

Background: Tyrosyl radicals react with superoxide to form bicyclic hydroperoxides that contains an α-β unsaturated carbonyl.
Results: GSH undergoes Michael addition with Tyr hydroperoxides in peptides and myoglobin.
Conclusion: Proteins that form hydroperoxides through radical-mediated oxidation should readily form GSH-Tyr adducts.
Significance: This is a novel mechanism of protein-glutathione adduct formation, and similar chemistry could result in Cys-Tyr cross-linking of proteins during oxidative stress.
Keywords: Free Radicals, Oxidative Stress, Superoxide Ion, Thiol, Tyrosine, Glutathione, Hydroperoxide, Michael Addition, Protein Oxidation
Abstract
Tyrosine residues are sensitive to oxidation and can be converted to hydroperoxides either by superoxide reacting with the Tyr radical or by singlet oxygen. These hydroperoxides rearrange to bicyclic derivatives that are readily reduced to more stable hydroxides. The aromatic character of tyrosine is lost, but the product contains an α-β unsaturated carbonyl group and is, therefore, an electrophile. We have generated hydroxide derivatives of several Tyr-containing peptides and shown using liquid chromatography/mass spectrometry that they undergo Michael addition with GSH. For Tyr-Gly, rate constants of 9.2 and 11.8 m−1min−1 were measured for the two chromatographically distinct isomers. Unusual for GSH addition to an electrophile, the reaction is reversible, with a half-life of many hours for the reverse reaction. These kinetics indicate that with a typical cellular concentration of 5 mm GSH, >95% Tyr-Gly hydroxide would become conjugated with a half-life of ∼15 min. Sperm whale myoglobin forms a hydroperoxide on Tyr-151 in a hydrogen peroxide/superoxide-dependent reaction. We show that its hydroxide derivative reacts with GSH to form a conjugate. Detection of the conjugate required stabilization by reduction; otherwise, the reverse reaction occurred during tryptic digestion and analysis. Our findings represent a novel mechanism for peptide or protein glutathionylation involving a carbon-sulfur cross-link between oxidized Tyr and Cys. As with other electrophiles, the oxidized Tyr should undergo a similar reaction with Cys residues in proteins to give intramolecular or intermolecular protein cross-links. This mechanism could give rise to protein cross-linking in conditions of oxidative stress.
Introduction
Proteins are critical biological targets for reactive oxygen species, and post-translational protein modification is a major mechanism for oxidative damage. It is also a regulatory mechanism for redox signaling and in many cases is structurally important for achieving correct polypeptide folding. Cys, Met, Trp, and Tyr residues are the most oxidant-sensitive, and a wide range of reversible and irreversible modifications have been characterized. One common modification is glutathionylation (1–5). Numerous thiol proteins have been shown to form mixed disulfides with GSH. This reversible process, termed S-glutathionylation, is a means for regulating their activity and is proposed to be one of the mechanisms involved in redox signaling. Glutathionylation through a carbon-sulfur bond can also occur if protein oxidation gives rise to electrophilic sites that could form adducts by Michael addition. The objective of this study was to determine whether tyrosine oxidation to its hydroperoxide could give rise to glutathionylation via this mechanism.
Tyr residues are particularly sensitive to one-electron (radical-mediated) oxidation to radicals. These radicals can combine to form dityrosine, which is an important cross-link in some structural proteins as well as a useful biomarker of oxidative stress (6). However, tyrosyl radicals react with superoxide even faster than with each other. This is the prevalent reaction under conditions where both radicals are generated (7–10), such as when tyrosine is oxidized by stimulated neutrophils (11, 12) or in the presence of GSH (13). The reaction between superoxide and Tyr radicals proceeds either by electron transfer (repair), which reduces the Tyr radical back to Tyr and generates oxygen, or by addition to give a hydroperoxide. When the tyrosine has a free terminal amine, the hydroperoxide is the predominant product (7, 9). Addition can theoretically occur at the ortho (as shown in Scheme 1, reaction 1) or para position of the Tyr. The repair reaction is more favored when there is no free amino group, but some hydroperoxide is formed in this case too (10). An alternative route to tyrosine hydroperoxides is the reaction of tyrosyl peptides or proteins with singlet oxygen (14).
SCHEME 1.

Proposed mechanism for the formation of the Tyr-hydroxide-GSH adduct. 1, the addition of superoxide to Tyr radicals results in the formation of hydroperoxide species (I). The scheme shows the addition at the ortho position but it could also occur at the para position. 2, Michael addition of the Tyr amine (or amide) nitrogen to the Tyr ring gives a bicyclic hydroperoxide derivative (II). 3, reduction of the hydroperoxide species by a nucleophile (Nu) gives the corresponding alcohol (III; the Tyr-hydroxide species was characterized for the parent Tyr amino acid as 3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid or 4-alanyl-4-hydroxy-cyclohexadienone for superoxide addition to the ortho positions; see Nagy et al. (10). 4, Michael addition of GSH to the α-β unsaturated carbonyl group of III gives IV.
Hydroperoxide formation on N-terminal Tyr residues is followed by conjugate addition of the Tyr nitrogen to the phenol ring (reaction 2), destroying its aromatic character and generating a bicyclic product (II). The product is less well characterized when there is no free N terminus. Our evidence favors the equivalent of (II) with conjugation to the amide nitrogen (10, 14, 15), but the unconjugated species (I) is also possible. The hydroperoxides readily undergo reduction or hydrolysis (reaction 3) to give the more stable hydroxyl derivative (III) (7). Alternatively, if a Met residue is in the vicinity, Tyr hydroxide is produced through internal transfer of one of the oxygens to give methionine sulfoxide (10).
The conjugate addition product (III) still contains an α-β unsaturated carbonyl group so could in theory undergo further Michael addition to thiols and other nucleophiles. This would result in a conjugate with GSH (Scheme 1, reaction 4), and the reaction is a potential mechanism for cross-linking between Tyr and Cys residues in proteins. We have investigated whether this is the case for glutathionylation using small Tyr-containing peptides as well as sperm whale myoglobin, which we have previously shown to form a tyrosyl hydroperoxide derivative when treated with superoxide and hydrogen peroxide (16). We show that GSH reduces the peptide hydroperoxide to hydroxide derivatives (Tyr hydroxide), which readily and efficiently form conjugates. We propose that this could be a physiological mechanism for protein glutathionylation or cross-linking.
