

Background: EmrE is a dual-topology membrane protein, but it is not clear whether the active form is composed of parallel or anti-parallel dimers.
Results: Antiparallel EmrE dimers are more stable than parallel dimers.
Conclusion: Antiparallel EmrE dimers most likely correspond to the functional form of the protein.
Significance: The results provide new information on the functional form of EmrE.
Keywords: Membrane Proteins, Membrane Transport, Multidrug Transporters, Protein Assembly, Protein-protein Interactions, EmrE, Membrane Protein Topology, Multidrug Resistance
Abstract
The bacterial multidrug transporter EmrE is a dual-topology membrane protein and as such is able to insert into the membrane in two opposite orientations. The functional form of EmrE is a homodimer; however, the relative orientation of the subunits in the dimer is under debate. Using EmrE variants with fixed, opposite orientations in the membrane, we now show that, although the proteins are able to form parallel dimers, an antiparallel organization of the subunits in the dimer is preferred. Blue-native PAGE analyses of intact oligomers and disulfide cross-linking demonstrate that in membranes, the proteins form parallel dimers only if no oppositely orientated partner is present. Co-expression of oppositely orientated proteins almost exclusively yields antiparallel dimers. Finally, parallel dimers can be disrupted and converted into antiparallel dimers by heating of detergent-solubilized protein. Importantly, in vivo function is correlated clearly to the presence of antiparallel dimers. Our results suggest that an antiparallel arrangement of the subunits in the dimer is more stable than a parallel organization and likely corresponds to the functional form of the protein.
Introduction
EmrE is a prototypical Escherichia coli inner membrane protein belonging to the small multidrug resistance family of secondary transporters (1). It imparts resistance to toxic compounds such as ethidium bromide (EtBr) and acriflavin (2). EmrE is 110 amino acids long and has four transmembrane helices. The minimal functional unit is a homodimer (3). Biochemical (4–7) and phylogenetic data (8) support a so-called dual topology for EmrE, i.e. the monomers have a mixed membrane orientation. Although it seems clear that the wild-type monomer is a dual-topology protein, the relative orientation of the subunits in the functional dimer is under debate. On the one hand, the available two-dimensional and three-dimensional crystal structures (9, 10) and recent FRET and NMR studies (11) show EmrE as an antiparallel dimer formed by two oppositely orientated monomers. On the other hand, parallel dimers have been detected by chemical cross-linking (12) and a model proposing that both parallel and antiparallel dimers are active has recently been put forward, together with the proposition that the affinity for parallel association is higher than for antiparallel association (7, 13).
In support of the antiparallel EmrE dimer being the more active species, we have shown previously that engineered EmrE variants with a predominant Cin or Cout orientation in the membrane (EmrE(Cin), EmrE(Cout)) impart no or very weak resistance to EtBr when expressed alone at low levels, whereas equally low level co-expression of the two variants provides robust resistance (4). When expressed at high levels in a ΔemrE strain, EmrE(Cin) imparts some resistance to EtBr; however, considerably less than what is seen when wild-type EmrE is expressed under the same conditions (7). It is not clear whether this activity is the result of weakly active parallel EmrE(Cin) dimers or if it is due to a small amount of EmrE(Cin) monomers that are in fact oppositely orientated, thereby allowing some formation of antiparallel dimers.
To better characterize the various dimer populations obtained with the different EmrE variants, we have now analyzed dimer formation in vivo and in vitro, using spontaneous disulfide bond formation and blue-native PAGE (BN-PAGE).3 We have also confirmed the predominant Cin and Cout topologies for EmrE(Cin) and EmrE(Cout) by cysteine-labeling of non-tagged proteins in intact cells.
Our results show that both EmrE(Cin) and EmrE(Cout) are able to form parallel homodimers when expressed alone, but that the antiparallel EmrE(Cin)/EmrE(Cout) heterodimer is the favored species when EmrE(Cin) and EmrE(Cout) are co-expressed in the same cell. This observation implies that in vivo, antiparallel dimers are more stable than parallel dimers. In agreement with this observation, when EmrE(Cin)/EmrE(Cout) heterodimers are solubilized in dodecylmaltoside (DDM) and heated to 60 °C to reversibly disassociate the dimer, the sample re-equilibrates into an approximate 1:2:1 mixture of EmrE(Cin) homodimer, EmrE(Cin)/EmrE(Cout) heterodimer, and EmrE(Cout) homodimer upon cooling. A similar ∼1:2:1 equilibrium is obtained when separately prepared samples of DDM-solubilized EmrE(Cin) and EmrE(Cout) homodimers are combined, heated to 60 °C, and cooled. The 1:2:1 distribution suggests that the three reassembled dimer forms are of nearly equal stabilities and correspond to antiparallel homo- and heterodimers. i.e. that, after dissociation of both parallel and antiparallel dimers in detergent, monomers reassemble preferentially into antiparallel dimers.
