

Background: Evidence suggests that fibroblast growth factor 21 (FGF21) inhibits longitudinal bone growth.
Results: FGF21 antagonizes the growth hormone (GH) stimulatory effects on thymidine incorporation and collagen X expression in chondrocytes.
Conclusion: FGF21 inhibits bone growth by antagonizing GH effects on chondrocyte proliferation and differentiation.
Significance: Increased FGF21 during food restriction may contribute to growth failure by directly inhibiting chondrogenesis.
Keywords: Bone, Chondrogenesis, Fibroblast Growth Factor (FGF), Growth Hormone, Growth Plate, Fasting
Abstract
Fibroblast growth factor 21 (FGF21) modulates glucose and lipid metabolism during fasting. In addition, previous evidence indicates that increased expression of FGF21 during chronic food restriction is associated with reduced bone growth and growth hormone (GH) insensitivity. In light of the inhibitory effects on growth plate chondrogenesis mediated by other FGFs, we hypothesized that FGF21 causes growth inhibition by acting directly at the long bones' growth plate. We first demonstrated the expression of FGF21, FGFR1 and FGFR3 (two receptors known to be activated by FGF21) and β-klotho (a co-receptor required for the FGF21-mediated receptor binding and activation) in fetal and 3-week-old mouse growth plate chondrocytes. We then cultured mouse growth plate chondrocytes in the presence of graded concentrations of rhFGF21 (0.01–10 μg/ml). Higher concentrations of FGF21 (5 and 10 μg/ml) inhibited chondrocyte thymidine incorporation and collagen X mRNA expression. 10 ng/ml GH stimulated chondrocyte thymidine incorporation and collagen X mRNA expression, with both effects prevented by the addition in the culture medium of FGF21 in a concentration-dependent manner. In addition, FGF21 reduced GH binding in cultured chondrocytes. In cells transfected with FGFR1 siRNA or ERK 1 siRNA, the antagonistic effects of FGF21 on GH action were all prevented, supporting a specific effect of this growth factor in chondrocytes. Our findings suggest that increased expression of FGF21 during food restriction causes growth attenuation by antagonizing the GH stimulatory effects on chondrogenesis directly at the growth plate. In addition, high concentrations of FGF21 may directly suppress growth plate chondrocyte proliferation and differentiation.
Introduction
Fibroblast growth factor 21 (FGF21) is a member of a subfamily of fibroblasts growth factors (FGFs) (with includes FGF15/19 and FGF23) that lacks the FGF heparin binding domain (1, 2). Thus, these FGFs can diffuse away from their tissue of synthesis and function as endocrine factors.
Previous evidence indicates that, in mammals, fasting induces the expression of FGF21 in the liver and raises its circulating levels; increased FGF21 expression and activity modulates metabolic adaptation to fasting by inducing increased gluconeogenesis, fatty acid oxidation, and ketogenesis (3–5). In addition, we have recently demonstrated (6) in mice that the increased expression of FGF21 during prolonged food restriction, both in the liver and in the long bones' growth plate, contributes to the attenuation of skeletal growth and growth plate chondrogenesis; one of the underlying mechanisms of growth attenuation is the FGF21-dependent systemic GH insensitivity. Additional evidence also indicates that transgenic mice overexpressing FGF21 are significantly smaller, have reduced tibial length, and display reduced hepatic GH insensitivity when compared with wild-type mice (7).
FGF21 has a specific affinity for FGFR12 but can also signal through all fibroblast growth factor receptors (FGFRs) (2, 8). FGF21-mediated signal transduction requires this growth factor to bind a FGFR complexed with β-klotho, a membrane-spanning protein (9–11). Although FGFRs are expressed in a variety of tissues, expression of FGF21 and β-klotho has been detected mostly in the liver, pancreas, and white adipose tissue (12, 13). The aim of our study was to determine whether during prolonged undernutrition FGF21 may also inhibit bone growth by directly suppressing chondrogenesis and GH action at the growth plate.
MATERIALS AND METHODS
Chondrocyte Culture
The cartilaginous portions of metatarsal bones isolated from C57BL/6N mouse embryos (19 days post-coitus) were dissected, rinsed in PBS, then incubated in 0.2% trypsin for 1 h and 0.2% collagenase for 3 h. Cell suspension was aspirated repeatedly and filtered through a 70-μm cell strainer, rinsed first in PBS then in serum-free DMEM, and counted. Chondrocytes were seeded in 100-mm dishes at a density of 5 × 104/cm2 in DMEM with 100 units/ml penicillin and 100 μg/ml streptomycin, 50 μg/ml ascorbic acid, and 10% FBS. The culture medium was changed at 72-h intervals. Upon confluence, cells were subcultured, and their chondrogenic phenotype was confirmed by studying the expression of type-I collagen, type-II collagen, and type X collagen by immunocytochemistry. In our culture conditions the percentage of cells expressing type-I, type-II, and type-X collagen was 3.9, 97.1, and 22.0%, respectively. Subsets of cells with confirmed chondrogenic phenotype (with at least 95% of the cells positive for type II collagen and negative for type I collagen) were treated without or with graded concentrations of rhFGF21 (0.01, 0.1, 1, 5, and 10 μg/ml, recombinant human, Lilly Research Laboratories) up to 14 days. In another series of experiments, cells were first cultured for 24 h without or with graded concentrations of rhFGF21 (0.01, 0.1, 1, 5, and 10μg/ml). After 24 h, culture medium containing FGF21 was removed and replaced with fresh serum-free DMEM without or with recombinant mouse growth hormone (rmGH; 10 ng/ml, National Hormone and Peptide Program, Harbor-UCLA Medical Center, Torrance, CA).
