

Background: DLC1 interacts with tensin2 PTB, playing a role as a tumor suppressor in many human cancers.
Results: We solved the tensin2 PTB-DLC1 complex structure and showed its importance for co-localization of DLC1 and tensin2 in cells.
Conclusion: The novel PTB-peptide binding mode provides a molecular basis for understanding the tumor suppression of DLC1 and tensin2.
Significance: A novel PTB-peptide binding mode was observed.
Keywords: Biophysics, NMR, Protein Complexes, Protein Structure, Structural Biology, Complex Structure, Deleted in Liver Cancer 1 (DLC1), Peptide Binding Mode, Phosphotyrosine Binding (PTB) Domain, Tensin2
Abstract
The protein deleted in liver cancer 1 (DLC1) interacts with the tensin family of focal adhesion proteins to play a role as a tumor suppressor in a wide spectrum of human cancers. This interaction has been proven to be crucial to the oncogenic inhibitory capacity and focal adhesion localization of DLC1. The phosphotyrosine binding (PTB) domain of tensin2 predominantly interacts with a novel site on DLC1, not the canonical NPXY motif. In this study, we characterized this interaction biochemically and determined the complex structure of tensin2 PTB domain with DLC1 peptide by NMR spectroscopy. Our HADDOCK-derived complex structure model elucidates the molecular mechanism by which tensin2 PTB domain recognizes DLC1 peptide and reveals a PTB-peptide binding mode that is unique in that peptide occupies the binding site opposite to the canonical NPXY motif interaction site with the peptide utilizing a non-canonical binding motif to bind in an extended conformation and that the N-terminal helix, which is unique to some Shc- and Dab-like PTB domains, is required for binding. Mutations of crucial residues defined for the PTB-DLC1 interaction affected the co-localization of DLC1 and tensin2 in cells and abolished DLC1-mediated growth suppression of hepatocellular carcinoma cells. This tensin2 PTB-DLC1 peptide complex with a novel binding mode extends the versatile binding repertoire of the PTB domains in mediating diverse cellular signaling pathways as well as provides a molecular and structural basis for better understanding the tumor-suppressive activity of DLC1 and tensin2.
Introduction
Deleted in liver cancer 1 (DLC1)5 is a tumor suppressor gene located on chromosome 8p21.3–22 first identified in primary human hepatocellular carcinoma (HCC) (1). The DLC1 protein contains an N-terminal sterile α motif domain, a GTPase activation protein domain for Rho family GTPase (RhoGAP), a C-terminal steroidogenic acute regulatory protein-related lipid transfer domain, and a serine-rich unstructured middle region. Loss or down-regulation of DLC1 expression mediated by genetic and epigenetic mechanisms has been associated with the development of many human cancers, including HCC; breast, lung, colon, and prostate cancers; and nasopharyngeal, esophageal, and cervical carcinomas (2–4). Restoration of DLC1 expression was found to suppress cell migration and invasion, inhibit growth, and induce apoptosis of various cancer cell line models in vivo and in vitro (5–8). Functional data regarding the loss of DLC1 in development of hepatocellular carcinoma cells have been shown in a mouse model (9). Accumulating evidence has well demonstrated that DLC1 is a bona fide tumor suppressor in diverse human malignancies. Different mechanisms regulate the tumor-suppressive function of DLC1 (10–12).
The tumor-suppressive function of DLC1 has been shown to be regulated by its association with tensins (13). The tensins are a family of proteins localized at integrin-mediated focal adhesions bridging the actin cytoskeleton and integrins (14). To date, four members, namely tensin1, tensin2/C1-TEN, tensin3, and tensin4/C-terminal tensin-like (cten), with structural similarity and high sequence homology have been identified (15, 16). All tensins contain C-terminal Src homology 2 and phosphotyrosine binding (PTB) domains, which confer interacting potentials with diverse scaffolding proteins and signaling molecules at focal adhesions (17–20). Identification of tensin2 as the novel interacting partner of DLC1 by our group has provided evidence about the focal adhesion localization of DLC1 (21, 22). Subsequent studies have further demonstrated the functional implication of the interactions between DLC and other tensin proteins in tumor suppression by DLC1 (13, 23). To date, association with tensin proteins is the best characterized regulatory event in the subcellular localization of DLC1 at the protein level. Different binding regions have been identified in DLC1 that interact with Src homology 2 (24) and/or PTB domains of tensins, and they localize to the focal adhesion-targeting region within amino acids 201–500 of DLC1 (25). More detailed biochemical analysis showed that DLC1 utilizes residues 375–385 to predominantly interact with the tensin2 PTB domain, and its growth inhibitory activity depends on this PTB domain interaction (26).
Overall, as a popular protein binding module, a large number of proteins contain the PTB domain, which exhibits a conserved core structure of a PH domain superfold containing a β-sandwich made of two antiparallel β-sheets capped by a C-terminal helix. Proteins containing PTB domains function as adaptors or scaffolds to organize signaling complexes involved in a wide range of physiological processes. The structure of PTB domains confers specificity for binding peptides having a NPXY motif via a structurally conserved mechanism with different requirements for the phosphorylation of tyrosine within this recognition sequence. Structurally, the C-terminal α helix and β5 strand of the PTB domain are involved in this interaction (27). Intriguingly, DLC1 does not contain an NPXY-like motif, indicating it may interact with the PTB domain of tensin2 through a different mechanism.
Despite the differential roles of tensins in different cellular contexts, association with DLC1 plays an unequivocal role in modulating the tumor-suppressive activity of DLC1. As mentioned, DLC1 predominantly interacts with the PTB domain of tensin2. To gain insight into the molecular mechanism of DLC1-tensin2-mediated tumor suppression, it is important to understand the binding mode between DLC1 and the PTB domain of tensin2. In this study, we characterized and verified the binding surface and key residues on the tensin2 PTB domain that are involved in the interaction with DLC1 peptide. We also determined the solution structure of tensin2 PTB in complex with the DLC1 peptide by NMR. The PTB domain of tensin2 binds to a non-phosphorylated peptide of DLC1 in a manner not reported before. The HADDOCK-derived complex structural model reported here reveals a novel peptide binding mode and expands the recognition modes of PTB domains beyond the canonical NPX(p)Y-like motif (where pY is phosphotyrosine) in mediating diverse cellular signaling pathways.