Taken together, our observations suggest that an antiparallel organization of the subunits in the EmrE dimer is more stable than either Cin- or Cout-orientated parallel dimers. In previous experiments, we demonstrated that maximum EtBr resistance is seen with cells either expressing the dual-topology EmrE(WT) or co-expressing EmrE(Cin) and EmrE(Cout) (4). We therefore conclude that the antiparallel dimer is not only the more active but also the more stable species.
EXPERIMENTAL PROCEDURES
Enzymes and Chemicals
All enzymes were from Fermentas (St. Leon-Rot, Germany), except for Pfu Turbo DNA polymerase from Stratagene (La Jolla, CA), complete supplement mixture of amino acids minus methionine (CSM Amino Acids, Met) from MP Biomedicals (Illkirch Cedex, France) and T4 DNA Ligase from Invitrogen. Oligonucleotides were from Eurofins MWG Operon (Ebersberg, Germany) and CyberGene (Stockholm, Sweden). l-[35S]methionine was from PerkinElmer Life Science. DDM and 2-(trimethylammonium)ethyl) methane thiosulfonate bromide (MTSET) were from Affymetrix-Anatrace (High Wycombe, UK). All other reagents were from Sigma-Aldrich.
Cloning and Mutagenesis
Cysteine-less EmrE variants (C39S, C41S, C95S), subsequent cysteine replacements, and Myc-His-tagged variants were generated by PCR mutagenesis of genes encoding EmrE, EmrE-Cin(R29G, R82S, S107K), and EmrE-Cout (T28R, L85R, R106A) (4). All constructs were cloned into the pET Duet-1 vector (Novagen), either solely or in pairs. The vector contains two multiple cloning sites (MCS), each preceded by a T7 promoter/lac operator and a ribosome binding site. Three combinations of primer-introduced restriction sites were used for cloning purposes: 5′-NcoI/3′-BamHI and 5′-NcoI/3′-SacI (EmrE-MycHis) for MCS1 and 5′-NdeI/3′-XhoI for MCS2. The 5′-restriction site in MCS1 results in an extra Gly residue following the initial Met; this does not affect the activity of the protein. All constructs were confirmed by DNA sequencing at Eurofins MWG Operon (Ebersberg, Germany).
Selective Radiolabeling and Spontaneous Cross-linking
Proteins were labeled selectively with [35S]methionine using the rifampicin-blocking technique (14). E. coli BL21(DE3) cells expressing single-cysteine EmrE variants were grown to an A600 of ∼0.5 at 37 °C. Cells were harvested by centrifugation at 1300 × g for 5 min in a tabletop Eppendorf centrifuge, resuspended, and starved for 90 min in minimal medium (M9 salts, 100 μg/ml thiamine, 0.1 mm CaCl2, 1 mm MgSO4, 0.4% glucose, 1 mg/ml CSM amino acids minus methionine, 100 μg/ml ampicillin). After inducing with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 10 min, 0.2 mg/ml rifampicin was added, and incubation was continued for 15 min. Proteins were labeled with 15 μCi [35S]Met for 3 min and put on ice for 2 min to stop the reaction. The samples were analyzed by 15% SDS-PAGE, and gels were imaged with Fujifilm FLA-3000. For reducing conditions, 50 mm dithiothreitol phosphate (DTT) was added prior to SDS-PAGE.
Topology Determination by Cysteine Labeling
For cysteine labeling, the protocol was adapted from Ref. 7 with some modifications. Selectively radiolabeled cells were harvested at 1300 × g for 4 min at 4 °C in a tabletop Eppendorf centrifuge, washed with potassium phosphate buffer (100 mm potassium phosphate, pH 7.0, 50 mm NaCl), resuspended in potassium phosphate buffer, and 0.14 mm MTSET, immediately harvested, and washed twice with potassium phosphate buffer. Cells were lysed in 30 mm Tris-Cl, pH 7.0, 50 μg/ml lysozyme, small amounts of DNase, and 5 mm MgCl for 30 min at 30 °C. The membrane fraction was collected by ultracentrifugation (20 min at 250,000 × g in a TLA-100.3 rotor) and incubated at 37 °C for one hour in 2% SDS, 6 m urea, 15 mm Tris-Cl, pH 7.5, and 25 mm methoxypolyethylene glycol maleimide (Mal-PEG). Samples were buffered with 100 mm Tris-HCl, pH 6.8, and analyzed by 15% SDS-PAGE and gels imaged with Fujifilm FLA-3000.