[3H]Thymidine Incorporation
To assess cell proliferation, we measured [3H]thymidine incorporation into newly synthesized DNA. At the indicated time points during the culture period, 2.5 μCi of [3H]thymidine/well (25 Ci/mmol; Amersham Biosciences) was added to the culture medium for an additional 3 h. Cells were released by trypsin and collected onto glass fiber filters. Incorporation of [3H]thymidine was measured by liquid scintillation counting. The data represent percentage of control from three independent experiments.
Western Blot
Whole cell lysates were solubilized with 1% SDS sample buffer and electrophoresed on a 4–15% SDS-PAGE gel (Bio-Rad). Proteins were transferred onto a nitrocellulose membrane and probed with the following primary antibodies: rabbit polyclonal against p-ERK 1/2 and ERK 1/2 (Cell Signaling Technology, Inc.), mouse beta Klotho Affinity-purified polyclonal antibody (R&D Systems Inc. Minneapolis, MN), goat polyclonal antibody against FGFR1 (Santa Cruz), mouse monoclonal antibody against FGFR3 (Santa Cruz), rabbit polyclonal antibody against GH receptor (GHR) (Santa Cruz), and rabbit polyclonal antibody against β-actin (Sigma). The blots were developed using a horseradish-peroxidase-conjugated polyclonal donkey-anti rabbit or mouse IgG antibody and enhanced chemiluminescence system (GE Healthcare). Quantitation of exposed x-ray films was performed with Image-J software (National Institutes of Health, Bethesda MD).
RNA Extraction and PCR
Total RNA was extracted from the 3-week-old mouse liver and tibial growth plates and from cultured chondrocytes isolated from fetal (20 days post-coitus) mouse metatarsal growth plates using the Qiagen RNeasy Mini kit (Qiagen Inc., Valencia CA). The recovered RNA was further processed using the 1st Strand cDNA synthesis kit for RT-PCR avian myeloblastosis virus (Roche Applied Science) to produce cDNA. 1 μg of total RNA and 1.6 μg of oligo-p(dT)15 primer were incubated for 10 min at 25 °C followed by incubation for 60 min at 42 °C in the presence of 20 units of avian myeloblastosis virus reverse transcriptase and 50 units of RNase inhibitor in a total 20-μl reaction. The cDNA products were directly used for PCR or stored at −80 °C for later analysis. Real-time quantitative PCR was carried out using the StepOne real time PCR system (Applied Biosystems, Foster City, CA) in a final volume of 25 μl containing 1 μl of cDNA, 12.5 μl of 2× SYBR Green master mix (Applied Biosystems), 0.1 μm primers (Applied Biosystems) in DNase-free water. Primers used were: mouse β-actin, 5′-TGT GAT GGT GGG AAT GGG TCA GAA-3′ and 5′-TGT GGT GCC AGA TCT TCT CCA TGT-3′ (PCR fragment 140 bp); mouse FGF21, 5′-AAC AGC CAT TCA CTT TGC CTG AGC-3′ and 5′-GGC AGC TGG AAT TGT GTT CTG ACT-3′ (PCR fragment 85 bp); mouse Collagen X, 5′-ATA AGA ACG GCA CGC CTA CGA TGT-3′ and 5′-CTG CAT TGG GCA ATT GGA GCC ATA-3′ (PCR fragment 128bp); FGFR1, 5′-CCA GTG CAT CCA TGA ACT CTG GGG TTC TCC-3′, 5′-GGT CAC ACG GTT GGG TTT GTC CTT ATC CAG-3′ (PCR fragment 230 bp); FGFR3, 5′-AGT GCT AAA TGC CTC CCA CGA AGA-3′, 5′-AGG ATG GAG CAT CTG TTA CAC GCA-3′(PCR fragment 107 bp); β-klotho, 5′-CTG GCT AAG GTT CAA GTA CGG AGA CCT CCC-3′ and 5′-GGA GCT GAG CGA TCA CTA AGT GAA TAC GCA-3′ (PCR fragment 198 bp); GHR, 5′-AGC CTC GAT TCA CCA AGT GTC GTT-3′ and 5′-CAG CTT GTC GTT GGC TTT CCC TTT-3′ (PCR fragment 138 bp); ERK 1, 5′-TCA ACC CAA ACA AGC GCA TCA CAG-3′ and 5′-TCC AGC TCC ATG TCG AAG GTG AAT-3′ (PCR fragment 121 bp); IGF-1, 5′-TGA GCT GGT GGA TGC TCT TCA GTT-3′ and 5′-TCA TCC ACA ATG CCT GTC TGA GGT-3′ (PCR fragment 114 bp). The real time PCR conditions were: 50 °C for 2 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Values were quantified using the comparative cycle threshold (CT) method (14), and samples were normalized to β-actin.
siRNA Transfection
Chondrocytes were transfected with pools of siRNAs targeted for IGF-1 (sc-37194, Santa Cruz), IGF-1R (sc-35638, Santa Cruz), ERK 1 (sc-29308, Santa Cruz), or for FGFR1 (sc-29317, Santa Cruz). A pool of siRNAs consisting of scrambled sequences was similarly transfected as control siRNA (Santa Cruz) and introduced into cells using Lipofectamine 2000 (Invitrogen) according to the procedure recommended by the manufacturer. One day before transfection, cells were plated in 500 μl of growth medium without antibiotics such that they were 30–50% confluent at the time of transfection. The transfected cells were cultured in DMEM containing 10% FCS for 72 h after transfection.