EXPERIMENTAL PROCEDURES
Materials
Tyr-Gly Met-enkephalin, Leu-enkephalin, sperm whale myoglobin, and other reagents and enzymes were purchased from Sigma unless otherwise indicated. Stock solutions of xanthine oxidase (XO)3 were prepared by dilution of the ammonium sulfate suspension with 50 mm phosphate buffer, pH 7.4, and spinning through a G25 Sephadex column. Enzyme and acetaldehyde solutions were prepared daily and stored on ice. The custom-synthesized peptide corresponding to the C-terminal end of myoglobin with the sequence ELGYQG was ordered from GenScript (Piscataway, NJ).
Peptide Tyr Hydroperoxides
Peptide hydroperoxides were prepared as in Nagy et al. (10) by treating the Tyr-containing peptide (typically 1–5 mm) in 50 mm sodium phosphate buffer, pH 7.4, containing 50 μm diethylenetriaminepenta-acetic acid and 3 or 10 mm acetaldehyde with 1–3 aliquots (over 30–90 min at 22 °C) of horseradish peroxidase (HRP; final concentration 0.5 μm) and XO (∼0.004 units/ml to give a superoxide generation rate of ∼10 μm/min). The concentration of the HRP stock was determined from its absorbance at 403 nm (ϵ = 112 mm−1cm−1). Superoxide production by XO was measured separately as cytochrome c reduction. The reaction was stopped by adding catalase (10–20 μg/ml), decreasing the volume to half using a vacuum concentrator, then removing the enzymes using a 10- or 50-kDa cut off Amicon Ultra Free-MC Biomax Polysulfone filter (Millipore, Bedford, MA). For ELGYQG, a similar protocol was used except that XO (typically 0.0006 units/ml; superoxide generation 1.5 μm/min) was added to a solution containing 0.2 mm peptide, 2.5 mm acetaldehyde, and 140 nm HRP (16). Reactions were run for 30 min at 22 °C.
Formation of Peptide Tyr-hydroxides and Reaction with GSH
Tyrosyl hydroperoxide derivatives were either reduced to the corresponding hydroxides by incubating with methionine (typically 0.7 mm) overnight at 20 °C or added directly to GSH (which also performs this reduction). For product analysis, the solutions were treated with GSH (2 mm in pH 7.4 phosphate buffer containing diethylenetriaminepenta-acetic acid). After 1–2 h, they were either analyzed immediately by liquid chromatography-mass spectrometry (LC/MS) or stored at −80 °C. For the experiment using purified Tyr-Gly hydroxide (YG-hydroxide), it was purified from the starting material by liquid chromatography using the LC/MS system. On reinjection of the collected peak, only the YG-hydroxide species were detected.
Kinetic Experiments
YG-hydroxide was prepared by reducing the hydroperoxide with methionine as above. LC/MS analysis confirmed that no YG-hydroperoxide remained. Phe (internal standard, final concentration 20 μm) plus an equal volume of a GSH solution in the same phosphate buffer was added, and after controlled times at 25 °C samples were injected into the LC/MS system. Controls without GSH were also run at intervals during the analysis period. For each sample, the peak integral for YG-hydroxide was calculated and expressed relative to the Phe peak (m/z = 166). GSH (m/z = 308), which was present in excess, was also monitored to ensure that it was not depleted over the course of the reaction. The loss of YG-hydroxide over time was followed at different GSH concentrations. Sigma Plot was used to fit the kinetic traces to an exponential decay (y = y0 + ae−bt) equation.
Glutathione Adduct Formation on Oxidized Myoglobin
Sperm whale myoglobin (50 μm in phosphate buffer containing diethylenetriaminepenta-acetic acid) was reacted with XO (typically 0.004 units/ml) plus acetaldehyde (1 mm) for 30 min at 22 °C. Total production of H2O2 over the period was ∼100 μm. It has previously been shown that a superoxide-tyrosyl radical addition product is formed on the myoglobin under these conditions (16). The reaction mixture was then incubated overnight with or without GSH, then treated with 20 mm sodium borohydride (NaBH4) for 30 min in the dark. Reactions were stopped, and heme was extracted from the myoglobin by adding 9 volumes of ice-cold acidified acetone (1% HCl v/v) while vortexing. Precipitated protein was separated, resuspended in 100 mm Tris-HCl buffer, pH 8.5, and incubated with a 50:1 substrate:trypsin ratio for 16 h at 37 °C. Digestion was stopped by the addition of 0.1% formic acid followed by injection of samples onto the LC/MS column.
LC/MS Analysis of Peptides and Product Identification
Most of the analyses were performed by liquid chromatography-electrospray ionization (ESI)-mass spectrometry and LC-ESI-MS/MS using a Thermo Finnigan LCQ Deca XP Plus ion trap mass spectrometer (San Jose, CA) in positive ion mode coupled to a Surveyor HPLC system and PDA detector under previously described conditions (10). Data were analyzed using Finnigan Xcalibur, Thermo Finnigan Qual Browser1.3, and HighChem Mass Frontier 3.0 programs. Identity of the peaks was established on the basis of characteristic MS/MS fragmentation patterns. Fragment ions of ELGYQG were assigned based on the Roepstorff-Fohlman nomenclature.
Some of the analyses of ELGYQG products and analysis of tryptic digests of myoglobin were performed with an Applied Biosystems 4000QTrap coupled to an Applied Biosystems HPLC system (Concord, Ontario, Canada). A Jupiter 4 μm Proteo 90A C12 150 × 2.0 mm column (Phenomenex, CA) set to 40 °C was used, and separation was achieved using an appropriate gradient of water and acetonitrile containing 0.1% formic acid. All samples were analyzed in the positive ion mode. The electrospray needle was held at 400 °C. Nitrogen was both the curtain and collision gas. The ion spray was 5 kV, and the declustering potential was 40 V. Samples were analyzed by selected ion monitoring, selected reaction monitoring (SRM), or recording the total ion chromatograms. MS/MS experiments were performed using helium as collision gas. Data acquisition and analysis were performed using Analyst 1.4.2 (AB Sciex).