Analysis of Dimer Formation by BN-PAGE
EmrE variants were labeled with [35S]Met as described above, and the preparation of the samples was carried out as described in Ref. 15. Cells were lysed in water and 0.4 mg/ml lysozyme (45 min at 30 °C and slow shaking), and the membrane fraction was collected by ultracentrifugation (40 min, 200,000 × g, 4 °C using TLA55 rotor). The pellet was resuspended in ACA buffer (750 mm amino-n-caproic acid, 50 mm Bis-Tris, 0.5 mm EDTA, pH 7.0), and DDM was added to a final concentration of 0.5% (w/v). After 1-h incubation on ice, non-solubilized material was removed by ultracentrifugation (40 min, 200,000 × g in TLA55 rotor at 4 °C) and G250 solution (5% (w/v) Coomassie Brilliant Blue G in ACA buffer) was added to the supernatant. The samples were loaded on a 5–15% BN gradient gel (14 cm × 20 cm × 1.5 mm) and run at 75 V, 10 mA/gel for 1 h, and 385 V, 10 mA/gel for 17 h at 4 °C. Gels were fixed, dried, and scanned with Fujifilm FLA-3000. Data were analyzed with Image Gauge (version 4.23, Fujifilm) and QtiPlot (version 0.9.8.3, ProIndep Serv S.r.l.). For some experiments, samples were incubated at 60 °C after solubilization in DDM and removal of non-solubilized material by ultracentrifugation. After cooling down, Coomassie Brilliant Blue G-solution was added to the samples, and samples were analyzed as described above.
Two-dimensional SDS-PAGE
The gel strip cut from the first dimension BN-PAGE was equilibrated in buffer containing 2% (w/v) SDS, 250 mm Tris-Cl, pH 6.8, and 100 mm DTT for 30 min. The strip was fixed onto a 15% and 14 cm × 20 cm × 1.5 mm SDS gel and run for 14 h at 250 V. Gels were fixed, dried, and scanned with Fujifilm FLA-3000.
RESULTS
Topology Mapping of EmrE(Cin) and EmrE(Cout) by Cysteine Labeling
As noted in the introduction, EmrE(WT) has a dual topology and functions as a homodimer. In earlier work (4, 5), we showed that the orientation of EmrE in the cytoplasmic membrane of E. coli is sensitive to the distribution of positively charged residues in the parts of the protein that flank the four transmembrane helices, and we exploited this sensitivity to engineer EmrE variants that are inserted into the membrane with a unique Cin or Cout orientation (Fig. 1A) (4).
FIGURE 1.

Topology mapping of EmrE variants by cysteine accessibility. A, schematic representations of the oppositely orientated EmrE variants, EmrE(Cin) and EmrE(Cout). The mutations in EmrE(Cin) are R29G, R82S, and S107K, and in EmrE(Cout) T28R, L85R, and R106A. Black dots represent positively charged residues. The white starburst represents the position of the single cysteine Cys108 used for topology determination by MTSET/Mal-PEG treatment of whole cells. B, cysteine labeling of EmrE(Cin)T108C and EmrE(Cout)T108C. Periplasmic cysteines were blocked by treatment of whole cells with membrane-impermeable MTSET, cells were lysed, and remaining free cysteines were reacted with Mal-PEG that causes a size shift (black dots). The disulfide-bonded dimer of EmrE(Cout)T108C is indicated by a white dot. C, SDS-PAGE gels showing spontaneous in vivo cysteine cross-linking of EmrE(Cout)T108C (lane 3, white dot) but not of EmrET108C, EmrE(Cin)T108C, or co-expressed EmrE(Cin)/EmrE(Cout)T108C (lanes 1, 2, and 4). Lanes 5–8 shows the same samples after reduction with DTT. Note that EmrE(Cin) migrates faster than EmrE(Cout) and EmrE(WT) on SDS-PAGE gels.
In these experiments, the topology of the engineered variants was determined using C-terminal fusions to the topology reporters PhoA and GFP. Because the membrane orientation of EmrE(WT) has been found to be sensitive to the addition of certain C-terminal tags (4, 7), we decided to adapt a cysteine-labeling procedure described by Nasie et al. (7) to obtain independent topological data for EmrE(Cin) and EmrE(Cout) without the use of tags. In this assay, cells expressing selectively radiolabeled EmrE constructs containing a single cysteine residue are treated with the small membrane-impermeable reagent (MTSET), which reacts with cysteines that are exposed to the periplasm without causing noticeable changes in the molecular weight of the protein. Following cell lysis, unreacted thiols are identified by reaction with Mal-PEG, a reagent that increases the molecular weight of the protein by ∼5 kDa, a shift in molecular mass that is detected readily by conventional SDS-PAGE. In this way, periplasmically exposed cysteines can be differentiated from cysteines not exposed to the periplasm.
Because no antibodies against EmrE are available, we visualized EmrE on SDS-PAGE by [35S]Met labeling of EmrE constructs expressed from a T7 promoter in cells expressing T7 RNA polymerase, using rifampicin to block endogenous transcription (14). A single cysteine residue was introduced close to the C terminus (T108C) in otherwise cysteine-less versions of EmrE(Cin) and EmrE(Cout) (Fig. 1A). PEGylation was efficient for EmrE(Cin)T108C (Fig. 1B, lanes 1 and 2), indicating that Cys108 faces the cytoplasm, whereas for EmrE(Cout)T108C, MTSET blocked the PEGylation reaction (Fig. 1B, lanes 5 and 6), as expected if Cys108 faces the periplasm. This confirms the orientations of EmrE(Cin) and EmrE(Cout) as determined by reporter fusions.