To validate siRNA-mediated inhibition of the target(s) expression, we analyzed the mRNA expression by real-time PCR. In the cells co-transfected with IGF-1 siRNAs and IGF-1R siRNAs, IGF-1 mRNA and IGF-1R mRNA were markedly reduced (13 and 20% of those measured in control siRNA-transfected cells, respectively). In cells transfected with ERK 1 siRNA or FGFR1 siRNA, the ERK 1 mRNA and FGFR1 mRNA were also significantly reduced (35 and 31% of those measured in control siRNA-transfected cells, respectively).
GH Binding Activity
Once reached 70–80% confluence, chondrocytes were washed with serum-free medium and treated for 24 h without or with graded concentrations of rhFGF21 (0.01, 0.1, 1, 5, and 10 μg/ml). After 24 h, culture medium containing FGF21 was removed and replaced with serum-free DMEM. Monolayer cultures set up in 100-mm culture (15, 16) dishes were then detached by scraping in 4 ml of PBS containing a mixture of protease inhibitors (10 mg/ml aprotinin, 10 mm benzamidine, and 0.2 mm phenylmethylsulfonyl fluoride). After centrifugation, the packed cells were resuspended in 2.5 ml of ice-cold inhibitor mixture and disrupted by sonication. Total cellular membranes were obtained by centrifugation of the cell sonicates at 150,000 × g for 15 min at 4 °C. To measure GH binding, the membrane pellets were resuspended at protein concentrations of 1 mg/ml in 300 μl of binding assay buffer (25 mm Tris-Cl (pH 7.4), 0.1% BSA, and 10 mm MgCl2) and incubated at 4 °C in triplicate with 105 cpm human 125I-labeled GH in the presence or absence of 10 μg of unlabeled GH. At the indicated time points (1, 6, and 24 h), the membrane suspensions were centrifuged and washed twice with 1 ml of ice-cold PBS/BSA, and radioactivity was counted using a scintillation counter.
To determine the soluble GH binding activity, conditioned media were collected, centrifuged for 5 min at 1500 × g to remove cell debris, and concentrated approximated 15-fold by ultracentrifugation using a Centricon (Millipore). Identical volumes of each supernatant were incubated for 8 h at 4 °C with 2 × 105 cpm of [125I]GH, and radioactivity was counted using a scintillation counter.
RESULTS
Expression of FGF21, FGFR1, FGFR3, and β-Klotho in Mouse Growth Plate Chondrocytes
To determine whether components of the FGF21 signaling pathway are expressed in growth plate chondrocytes, we used real-time PCR and Western blot. In cultured chondrocytes isolated from 3-week-old mouse tibial growth plates and from fetal (20 days post-coitus) mouse metatarsal growth plates, we demonstrated mRNA (FGF21, FGFR1, FGFR3, and β-klotho) and protein (FGFR1, FGFR3, and β-klotho) expression (Figs. 1 and 2).
FIGURE 1.
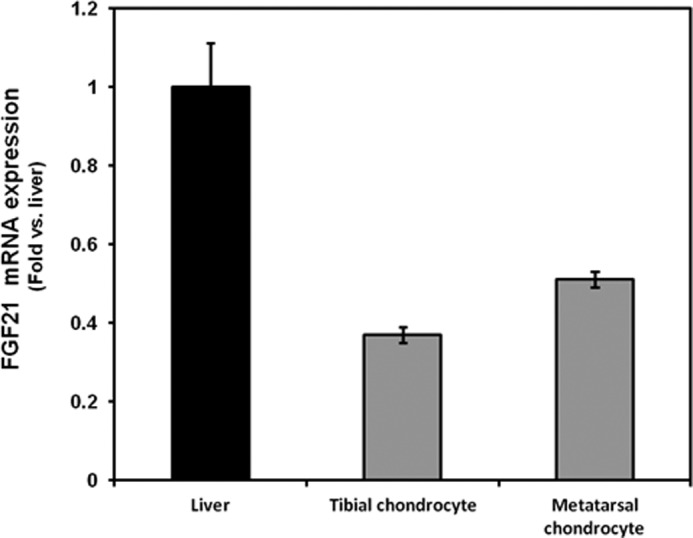
FGF21 mRNA expression in the mouse liver and growth plate chondrocytes. Liver, tibial, and metatarsal chondrocytes were isolated, and total RNA was extracted and processed as described under “Materials and Methods.” FGF21 mRNA expression was determined by real-time PCR.
FIGURE 2.