EXPERIMENTAL PROCEDURES
Plasmid Construction
The PCR-amplified cDNA fragment encoding residues 1262–1409 of PTB domain of human tensin2 (GI 38787956) was cloned into the pET vector (Novagen) by standard molecular cloning techniques with a His6 tag-thrombin cleavage sequence at the N terminus. The coding sequence of DLC1 peptide, corresponding to residues 374–388 of human DLC1 (GI 33188437) with an additional tryptophan residue at the C terminus for protein quantification, was PCR-amplified, purified using a QIAEX II gel extraction kit, and cloned into a pET-derived expression vector. The resulting construct was tagged with a 56-residue B1 immunoglobulin binding domain of streptococcal protein (GB1) and a protein 3C protease cleavage site at the N terminus. For in vitro biochemical analysis, the DLC1 peptide was constructed as a GST fusion protein using pGEX-4T-1 vector (GE Healthcare).
Site-directed Mutagenesis
Alanine point mutations and deletions in the PTB domain were performed by QuikChange site-directed mutagenesis using Pfu DNA polymerase (Sangon Biotech Shanghai) with suitable primer sets. The presence of appropriate mutations was confirmed by DNA sequencing. For immunofluorescence imaging and the colony formation assay, GFP-tagged full-length tensin2 and its mutants were constructed on pEGFPC1. The number in the name of full-length tensin2 alanine mutants is the residue's sequential number in our PTB complex structure. The “Δα” mutant refers to the full-length tensin2 mutant deleted of the N-terminal α1 helix (residues 15–25 of the PTB domain).
Protein Expression and Purification
The recombinant proteins were expressed in Escherichia coli strain BL21(DE3) at 18 °C overnight. For PTB domain purification, the proteins were purified from the bacterial lysates by sequential chromatography on nickel-nitrilotriacetic acid and Superdex 75 columns (GE Healthcare). For DLC1 peptide purification, proteins were expressed and purified with nickel-nitrilotriacetic acid resin as described above. The N-terminal GB1 tag was cleaved by protein 3C protease and separated by gel filtration. The peak fraction containing DLC1 peptide was desalted in H2O using a HiTrap Desalting column (GE Healthcare) and lyophilized for further study. The molecular weight of purified DLC1 peptide was confirmed by mass spectrometry. For the NMR studies, uniformly isotope-labeled proteins were expressed and purified as described above except that the bacteria were grown in M9 minimal medium using 15NH4Cl and/or [13C]glucose (Cambridge Isotope Laboratories Inc.) as the sole nitrogen and/or carbon sources. The NMR samples were concentrated to ∼0.2 (for HSQC-based titration experiments) or ∼0.8 mm (for structural determinations) in optimized NMR sample buffer (50 mm sodium phosphate, 100 mm NaCl, 1 mm EDTA, pH 7.0).
GST Pulldown Assay
For coupling, prewashed glutathione-Sepharose 4B slurry beads were incubated with bacterial lysates of GST or GST-DLC1(374–392) in an assay buffer (50 mm sodium phosphate, 150 mm NaCl, 1 mm EDTA, 0.1% Nonidet P-40, pH 7.0) at 4 °C for 1 h and washed three times. Then the beads were incubated with bacterial lysates of wild-type or mutated PTB domains for another 2 h at 4 °C and washed three times. The captured proteins were eluted in 50 mm Tris, pH 7.9 containing 10 mm glutathione. The samples were then resolved by 15% SDS-PAGE and subjected to Western blot analysis with an anti-His tag antibody (His-probe, H-3, Santa Cruz Biotechnology).
NMR Binding Studies
To investigate the ligand binding, the two-dimensional 1H-15N HSQC spectra were recorded on uniformly 15N-labeled PTB domain (∼0.2 mm) in the presence of different concentrations of DLC1 peptide ranging from 0 to 1.5 mm. Both the PTB sample and the stock solutions of DLC1 were prepared in the NMR buffer (50 mm sodium phosphate, 100 mm NaCl, 1 mm EDTA, pH 7.0). The chemical shift perturbation between the free form and DLC1-bound PTB domain was normalized by the following formula and expressed in ppm.
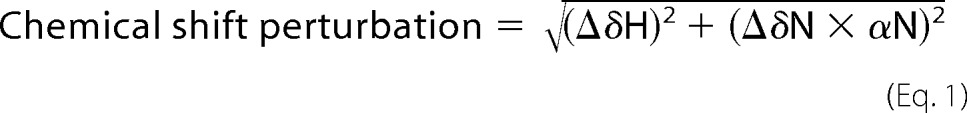 |
In the equation, ΔδH and ΔδN are the differences in chemical shifts of amide protons and nitrogen between the initial and final data points of the titration, respectively. The nitrogen frequency is normalized for proton with the scale factor αN = 0.17, established from estimates of atom-specific chemical shift ranges in a protein environment, in the equation (28).
Fluorescence Polarization Assay
The fluorescence polarization assay was performed on a PerkinElmer Life Sciences LS-55 fluorometer equipped with an automated polarizer at 20 °C. Fluorescence titration was performed by adding increasing amounts of purified PTB domains to a fixed amount of fluorescein 5-isothiocynate (FITC; Molecular Probes)-labeled DLC1 peptide in 50 mm sodium phosphate, 100 mm NaCl, 1 mm EDTA, pH 7.0. The polarization value of the FITC-labeled DLC1 peptide was measured at each titration point. The Kd values were obtained by fitting the titration curve with the classical one-site binding model.
NMR Spectroscopy
NMR spectra were acquired at 37 °C on 750- and 500-MHz Varian NMR spectrometers with self-shielded z axis gradients. All spectra were processed using NMRPipe (29, 30) and analyzed using SPARKY 3.6 The 1H, 15N, and 13C resonances of backbone and side chain atoms were assigned by using a standard set of triple resonance experiments on either uniformly 15N, 13C-labeled PTB domain with/without unlabeled DLC1 or uniformly 15N, 13C-labeled DLC1 with/without unlabeled PTB domain at protein concentrations of ∼0.8 mm (31–33). The PTB-DLC1 complex was prepared at a ratio of 1:2 between 15N, 13C-labeled and unlabeled components. The initial backbone assignment was generated by Mars (34) and manually checked in combination with 15N-edited three-dimensional NOESY. NOE-derived distance restraints were obtained from 15N- or 13C-edited three-dimensional NOESY spectra, each with a mixing time of 120 ms, complemented by 13C-edited, 13C/15N-filtered three-dimensional NOESY spectra for the intermolecular contact (mixing time, 150 ms) (35). Steady-state heteronuclear {1H}-15N NOE values were determined from spectra recorded with 5-s relaxation delay and the presence and absence of a proton presaturation period of 3 s in the 500-MHz spectrometer.