EmrE(Cout)T108C Forms Disulfide-bonded Dimers in Vivo
Interestingly, for EmrE(Cout)T108C, a band of the size expected for a disulfide-bonded dimer was observed in the absence but not in the presence of the reductant DTT (Fig. 1B, lanes 5 and 7; Fig. 1C, lanes 3 and 7). Incubation with MTSET/Mal-PEG after cell lysis does not prevent dimer formation (Fig. 1B, lane 6), indicating that EmrE(Cout) can form a parallel homodimer already in the inner membrane of intact cells.
In contrast, no dimer was seen when EmrE(Cout)T108C was co-expressed with Cys-less EmrE(Cin) (Fig. 1C, lane 4), suggesting that formation of the antiparallel EmrE(Cout)/EmrE(Cin) heterodimer may outcompete formation of the EmrE(Cout) homodimer. The dual-topology EmrE(WT)T108C does not form a disulfide-bonded dimer, as expected if the EmrE(WT) homodimer is antiparallel (Fig. 1C, lane 1). No disulfide-bonded dimer is seen for EmrE(Cin)T108C when it is expressed on its own (Fig. 1B, lane 1; Fig. 1C, lane 2) because Cys108 faces the cytoplasm in this case. We conclude that EmrE(Cout) can form parallel homodimers in the inner membrane when expressed on its own but not when co-expressed with EmrE(Cin), providing a first indication that the antiparallel EmrE(Cout)/EmrE(Cin) heterodimer may be more stable than the parallel EmrE(Cin) homodimer.
Analysis of EmrE Homo- and Heterodimers by BN-PAGE
To visualize all homo- and heterodimers formed by EmrE(WT), EmrE(Cin), and EmrE(Cout), we used BN-PAGE to separate protein complexes under non-denaturing conditions. All three proteins migrate predominantly as dimers when extracted into DDM and analyzed by BN-PAGE (Fig. 2A, lanes 1, 3, and 6). Some higher oligomers, most likely tetramers, are also seen in all samples. A notable amount of monomer is seen for EmrE(Cout) but not for EmrE(WT) or EmrE(Cin). Fortuitously, the EmrE(Cout) dimer migrates noticeably faster than do the EmrE(WT) and EmrE(Cin) dimers, making it possible to distinguish homo- and heterodimers when the different EmrE versions are co-expressed.
FIGURE 2.

Analysis of dimer formation by BN-PAGE. A, BN-PAGE of EmrE(WT) (○), EmrE(Cin) (△), EmrE(Cout) (▿), and co-expressed versions. Monomeric (M), dimeric (D), and tetrameric (T) forms and their composition are indicated. Note that EmrE(Cout) migrates faster than EmrE(Cin) and EmrE(WT) on BN-PAGE, opposite to the situation for SDS-PAGE. B, two-dimensional SDS-gel of BN-PAGE separated samples of EmrE(Cin), EmrE(Cout), co-expressed EmrE(Cin)/EmrE(Cout), and co-expressed EmrE(Cout)/EmrE(Cin). C, SDS-PAGE of the same samples as in A and B (co-expressed EmrE(Cin)/EmrE(Cout) and co-expressed EmrE(Cout)/EmrE(Cin)), illustrating that the relative expression levels depend on the cloning site (MCS1, MCS2) in the vector. The protein encoded by the gene in MCS1 tends to be more highly expressed than the gene in MCS2.
Co-expression of EmrE(Cin) and EmrE(Cout) from the same plasmid yields a heterodimer with a mobility that is intermediate between that of the EmrE(Cin) and EmrE(Cout) homodimers (Fig. 2A, see inset). Only small amounts of EmrE(Cin) or EmrE(Cout) homodimer are present in this case. Notably, the two cloning sites in the vector yield somewhat different amounts of protein (Fig. 2C) and the remaining homodimer that is seen correlates with the monomer that is present in excess. Analysis by two-dimensional SDS-PAGE confirms that the intermediate mobility dimer bands contain EmrE(Cin) and EmrE(Cout) in amounts that reflect their expression levels (Fig. 2B). These results imply that the formation of the antiparallel EmrE(Cin)/EmrE(Cout) heterodimer is favored over either of the two parallel homodimers.
Co-expression of EmrE(WT) and EmrE(Cout) (Fig. 2A, lane 8) yields two bands: one corresponding to the EmrE(WT) homodimer (upper band) and one of intermediate mobility that presumably corresponds to EmrE(WT)/EmrE(Cout) heterodimers. No EmrE(Cout) homodimer and very little EmrE(Cout) monomer is seen, most likely because EmrE(WT) is produced at somewhat higher levels than EmrE(Cout) in this case. We conclude that the EmrE(WT) homodimer and the EmrE(WT)/EmrE(Cout) heterodimer have comparable stabilities.