RESULTS
Formation of a Glutathione Adduct on Tyr-Gly Hydroxide
Tyr hydroperoxide formation from Tyr radicals and superoxide requires the simultaneous generation of both radicals. This can be achieved using XO/acetaldehyde to generate superoxide and H2O2 plus a peroxidase to catalyze oxidation of the Tyr by the H2O2 (11). An alternative is to generate Tyr radicals in the presence of GSH (13). In the latter case the glutathionyl radical is formed; this reacts with more GSH to give the glutathione disulfide anion radical (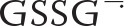 ), and superoxide is generated in the reaction of with oxygen. When we used the GSH system with YG and examined the products by LC/MS, we observed the hydroperoxide, but it was short lived. Isomers of YG-hydroxide were formed, but these also disappeared over time, and a peak corresponding to an increase in mass of 307 Da was observed (data not shown). This is consistent with previous studies showing that Tyr hydroperoxides are reduced to hydroxides by GSH (11, 17) and also suggested that the hydroxides react with GSH. These reactions were further investigated by adding GSH to preformed YG-hydroxide.
), and superoxide is generated in the reaction of with oxygen. When we used the GSH system with YG and examined the products by LC/MS, we observed the hydroperoxide, but it was short lived. Isomers of YG-hydroxide were formed, but these also disappeared over time, and a peak corresponding to an increase in mass of 307 Da was observed (data not shown). This is consistent with previous studies showing that Tyr hydroperoxides are reduced to hydroxides by GSH (11, 17) and also suggested that the hydroxides react with GSH. These reactions were further investigated by adding GSH to preformed YG-hydroxide.
YG-OH was prepared by treating the dipeptide with the XO/HRP system then reduction of the hydroperoxide with methionine. As shown in Fig. 1a, LC/MS with selected ion monitoring of the reaction mixture at m/z of 255 (the theoretical value for the hydroxide; III) showed two isomeric peaks. The two peaks had very similar fragmentation patterns that are consistent with the isomeric structures of III, as established previously (10). After treatment with 5 mm GSH, these peaks disappeared, and a peak with two unresolved shoulders appeared (Fig. 1b). This peak showed the same mass throughout, corresponding to the addition of GSH (+307 Da) to YG-hydroxide. This mass is consistent with a Michael addition reaction, and the fragmentation pattern (Fig. 1c) indicates that the mass increase is due to the addition of GSH to Tyr-hydroxide. The unresolved peaks are likely to correspond to structural isomers, more of which are theoretically possible for the GSH adduct than for the hydroxide alone. The comparable total areas of the peaks in Figs. 1, a and b, suggest efficient conversion. To exclude the possibility that any of the reagents used to prepare YG-hydroxide were involved in the formation of the GSH adduct, YG-hydroxide was purified and reacted with GSH. Analysis by direct infusion mass spectrometry again showed conversion to the same GSH adduct as in Fig. 1c, indicating a direct reaction between GSH and YG-hydroxide.
FIGURE 1.

LC/MS detection of glutathione adducts of YG-hydroxide. Selected ion monitoring detection is shown of YG-hydroxide isomers at m/z = 255 (dotted line) without the addition of GSH (a) and GSH adducts of YG-OH at m/z = 562 (solid line) with no YG-hydroxide remaining (dotted line) after 6 h of incubation with 5 mm GSH (b). c, shown is a fragmentation pattern of YG-hydroxide-GSH. The proposed structure of the adduct is shown in the inset.
Michael Addition of GSH or Cys to Different Peptide-Tyr Hydroxides
Gly-Tyr, Met-enkephalin, and Leu-enkephalin all form radical addition products with superoxide (10, 12). The hydroxide products from these peptides also reacted with GSH to form adducts. LC/MS analysis of the products gave masses corresponding to the addition of GSH, and the major fragments identify them as Michael addition products equivalent to that for YG. The fragmentation patterns indicate that the GSH is localized on the modified Tyr-hydroxide residues (see Table 1 and Fig. 2). As shown for YG-hydroxide, reaction with Cys formed the equivalent Cys conjugate.
TABLE 1.
Identification of Michael addition products formed from superoxide-modified tyrosyl peptides
Peptide hydroxides were formed as described under “Experimental Procedures” and analyzed as in Fig. 1. Fragmentation patterns and characterization of parent peptides are given in Nagy et al. (10). Assignments of the major fragments of the glutathionylated derivatives are shown in Fig. 2.
| Peptide | m/z [peptide-OH +H]+ | Thiol (mass) | Observed product m/z [peptide-OH + thiol+H]+ | Major fragments m/z |
|---|---|---|---|---|
| Da | ||||
| YG | 255 | Cys (122) | 376 | 237, 255, 358 |
| GY | 255 | GSH (307) | 562 | 415, 433, 544 |
| Met-enkephalin YGGFMa | 606 (dioxide) | GSH (307) | 913 | 606, 784 |
| Leu-enkephalin YGGFL | 572 | GSH (307) | 879 | 750, 804 |
a The initial Tyr-hydroperoxide spontaneously undergoes intramolecular oxygen transfer to give a dioxide with the extra oxygens on the Met and modified Tyr residues Nagy et al. (10).
FIGURE 2.

Proposed structures of major fragments of GSH adducts of Tyr peptides in Table 1. The proposed structures of the two or three most prominent fragments observed on MS/MS of the products are shown. MetEnk-OH-GSH, GSH adduct of Met-enkephalin-OH; LeuEnk-OH-GSH, GSH adduct of Leu-enkephlin-OH.
Kinetics and Reversibility of Adduct Formation
Kinetic experiments were carried out with YG-hydroxide using LC/MS to follow its consumption over time. Loss of both hydroxide isomers (shown in Fig. 3a for the peak eluting at 4 min) occurred over minutes or hours depending on the GSH concentration. GSH conjugate peaks increased in parallel with the decrease of the hydroxides, but product formation was not analyzed kinetically. At the lower GSH concentrations, the reactions did not go to completion, suggesting that reaction 4 (Scheme 1) should be represented as an equilibrium,
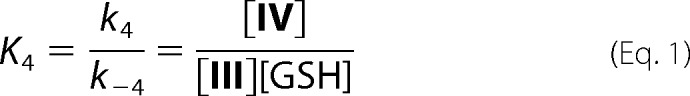 |
As all kinetic experiments were conducted under pseudo first order conditions using an excess of GSH, the GSH concentration can be treated as constant, and Equation 1 was simplified to,
 |
where K4′ = K4[GSH] and k4′ = k4[GSH]. Kinetic analysis needs to take into account both the forward and the reverse reactions of an equilibrium, and for reaction 4, the following rate law can be derived (18),
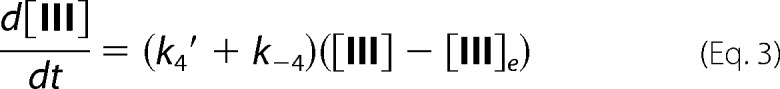 |
where [III]e is the concentration of YG-hydroxide at equilibrium. Integration of Equation 3 gives,
where [III]t is the concentration of YG-hydroxide at a given time point. Therefore, from the y = y0 + ae−bt) equation that was used to fit the experimental data sets, the following parameters were calculated for each kinetic run.