NOE Analysis and Structure Calculations
NOE assignment and structure calculations were performed using the CANDID (36) module of the program CYANA2.1 (37). We first calculated the structures of the PTB domain in its free and DLC1-bound forms and vice versa for DLC1 peptide. Next, the initial 200 complex structures of the PTB domain and DLC1 were generated with manually assigned unambiguous intermolecular NOE distance restraints by CNS 1.1 (38). Then the 120 top scoring complex structures from CNS were further refined with additional chemical shift perturbation data using a HADDOCK-type protocol without rigid body docking (39). The calculation details are described in the supplemental Materials and Methods. A cutoff value of 1.4 Å was defined to select the well resolved ensembles of structures. The ten lowest scored structures from each cluster were analyzed further. A family of 10 lowest energy structures in which none showed any restraint violation over 0.5 Å was used for statistical analysis. The dihedral restraints were generated from TALOS (40). The quality of the HADDOCK-derived structural models was assessed using PROCHECK (41) and analyzed by MOLMOL (42). All of the figures representing the structures were generated by PyMOL.
Paramagnetic Labeling
To produce spin-labeled DLC1 peptides, a single cysteine was added to the N- or C-terminal ends, respectively. After purification, the protein was exchanged with phosphate buffer with no reducing agent. A 5-fold molar excess of spin labeling reagent MTSL was added, and the reaction proceeded at 25 °C for 12 h (Toronto Research Chemicals). Excess reagent was removed with a desalting column. A series of 1H-15N HSQC spectra were obtained to monitor the disappearance of a subset of resonances of 15N-labeled tensin2 PTB in the presence of increasing molar ratios of unlabeled, MTSL-coupled GB1-DLC1. A control spectrum was collected at the end of the titration after the addition of 10 mm ascorbic acid to remove the spin label MTSL.
Immunofluorescence Microscopy
To reveal the co-localization of tensin2 and DLC1 in cells, HEK293T cells seeded on coverslips were transfected with Myc-tagged DLC1 full length (FL) and GFP-tagged tensin2 FL or mutants. Transfected cells were fixed with 4% paraformaldehyde, PBS and permeabilized with 0.5% Triton X-100, PBS. Cells were then blocked with 5% bovine serum albumin. Myc-DLC1 was stained by anti-Myc antibody followed by rhodamine-conjugated secondary antibody. Cells were mounted in Vectashield antifade mountant (Vector Laboratories, Burlingame, CA). Images were captured by a Leica Q550CW fluorescence microscope (Leica, Wetzler, Germany).
Colony Formation Assay
The DLC1-deficient human HCC cell line BEL7402 was obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. BEL7402 has been used for the functional characterization of DLC1 and tensin2 elsewhere (5, 21). Cells at 2.5 × 104/well were seeded into 12-well tissue culture plates 2 days before transfection. Each of 0.25 μg of GFP-tagged tensin2 and its mutant expression vector (wild-type tensin2, Δα, L139A, D140A, F151A, and ΔPTB) was co-transfected with 1 μg of DLC1-pCS2+MT into cells. One day after transfection, cells were trypsinized and replated at a 1:20 dilution in triplicates into 6-well tissue culture plates. Cells were selected in 700 μg/ml G418 (Merck) for 3 weeks. Colonies formed were fixed with 3.7% formaldehyde and stained with crystal violet solution.
RESULTS
Binding Studies of Tensin2 PTB Domain and the DLC1 Peptide
In a previous study, we identified that amino acids 375–385 of DLC1 were responsible for binding the PTB domain of tensin2 (26) (Fig. 1A). Before the structural study, we first performed an in vitro pulldown assay to verify the binding. The GST-tagged peptide (EDHKPGTFPKALTNG) comprising DLC1 amino acids 374–388 was able to pull down tensin2 PTB domain from bacterial lysate (Fig. 1B), confirming their direct physical interaction. Subsequently, the DLC1 peptide was labeled with FITC to examine its affinity for tensin2 PTB domain in a fluorescence polarization study. As shown in Fig. 1C, tensin2 PTB domain bound to the DLC1 peptide in a concentration-dependent manner. The measured dissociation constant (Kd) of 86.5 μm for tensin2 PTB-DLC1 peptide complex was obtained from repetitive measurements with deviations less than 5% from each other.
FIGURE 1.

Binding studies of tensin2 PTB with DLC1 peptide. A, schematic diagram of DLC1 and tensin2 domain structures. The domain length and position are in proportion to the real situation. ABD, actin-binding domain; FAB, focal adhesion binding. START, steroidogenic acute regulatory protein-related lipid transfer domain; SH2, Src homology 2 domain; SAM, sterile α motif. The interaction between DLC1 and tensin2 PTB domain is outlined. B, GST pulldown (PD) assay. Bacterial lysate of wild-type PTB domain was incubated with immobilized GST or GST-DLC1 peptide. The associated proteins were analyzed by immunoblotting (IB) with anti-His6 tag antibody. Gluta, glutathione; pep, peptide. C, interaction of PTB and DLC1 peptide measured by fluorescence polarization. In this assay, the FITC-labeled DLC1 peptide was titrated with increasing amounts of unlabeled PTB, and the polarization value of the FITC-labeled DLC1 peptide was measured at each titration point. D, a section of the overlaid 1H-15N HSQC spectra of 15N-labeled PTB in free form (black) and titrated with non-labeled DLC1 peptide at different molar ratios, 1:1 (blue), 1:2 (magenta), 1:3 (green), 1:4 (yellow), 1:5 (maroon), and 1:6 (red). Residues that undergo significant changes in chemical shifts upon formation of the complex with DLC1 are highlighted by arrows and labeled with peak assignments. E, the weighted chemical shift changes between the free form and DLC1 peptide-saturated PTB obtained from NMR titration experiments are plotted as a function of residue number.
The DLC1 peptide binding properties were further investigated by NMR titration using chemical shift perturbation analyses. A 15N-labeled tensin2 PTB domain was titrated with non-labeled GB1-tagged DLC1 peptide (amino acids 374–388). The residues with chemical shift changes were monitored by two-dimensional 1H-15N HSQC spectra. As shown in Fig. 1D and supplemental Fig. 4, the addition of DLC1 peptide induced significant chemical shift perturbations and resulted in a step-by-step peak shift pattern at many positions in the spectra, indicating a fast exchange regime on the NMR time scale. The 1H-15N HSQC spectra of tensin2 PTB domain showed no change when titrated with GB1 protein alone (data not shown). As shown in Fig. 1E, the weighted chemical shift changes of the amide 1H and 15N obtained from NMR titration experiments were plotted as a function of the residue number. The signals having large chemical shift changes (Δδ > 0.038 ppm) corresponded to the region formed by the helices α1 and α3; β strands β1, β2, and β3; and the loop connecting β7 to the helix α3. The titration results entail the discovery of a new binding region different from the well known PTB substrate binding site reviewed by Uhlik et al. (27).