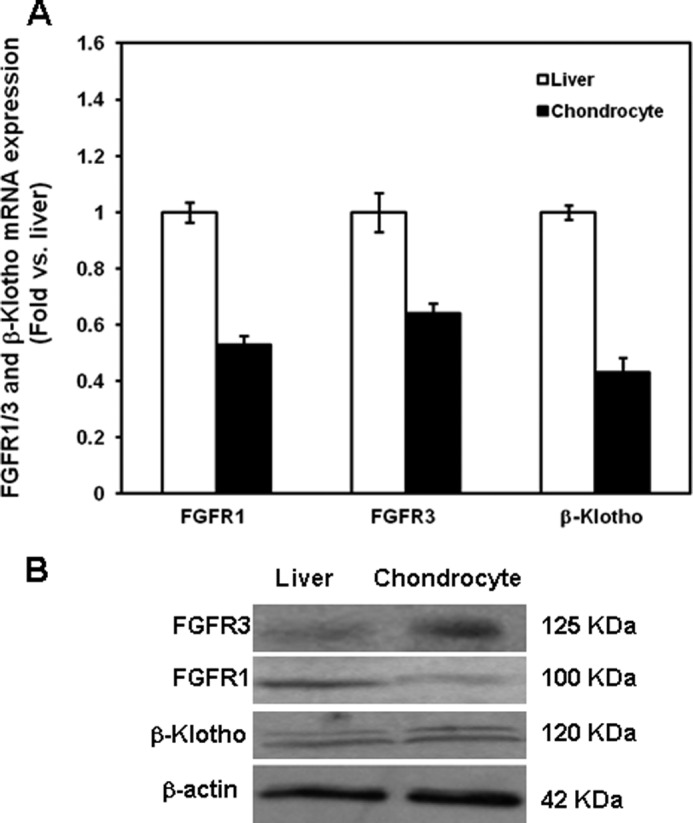
mRNA and protein expression of FGFR1, FGFR3, and β-klotho in the mouse liver and fetal metatarsal chondrocytes. A, FGFR1, FGFR3, and β-klotho mRNA expression in chondrocytes and liver was assessed by real time PCR. Total RNA was extracted from chondrocytes isolated from fetal (20 days post-coitus) mouse metatarsal growth plates and from the liver of 3-week-old mice. It was then processed as described under “Materials and Methods.” The mRNA expression of FGFR1, FGFR3, and β-klotho mRNA was normalized by β-actin in the same samples. B, protein extracted from liver and chondrocytes was immunoblotted for FGFR1, FGFR3, and β-klotho. β-Actin was used as a loading control. A representative blot from three independent experiments is presented.
Effects of FGF21 on Chondrocyte Proliferation and Differentiation
To evaluate the direct effects of FGF21 on chondrocyte proliferation and differentiation, we added graded concentrations of rhFGF21 (0.01, 0.1, 1, 5, and 10 μg/ml) to the culture medium. After 24 h, higher concentrations of rhFGF21 (5–10 μg/ml) significantly reduced total thymidine incorporation (Fig. 3A, p < 0.01 versus control) and decreased the mRNA expression of collagen X (assessed by real-time PCR, Fig. 3B, p < 0.01 versus control), a marker of chondrocyte differentiation. The inhibitory effects of the higher concentrations of FGF21 on both total thymidine incorporation (supplemental Fig. 1A) and collagen X mRNA expression (supplemental Fig. 1B) persisted at subsequent time points (days 3, 7, and 14). These findings suggest that the FGF21-dependent decreased chondrocyte differentiation is, at least in part, independent from the inhibitory effects of FGF21 on chondrocyte proliferation.
FIGURE 3.

Effects of FGF21 on total [3H]thymidine incorporation and collagen X expression in chondrocytes. Chondrocytes were washed with fresh serum-free DMEM, seeded in 24-well plate, and cultured in the absence or presence of graded rhFGF21 (0.01–10 μg/ml) for 24 h. A, at the end of the culture period, 2.5 μCi of [3H]thymidine/well (Amersham Biosciences) was added to the culture medium for an additional 3 h. Chondrocytes were released by trypsin and collected onto glass fiber filters. Incorporation of [3H]thymidine was measured by liquid scintillation counting. Results are expressed as % of control, and represent mean values obtained from three independent experiments. B, collagen X mRNA expression was determined by real time PCR. Total RNA was extracted from chondrocytes and then processed as described under “Materials and Methods.” The relative expression levels of mRNA were normalized by β-actin in the same samples. Results were expressed as -fold change compared with control chondrocytes (mean ± S.E.).
Effects of FGF21 on the FGF Signaling Pathway
To determine whether FGF21 directly modulates the expression and activation of the FGF-dependent signaling pathway in chondrocytes, we first studied the effects of graded concentrations of FGF21 on the expression of FGFR1, FGFR3, and β-klotho. None of the concentrations of rhFGF21 used induced the expression of FGFR3, whereas the highest concentration (10 μg/ml) significantly increased the mRNA expression of FGFR1 and β-klotho (Fig. 4A, p < 0.05 versus control). We then evaluated the effects of rhFGF21 on the phosphorylation of ERK 1/2, members of the FGFR-dependent intracellular signaling cascade. After 24 h, rhFGF21 (0.1, 1, 5, and 10 μg/ml) increased ERK 1/2 phosphorylation (Fig. 4, B and C, p < 0.01 or p < 0.05 versus control).
FIGURE 4.

Effects of FGF21 on FGFR1, FGFR3, and β-klotho mRNA expression and ERK 1/2 phosphorylation in chondrocytes. Chondrocytes were washed with fresh serum-free DMEM, seeded in 24-well plate, and cultured in the absence or presence of graded rhFGF21 (0.01–10 μg/ml) for 24 h. A, at the end of culture period, total RNA was extracted from chondrocytes and then processed as described under “Materials and Methods.” The relative expression levels of mRNA were normalized by β-actin in the same samples. Results were expressed as -fold change compared with control chondrocytes (mean ± S.E.). B, chondrocytes were harvested, lysed, electrophoresed, and immunoblotted for phosphorylated Erk1/2 and total Erk1/2. A representative blot from three independent experiments is presented. C, the intensity of the bands on Western blots of phosphorylated ERK 1/2 and total ERK 1/2 was analyzed by Image J (software from NCBI). Results are expressed as ratio of phosphorylated ERK 1/2 to total ERK 1/2 (mean ± S.E.).
