of catalytically inactive glucosyltransferase with its enzymatically incompetent Tef1-S53A substrate leading to restraint of eEF1A functions in yeast cells.
Elongation factor 1A is necessary for delivery of aminoacylated tRNA molecules to the A-site of a translating ribosome (32). Despite thorough and long lasting investigations, many aspects of mechanisms of its participation in protein synthesis and in other processes in eukaryotic cells remain unanswered. eEF1A has undergone thorough mutational analysis to discover amino acid residues important for its functioning in yeast (20, 33, 35–38). These studies were successful in identifying mutations that affected fidelity of translation, dependence upon nucleotide exchange factor, rate of GTP binding, and GTP hydrolysis. Some of the obtained mutations resulted in non-functional eEF1A, producing non-viable phenotype of S. cerevisiae. Although the mechanisms of lethality remain unexplained, the obtained results allow identification of vitally important regions of the protein.
We studied the decapeptide Gly50–Val59 of Tef1, which includes Lgt1-modifiable serine 53. This region remained a largely unexplored segment of eEF1A. By using yeast strains with mutated Tef1 as the only elongation factor 1A, we identified various amino acid residues within this area, which appear to be essential, as their mutation to alanine resulted in severely affected or even non-viable yeast. Such essential amino acids include Phe54, Tyr56, and Trp58.
Currently, we are not able to explain the mechanisms of the functional incompetence of the mutated forms of elongation factor 1A. Strikingly, in previous studies, we identified the very similar aromatic residues as absolutely important for substrate recognition of eEF1A by Lgt1 (10). Thus, it is likely that the mutations in Phe54, Tyr56, and Trp58 result in structural changes or alterations, which similarly affect cellular function of eEF1A in translation and in recognition by Lgt1. Thus, this region around serine 53 appears to be a hot spot for functional efficiency of eEF1A.
Available genome sequencing data allow computer analysis of this area (supplemental Table S5). Interestingly, representatives of >40 species of mammals, fish, insects, and nematodes demonstrated strict sequence conservation in this region and contained the typical decapeptide 50GKGSFKYAWV59. Sequences of eEF1A of plant origin also demonstrated considerable homology. However, the structure of this region contained G50N and G52R substitutions. Archaeal phyla displayed considerable variations in elongation factor 1A sequences. Although in a few examples Phe54 was substituted by non-aromatic amino acid residues, positions 56 and 58 of the protein were occupied exclusively by phenylalanine/tyrosine/tryptophan residues. Thus, in silico data demonstrated evolutionary conservation of the decapeptide structure and supported in general our yeast mutational experiments on essential roles of aromatic amino acid residues at positions 54, 56, and 58 in elongation factor 1A.
Of interest is also the finding that substitution of serine 53 with alanine, cysteine, or lysine residues did not considerably affect the growth phenotype of engineered yeast. However, substitution S53E resulted in non-functional Tef1 failing to support growth of S. cerevisiae.
In numerous published papers, substitution of serine in a protein by negatively charged glutamate residue has been attributed to as “pseudophosphorylation” (39–41). Thus, our data leads to the assumption that the modification of the serine 53 residue by phosphorylation can shut down protein synthesis. Interestingly, in a recent report, phosphorylation of serine 53 has been demonstrated by a mass spectrometry approach, although physiological consequences of such modification remain to be determined (42).
This work was supported by the Deutsche Forschungsgemeinschaft and the Center for Biological Signaling Studies (BIOSS).

This article contains supplemental “Results,” Tables S1–S5, and Figs. S1–S4.
- eEF1A
- eukaryotic elongation factor 1A
- Hbs1
- Hsp70 subfamily B suppressor 1
- SD
- yeast synthetic medium with glucose
- SGal
- yeast synthetic medium with galactose
- 5-FOA
- 5-fluoroorotic acid.