Overall Structure of the Complex
As our binding studies indicated a new binding mode in which DLC1 peptide occupies the non-canonical binding site of PTB domain, we went further to fully characterize this interaction at the atomic level by multidimensional heteronuclear NMR spectroscopy as described under “Experimental Procedures.” Combining the NMR and mutagenesis data, we determined a HADDOCK-derived structural model for the tensin2 PTB and DLC1 peptide complex. A superimposed ensemble of 10 structures and the ribbon plot of the lowest energy structure are shown in Fig. 2A. The structure ensemble had good backbone geometry, no significant restraint violation, and low pairwise root mean square deviation values (Table 1). The structure of tensin2 PTB domain in the complex presents a typical PH domain superfold, a seven-stranded β-sandwich composed of two antiparallel β-sheets capped by a C-terminal α-helix, α3. Moreover, it contains two additional α-helices, N-terminal α1 and α2, connected to strands β1 and β2, respectively. In the complex, DLC1 peptide is unstructured and interacts with the surface of PTB domain formed by N-terminal helix α1; β-strands β1, β2, and β3; and the loop connecting β7 to C-terminal helix α3, consistent with the titration data (Fig. 2A). Intermolecular NOEs defined the orientation of the DLC1 peptide in the complex (supplemental Fig. 1), which was later independently confirmed by paramagnetic relaxation enhancement experiments as shown in Fig. 3. The backbone amide resonances of Asn-80 and Gln-143 disappear in the presence of the N-terminal spin-labeled DLC1 peptide (Fig. 3A, red), whereas they remain when titrated by C-terminal spin-labeled DLC1 peptide (Fig. 3B, red). This result indicates that residues Asn-80 and Gln-143 are closer to the N terminus of DLC1 peptide than to the C terminus (Fig. 3C). Ascorbic acid can reduce MTSL spin label. The cross-peaks of Asn-80 and Gln-143 recovered after addition of ascorbic acid, indicating that the intensity loss of the two peaks was caused specifically by the paramagnetic spin label (Fig. 3A, black). The paramagnetic relaxation enhancement experiments independently confirmed the orientation of the DLC1 peptide.
FIGURE 2.

The solution structure of PTB-DLC1 peptide complex. A, left panel, backbone superposition of the 10 lowest energy NMR structures of PTB-DLC1 peptide complex. Secondary structural elements of PTB are indicated by color-coding: α-helices, blue; β-strands, green; and loops, gray. DLC1 is shown in red. N-terminal and C-terminal ends are indicated as N and C. Right panel, ribbon diagram of the complex using the coordinates of the lowest energy structure. B, detailed view of the complex binding surface. Left panel, surface representation of PTB colored by residue type: acidic, red; basic, blue; hydrophobic, yellow; polar, cyan; non-interacting, gray. Right panel, residues having intermolecular NOEs are shown in sticks, green for PTB and orange for DLC1. The backbone of PTB is colored gray. DLC1 is indicated as in the left panel.
TABLE 1.
The statistics of tensin2 PTB/DLC
r.m.s., root mean square; r.m.s.d., root mean square deviation; bb, backbone.
| NMR restraints | |
| Total experimental restraints | 2206 |
| Total NOE distance restraints | 1828 |
| Short range, |i − j| ≤ 1 | 873 |
| Medium range, 1 <|i − j| < 5 | 272 |
| Long range, |i − j| ≥ 5 | 657 |
| Intra-DLC1 | 18 |
| Inter-PTB-DLC1 | 8 |
| Dihedral angle restraints | |
| φ | 169 |
| ψ | 169 |
| Statistics for structures | |
| Final energies (kcal/mol) | |
| van der Waals (kcal/mol) | −621.89 ± 34.79 |
| NOE (kcal/mol) | 0.9765 ± 0.2475 |
| Violations | |
| Number of NOE violations >0.5 Å | 0 ± 0 |
| r.m.s. deviation (Å) from experimental distance restraints | 0.0229 ± 0.0028 |
| Number of dihedral angle constraint violations >5° | 0 ± 0 |
| R.m.s. deviation (°) from experimental torsion restraints | 0.5547 ± 0.0607 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.0037 ± 0.0001 |
| Angles (°) | 0.4928 ± 0.0294 |
| Improper (°) | 0.4766 ± 0.0383 |
| Structural r.m.s.d. to the mean coordinate, bb/heavy (Å) | |
| Regions (residue numbers) 15–161, 367–380 | 1.15/1.73 |
| Regions (residue numbers) 20–23, 27–37, 43–56, 64–71, 74–78, 90–91, 95–99, 106–108, 114–124, 127–137, 145–155, 370–377 | 0.80/1.23 |
| Ramachandran plot (% of residues) | |
| Residues in most favored regions | 80.5 |
| Residues in additional allowed regions | 17.1 |
| Residues in generously allowed regions | 1.4 |
| Residues in disallowed regions | 1 |
FIGURE 3.

DLC1 peptide orientation determined by paramagnetic relaxation enhancement experiments. A cysteine residue was added to the N and C termini of the DLC1 peptide by site-directed mutagenesis. The paramagnetic spin label MTSL was introduced to the cysteine residue through the formation of a thioester bond. Sections of 1H-15N HSQC spectra of PTB domain are shown here. A, a section of the overlaid 1H-15N HSQC spectra of 15N-labeled PTB in the presence of the N-terminal spin-labeled GB1-tagged DLC1 peptide (red) and after addition of 10 mm ascorbic acid (black). B, a section of the overlaid 1H-15N HSQC spectra of 15N-labeled PTB in the presence of the C-terminal spin-labeled DLC1 peptide (red) and after addition of 10 mm ascorbic acid (black). Note that the backbone amide resonances of Asn-80 and Gln-143 disappear in the presence of the N-terminal spin-labeled GB1-tagged DLC1 peptide. The paramagnetic relaxation enhancement experiments independently confirmed the orientation of the DLC1 peptide. C, ribbon diagrams of the complex showing the position of the residues Asn-80 and Gln-143, which are in closer proximity to the N terminus of DLC1 peptide than to the C terminus. The backbone of PTB is colored gray. DLC1 is indicated as line mode and colored red. The residues Asn-80 and Gln-143 are shown in stick mode and colored blue and pink, respectively. The middle and right panels are the same as the left panel after a 90° and 180° rotation, respectively, about the z axis.