Functional Interaction between FGF21 and GH in Chondrocytes
To determine whether FGF21 antagonizes the effects of GH on chondrocyte function, we cultured mouse growth plate chondrocytes without or with graded concentrations of rhFGF21 (0.01, 0.1, 1, 5, and 10 μg/ml) for 24 h. After removal of FGF21 from the culture medium, chondrocytes were incubated with 10 ng/ml rmGH. In chondrocytes previously cultured without rhFGF21, rmGH significantly induced total thymidine incorporation (Fig. 5A) and collagen X mRNA expression (Fig. 5B). Both effects were prevented in chondrocytes previously treated with rhFGF21, with the lowest effective concentration being 0.1 μg/ml. We then evaluated the effects of rhFGF21 on GH binding; in chondrocytes previously cultured in the presence of rhFGF21, 125I-labeled GH binding (Fig. 6) was significantly decreased when compared with that in chondrocytes not previously cultured in the presence of FGF21. Such effect was detected as early as 1 h after 125I-labeled GH incubation. To determine whether the FGF21-dependent decreased GH binding was due to increased proteolysis at the cell surface, we measured soluble growth hormone binding protein by analyzing 125I-labeled GH binding in transfected cell culture medium. The GH binding in the medium of FGF21-treated cells was not significantly different from that in the medium of untreated cells (supplemental Fig. 2). We then evaluated the GHR expression in mouse growth plate chondrocytes cultured in the presence of graded concentrations of FGF21. None of the FGF21 concentrations significantly modified GHR mRNA expression (assessed by real-time PCR, Fig. 7A) or GHR protein expression (assessed by Western, Fig. 7, B and C) when compared with untreated chondrocytes. These findings indicate that FGF21 specifically prevents the GH stimulatory effects on chondrocyte function by reducing GH binding. Such reduced binding is not due to either changes in GHR expression or increased proteolysis of the cell surface-bound GHR.
FIGURE 5.

Effects of GH and FGF21 on total [3H]thymidine incorporation and collagen X expression in chondrocytes. Chondrocytes were washed with fresh serum-free DMEM, seeded in 24-well plate, and cultured for 24 h in the absence or presence of rmGH (10 ng/ml) without or with graded concentrations of rhFGF21 (0.01–10 μg/ml). A, at the end of culture period, 2.5 μCi/well of [3H]thymidine (Amersham Biosciences) was added to the culture medium for an additional 3 h. Chondrocytes were released by trypsin and collected onto glass fiber filters. Incorporation of [3H]thymidine was measured by liquid scintillation counting. Results are expressed as % of control and represent mean values obtained from three independent experiments. B, collagen X mRNA expression was determined by real time PCR. Total RNA was extracted from chondrocytes and then processed as described under “Materials and Methods.” The relative expression levels of mRNA were normalized by β-actin in the same samples. Results were expressed as -fold change compared with control chondrocytes (mean ± S.E.).
FIGURE 6.
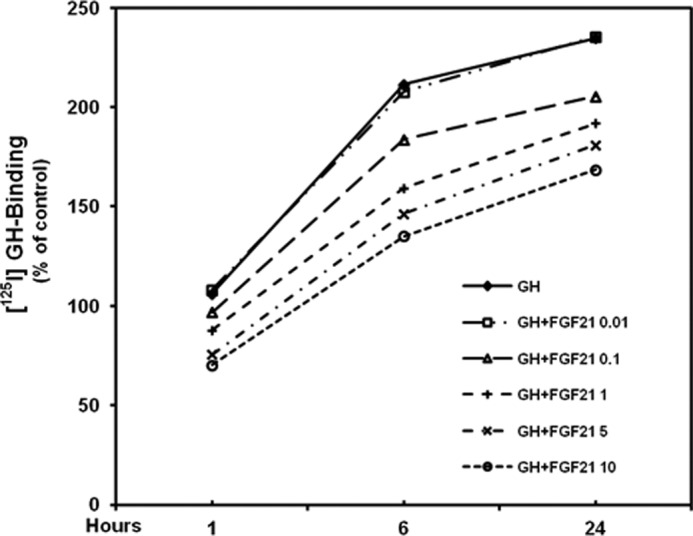
Time-dependent and concentration-dependent effects of FGF21 on GH receptor binding activity in chondrocytes. [125I]GH binding activity was measured at the indicated time points in chondrocytes previously cultured without or with graded concentrations of rhFGF21. Results are expressed as % of control and represent mean values obtained from three independent experiments.
FIGURE 7.

Effects of FGF21 on GHR expression in chondrocytes. Chondrocytes were washed with fresh serum-free DMEM, seeded in 24-well plate, and cultured for 24 h in the absence or presence of graded concentrations of rhFGF21 (0.01–10 μg/ml). A, total RNA was extracted from chondrocytes and then processed as described under “Materials and Methods.” GHR mRNA expression was evaluated by real time PCR. The relative expression levels of mRNA were normalized by β-actin in the same samples. Results were expressed as -fold change compared with control (untreated) chondrocytes (mean ± S.E.). B, at the end of culture period chondrocytes were harvested, lysed, electrophoresed, and immunoblotted for GHR protein. A representative blot from three independent experiments is presented. C, the intensity of the bands on Western blots was analyzed by Image J (software from NCBI). Results are expressed as ratio of GHR to β-actin (mean ± S.E.).