REFERENCES
- 1. McDade J. E., Shepard C. C., Fraser D. W., Tsai T. R., Redus M. A., Dowdle W. R. (1977) Legionnaires' disease: Isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297, 1197–1203 [DOI] [PubMed] [Google Scholar]
- 2. Fraser D. W., Tsai T. R., Orenstein W., Parkin W. E., Beecham H. J., Sharrar R. G., Harris J., Mallison G. F., Martin S. M., McDade J. E., Shepard C. C., Brachman P. S. (1977) Legionnaires' disease: Description of an epidemic of pneumonia. N. Engl. J. Med. 297, 1189–1197 [DOI] [PubMed] [Google Scholar]
- 3. Jules M., Buchrieser C. (2007) Legionella pneumophila adaptation to intracellular life and the host response: Clues from genomics and transcriptomics. FEBS Lett. 581, 2829–2838 [DOI] [PubMed] [Google Scholar]
- 4. Ensminger A. W., Isberg R. R. (2009) Legionella pneumophila Dot/Icm translocated substrates: A sum of parts. Curr. Opin. Microbiol. 12, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hubber A., Roy C. R. (2010) Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283 [DOI] [PubMed] [Google Scholar]
- 6. Belyi Y., Jank T., Aktories K. (2011) Effector glycosyltransferases in Legionella. Front Microbiol. 2, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belyi I., Popoff M. R., Cianciotto N. P. (2003) Purification and characterization of a UDP-glucosyltransferase produced by Legionella pneumophila. Infect. Immun. 71, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hurtado-Guerrero R., Zusman T., Pathak S., Ibrahim A. F., Shepherd S., Prescott A., Segal G., van Aalten D. M. (2010) Molecular mechanism of elongation factor 1A inhibition by a Legionella pneumophila glycosyltransferase. Biochem. J. 426, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lü W., Du J., Stahl M., Tzivelekidis T., Belyi Y., Gerhardt S., Aktories K., Einsle O. (2010) Structural basis of the action of glucosyltransferase Lgt1 from Legionella pneumophila. J. Mol. Biol. 396, 321–331 [DOI] [PubMed] [Google Scholar]
- 10. Belyi Y., Stahl M., Sovkova I., Kaden P., Luy B., Aktories K. (2009) Region of elongation factor 1A1 involved in substrate recognition by Legionella pneumophila glucosyltransferase Lgt1: Identification of Lgt1 as a retaining glucosyltransferase. J. Biol. Chem. 284, 20167–20174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tzivelekidis T., Jank T., Pohl C., Schlosser A., Rospert S., Knudsen C. R., Rodnina M. V., Belyi Y., Aktories K. (2011) Aminoacyl-tRNA-charged eukaryotic elongation factor 1A is the bona fide substrate for Legionella pneumophila effector glucosyltransferases. PLoS. One. 6, e29525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belyi Y., Niggeweg R., Opitz B., Vogelsgesang M., Hippenstiel S., Wilm M., Aktories K. (2006) Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U.S.A. 103, 16953–16958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belyi Y., Tabakova I., Stahl M., Aktories K. (2008) Lgt: A family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190, 3026–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heitman J., Movva N. R., Hiestand P. C., Hall M. N. (1991) FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 88, 1948–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gietz R. D., Sugino A. (1988) New yeast Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 [DOI] [PubMed] [Google Scholar]
- 17. Conz C., Otto H., Peisker K., Gautschi M., Wölfle T., Mayer M. P., Rospert S. (2007) Functional characterization of the atypical Hsp70 subunit of yeast ribosome-associated complex. J. Biol. Chem. 282, 33977–33984 [DOI] [PubMed] [Google Scholar]
- 18. Sherman F. (2002) Getting started with yeast. Methods Enzymol. 350, 3–41 [DOI] [PubMed] [Google Scholar]
- 19. Brenner D. J., Steigerwalt A. G., McDade J. E. (1979) Classification of the Legionnaires' disease bacterium: Legionella pneumophila, genus novum, species nova, of the family Legionellaceae, familia nova. Ann. Intern. Med. 90, 656–658 [DOI] [PubMed] [Google Scholar]
- 20. Cavallius J., Merrick W. C. (1998) Site-directed mutagenesis of yeast eEF1A. Viable mutants with altered nucleotide specificity. J. Biol. Chem. 273, 28752–28758 [DOI] [PubMed] [Google Scholar]
- 21. Boeke J. D., Trueheart J., Natsoulis G., Fink G. R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154, 164–175 [DOI] [PubMed] [Google Scholar]