Interactions at the Tensin2 PTB Domain and DLC1 Peptide Interface
The binding interface of the tensin2 PTB-DLC1 complex comprises the strands β1–β3, helices α1 and α3, and the loop between β7 and helix α3 (Fig. 2B). The binding site accommodates nearly all the residues of DLC1 peptide in an extended conformation. The DLC1 binding site on PTB domain is characterized by a hydrophobic surface formed by Leu-21, Leu-22, Gln-24, Ala-27, Ser-71, Ala-72, Gln-73, and Thr-76 that helps to occupy the Pro-Gly-Thr-Phe-Pro segment of DLC1. Additionally, the aromatic side chain of DLC1 Phe-381 fills a shallow pocket formed by Lys-69, Val-70, Ser-71, Gln-73, Gly-74, and Thr-76 of PTB domain and is nearly perpendicular to the β2 and β3 strands. Another key residue of DLC1 at the binding interface is His-376. The imidazole ring of His-376 is stabilized by stacking against the side chains of Cys-28, Ser-29, Leu-139, and Pro-141. The hydrophobic residues Ala-384 and Leu-385 of DLC1 are involved in the complex formation, making van der Waals contacts with a narrow cleft formed by the side chains of Arg-23 and Gln-73 of PTB domain.
Structure-based Mutational Studies of the Interaction between Tensin2 PTB and DLC1 Peptide
Based on the complex structure and the chemical shift perturbations observed, a series of point mutants in PTB domain were constructed to evaluate the contributions of these residues to the tensin2 PTB-DLC1 interaction. These point mutants (D20A, L21A, R23A, Q24A, C28A, S29A, V30A, H67A, F68A, K69A, V70A, S71A, R82A, S99A, L139A, D140A, F151A, K154A, and V155A) were expressed and purified to homogeneity. The results of a GST pulldown assay (Fig. 4A) indicate that Q24A, C28A, S29A, and V70A mutants partially reduced binding of the DLC1 peptide to PTB domain, whereas L139A and D140A significantly decreased binding, and F68A and F151A abolished binding. These residues have major contributions to the interaction. The F151A mutant might alter the structure of the binding pocket of the PTB domain, although the overall tertiary structure of the F151A mutant was not substantially affected as judged by the similarity of its two-dimensional 1H-15N HSQC spectrum to that of wild-type PTB (supplemental Fig. 2). However, the F68A mutation might affect the folding of the PTB domain as the mutant proteins were unstable during the purification process. Previously reported mutational studies on DLC1 peptide showed that His-376, Pro-378, Gly-379, Thr-380, Phe-381, Pro-382, and Leu-395 are crucial for the interaction (22). This is consistent with the DLC1 fragment range in our complex structure.
FIGURE 4.
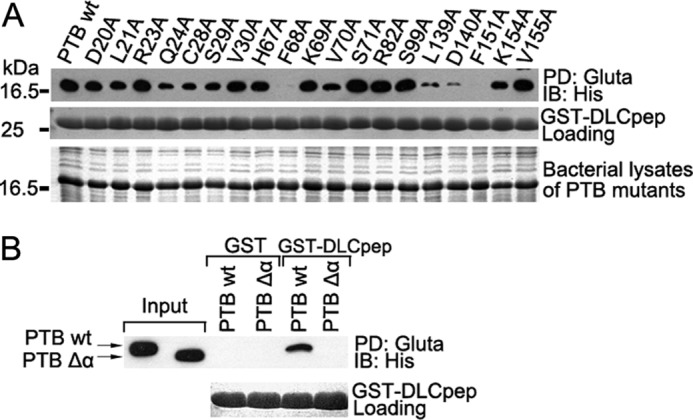
Mutational studies of the interaction between tensin2 PTB and DLC1 peptide in vitro. A, bacterial lysates of wild type or a series of PTB mutants were incubated with immobilized GST or GST-DLC1 peptide. The associated proteins were analyzed by immunoblotting (IB) with anti-His6 tag antibody (upper panel). The middle and lower panels indicate a relatively equal amount of GST/GST-DLC1 peptide (DLCpep) and bacterial lysates of PTB mutants used in the GST pulldown (PD) assay. Gluta, glutathione. B, the effect of N-terminal helix α1 of PTB on DLC1 peptide interaction. The wild type (wt) or PTB Δα were pulled down by GST/GST-DLC1 peptide from bacterial lysates and visualized by anti-His antibody (upper panel). The lower panel indicates a relatively equal amount of GST/GST-DLC1 peptide used in the GST pulldown assay.
We also tested the role of N-terminal helix α1 (residues 15–25 of PTB domain) in the binding. Deletion of the helix α1, which is only seen in Shc- and Dab-like PTB domains, abolished binding of the DLC1 peptide (Fig. 4B). Examination of a two-dimensional 1H-15N HSQC spectrum of PTB Δα showed little change from the whole PTB domain, indicating that the core structure was not perturbed. No chemical shift changes could be seen in PTB Δα when titrated with DLC1 peptide. These results indicate that the novel binding mode would only apply to Shc- or Dab-like PTB domains that contain an N-terminal helix.
Effects of PTB Mutants on the Co-localization of Tensin2 and DLC1 in Cells
To further confirm the importance of the defined residues in the tensin2-DLC1 interaction, we examined their effect on the co-localization of tensin2 and DLC1 in cells. GFP-tagged wild-type or alanine substitution mutants of full-length tensin2 (GFP-tensin2 FL) were co-transfected with Myc-tagged full-length DLC1 (Myc-DLC1 FL) into HEK293T cells, and the proteins were visualized by immunofluorescence microscopy. As shown in Fig. 5A, GFP-tagged full-length tensin2 co-localized well with Myc-DLC1 FL, whereas the GFP-tensin2 FL mutant (PTB L139A) did so less frequently. In addition, GFP-tensin2 FL mutants (PTB D140A and PTB F151A) failed to co-localize with Myc-DLC1 FL. Similar results were obtained in the DLC1-deficient human BEL7402 HCC cells (data not shown), indicating the role of Leu-139, Asp-140, and Phe-151 in the PTB domain in formation of the tensin2-DLC1 complex.
FIGURE 5.

Effects of PTB mutations in full-length tensin2 on the co-localization with DLC1 in cells. A, HEK293T cells were co-transfected with GFP-tensin2 FL or mutants (green) and Myc-DLC1 (red). Myc-DLC1 was stained by anti-Myc antibody followed by rhodamine-conjugated secondary antibody. Fluorescence microscopy revealed co-localization of Myc-DLC1 FL and GFP-tensin2 FL and partially co-localized Myc-DLC1 FL and GFP-tensin2 L139A. GFP-tensin2 D140A and F151A failed to co-localize with Myc-DLC1 FL. B, colony formation assay. GFP-tensin2 (WT) or mutants (PTB L139A, D140A, F151A, ΔPTB, and Δα) were co-transfected with DLC1 into the DLC1-deficient BEL7402 HCC cell line. After G418 selection, the colonies formed were fixed and stained. The number of colonies was counted and plotted. The error bars indicate the standard deviations of triplicate plates for each construct. C, representative images of the colony formation assay for each construct are shown. Significant inhibition of colony formation was observed in tensin2- and DLC1-co-transfected cells as compared with the vector control (p = 0.015). Growth suppression activity was lost in all tensin2 mutant-DLC1 co-transfection groups.