To determine whether the effects of GH and/or FGF21 depend on the intracellular activity of IGF-1 in chondrocytes, we transfected cells with both IGF-1 siRNAs and IGF-1R siRNAs. Total thymidine incorporation and collagen X mRNA expression in GH-treated, siRNA-transfected chondrocytes remained significantly greater than in untreated transfected chondrocytes (supplemental Figs. 3 and 4, respectively). In addition, the antagonistic effects of rhFGF21 on GH activity were confirmed in IGF-1 siRNA + IGF-1R siRNA-transfected cells.
Last, we evaluated the effects of FGF21 and GH in chondrocytes transfected with FGFR1 siRNA or ERK 1 siRNA. As previously shown, FGF21 dose-dependently (0.1, 1, 5, and 10 μg/ml) prevented the GH stimulatory effects on total thymidine incorporation (Fig. 8, panel A) and collagen X expression (Fig. 8, panel B) in control siRNA-transfected chondrocytes. In contrast, in FGFR1 siRNA-transfected or ERK 1 siRNA-transfected cells only the highest concentration of FGF21 (10 μg/ml) prevented the GH effects in chondrocytes. These findings suggest that the FGF21 effects on chondrocyte function are specific and depend on the normal activity of the FGFR1 and ERK 1.
FIGURE 8.

Effects of GH and FGF21 on total [3H]thymidine incorporation and collagen X expression in FGFR1-siRNA- and ERK 1 siRNA-transfected chondrocytes. After being previously cultured without or with graded concentrations of FGF21, siRNA-transfected chondrocytes were cultured for 24 h in the absence or presence of rmGH (10 ng/ml). A, at the end of culture period, 2.5 μCi/well of [3H]thymidine (Amersham Biosciences) was added to the culture medium for an additional 3 h. Incorporation of [3H]thymidine was measured by liquid scintillation counting. Results are expressed as % of untreated and represent mean values obtained from three independent experiments. B, collagen X mRNA expression was determined by real time PCR. Total RNA was extracted from transfected chondrocytes and then processed as described under “Materials and Methods.” The relative expression levels of mRNA were normalized by β-actin in the same samples. Results were expressed as -fold change compared with control chondrocytes (mean ± S.E.).
DISCUSSION
Longitudinal bone growth occurs at the growth plate by a process called endochondral ossification (17) in which cartilage is first formed and then remodeled into bone tissue. Cartilage formation primarily results from growth plate chondrocyte proliferation and hypertrophy (18) and extracellular matrix secretion. The newly formed cartilage is invaded by blood vessels and bone cell precursors from the metaphysis, which remodel the cartilaginous hypertrophic zone into bone (19). The net effect of these events is that new bone tissue is progressively laid down at the bottom of the growth plate, resulting in bone elongation. Longitudinal bone growth is tightly governed by a complex network of endocrine and paracrine growth factors, the latter of which includes the FGFs.
The FGFs is a family of proteins known to be implicated in the regulation of growth plate chondrogenesis (20, 21). The murine growth plate expresses several members of this family, including FGF21; it has been shown to be expressed in the perichondrium surrounding the rat postnatal growth plate (22). In addition, all four FGFRs are also expressed in the growth plate (22); evidence in humans and in experimental animals indicates that FGFR1 and FGFR3 act as negative regulators of growth plate chondrogenesis (23, 24) and share a remarkably similar downstream signaling pathway (25). In contrast, the activation of FGFR2 and FGFR4 appear to facilitate growth plate chondrogenesis (26, 27). With respect to the functional role of the FGFs expressed in the growth plate, mice overexpressing FGF2 or FGF9 exhibit suppressed growth plate chondrocyte hypertrophy, shortened long bones, and dwarfism (28, 29). Mice with a targeted disruption of FGF18 display expanded growth plate proliferative and hypertrophic zones, thus suggesting that FGF18 (similarly to FGF2 and FGF9) inhibits growth plate chondrogenesis, likely by binding and activating FGFR3 (30, 31).
FGF21 is a member of a subfamily of atypical FGFs that includes FGF15/19 (FGF15 and FGF19 are mouse and human orthologs, respectively) and FGF23 (1, 2). Because these FGFs lack the typical FGF heparin binding domain, they can be released from the tissue of synthesis and act on distant sites as systemic or endocrine factors. On the other hand, recent evidence suggests that FGF21 may also function in a paracrine/autocrine manner (32, 33). To activate its intracellular signaling pathway, FGF21 binds one of the classic, cell-bound, FGF receptors complexed to β-klotho (9–11), a recently identified membrane-spanning protein. Although some studies indicate that FGF21 may preferentially activate the FGFR1/β-klotho complex, other evidence indicates that it can bind all the four FGFRs (2, 8). FGF21 is predominantly expressed in the exocrine and endocrine pancreas, liver, muscle, and white adipose tissue. The co-localization of the FGFRs and β-klotho in these tissues suggest that that FGF21 may exert its function in the same sites (12, 13).
Although FGF21 is most known as an important metabolic regulator during fasting, recent evidence suggests that it may be implicated in the regulation of skeletal growth. In 2008, Inagaki et al. (7) showed that transgenic mice overexpressing FGF21 are smaller and exhibit shorter tibiae when compared with WT mice. In addition, the decreased expression of phosphorylated Stat5 and IGF-1 in the liver of FGF21 transgenic mice suggests that overexpression of FGF21 may cause GH insensitivity. These findings and the fact that FGF21 expression is increased during fasting, led us to hypothesize that increased expression of FGF21 during chronic undernutrition may cause undernutrition-associated growth failure in mammals.