FIGURE 3.

Kinetics of the addition reaction of YG-hydroxide with GSH. The kinetic runs were obtained by monitoring the loss of YG-hydroxide using selected ion monitoring detection (for details, see “Experimental Procedures”). a, kinetic traces show the loss of the YG-hydroxide peak integral for the 4-min peak (shown in Fig. 1) as a function of time for GSH concentrations of 0.5 mm (●), 1 mm (△), 3 mm (○), and 5 mm (▴) with fitted exponential curves. b, shown are changes in observed k4′ (equation 6) as a function of GSH for the YG-hydroxide isomers with a retention time of 4 min (●, dashed line) or 11 min (○, solid line). Data are from exponential curves (equation 4) constructed as in a. c, K4′ (equation 5) was plotted against [GSH]; symbols are as for b. lines represent linear fits of the data with the calculated slopes giving the corresponding k4 or K4 values.
For each YG-hydroxide isomer, both K4′ and k4′ gave linear dependences on GSH concentration (Fig. 3, b and c). From the slopes in Fig. 3b, the obtained second order rate constants (k4) for the forward reactions are 11.8 ± 0.7 m−1min−1 for the isomer eluting at 4 min and 9.2 ± 0.2 m−1min−1 for the 11-min isomer. The respective equilibrium constants (K4) calculated from the plots in Fig. 3c are (7.5 ± 1.2) × 103 and (21 ± 4) × 103 m−1. From Equation 1, these give reverse rate constants (k−4) of 0.0016 and 0.0004 min−1, respectively.
As further evidence of reversibility, the addition of excess Cys to the GSH conjugate of YG-hydroxide resulted in replacement of the conjugated GSH with Cys. The product had similar chromatographic mobility, mass, and MS/MS fragmentation patterns as the YG-hydroxide-Cys adduct (see Table 1). Formation of this product was accompanied by a decrease of ∼60% in the integral of the YG-hydroxide-GSH peaks.
Conjugation of GSH to a Protein Tyr Hydroxide
We next investigated whether GSH could conjugate to superoxide-modified Tyr on a protein. Sperm whale myoglobin was chosen because it reacts with H2O2 to form a radical on Tyr-151 (19), and this radical can combine with superoxide to form an addition product (16). The synthetic peptide ELGYQG, an analog of the tryptic peptide 148–153 that contains Tyr-151, was initially examined. As observed previously (16), treatment with the HRP/XO system produced a hydroperoxide (not shown), which decomposed at 37 °C overnight and gave rise to peaks of m/z 682.3 corresponding to the hydroxide (Fig. 4a). The integrals of the LC/MS peaks of the hydroxides disappeared when the reaction mixture was treated with GSH along with the formation of several peaks of m/z 495.2. This corresponds in mass to the double-charged ion of the hydroxide plus 307 Da (Fig. 4b). The MS/MS spectra of all the peaks were similar and indicate that they are isomers of the glutathionylated form of the ELGYQG peptide (Fig. 4, c and d). The fact that GSH is conjugated to the modified Tyr-hydroxide residue is clear from the mass increase of 307 Da from the corresponding y2 to y3 fragments.
FIGURE 4.

LC/MS characterization of a GSH adduct generated on the Tyr-hydroxide of ELGYQG. The peptide was reacted with HRP/XO as described under “Experimental Procedures” then incubated overnight at 37 °C either without (a) or with GSH (10 mm) (b). LC/MS/MS SRM traces are shown for the hydroxide (dotted line, 682.3/589.4) and the GSH adduct (solid line, 495.2/449.3). Results are expressed as relative abundance with the tallest peak in a set at 100%. No GSH adduct was detected in a, and no hydroxide was detected in b. c, shown is a fragmentation pattern of the GSH adduct and assignment of peptide fragments for the peak eluting at 8.6 min. The MS/MS spectrum was acquired from the molecular ion of m/z 495.2 (double-charged ion), which corresponds in mass to ELGYQG + 16 Da + GSH. The peak eluting at 7.5 min had an identical fragmentation pattern. The loss of water is denoted by °, where y°x is yx − H2O. Fragments labeled −(H2O correspond to the loss of water from the parent peptide. d, a structure of GSH adduct of ELGYQG shows fragmentation.
Myoglobin was then reacted with the XO system (16) to generate the Tyr-hydroxide derivative. When this sample was treated with GSH and digested with trypsin, only minimal conversion of the ELGYQG hydroxide species to its GSH conjugate was detected (not shown). However, the tryptic digest protocol involved removal of the residual GSH by acetone precipitation and incubating the protein with trypsin in Tris-HCl buffer. It is possible that the GSH adduct could have formed on the protein, but subsequent incubation in the absence of GSH could have shifted the equilibrium toward decomposition and regeneration of the hydroxide derivative.
This was confirmed by adding NaBH4 after the GSH, on the rationale that it would reduce the carbonyl bond present on the bicyclic ring (IV) and stabilize the adduct species. Initial experiments were carried out with the GSH adduct of the synthetic peptide. The addition of NaBH4 led to the disappearance of the m/z 495.2 peaks (shown in Fig. 4b) and formation of major and several minor peaks at m/z 496.2 (Fig. 5a). It is important to note that these are double-charged species, so their singly charged counterparts will be 2 Da apart. These species all had a similar fragmentation pattern that is consistent with the 2-Da increase corresponding to reduction of the carbonyl group of the modified Tyr residue (Fig. 5, b and c). After blocking the residual GSH with iodoacetamide, the stability of the GSH adducts was compared. In contrast to the time-dependent disappearance of the adduct (Fig. 6a) and recovery of the hydroxide (Fig. 6b) observed with non-reduced species, there was no loss of the reduced GSH adduct. Therefore, reduction of the GSH adduct with NaBH4 was successful in producing a stable, well characterized species.
FIGURE 5.