In a previous colony formation assay, the DLC1 mutant of which the PTB binding region was deleted partially lost the growth-suppressive activity (26). To further address the requirement of tensin2 PTB/DLC1 binding for the tumor-suppressive function of DLC1, we carried out the same colony formation assay using tensin2 mutants derived from our structural and mutational studies in DLC1-deficient BEL7402 HCC cells. The co-transfection of tensin2 with DLC1 significantly suppressed colony formation (p = 0.009) when compared with the transfection of tensin2 only (data not shown). In contrast, growth suppression was abolished when DLC1 was co-transfected with tensin2 PTB mutants (Fig. 5, B and C). These results indicate that mutations in the DLC1 binding region of tensin2 PTB domain reduced the growth suppression activity of tensin2 and DLC1 probably due to the loss of efficient tensin2-DLC1 interaction and that our PTB-peptide complex structure provides a molecular and structural basis for the tumor-suppressive activity of DLC1 and tensin2.
DISCUSSION
The tumor suppressor DLC1 utilized a novel binding site for tensin2 PTB domain interaction, and this binding is required for the tumor-suppressive function of DLC1 (26). To obtain structural insight into the molecular mechanism of the above mentioned binding, we performed binding assays with tensin2 PTB and the DLC1 peptide using GST pulldown, fluorescence polarization, and NMR chemical shift perturbation. We also determined the solution structure of tensin2 PTB domain and provided a HADDOCK-derived structural model for the tensin2 PTB-DLC1 peptide complex by magnetic resonance spectroscopy (NMR), which was the first complex structure model for tensin2 PTB domain. In the complex structure, the binding features revealed a novel peptide binding mode that has not been observed in PTB-peptide complexes. Our complex structural model is consistent with the binding study, mutagenesis, and cell assay data. However, a future co-crystal structure would be more precise than the present HADDOCK-derived structural model.
Comparison with Representative PTB-Peptide Complexes
As adapter or scaffold proteins, PTB domain-containing proteins play a critical role in regulating the spatial and temporal organization of signaling networks in a wide range of physiological and pathological processes. The structure and function of PTB-peptide complexes have always been a research focus. Most of the complexes reported share a general mode of peptide binding, a conserved location of the peptide binding pocket and a consensus binding motif. In this canonical binding mode, the PTB peptide ligands are bound as an antiparallel pseudo-β-sheet, forming contacts with the β5 strand and the C-terminal α-helix (Fig. 6, pink region), and the consensus NPXY motifs form a type I β-turn toward their C termini. Although this canonical peptide binding mode does not depend on the phosphorylation state of the tyrosine in the peptide, the phosphorylated tyrosine may greatly increase the binding affinity. In light of the structure/function relationships, PTB domains are divided into Shc-like, IRS-like, and Dab-like subgroups (27). The canonical peptide binding mode is seen in the representative complexes structures Shc and TrkA complex (Fig. 6B), IRS-1 PTB and IL-4 peptide complex (Fig. 6C), and Dab1 PTB and ApoER2 complex (43) (Fig. 6E). On the other hand, some peptides bind to the canonical site on the PTB with small differences. As shown in Fig. 6D, which shows the SNT-1 PTB domain in complex with peptide derived from fibroblast growth factor receptor 1, the C-terminal region of the peptide forms antiparallel β-sheets with a unique C-terminal β8 strand and the β5 strand, whereas the N-terminal region of FGFR1 peptide wraps around the β-sandwich structure of the PTB domain (44). Another example is the Numb PTB domain in complex with GPpY-containing peptide (Fig. 6F). The peptide is in a helical turn conformation that does not hydrogen bond to the PTB domain β-sheet (45).
FIGURE 6.

Representative PTB domains in complex with target peptides. A, tensin2 PTB domain in complex with DLC1 peptide. B, Shc PTB domain in complex with TrkA receptor peptide (Protein Data Bank code 1SHC). C, IRS-1 PTB domain in complex with IL-4 peptide (Protein Data Bank code 1IRS). D, SNT-1 PTB domain in complex with fibroblast growth factor receptor 1 peptide (Protein Data Bank code 1XR0). E, Dab1 PTB domain in complex with ApoER2 receptor (Protein Data Bank code 1NU2). F, Numb PTB domain in complex with GPpY-containing peptide (Protein Data Bank code 2NMB). PTB domains are shown in green. The C-terminal α-helix and the β5 strand, which form the canonical binding groove, are labeled as “αc” and “β5” and colored pink. The peptides are shown in blue except the DLC1 peptide, which is shown in red. In the peptide, the NPX(p)Y motif is shown in red, and the tyrosine or phosphorylated tyrosine residue is indicated as orange sticks.
Novel Peptide Binding Mode of PTB Domain
Our NMR solution structure provides the first view of the tensin2 PTB domain in complex with its peptide ligand in a non-canonical fashion that has not been observed before. First, the DLC1 peptide occupies a novel binding site on tensin2 PTB domain. The complete solution structure of the tensin2 PTB domain with the binding peptide from DLC1 revealed that the binding cleft is formed by the N-terminal helix α1; β-strands β1, β2, and β3; and the loop connecting β7 to C-terminal helix α3. This region is on the opposite side of the PTB domain from the conserved peptide binding location formed by the β5 strand and the C-terminal α-helix in the canonical binding mode. Second, DLC1 peptide utilizes a non-canonical binding motif for the tensin2 PTB domain interaction. DLC1 peptide does not have the consensus NPXY motif. The binding site on PTB accommodates nearly all the residues of DLC1 peptide in an extended conformation.
In addition, the N-terminal helix α1 is required for this binding, whereas it is either misfolded or not present in many PTB structures obtained. Deletion of helix α1 (amino acids 15–24) prevented pulldown of the PTB domain by the DLC1 peptide, and no chemical shift perturbations indicating binding could be observed in an NMR titration experiment although this deletion did not make noticeable structural changes to the PTB domain as judged by the two-dimensional 1H-15N HSQC spectrum of PTB Δα. Comparison of the tensin2 PTB domain in complex with the DLC1 peptide and other tensin PTB domain structures (Protein Data Bank codes 2GJY (46), 1WVH (47), 2DKQ, and 3HQC) showed that binding of DLC1 does not substantially alter the structure (see supplemental Fig. 3). However, there is a difference in the orientation of helix α1 in the tensin2 PTB complex structures. In the x-ray crystal structure of tensin2 PTB (Protein Data Bank code 3HQC), this helix protrudes out from the core of the molecule, whereas in the NMR structure (Protein Data Bank code 2DKQ), there is a difference of about 60° in the relative orientation of helices α1 and α3 from that in the bound form. In the complex structure, helix α1 makes close contacts with the C-terminal helix α3 as a network of NOEs between the residues in these helices (Leu-21 to Phe-151 and Leu-22 to Val-155) was observed in extensive analysis of NOESY spectra. This is consistent with the result of the mutational study that the F151A mutant lost binding with no change to the overall structure fold. The loss of binding may be due to the release of helix α1 from α3 caused by Phe-151 mutation. The requirement of the N-terminal helix α1 is a new feature of PTB-peptide binding found in the tensin2 PTB-DLC1 interaction. This indicates that the binding mode shown by DLC1 may only apply to Shc- or Dab-like PTB domains, which have this N-terminal helix.