Indeed, we have recently demonstrated that mice undergoing food restriction experience reduced body and tibial growth along with increased FGF21 expression in the liver and in the tibial growth plate (6). Food-restricted Fgf21 knock-out mice exhibit greater body and tibial growth and spared growth plate chondrogenesis compared with food-restricted wild-type littermates. In addition, food-restricted Fgf21 knock-out mice are more GH-sensitive than the wild-type mice, with such difference resulting from greater GH receptor abundance in hepatocytes. These findings suggest that increased expression of FGF21 during prolonged food restriction causes growth attenuation by inducing systemic GH insensitivity. Interestingly, food-restricted Fgf21 knock-out mice also show greater GH receptor abundance and reduced Stat5 phosphorylation and IGF-1 expression in the tibial growth plate chondrocytes. Although it is plausible to hypothesize that FGF21 may also cause GH insensitivity directly at the growth plate, these previous findings do not confirm a direct effect of FGF21 on growth plate chondrocytes.
In this study, we have demonstrated that FGF21 directly antagonizes GH stimulatory effects on proliferation and differentiation of growth plate chondrocytes. Of note, the GH effects on chondrocyte function (as well as the antagonistic effects of FGF21) persist in IGF-1 siRNA-transfected chondrocytes. Thus, it is plausible to speculate that these effects are, at least in part, non-IGF-1-dependent. The direct effects of FGF21 on the growth plate are supported by the expression of FGFR1, FGFR3, and β-klotho in growth plate chondrocytes as well as by the FGF21-mediated induction of ERK 1/2 phosphorylation (ERK 1/2 are members of the FGFR-dependent intracellular signaling cascade). The fact that the antagonistic properties of FGF21 on GH binding and GH-stimulated chondrocyte proliferation and differentiation are prevented in FGFR1 siRNA- or ERK 1 siRNA-transfected chondrocytes further support the conclusion that the effects of FGF21 are specific. In our study high concentrations of rhFGF21 have been found to directly inhibit chondrocyte proliferation and differentiation; such effects may be explained by the increased expression of FGFR1 and β-klotho induced by the highest concentration of FGF21, which could potentiate the intracellular transcriptional changes elicited by FGF21. However, we do not know whether these high concentrations used in vitro reflect the levels of expression in tissues or the serum levels of FGF21 during prolonged food restriction.
In our study the lack of any direct effect of FGF21 on GHR expression or on the proteolysis of the cell membrane-bound GHR suggests that the FGF21-dependent decreased GH binding likely results from a reduced GHR intracellular recycling. Interestingly, a recent study has demonstrated the increased hepatic expression of LEPROT and LEPROTL1 (two small proteins known to regulate intracellular protein trafficking) during fasting in rodents. The increased activity of LEPROT and LEPROTL1 causes GH insensitivity by inhibiting GHR intracellular trafficking and, in turn, GHR cell-surface abundance without affecting GHR expression (16). Ongoing studies in our laboratory are evaluating the functional interaction between FGF21 and LEPROT/LEPROTL1 in the liver and in chondrocytes.
Although our study demonstrates its direct effects on chondrocytes, it is still unclear whether FGF21 antagonizes GH effects on growth plate chondrogenesis as a systemic (endocrine) and/or as a paracrine growth factor. Indeed, its food restriction-related increased expression in the liver (and plausibly in other tissues) as well as in the mouse tibial growth plate would suggest both (or either) modalities of action.
In conclusion, our study provides evidence for a specific, direct effect of FGF21 on growth plate chondrocytes; by binding FGFR1 and/or FGFR3 (negative regulators of growth plate chondrogenesis), FGF21 prevents GH-mediated stimulation of chondrocyte proliferation and differentiation. In addition, high concentrations of FGF21 directly inhibit cell proliferation and differentiation. Thus, our findings contribute to elucidate the cellular and molecular mechanisms underlying the growth attenuation associated with the increased expression of FGF21 during food restriction in rodents.
Supplementary Material

This article contains supplemental Figs. 1–4.
- FGFR
- FGF receptor
- GH
- growth hormone
- rmGH
- recombinant mouse GH
- GHR
- GH receptor
- rhFGF21
- recombinant human FGF21.