- 22. Garcia P. D., Hansen W., Walter P. (1991) In vitro protein translocation across microsomal membranes of Saccharomyces cerevisiae. Methods Enzymol. 194, 675–682 [DOI] [PubMed] [Google Scholar]
- 23. Fünfschilling U., Rospert S. (1999) Nascent polypeptide-associated complex stimulates protein import into yeast mitochondria. Mol. Biol. Cell 10, 3289–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berndt U., Oellerer S., Zhang Y., Johnson A. E., Rospert S. (2009) A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc. Natl. Acad. Sci. U.S.A. 106, 1398–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rakwalska M., Rospert S. (2004) The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol. Cell. Biol. 24, 9186–9197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 27. Towbin H., Staehelin T., Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yaffe M. P., Schatz G. (1984) Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. U.S.A. 81, 4819–4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson R. J., Ziegelhoffer T., Nicolet C., Werner-Washburne M., Craig E. A. (1992) The translation machinery and 70-kDa heat shock protein cooperate in protein synthesis. Cell 71, 97–105 [DOI] [PubMed] [Google Scholar]
- 30. Chen L., Muhlrad D., Hauryliuk V., Cheng Z., Lim M. K., Shyp V., Parker R., Song H. (2010) Structure of the Dom34-Hbs1 complex and implications for no-go decay. Nat. Struct. Mol. Biol. 17, 1233–1240 [DOI] [PubMed] [Google Scholar]
- 31. Cole S. E., LaRiviere F. J., Merrikh C. N., Moore M. J. (2009) A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol. Cell 34, 440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramakrishnan V. (2002) Ribosome structure and the mechanism of translation. Cell 108, 557–572 [DOI] [PubMed] [Google Scholar]
- 33. Carr-Schmid A., Pfund C., Craig E. A., Kinzy T. G. (2002) Novel G-protein complex whose requirement is linked to the translational status of the cell. Mol. Cell. Biol. 22, 2564–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deleted in proof
- 35. Dinman J. D., Kinzy T. G. (1997) Translational misreading: mutations in translation elongation factor 1α differentially affect programmed ribosomal frameshifting and drug sensitivity. RNA 3, 870–881 [PMC free article] [PubMed] [Google Scholar]
- 36. Magazinnik T., Anand M., Sattlegger E., Hinnebusch A. G., Kinzy T. G. (2005) Interplay between GCN2 and GCN4 expression, translation elongation factor 1 mutations, and translational fidelity in yeast. Nucleic Acids Res. 33, 4584–4592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ozturk S. B., Vishnu M. R., Olarewaju O., Starita L. M., Masison D. C., Kinzy T. G. (2006) Unique classes of mutations in the Saccharomyces cerevisiae G-protein translation elongation factor 1A suppress the requirement for guanine nucleotide exchange. Genetics 174, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ozturk S. B., Kinzy T. G. (2008) Guanine nucleotide exchange factor independence of the G-protein eEF1A through novel mutant forms and biochemical properties. J. Biol. Chem. 283, 23244–23253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang Y., Craig T. J., Chen X., Ciufo L. F., Takahashi M., Morgan A., Gillis K. D. (2007) Phosphomimetic mutation of Ser-187 of SNAP-25 increases both syntaxin binding and highly Ca2+-sensitive exocytosis. J. Gen. Physiol. 129, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rankin C. A., Sun Q., Gamblin T. C. (2005) Pseudo-phosphorylation of tau at Ser202 and Thr205 affects Tau filament formation. Brain Res. Mol. Brain Res. 138, 84–93 [DOI] [PubMed] [Google Scholar]
- 41. Eidenmüller J., Fath T., Maas T., Pool M., Sontag E., Brandt R. (2001) Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated Tau protein. Biochem. J. 357, 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Piazzi M., Bavelloni A., Faenza I., Blalock W., Urbani A., D'Aguanno S., Fiume R., Ramazzotti G., Maraldi N. M., Cocco L. (2010) eEF1A phosphorylation in the nucleus of insulin-stimulated C2C12 myoblasts: Ser53 is a novel substrate for protein kinase C βI. Mol. Cell Proteomics 9, 2719–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]