Characterization of the reduced GSH adduct of ELGYQG. a, LC/MS/MS SRM traces for the GSH adduct prepared as in Fig. 3b and reduced with NaBH4 (20 mm) for 30 min (solid line m/z 496.2/450.3). The reduced adduct here has an m/z increase of 1 because it is double-charged. Results are expressed as relative abundance with the tallest peak set to 100%. No unreacted GSH adduct (dashed line, m/z 495.2/449.3) was detected. b, shown is a fragmentation pattern of the reduced GSH adduct. The MS/MS spectrum was acquired from the molecular ion of m/z 496.2 eluting at 4.5 min. c, the structure of the reduced GSH adduct of ELGYQG shows fragmentation.
FIGURE 6.
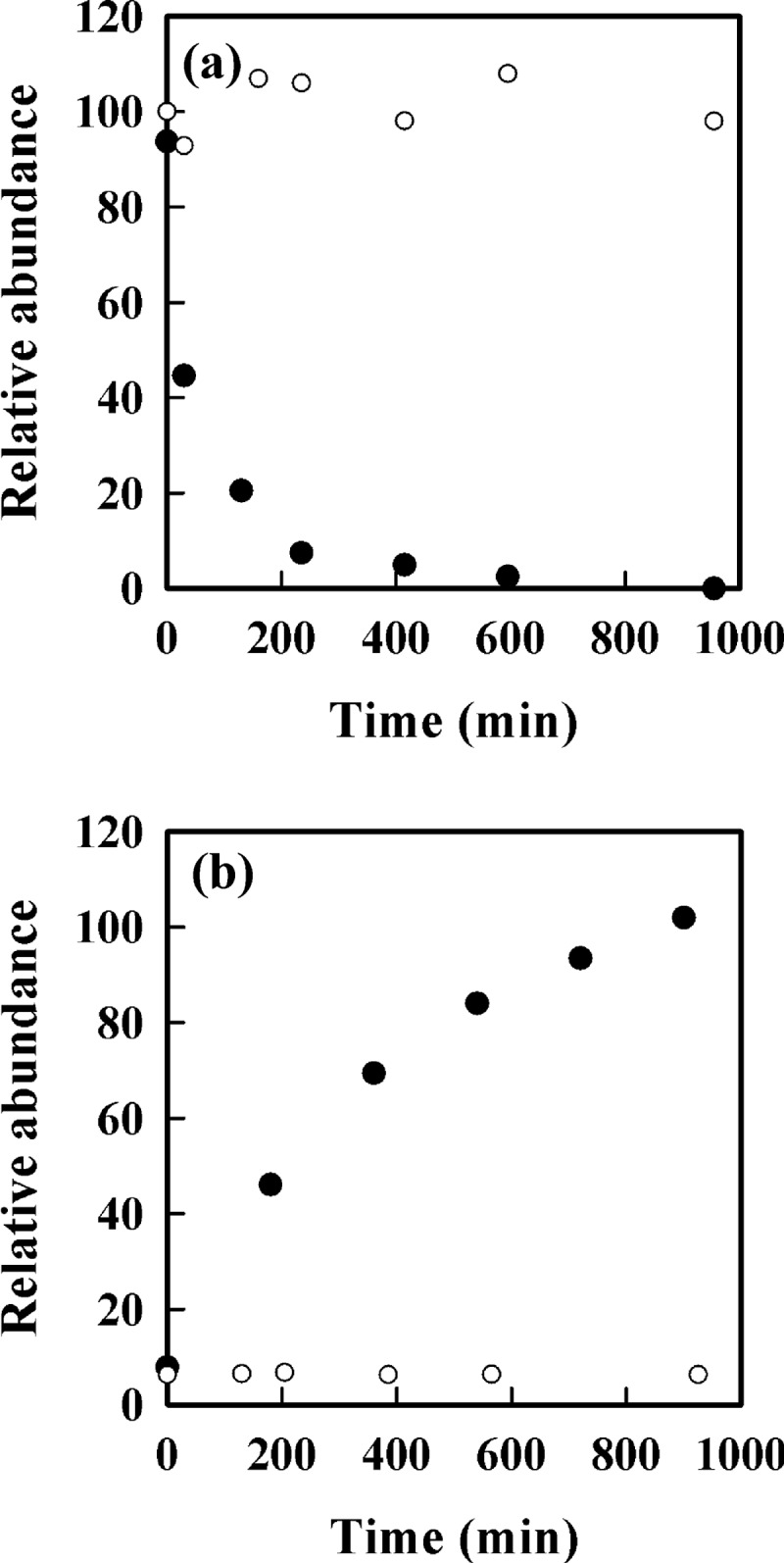
Time course for dissociation of the GSH adduct of ELGYQG and prevention by NaBH4 reduction. Loss of GSH adduct without (●) or with NaBH4 reduction (○) is shown. a, the GSH adduct was prepared with and without NaBH4 reduction as in Fig. 4 then reacted with iodoacetamide (5 mm). Samples were removed at intervals and analyzed by LC/MS with SRM at m/z 495.2/449.3 for the non-reduced and m/z 496.2/450.3 for the reduced GSH adduct. The amount of adduct at time zero is set at 100%. b, shown is the time course for recovery of the hydroxide (●), monitored by SRM at m/z 682.3/589.4, and the reduced hydroxide (○) monitored by SRM at m/z 684.3/590.4 under the same conditions. The amount of hydroxide measured at 900 min was set at 100%.
Myoglobin was, therefore, treated with the XO system followed by GSH then NaBH4. The mixture was reduced and alkylated and subjected to tryptic digestion. Without the GSH treatment of the XO-treated myoglobin, LC/MS with selected ion monitoring showed the presence of the hydroxide derivative of tryptic peptide 148–153 (ELGYQG-hydroxide) at m/z 682.3 (Fig. 7a). After treatment with GSH and NaBH4, this peak disappeared, and a new peak at m/z 496.2 was clearly detectable (Fig. 7b). The new peak had the same retention time and fragmentation pattern as the reduced GSH adduct of the synthetic peptide (shown in Fig. 5). There was no peak at m/z 684.3 (which would correspond to the hydroxide simply being reduced). We conclude, therefore, that the addition product formed between superoxide and the Tyr-151 radical of sperm whale myoglobin reacts further with GSH to form a Michael addition product.
FIGURE 7.