Unlike EmrE(Cout), both EmrE(WT) and EmrE(Cin) homodimers have the same mobility in BN-PAGE. Therefore, co-expression yields only a single dimer band (Fig. 2A, lanes 1, 3, and 7). In an attempt to detect EmrE(WT)/EmrE(Cin) heterodimers, EmrE(WT) was modified by the addition of a 27-residue C-terminal Myc-His tag; incidentally, addition of this tag has been found to lead to a partial shift in the topology (7). As shown in Fig. 3, the EmrE(WT)-Myc-His construct (lane 2) migrates mainly as a homodimer on BN-PAGE. Co-expression with either EmrE(Cin) or EmrE(Cout) leads to formation of heterodimers (Fig. 3, lanes 3 and 4), although the efficiency of EmrE(WT)-Myc-His/EmrE(Cout) heterodimer formation is lower than that seen with untagged EmrE(WT)/EmrE(Cout) (Fig. 2A, lane 8). We conclude that both EmrE(Cin) and EmrE(Cout) form heterodimers with EmrE(wt)-MycHis, and, seeing that EmrE(WT)/EmrE(Cout) heterodimers can be detected on BN-PAGE, that EmrE(Cin) also forms heterodimers with untagged EmrE(WT).
FIGURE 3.
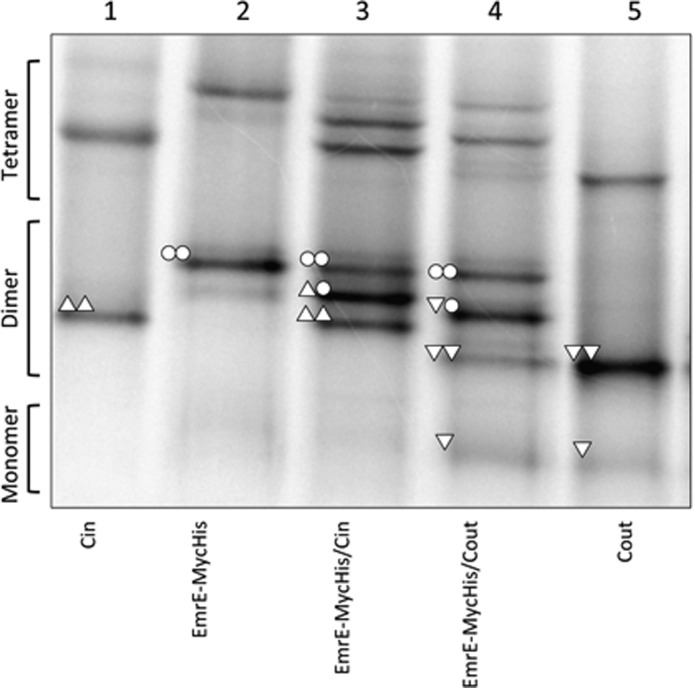
BN-PAGE of EmrE(WT)-MycHis, co-expressed EmrE(WT)-Myc-His/EmrE(Cin), and co-expressed EmrE(WT)-Myc-His/EmrE(Cout). The compositions of the different dimers are indicated by the same symbols as described in the legend to Fig. 2. EmrE(Cin) and EmrE(Cout) are included for comparison.
Heat-induced Disruption and Reassembly of EmrE Homo- and Heterodimers
The EmrE(WT) homodimer is exceptionally stable in DDM but can be disrupted by heating to 60 °C (16). We took advantage of this observation to further assess the stabilities of the EmrE(Cin) and EmrE(Cout) homodimers relative to the EmrE(Cin)/EmrE(Cout) heterodimer.
First, we confirmed that the DDM-solubilized EmrE(Cin) and EmrE(Cout) homodimers are stable when mixed without heating. As seen in Fig. 4A, if cells expressing either EmrE(Cin) or EmrE(Cout) (lanes 1 and 6) are mixed immediately after harvesting (lane 3), or if purified membrane fractions from such cells are mixed (lane 4), no EmrE(Cin)/EmrE(Cout) heterodimer is seen by BN-PAGE after DDM solubilization. The same is true if DDM-solubilized EmrE(Cin) and EmrE(Cout) are mixed and kept at 4 °C prior to BN-PAGE (Fig. 4B, lane 3). In contrast, when roughly equal amounts of DDM-solubilized EmrE(Cin) and EmrE(Cout) are mixed and then incubated at 60 °C for at least 10 min prior to analysis by BN-PAGE, a substantial amount of EmrE(Cin)/EmrE(Cout) heterodimer is formed (Fig. 4B, lanes 4–6). Heat treatment of DDM-solubilized EmrE(WT)-Myc-His mixed with either EmrE(Cin) or EmrE(Cout) gives similar results (Fig. 4C) except that the relative amount of heterodimer versus homodimer is lower, as seen also in the co-expression experiments (Fig. 3).
FIGURE 4.