REFERENCES
- 1. Nishimura T., Nakatake Y., Konishi M., Itoh N. (2000) Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 1492, 203–206 [DOI] [PubMed] [Google Scholar]
- 2. Kharitonenkov A., Shiyanova T. L., Koester A., Ford A. M., Micanovic R., Galbreath E. J., Sandusky G. E., Hammond L. J., Moyers J. S., Owens R. A., Gromada J., Brozinick J. T., Hawkins E. D., Wroblewski V. J., Li D. S., Mehrbod F., Jaskunas S. R., Shanafelt A. B. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 4. Gälman C., Lundåsen T., Kharitonenkov A., Bina H. A., Eriksson M., Hafström I., Dahlin M., Amark P., Angelin B., Rudling M. (2008) The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARα activation in man. Cell Metab. 8, 169–174 [DOI] [PubMed] [Google Scholar]
- 5. Reitman M. L. (2007) FGF21. A missing link in the biology of fasting. Cell Metab. 5, 405–407 [DOI] [PubMed] [Google Scholar]
- 6. Kubicky R. A., Wu S., Kharitonenkov A., De Luca F. (2012) Role of fibroblast growth factor 21 (FGF21) in undernutrition-related attenuation of growth in mice. Endocrinology 153, 2287–2295 [DOI] [PubMed] [Google Scholar]
- 7. Inagaki T., Lin V. Y., Goetz R., Mohammadi M., Mangelsdorf D. J., Kliewer S. A. (2008) Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 8, 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kliewer S. A., Mangelsdorf D. J. (2010) Fibroblast growth factor 21. From pharmacology to physiology. Am. J. Clin. Nutr. 91, 254S–257S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kharitonenkov A., Dunbar J. D., Bina H. A., Bright S., Moyers J. S., Zhang C., Ding L., Micanovic R., Mehrbod S. F., Knierman M. D., Hale J. E., Coskun T., Shanafelt A. B. (2008) FGF-21/FGF-21 receptor interaction and activation is determined by β-klotho. J. Cell Physiol. 215, 1–7 [DOI] [PubMed] [Google Scholar]
- 10. Ogawa Y., Kurosu H., Yamamoto M., Nandi A., Rosenblatt K. P., Goetz R., Eliseenkova A. V., Mohammadi M., Kuro-o M. (2007) β-Klotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. U.S.A. 104, 7432–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki M., Uehara Y., Motomura-Matsuzaka K., Oki J., Koyama Y., Kimura M., Asada M., Komi-Kuramochi A., Oka S., Imamura T. (2008) β-Klotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 22, 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurosu H., Choi M., Ogawa Y., Dickson A. S., Goetz R., Eliseenkova A. V., Mohammadi M., Rosenblatt K. P., Kliewer S. A., Kuro-o M. (2007) Tissue-specific expression of β-klotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282, 26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fon Tacer K., Bookout A. L., Ding X., Kurosu H., John G. B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D. J., Kliewer S. A. (2010) Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 24, 2050–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 15. Leung K. C., Doyle N., Ballesteros M., Waters M. J., Ho K. K. (2000) Insulin regulation of human hepatic growth hormone receptors. Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 85, 4712–4720 [DOI] [PubMed] [Google Scholar]
- 16. Touvier T., Conte-Auriol F., Briand O., Cudejko C., Paumelle R., Caron S., Baugé E., Rouillé Y., Salles J. P., Staels B., Bailleul B. (2009) LEPROT and LEPROTL1 cooperatively decrease hepatic growth hormone action in mice. J. Clin. Invest. 119, 3830–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hunziker E. B. (1994) Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc. Res. Tech. 28, 505–519 [DOI] [PubMed] [Google Scholar]
- 18. Farnum C. E., Wilshan N. J. (1987) Morphologic stages of terminal hypertrophic chondrocyte of growth plate cartilage. Anat. Rec. 219, 221–232 [DOI] [PubMed] [Google Scholar]
- 19. Aharinejad S., Marks S. C., Jr., Böck P., MacKay C. A., Larson E. K., Tahamtani A., Mason-Savas A, Firbas W. (1995) Microvascular pattern in the metaphysis during bone growth. Anat. Rec. 242, 111–122 [DOI] [PubMed] [Google Scholar]
- 20. De Luca F., Baron J. (1999) Control of bone growth by fibroblast growth factors. Trends. Endocrinol. Metab. 10, 61–65 [DOI] [PubMed] [Google Scholar]
- 21. Horton W. A., Degnin C. R. (2009) FGFs in endochondral skeletal development. Trends. Endocrinol. Metab. 20, 341–348 [DOI] [PubMed] [Google Scholar]
- 22. Lazarus J. E., Hegde A., Andrade A. C., Nilsson O., Baron J. (2007) Fibroblast growth factor expression in the postnatal growth plate. Bone 40, 577–586 [DOI] [PubMed] [Google Scholar]
- 23. Deng C., Wynshaw-Boris A., Zhou F., Kuo A., Leder P. (1996) Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911–921 [DOI] [PubMed] [Google Scholar]
- 24. White K. E., Cabral J. M., Davis S. I., Fishburn T., Evans W. E., Ichikawa S., Fields J., Yu X., Shaw N. J., McLellan N. J., McKeown C., Fitzpatrick D., Yu K., Ornitz D. M., Econs M. J. (2005) Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am. J. Hum. Genet. 76, 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q., Green R. P., Zhao G., Ornitz D. M. (2001) Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3-tyrosine kinase domains. Development 128, 3867–3876 [DOI] [PubMed] [Google Scholar]
- 26. Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A., Ornitz D. M. (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074 [DOI] [PubMed] [Google Scholar]
- 27. Weinstein M., Xu X., Ohyama K., Deng C. X. (1998) FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125, 3615–3623 [DOI] [PubMed] [Google Scholar]
- 28. Coffin J. D., Florkiewicz R. Z., Neumann J., Mort-Hopkins T., Dorn G. W., 2nd, Lightfoot P., German R., Howles P. N., Kier A., O'Toole B. A. (1995) Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol. Biol. Cell 6, 1861–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garofalo S., Kliger-Spatz M., Cooke J. L., Wolstin O., Lunstrum G. P., Moshkovitz S. M., Horton W. A., Yayon A. (1999) Skeletal dysplasia and defective chondrocyte differentiation by targeted overexpression of fibroblast growth factor 9 in transgenic mice. J. Bone. Miner. Res. 14, 1909–1915 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z., Xu J., Colvin J. S., Ornitz D. M. (2002) Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16, 859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohbayashi N., Shibayama M., Kurotaki Y., Imanishi M., Fujimori T., Itoh N., Takada S. (2002) FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 16, 870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson C. L., Weston J. Y., Chadi S. A., Fazio E. N., Huff M. W., Kharitonenkov A., Köester A., Pin C. L. (2009) Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology 137, 1795–1804 [DOI] [PubMed] [Google Scholar]
- 33. Fisher F. M., Chui P. C., Antonellis P. J., Bina H. A., Kharitonenkov A., Flier J. S., Maratos-Flier E. (2010) Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59, 2781–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.