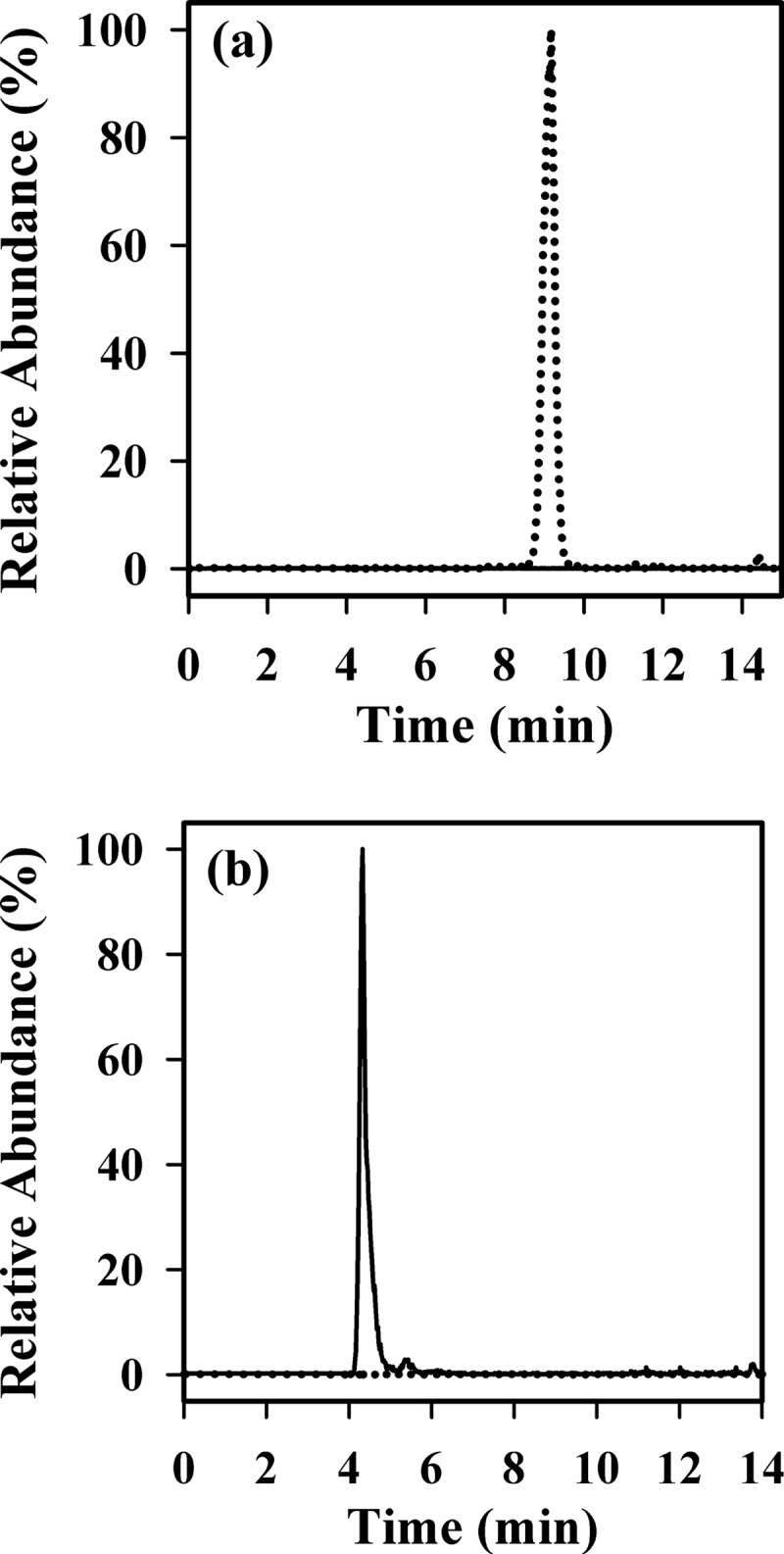
Detection of the reduced GSH adduct on myoglobin tryptic peptide 148–153 (ELGYQG). Myoglobin was treated with the XO system without (a) and with GSH treatment (b) and NaBH4 reduction. Both samples then underwent tryptic digestion and analysis by LC/MS as described under “Experimental Procedures”. LC/MS/MS was performed, and the SRM traces for the reduced GSH adduct (m/z 495.2/449.3; solid line) and hydroxide (m/z 682.3/589.4; dotted line) are shown. Results are expressed as relative abundance with the tallest peak set to 100%.
DISCUSSION
Hydroperoxide formation is a common oxidative post-translational modification of peptides or proteins (20). It occurs readily on Tyr residues either by rapid addition of superoxide to Tyr radicals, or by singlet oxygen addition to Tyr residues, The hydroperoxide group itself is a reactive species, capable of oxidizing thiol enzymes and modifying macromolecules including DNA (17, 20, 21). When formed on a Tyr, it will also cause substantial change to the structure of a peptide or protein by destroying its aromatic character and by forming bicyclic conjugates with N-terminal amine or amide nitrogens. Reduction of the hydroperoxide, for example by GSH, eliminates its oxidizing capacity, but structural modifications remain. With Tyr, the resultant hydroxide derivatives contain an α-β unsaturated carbonyl group and should be reactive electrophiles. We have demonstrated here that this is the case, and they readily undergo Michael addition reactions with thiols to give cross-linked Tyr-Cys adducts (IV, reaction 4). Product analyses by mass spectrometry showed that the reaction occurs with a range of Tyr-containing peptides and with Cys as well as GSH.
Our kinetic investigation showed that the two chromatographically distinct isomers of YG-hydroxide reacted with GSH at similar (although not identical) rates. GSH also reduces hydroperoxides (reaction 3). Our finding that YG-hydroxide was observed transiently when YG-hydroperoxide was treated with GSH implies that this reaction is faster than addition to the hydroxide. Therefore, any Tyr hydroperoxides formed in the presence of GSH should be converted in two steps to the GSH adduct, with the addition reaction being rate-determining. The rate constants of 9.2 and 11.8 m−1min−1 measured for GSH addition to YG-hydroxide are ∼10 times less than for hydroxynonenal (22), which is considered a good biological electrophile. They indicate that at a typical intracellular GSH concentration of 5 mm, hydroperoxides with similar reactivity to YG-hydroxide would have half-lives of ∼15 min and become more than 95% GSH-conjugated.