EmrE(Cin) and EmrE(Cout) homodimers reassemble into a mixture of EmrE(Cin)/EmrE(Cout) heterodimers, EmrE(Cin) homodimers, and EmrE(Cout) homodimers after heating to 60 °C and cooling. A, BN-PAGE of cells expressing EmrE(Cin) and cells expressing EmrE(Cout) that were mixed before lysis (lane 3) or mixed after membrane collection but before solubilization in DDM (lane 4). EmrE(Cin) (lane 1), co-expressed EmrE(Cin)/EmrE(Cout) (lane 2), co-expressed EmrE(Cout)/EmrE(Cin) (lane 5), and EmrE(Cout) (lane 6) are included for comparison. B, BN-PAGE of DDM-solubilized EmrE(Cin) mixed with DDM-solubilized EmrE(Cout) and incubated at either 4 °C or 60 °C for different times as indicated (lanes 3–6). EmrE(Cin) (lane 1), co-expressed EmrE(Cin)/EmrE(Cout) (lane 2), and EmrE(Cout) (lane 7) are included for comparison. C, BN-PAGE of DDM-solubilized EmrE(Cin) mixed with DDM-solubilized EmrE(WT)-Myc-His (lanes 1–4) and of EmrE(Cout) mixed with DDM-solubilized EmrE(WT)-Myc-His (lanes 5–8) incubated at either 4 or 60 °C for different times as indicated. The composition of the different dimers are indicated by the same symbols as described in the legend to Fig. 2.
Comparing the relative amounts of homodimer and heterodimer seen after mixing and heating of DDM-solubilized EmrE(Cin) and EmrE(Cout) (Fig. 4B) to those seen upon co-expression of EmrE(Cin) and EmrE(Cout) in the same cell (Fig. 2A), it is clear that the amount of remaining homodimer is higher in the mixed sample. A possible explanation for this discrepancy is that, following heat disruption of the DDM-solubilized, parallel EmrE(Cin) and EmrE(Cout) homodimers, the monomers, no longer being constrained by a lipid bilayer, reassemble preferentially into antiparallel homodimers and EmrE(Cin)/EmrE(Cout) heterodimers of roughly equal stability (Fig. 5A).
FIGURE 5.

EmrE(Cin)/EmrE(Cout) heterodimers reassemble into a mixture of EmrE(Cin)/EmrE(Cout) heterodimers, EmrE(Cin) homodimers, and EmrE(Cout) homodimers after heating to 60 °C and cooling. A, schematic figure summarizing the proposed rearrangement of homodimers and the formation of heterodimers after mixing and heating DDM-solubilized EmrE(Cin) and EmrE(Cout). The starting point (I) indicates cells expressing EmrE(Cin) (triangles) and cells expressing EmrE(Cout) (inverted triangles). Solubilization (II) preserves the native organization of the dimers. After mixing of micelles (III), the native organization is preserved (IV), unless the micelles are heated and then cooled (V). Heat treatment results in a 1:2:1 mixture of antiparallel EmrE(Cin) homodimers, EmrE(Cin)/EmrE(Cout) heterodimers, and EmrE(Cout) homodimers. B, BN-PAGE of DDM-solubilized co-expressed EmrE(Cin)/EmrE(Cout) (lanes 2–4) and co-expressed EmrE(Cout)/EmrE(Cin) (lanes 5–7) incubated at 60 °C for the indicated times. Co-expressed but unheated EmrE(Cin)/EmrE(Cout) (lane 1) and co-expressed EmrE(Cout)/EmrE(Cin) (lane 8) samples are included for comparison. C, quantitation of dimer disruption and reassembly of DDM-solubilized samples of separately expressed EmrE(Cin) and EmrE(Cout) (left panel; cf. Fig. 4B) and of co-expressed EmrE(Cin)/EmrE(Cout) (middle panel; cf. Fig. 5B) after incubation at 60 °C for the indicated times and cooling. The fractions of the different dimeric forms are shown as a function of incubation time at 60 °C. Quantitation of the three dimer peaks was done by fitting of three Gaussians to the scanned density in this region of the gel using the QtiPlot software, as in the example shown (right panel); au, arbitrary units. Because of the rather large peak overlaps, the width of each peak was set manually to the same constant value for the three peaks before the fitting to ensure a reproducible fit. 10′, 10 min; 20′, 20 min; 30′, 30 min.
If this explanation is correct, heat treatment of DDM-solubilized EmrE(Cin)/EmrE(Cout) heterodimers obtained by co-expression should give the same final distribution of homo- and heterodimers as that obtained after heat treatment of separately produced EmrE(Cin) and EmrE(Cout) homodimers. This is indeed the case (Fig. 5B). For both EmrE(Cin)+EmrE(Cout) and EmrE(Cin)/EmrE(Cout), the distributions converge on a distribution composed of ∼45% EmrE(Cin)/EmrE(Cout) heterodimer and ∼27% each of the EmrE(Cin) and EmrE(Cout) homodimers, i.e. a 1:1.7:1 distribution of EmrE(Cin) homodimer:EmrE(Cin)/EmrE(Cout) heterodimer:EmrE(Cout) homodimer (Fig. 5C). This is remarkably close to the 1:2:1 distribution that would be expected if all three kinds of dimers were of exactly equal stability. It thus appears that the fixed topology EmrE(Cin) and EmrE(Cout) constructs reassemble into antiparallel rather than parallel homodimers and that the antiparallel homodimers and the antiparallel heterodimers are of comparable stabilities.