Biological Implications
Although the role of DLC1 as a bona fide tumor suppressor has become clearer recently, its molecular mechanism of biological regulation remains unclear (48). Tensin proteins are the first identified interacting partners of the DLC family. Interaction with tensin proteins not only determines the subcellular localization of DLC1 but more importantly influences the biological activities of both DLC1 and the tensin family. These reported studies have provided compelling evidence about the impact of the regulatory mechanism of DLC1 at the protein level with its interaction with tensin family proteins (13, 21, 23, 49). We have previously documented the interactions between DLC1 and the tensin2 PTB domain and identified the binding site on DLC1. In this study, we further identified the important binding residues in tensin2 PTB domain by structural analysis. Residues Leu-139, Asp-140, and Phe-151 are critical in mediating the association with DLC1 peptide comprising amino acids 374–388. Mutations of these residues in FL tensin2 affected the formation of the DLC1-tensin2 binding complex and further reduced the growth-suppressive activity of the DLC1-tensin2 complex. Our findings have provided evidence that proper co-localization of tensin2-DLC1 through PTB-peptide interaction and their biological activities are tightly associated. These findings substantiate the biological regulation of the tumor suppressor DLC1 by the tensin family and provide a better understanding of DLC1-tensin2 PTB domain interaction at the structural and molecular levels.
In conclusion, we determined the NMR solution structure of tensin2 PTB in complex with the DLC1 peptide. Our HADDOCK-derived complex structural model and binding studies have elucidated the molecular mechanism by which tensin2 PTB domain recognizes DLC1 peptide and revealed a novel PTB-peptide binding mode not reported before that extends the versatile recognition modes of the PTB domains in mediating diverse cellular signaling pathways. This PTB-peptide binding mode is unique in that peptide occupies a novel binding site separate from the canonical NPXY motif interaction site and that the N-terminal helix of PTB is required for binding. Meanwhile, this PTB-peptide complex structure defines the residues essential for the formation of the tensin2 and DLC1 complex and provides a molecular and structural basis for better understanding the tumor-suppressive activity of DLC1 and tensin2.
Supplementary Material
Acknowledgment
We thank Feng Rui for helping to set up the NMR experiments.
This work was supported by Hong Kong Research Grants Council Grants RGC664109 (to G. Z. and J. W. P. Y.), SEG-HKUST06, and TUYF10SC03 (to G. Z).

This article contains supplemental Materials and Methods and Figs. 1–4.
The atomic coordinates and structure factors (code 2LOZ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
T. D. Goddard and D. G. Kneller, University of California, San Francisco, unpublished software.
- DLC
- deleted in liver cancer
- PTB
- phosphotyrosine binding
- HCC
- human hepatocellular carcinoma
- PH
- pleckstrin homology
- HSQC
- heteronuclear single quantum correlation
- MTSL
- S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate
- FL
- full length
- IRS
- insulin receptor substrate.
REFERENCES
- 1. Yuan B. Z., Miller M. J., Keck C. L., Zimonjic D. B., Thorgeirsson S. S., Popescu N. C. (1998) Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 58, 2196–2199 [PubMed] [Google Scholar]
- 2. Seng T. J., Low J. S., Li H., Cui Y., Goh H. K., Wong M. L., Srivastava G., Sidransky D., Califano J., Steenbergen R. D., Rha S. Y., Tan J., Hsieh W. S., Ambinder R. F., Lin X., Chan A. T., Tao Q. (2007) The major 8p22 tumor suppressor DLC1 is frequently silenced by methylation in both endemic and sporadic nasopharyngeal, esophageal, and cervical carcinomas, and inhibits tumor cell colony formation. Oncogene 26, 934–944 [DOI] [PubMed] [Google Scholar]
- 3. Wong C. M., Lee J. M., Ching Y. P., Jin D. Y., Ng I. O. (2003) Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Res. 63, 7646–7651 [PubMed] [Google Scholar]
- 4. Yuan B. Z., Durkin M. E., Popescu N. C. (2003) Promoter hypermethylation of DLC-1, a candidate tumor suppressor gene, in several common human cancers. Cancer Genet. Cytogenet. 140, 113–117 [DOI] [PubMed] [Google Scholar]
- 5. Ng I. O., Liang Z. D., Cao L., Lee T. K. (2000) DLC-1 is deleted in primary hepatocellular carcinoma and exerts inhibitory effects on the proliferation of hepatoma cell lines with deleted DLC-1. Cancer Res. 60, 6581–6584 [PubMed] [Google Scholar]
- 6. Yuan B. Z., Zhou X., Durkin M. E., Zimonjic D. B., Gumundsdottir K., Eyfjord J. E., Thorgeirsson S. S., Popescu N. C. (2003) DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene 22, 445–450 [DOI] [PubMed] [Google Scholar]
- 7. Yuan B. Z., Jefferson A. M., Baldwin K. T., Thorgeirsson S. S., Popescu N. C., Reynolds S. H. (2004) DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene 23, 1405–1411 [DOI] [PubMed] [Google Scholar]
- 8. Wong C. M., Yam J. W., Ching Y. P., Yau T. O., Leung T. H., Jin D. Y., Ng I. O. (2005) Rho GTPase-Activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Res. 65, 8861–8868 [DOI] [PubMed] [Google Scholar]
- 9. Xue W., Krasnitz A., Lucito R., Sordella R., Vanaelst L., Cordon-Cardo C., Singer S., Kuehnel F., Wigler M., Powers S., Zender L., Lowe S. W. (2008) DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 22, 1439–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li G., Du X., Vass W. C., Papageorge A. G., Lowy D. R., Qian X. (2011) Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK). Proc. Natl. Acad. Sci. U.S.A. 108, 17129–17134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X., Popescu N. C., Zimonjic D. B. (2011) DLC1 interaction with S100A10 mediates inhibition of in vitro cell invasion and tumorigenicity of lung cancer cells through a RhoGAP-independent mechanism. Cancer Res. 71, 2916–2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erlmann P., Schmid S., Horenkamp F. A., Geyer M., Pomorski T. G., Olayioye M. A. (2009) DLC1 activation requires lipid interaction through a polybasic region preceding the RhoGAP domain. Mol. Biol. Cell 20, 4400–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qian X., Li G., Asmussen H. K., Asnaghi L., Vass W. C., Braverman R., Yamada K. M., Popescu N. C., Papageorge A. G., Lowy D. R. (2007) Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc. Natl. Acad. Sci. U.S.A. 104, 9012–9017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lo S. H. (2004) Tensin. Int. J. Biochem. Cell Biol. 36, 31–34 [DOI] [PubMed] [Google Scholar]