Interestingly, the addition reaction was found to be reversible. Several observations led to this conclusion; at low GSH concentrations the reaction did not go to completion, Cys was able to displace GSH from a preformed conjugate with YG-hydroxide, and the myoglobin peptide conjugate reverted to the hydroxide once GSH was removed. This is perhaps a surprising result as it is widely regarded that formation of GSH adducts with electrophiles (as distinct from disulfide formation) is not readily reversed (23). Esterbauer et al. (22), for example, measured half-lives in days for GSH adducts of acrolein and hydroxyalkenals. However, there is a precedent for reversibility with nitrolipids (24), and quantum mechanical calculations also favor a reversible reaction that proceeds through an enol intermediate (25). The k−4 values for the GSH adducts of YG correspond to half-lives of the order of 8–24 h. The faster decay of the myoglobin peptide conjugate (Fig. 6) implies variable stability of these species. However, the apparent lag between loss of the parent and formation of product suggests that the mechanism requires further elucidation. Nevertheless, it appears that glutathione adducts of modified Tyr residues are more labile than adducts with hydroxyalkenals, and the reaction may be slowly reversible under physiological conditions.
As well as peptides, we have shown that a tyrosyl hydroperoxide formed on a protein (Tyr-151 of sperm whale myoglobin) can form a Michael addition product with GSH. To observe the protein adduct, it was necessary to reduce the carbonyl group of the modified Tyr residue so that the reverse reaction did not occur during digestion and analysis. We were then able to characterize the product by MS analysis as identical to the glutathione adduct of the synthetic tryptic peptide. Essentially all the superoxide-modified protein Tyr-151 became conjugated at a physiological concentration of GSH, indicating that it is readily accessible and that GSH addition is efficient.
These results suggest that any tyrosyl hydroperoxide formed on cellular proteins, either by superoxide or singlet oxygen addition, is likely to become glutathionylated. Even after reduction, for example by peroxiredoxins (26), the α-β unsaturated carbonyl group is still present, and the reaction should proceed. Although it has not been established whether glutathionylated proteins are formed in vivo by this mechanism, they would not have been detected by methods normally employed to look for post-translational modification due to the reversibility of the reaction. Further investigation using an approach that includes a reduction step to stabilize the GSH adducts is required.
Protein thiols also have the potential to form Michael addition products with Tyr-hydroxide derivatives to produce Cys-Tyr cross-links either within or between polypeptide chains. Analogous adducts are formed between protein thiols and other electrophiles such as 4-hydroxynonenal (27–29), and susceptible targets have been identified in proteomic studies (30, 31). One example is the Cys residues of KEAP1, which readily undergoes electrophilic addition. This enables the transcription factor Nrf2 to dissociate from KEAP1, translocate to the nucleus, and activate Phase 2 stress response genes (32, 33). Michael addition is faster when the thiol is deprotonated, so low pKa Cys residues should be more reactive than GSH. Conjugation to modified Tyr residues could, therefore, be a mechanism for oxidatively inactivating enzymes such as protein-tyrosine phosphatases and cysteine proteases that have low pKa thiols at their active site. Intermolecular Cys-Tyr cross-linking via this mechanism could have other deleterious effects such as protein aggregation.
The glutathionylation mechanism we describe is distinct from the better known S-glutathionylation, which involves a disulfide linkage and is readily reversible. It involves formation of a carbon-sulfur bond, which dissociates only slowly and which is likely to persist in a cellular environment. It also differs from the Cys-Tyr linkage that exists in a number of metalloenzymes including galactose oxidase (34) and cysteine dioxygenase (35). This is an intramolecular thioether bond between a Cys residue and the ortho position of Tyr, which is formed during protein synthesis in a reaction that requires oxygen plus either copper or iron (34, 36).
Further investigation is required to establish whether the mechanism characterized here gives rise to glutathione adducts or cross-linking proteins in cells or tissues subjected to oxidative stress. However, hydroperoxides are formed in cells exposed to a radical generating system or singlet oxygen (37, 38) and on tyrosyl peptides exposed to stimulated neutrophils (11, 12). Considering the ease with which they react with GSH, it would seem highly likely that conjugates with GSH or other thiol compounds would be formed in a cellular environment.
Acknowledgment
We grateful to Nick Magon for helpful advice on the myoglobin peptide mass spectrometry.
This work was supported by the New Zealand Marsden Fund.
- XO
- xanthine oxidase
- HRP
- horseradish peroxidase
- SRM
- selected reaction monitoring
- Tyr-hydroxide
- tyrosine hydroxide
- YG-hydroxide
- Tyr-Gly hydroxide.
REFERENCES
- 1. Schuppe I., Moldéus P., Cotgreave I. A. (1992) Protein-specific S-thiolation in human endothelial cells during oxidative stress. Biochem. Pharmacol. 44, 1757–1764 [DOI] [PubMed] [Google Scholar]
- 2. Gallogly M. M., Mieyal J. J. (2007) Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391 [DOI] [PubMed] [Google Scholar]
- 3. Ghezzi P. (2005) Regulation of protein function by glutathionylation. Free Radic. Res. 39, 573–580 [DOI] [PubMed] [Google Scholar]
- 4. Dalle-Donne I., Rossi R., Colombo G., Giustarini D., Milzani A. (2009) Protein S-glutathionylation. A regulatory device from bacteria to humans. Trends Biochem. Sci. 34, 85–96 [DOI] [PubMed] [Google Scholar]
- 5. Hill B. G., Bhatnagar A. (2012) Protein S-glutathiolation. Redox-sensitive regulation of protein function. J. Mol. Cell Cardiol. 52, 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heinecke J. W. (2002) Tyrosyl radical production by myeloperoxidase. A phagocyte pathway for lipid peroxidation and dityrosine cross-linking of proteins. Toxicology 177, 11–22 [DOI] [PubMed] [Google Scholar]
- 7. Jin F., Leitich J., von Sonntag C. (1993) The superoxide radical reacts with tyrosine-derived phenoxyl radicals by addition rather than by electron transfer. J. Chem. Soc. Perkin Trans. I 15, 1583–1588 [Google Scholar]