DISCUSSION
The EmrE multidrug resistance protein is the best characterized of a class of membrane proteins that have a dual topology, i.e. the polypeptide chains insert into the membrane in such a way that the final population is roughly a 1:1 mixture of molecules with a Cin and a Cout orientation (7, 8). In most, if not all, cases the active form of a dual-topology protein is expected to be a homodimer or a higher oligomer. A priori, dimers of dual-topology proteins could be formed between monomers that have the same orientation in the membrane (parallel dimers) or opposite orientations (antiparallel dimers), and higher-order oligomers could also be formed with varying numbers of parallel and antiparallel monomers.
Despite a multitude of studies, the active conformation of EmrE is still contentious: although the arguments for a dual topology of the monomer now seem compelling (4–10) and the case for a homodimer being the minimal functional unit is strong (3), there are data favoring both an antiparallel and a parallel arrangement of the monomers in the active dimer (7, 11, 12, 17). In fact, a recent model proposes that both the parallel and antiparallel dimers are active but that the affinity for parallel association is higher than for antiparallel association (13). Here, we have revisited the conformation of EmrE dimers, using both spontaneous cross-linking of single-Cys EmrE mutants and BN-PAGE analysis of singly expressed or co-expressed EmrE(WT), EmrE(Cin), and EmrE(Cout) constructs.
EmrE(Cin) and EmrE(Cout) were originally designed to have a strong preference for either the Cin or the Cout topology, in contrast to EmrE(WT) that has a dual topology (4). Already at very low expression levels, co-expression of EmrE(Cin) and EmrE(Cout) imparts the same level of EtBr resistance to cells as does low level expression of EmrE(WT), whereas when expressed on their own at low levels, neither EmrE(Cin) nor EmrE(Cout) impart significant resistance (4). Because the two constructs have been shown by reporter fusion analysis (4) and by cysteine labeling (Fig. 1B) to have opposite membrane orientations in the inner membrane of E. coli, these results suggested that an antiparallel dimer is the more active species.
Because EmrE(Cin) and EmrE(Cout) have slightly different mobilities both during SDS-PAGE and BN-PAGE, we can readily differentiate between homo- and heterodimers of these molecules by gel electrophoresis, and we can also detect heterodimers between EmrE(Cin) or EmrE(Cout) and EmrE(WT) in this way. Strikingly, both spontaneous cysteine cross-linking and BN-PAGE show efficient formation of homodimers when EmrE(WT), EmrE(Cin), and EmrE(Cout) are expressed alone (Figs. 1C and 2). Because EmrE(Cin) and EmrE(Cout) both have unique membrane topologies, this strongly suggests that parallel EmrE(Cin) and EmrE(Cout) homodimers can form in the inner membrane. However, only the dual-topology protein EmrE(WT) imparts robust EtBr resistance when expressed at low levels in standard strains (4), suggesting that parallel EmrE(Cin) and EmrE(Cout) homodimers have at best a marginal activity compared with EmrE(WT).
A different picture emerges when EmrE(Cin) and EmrE(Cout) are co-expressed in the same cell. In this case, the heterodimer is the dominant species, as seen both by the inhibition of spontaneous disulfide bond formation and BN-PAGE. The antiparallel EmrE(Cin)/EmrE(Cout) heterodimer is thus considerably more stable in vivo than either of the two parallel homodimers, and the EtBr resistance of cells co-expressing EmrE(Cin) and EmrE(Cout) (4) clearly correlates with the presence of the antiparallel heterodimer.
Finally, when DDM-solubilized EmrE(Cin)/EmrE(Cout) heterodimer is heat-treated at 60 °C and then cooled, the molecules reassemble into an ∼1:2:1 mixture of EmrE(Cin) homodimer, EmrE(Cin)/EmrE(Cout) heterodimer, and EmrE(Cout) homodimer; the same final mixture is obtained if separate samples of DDM-solubilized EmrE(Cin) and EmrE(Cout) parallel homodimers are mixed and heated. Consistent with the idea that antiparallel dimers are more stable than parallel dimers, we propose that heating leads to dimer disassembly and micelle mixing, and that upon cooling the monomers reassemble into antiparallel homo- and heterodimers of comparable stability. This kind of heat-induced rearrangement can only happen in detergent solution and not in an intact membrane.