- 15. Chen H., Duncan I. C., Bozorgchami H., Lo S. H. (2002) Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc. Natl. Acad. Sci. U.S.A. 99, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui Y., Liao Y. C., Lo S. H. (2004) Epidermal growth factor modulates tyrosine phosphorylation of a novel tensin family member, tensin3. Mol. Cancer Res. 2, 225–232 [PubMed] [Google Scholar]
- 17. Lo S. H., Janmey P. A., Hartwig J. H., Chen L. B. (1994) Interactions of tensin with actin and identification of its three distinct actin-binding domains. J. Cell Biol. 125, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen H., Lo S. H. (2003) Regulation of tensin-promoted cell migration by its focal adhesion binding and Src homology domain 2. Biochem. J. 370, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katz M., Amit I., Citri A., Shay T., Carvalho S., Lavi S., Milanezi F., Lyass L., Amariglio N., Jacob-Hirsch J., Ben-Chetrit N., Tarcic G., Lindzen M., Avraham R., Liao Y. C., Trusk P., Lyass A., Rechavi G., Spector N. L., Lo S. H., Schmitt F., Bacus S. S., Yarden Y. (2007) A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat. Cell Biol. 9, 961–969 [DOI] [PubMed] [Google Scholar]
- 20. Hafizi S., Sernstad E., Swinny J. D., Gomez M. F., Dahlbäck B. (2010) Individual domains of Tensin2 exhibit distinct subcellular localisations and migratory effects. Int. J. Biochem. Cell Biol. 42, 52–61 [DOI] [PubMed] [Google Scholar]
- 21. Yam J. W., Ko F. C., Chan C. Y., Jin D. Y., Ng I. O. (2006) Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res. 66, 8367–8372 [DOI] [PubMed] [Google Scholar]
- 22. Kawai K., Kitamura S. Y., Maehira K., Seike J., Yagisawa H. (2010) START-GAP1/DLC1 is localized in focal adhesions through interaction with the PTB domain of tensin2. Adv. Enzyme Regul. 50, 202–215 [DOI] [PubMed] [Google Scholar]
- 23. Liao Y. C., Si L., deVere White R. W., Lo S. H. (2007) The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J. Cell Biol. 176, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dai K., Liao S., Zhang J., Zhang X., Tu X. (2011) Solution structure of tensin2 SH2 domain and its phosphotyrosine-independent interaction with DLC-1. PLoS One 6, e21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao Y. C., Shih Y. P., Lo S. H. (2008) Mutations in the focal adhesion targeting region of deleted in liver cancer-1 attenuate their expression and function. Cancer Res. 68, 7718–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan L. K., Ko F. C., Ng I. O., Yam J. W. (2009) Deleted in liver cancer 1 (DLC1) utilizes a novel binding site for tensin2 PTB domain interaction and is required for tumor-suppressive function. PLoS One 4, e5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uhlik M. T., Temple B., Bencharit S., Kimple A. J., Siderovski D. P., Johnson G. L. (2005) Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J. Mol. Biol. 345, 1–20 [DOI] [PubMed] [Google Scholar]
- 28. Farmer B. T., 2nd, Constantine K. L., Goldfarb V., Friedrichs M. S., Wittekind M., Yanchunas J., Jr., Robertson J. G., Mueller L. (1996) Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat. Struct. Biol. 3, 995–997 [DOI] [PubMed] [Google Scholar]
- 29. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe—a multidimensional spectral processing system based on Unix Pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 30. Zhu G., Bax A. (1992) Improved linear prediction of damped NMR signals using modified forward backward linear prediction. J. Magn. Reson. 100, 202–207 [Google Scholar]
- 31. Bax A., Grzesiek S. (1993) Methodological advances in protein NMR. Acc. Chem. Res. 26, 131–138 [Google Scholar]
- 32. Clore G. M., Gronenborn A. M. (1998) Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 16, 22–34 [DOI] [PubMed] [Google Scholar]
- 33. Sattler M., Schleucher J., Griesinger C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34, 93–158 [Google Scholar]
- 34. Jung Y. S., Zweckstetter M. (2004) Mars—robust automatic backbone assignment of proteins. J. Biomol. NMR 30, 11–23 [DOI] [PubMed] [Google Scholar]
- 35. Zwahlen C., Legault P., Vincent S. J., Greenblatt J., Konrat R., Kay L. E. (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-peptide/boxB RNA complex. J. Am. Chem. Soc. 119, 6711–6721 [Google Scholar]
- 36. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 37. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 38. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 39. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 40. Cornilescu G., Delaglio F., Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 41. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 42. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 43. Stolt P. C., Jeon H., Song H. K., Herz J., Eck M. J., Blacklow S. C. (2003) Origins of peptide selectivity and phosphoinositide binding revealed by structures of disabled-1 PTB domain complexes. Structure 11, 569–579 [DOI] [PubMed] [Google Scholar]
- 44. Dhalluin C., Yan K. S., Plotnikova O., Lee K. W., Zeng L., Kuti M., Mujtaba S., Goldfarb M. P., Zhou M. M. (2000) Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Mol. Cell 6, 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li S. C., Zwahlen C., Vincent S. J., McGlade C. J., Kay L. E., Pawson T., Forman-Kay J. D. (1998) Structure of a Numb PTB domain-peptide complex suggests a basis for diverse binding specificity. Nat. Struct. Biol. 5, 1075–1083 [DOI] [PubMed] [Google Scholar]
- 46. Leone M., Yu E. C., Liddington R. C., Pasquale E. B., Pellecchia M. (2008) The PTB domain of tensin: NMR solution structure and phosphoinositides binding studies. Biopolymers 89, 86–92 [DOI] [PubMed] [Google Scholar]
- 47. McCleverty C. J., Lin D. C., Liddington R. C. (2007) Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions. Protein Sci. 16, 1223–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lahoz A., Hall A., (2008) DLC1: a significant GAP in the cancer genome. Genes Dev. 22, 1724–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cao X., Voss C., Zhao B., Kaneko T., Li S. S. (2012) Differential regulation of the activity of deleted in liver cancer 1 (DLC1) by tensins controls cell migration and transformation. Proc. Natl. Acad. Sci. U.S.A. 109, 1455–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.