- 8. Jonsson M., Lind T., Reitberger T. E., Eriksen T. E., Merenyi G. (1993) Free radical combination reactions involving phenoxyl radicals. J. Phys. Chem. 97, 8229–8233 [Google Scholar]
- 9. Winterbourn C. C., Parsons-Mair H. N., Gebicki S., Gebicki J. M., Davies M. J. (2004) Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides. Biochem. J. 381, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagy P., Kettle A. J., Winterbourn C. C. (2009) Superoxide-mediated formation of tyrosine hydroperoxides and methionine sulfoxide in peptides through radical addition and intramolecular oxygen transfer. J. Biol. Chem. 284, 14723–14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Winterbourn C. C., Pichorner H., Kettle A. J. (1997) Myeloperoxidase-dependent generation of a tyrosine peroxide by neutrophils. Arch. Biochem. Biophys. 338, 15–21 [DOI] [PubMed] [Google Scholar]
- 12. Nagy P., Kettle A. J., Winterbourn C. C. (2010) Neutrophil-mediated oxidation of enkephalins via myeloperoxidase-dependent addition of superoxide. Free Radic. Biol. Med. 49, 792–799 [DOI] [PubMed] [Google Scholar]
- 13. Pichorner H., Metodiewa D., Winterbourn C. C. (1995) Generation of superoxide and tyrosine peroxide as a result of tyrosyl radical scavenging by glutathione. Arch. Biochem. Biophys. 323, 429–437 [DOI] [PubMed] [Google Scholar]
- 14. Wright A., Bubb W. A., Hawkins C. L., Davies M. J. (2002) Singlet oxygen-mediated protein oxidation. Evidence for the formation of reactive side chain peroxides on tyrosine residues. Photochem. Photobiol. 76, 35–46 [DOI] [PubMed] [Google Scholar]
- 15. Saito I., Chujo Y., Shimazu H., Yamane M., Matsuura T. (1975) Nonenzymic oxidation of p-hydroxyphenylpyruvic acid with singlet oxygen to homogentisic acid. A model for the action of p-hydroxyphenylpyruvate hydroxylase. J. Am. Chem. Soc. 97, 5272–5277 [DOI] [PubMed] [Google Scholar]
- 16. Das A. B., Nagy P., Abbott H. F., Winterbourn C. C., Kettle A. J. (2010) Reactions of superoxide with the myoglobin tyrosyl radical. Free Radic. Biol. Med. 48, 1540–1547 [DOI] [PubMed] [Google Scholar]
- 17. Morgan P. E., Dean R. T., Davies M. J. (2004) Protective mechanisms against peptide and protein peroxides generated by singlet oxygen. Free Radic. Biol. Med. 36, 484–496 [DOI] [PubMed] [Google Scholar]
- 18. Espenson J. H. (1995) Chemical Kinetics and Reaction Mechanisms, pp. 46–69, McGraw-Hill Inc., New York [Google Scholar]
- 19. Lardinois O. M., Ortiz de Montellano P. R. (2003) Intra- and intermolecular transfers of protein radicals in the reactions of sperm whale myoglobin with hydrogen peroxide. J. Biol. Chem. 278, 36214–36226 [DOI] [PubMed] [Google Scholar]
- 20. Davies M. J. (2005) The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 [DOI] [PubMed] [Google Scholar]
- 21. Morgan P. E., Dean R. T., Davies M. J. (2002) Inhibition of glyceraldehyde-3-phosphate dehydrogenase by peptide and protein peroxides generated by singlet oxygen attack. Eur. J. Biochem. 269, 1916–1925 [DOI] [PubMed] [Google Scholar]
- 22. Esterbauer H., Zollner H., Scholz N. (1975) Reaction of glutathione with conjugated carbonyls. Z. Naturforsch. C 30, 466–473 [DOI] [PubMed] [Google Scholar]
- 23. Lin D., Saleh S., Liebler D. C. (2008) Reversibility of covalent electrophile-protein adducts and chemical toxicity. Chem. Res. Toxicol. 21, 2361–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker L. M., Baker P. R., Golin-Bisello F., Schopfer F. J., Fink M., Woodcock S. R., Branchaud B. P., Radi R., Freeman B. A. (2007) Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J. Biol. Chem. 282, 31085–31093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwobel J. A., Madden J. C., Cronin M. T. (2010) Examination of Michael addition reactivity towardglutathione by transition-state calculations. SAR QSAR Environ. Res. 21, 693–710 [DOI] [PubMed] [Google Scholar]
- 26. Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., Winterbourn C. C. (2010) Removal of amino acid, peptide, and protein hydroperoxides by reaction with peroxiredoxins 2 and 3. Biochem. J. 432, 313–321 [DOI] [PubMed] [Google Scholar]
- 27. Liebler D. C. (2008) Protein damage by reactive electrophiles. Targets and consequences. Chem. Res. Toxicol. 21, 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sayre L. M., Lin D., Yuan Q., Zhu X., Tang X. (2006) Protein adducts generated from products of lipid oxidation. Focus on HNE and ONE. Drug Metab. Rev. 38, 651–675 [DOI] [PubMed] [Google Scholar]
- 29. Rudolph T. K., Freeman B. A. (2009) Transduction of redox signaling by electrophile-protein reactions. Sci. Signal. 2, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Codreanu S. G., Zhang B., Sobecki S. M., Billheimer D. D., Liebler D. C. (2009) Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteomics 8, 670–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Higdon A., Landar A., Barnes S., Darley-Usmar V. M. (2012) The electrophile-responsive proteome. Integrating proteomics and lipidomics with cellular function. Antioxid. Redox. Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hong F., Sekhar K. R., Freeman M. L., Liebler D. C. (2005) Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 280, 31768–31775 [DOI] [PubMed] [Google Scholar]
- 33. Holland R., Fishbein J. C. (2010) Chemistry of the cysteine sensors in Kelch-like ECH-associated protein 1. Antioxid. Redox. Signal. 13, 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ito N., Phillips S. E., Stevens C., Ogel Z. B., McPherson M. J., Keen J. N., Yadav K. D., Knowles P. F. (1991) Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 350, 87–90 [DOI] [PubMed] [Google Scholar]
- 35. Simmons C. R., Liu Q., Huang Q., Hao Q., Begley T. P., Karplus P. A., Stipanuk M. H. (2006) Crystal structure of mammalian cysteine dioxygenase. A novel mononuclear iron center for cysteine thiol oxidation. J. Biol. Chem. 281, 18723–18733 [DOI] [PubMed] [Google Scholar]
- 36. Siakkou E., Rutledge M. T., Wilbanks S. M., Jameson G. N. (2011) Correlating cross-link formation with enzymatic activity in cysteine dioxygenase. Biochim. Biophys. Acta 1814, 2003–2009 [DOI] [PubMed] [Google Scholar]
- 37. Gieseg S., Duggan S., Gebicki J. M. (2000) Peroxidation of proteins before lipids in U937 cells exposed to peroxyl radicals. Biochem. J. 350, 215–218 [PMC free article] [PubMed] [Google Scholar]
- 38. Rahmanto A. S., Morgan P. E., Hawkins C. L., Davies M. J. (2010) Cellular effects of photogenerated oxidants and long-lived, reactive, hydroperoxide photoproducts. Free Radic. Biol. Med. 49, 1505–1515 [DOI] [PubMed] [Google Scholar]