Taken together, our results suggest that an antiparallel arrangement of the subunits in the EmrE(WT) dimer is more stable than a parallel organization and likely corresponds to the active form of the protein in vivo. However, unless EmrE(WT) is produced as a precisely balanced mixture of Cin- and Cout-orientated monomers, some parallel dimers will inevitably form, in addition to the dominating antiparallel dimers. Are such parallel dimers of some functional relevance, and are the critical monomer-monomer interactions that drive formation of parallel dimers maintained by selection? As pointed out by Schuldiner (13), it will be interesting to try to understand how a single protein can form both antiparallel and parallel dimers, albeit of somewhat different stabilities.
This work was supported by European Research Council Grant ERC-2008-AdG 232648, the Swedish Foundation for Strategic Research, the Swedish Research Council, and the Swedish Cancer Foundation (to G.v. H.); the European Communities TranSys Grant PITN-2008-215524 (to P. L. G.); and the International Human Frontier Science Program Organization (to J. S. G. S.).
- BN-PAGE
- blue native PAGE
- DDM
- dodecylmaltoside
- MTSET
- (2-(trimethylammonium) ethyl) methane thiosulfonate bromide)
- MCS
- multiple cloning sites
- Mal-PEG
- methoxypolyethylene glycol maleimide 5000.
REFERENCES
- 1. Bay D. C., Rommens K. L., Turner R. J. (2008) Small multidrug resistance proteins: A multidrug transporter family that continues to grow. Biochim. Biophys. Acta 1778, 1814–1838 [DOI] [PubMed] [Google Scholar]
- 2. Schuldiner S. (2009) EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim. Biophys. Acta 1794, 748–762 [DOI] [PubMed] [Google Scholar]
- 3. Tate C. G., Ubarretxena-Belandia I., Baldwin J. M. (2003) Conformational changes in the multidrug transporter EmrE associated with substrate binding. J. Mol. Biol. 332, 229–242 [DOI] [PubMed] [Google Scholar]
- 4. Rapp M., Seppälä S., Granseth E., von Heijne G. (2007) Emulating membrane protein evolution by rational design. Science 315, 1282–1284 [DOI] [PubMed] [Google Scholar]
- 5. Seppälä S., Slusky J. S., Lloris-Garcerá P., Rapp M., von Heijne G. (2010) Control of membrane protein topology by a single C-terminal residue. Science 328, 1698–1700 [DOI] [PubMed] [Google Scholar]
- 6. Nara T., Kouyama T., Kurata Y., Kikukawa T., Miyauchi S., Kamo N. (2007) Anti-parallel membrane topology of a homodimeric multidrug transporter, EmrE. J Biochem. 142, 621–625 [DOI] [PubMed] [Google Scholar]
- 7. Nasie I., Steiner-Mordoch S., Gold A., Schuldiner S. (2010) Topologically random insertion of EmrE supports a pathway for evolution of inverted repeats in ion-coupled transporters. J. Biol. Chem. 285, 15234–15244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rapp M., Granseth E., Seppälä S., von Heijne G. (2006) Identification and evolution of dual-topology membrane proteins. Nat. Struct. Mol. Biol. 13, 112–116 [DOI] [PubMed] [Google Scholar]
- 9. Ubarretxena-Belandia I., Baldwin J. M., Schuldiner S., Tate C. G. (2003) Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 22, 6175–6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y. J., Pornillos O., Lieu S., Ma C., Chen A. P., Chang G. (2007) X-ray structure of EmrE supports dual-topology model. Proc. Natl. Acad. Sci. U.S.A. 104, 18999–19004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ito K., Chiba S., Pogliano K. (2010) Divergent stalling sequences sense and control cellular physiology. Biochem. Biophys. Res. Comm. 393, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soskine M., Mark S., Tayer N., Mizrachi R., Schuldiner S. (2006) On parallel and antiparallel topology of a homodimeric multidrug transporter. J. Biol. Chem. 281, 36205–36212 [DOI] [PubMed] [Google Scholar]
- 13. Schuldiner S. (2012) Undecided membrane proteins insert in random topologies. Up, down and sideways: It does not really matter. Trends Biochem. Sci. 37, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Studier F. W., Rosenberg A. H., Dunn J. J., Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89 [DOI] [PubMed] [Google Scholar]
- 15. Stenberg F., Chovanec P., Maslen S. L., Robinson C. V., Ilag L. L., von Heijne G., Daley D. O. (2005) Protein complexes of the Escherichia coli cell envelope. J. Biol. Chem. 280, 34409–34419 [DOI] [PubMed] [Google Scholar]
- 16. Rotem D., Sal-man N., Schuldiner S. (2001) In vitro monomer swapping in EmrE, a multidrug transporter from Escherichia coli, reveals that the oligomer is the functional unit. J. Biol. Chem. 276, 48243–48249 [DOI] [PubMed] [Google Scholar]
- 17. Steiner-Mordoch S., Soskine M., Solomon D., Rotem D., Gold A., Yechieli M., Adam Y., Schuldiner S. (2008) Parallel topology of genetically fused EmrE homodimers. EMBO J. 27, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]